
Abstract
The unique low temperature synthesis of zinc pyrovanadate from oxides was proposed. Zn3V2O7(OH)2·2(H2O) was synthesized by ultrasonic (US) method using ZnO and V2O5 as raw materials. It was established using SEM and TEM methods that Zn3V2O7(OH)2·2(H2O) has the structure of nanosheets. The DTA method and XRD analysis showed the formation of the Zn3V2O8 phase after the removal of crystallization water from Zn3V2O7(OH)2·2(H2O). Ultrasonic treatment of oxides as initial reagents allows obtaining Zn3V2O8 with the specific surface area of 14 m2/g. For comparison, Zn3V2O8 was synthesized by solid-state (SS) synthesis from oxides. The properties of zinc pyrovanadate obtained by US synthesis and conventional SS synthesis were compared. The advantages of US method over conventional SS synthesis were noted.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Vanadium compounds are widely used in chemical industry as catalysts for the production of maleic acid, sulfuric acid, purification of industrial gases from NOx [1, 2]. Moreover, vanadium compounds seem to be promising as novel potential drugs in the treatment of diabetes, atherosclerosis, and cancer [3, 4].
In the past few decades, Zn3V2O8 has attracted significant attention due to its unique properties and perspective for wide applications in industry, ecology, and medicine.
Zn3V2O7(OH)2·2(H2O) and Zn3V2O8 are considered to be perspective for the creation of photoluminescence materials and can be used as a phosphor for white light emitting diodes [5]. Zn3V2O8 with nanosheet structure demonstrates high performance as an anode material for lithium-ion batteries with good rate capacity, high cycling stability, and excellent discharge capacity as reported in [6]. Moreover, the zinc pyrovanadate nanowires show significantly improved electrochemical performance as an intercalation cathode for an aqueous zinc-ion battery [7]. It can be used for effective catalytic delignification and fractionation of lignocellulosic biomass [8], selective oxidation of sugars to carboxylic acids [9], and as photocatalysts [10,11,12]. It can be also applied as material for the sensing electrode to detect acetone (to 100 ppm) [13] and as material for an ethanol gas sensor [14].
However, Zn3V2O8 is usually obtained from soluble salts by a co-precipitation method [6, 11, 12, 15, 16] or from oxides by solid-state synthesis [10, 11, 17,18,19,20]. Both of these methods are not eco-friendly and require a long time. Usually for the obtaining Zn3V2O7(OH)2·2(H2O) with subsequent formation of Zn3V2O8, typical synthesis from soluble salts is applied. The salts, Zn(NO3)2·6H2O and NH4VO3, are used more often for the synthesis of Zn3V2O7(OH)2·2(H2O) [13, 15, 16, 21, 22]. ZnCl2 or ZnSO4 is used less often [6, 12, 23] but for all the experiments the molar ratio of Zn/V = 3:2 was used. However, using highly diluted solutions due to the low solubility of NH4VO3 should be noted. In addition, after the synthesis, the product must be washed several times to remove by-products. The superiority of this synthesis is possible formation of 2D nanosheets [6, 15] or 3D microspheres [11, 15] or even nanobelts [11, 16] structures of Zn3V2O7(OH)2·2(H2O). The most of these references mention the use of an autoclave for the successful synthesis [6, 11, 16, 21, 23, 24]. An alternative is a solid-state method of syntheses leading to the direct formation of Zn3V2O8. However, this method results in the formation of Zn3V2O8 with the grain shape possessing low SBET of ca. 0.5 m2/g [5]. Only conventional SS synthesis as the way to the formation of Zn3V2O8 from oxides was described in scientific literature. Although the SS synthesis can lead to the complete transformation of starting reagents without any pollution by by-products, it is impossible to obtain nanostructures by this method.
Therefore, the first method requires a lot of water for washing while the second one demands high temperature and doesn’t lead to the formation of nanostructures. The possibility of the formation of zinc pyrovanadate from oxides under ultrasonic treatment has been studied in the current work. In addition, zinc pyrovanadate was synthesized by conventional solid-state method for comparison.
2 Experimental
The samples preparation
Ultrasonic synthesis was carried out according to the procedure described in the literature [25] with some modifications. ZnO 2.86 g and V2O5 2.14 g powders (3:1 molar ratio) were mixed together, transferred into a beaker and de-ionized water (80 ml) was added. The weight ratio of initial oxides/water is 1/16. Ultrasonic treatment (UST) of the oxides mixture was carried out for 40 min at room temperature using an UZDN-A ultrasonic dispersant operating in an acoustic cavitations mode at a frequency of 22 kHz. A yellow suspension was obtained. The resulting product was filtered off, dried in air at 100 °C, and labeled as ZnVO_US_100. The sample ZnVO_US_100 was calcined at 500 °C in an alumina crucible in air for 4 h and labeled as ZnVO_US_500.
Additionally, Zn3V2O8 was synthesized by a conventional solid-state method according to the procedure used in [10] with some modifications. ZnO 2.86 g and V2O5 2.14 g powders (3:1 molar ratio of oxides) were grinded using an agate mortar for 3 min, then transferred in an alumina crucible and calcined at 600 °C in air for 5 h. Afterward, the sample was cooled and grinded again using an agate mortar for 3 min. Finally, the sample was calcined at 750 °C in air for 6 h and labeled as ZnVO_SS.
Characterization of the samples
To analyze the samples composition and morphology, a scanning electron microscope (SEM JSM6490 LV, JEOL, Japan) with an integrated system for electron microprobe analysis INCA Energy based on energy-dispersive and wavelength-dispersive spectrometers (EDS + WDS, OXFORD, UK) with HKL Channel system was used.
Transmission electron microscopy (TEM) JEM-1200 EX (JEOL, Japan) was applied to study the materials morphology.
Nitrogen ad(de)sorption isotherms at –196 °C were obtained on a Quantachrome NOVA-220e Gas Sorption Analyzer. Specific surface area SBET was estimated using the BET equation.
Thermogravimetric analysis (DTA/TG) was carried out on a Derivatograph-Q apparatus with a heating rate of 10 °C/min under air atmosphere from room temperature to 700 °C. The sample mass was 200 mg.
3 Results and Discussion
XRD
Figure 1a shows the X-ray diffraction (XRD) patterns of the initial oxides mixture: ZnO (JCPDS card no. 00-080-0075) and V2O5 (JCPDS card no. 00-041-1426).

XRD patterns of a the initial mixture ZnO and V2O5, b Zn3V2O7(OH)2·2(H2O), and c Zn3V2O8 where ■—ZnO, □—V2O5, #—Zn3V2O7(OH)2·2(H2O), and *—Zn3V2O8
In XRD pattern of ZnVO_US_100 (Fig. 1b), all the reflexes can be attributed to hexagonal structure Zn3V2O7(OH)2·2(H2O) (JCPDS card no. 00-087-0417) indicating high sample purity and crystallinity. ZnVO_US_500 has the diffraction signals corresponded to Zn3V2O8 (JCPDS card no. 00-034-0378) (Fig. 1c). Any peaks from others phases were not detected in the XRD patterns both for ZnVO_US_100 and ZnVO_US_500 testifying the high purity of Zn3V2O7(OH)2·2(H2O) and Zn3V2O8 synthesized by ultrasonic treatment.
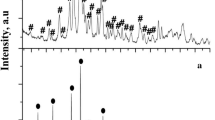
Figure 2 shows the XRD pattern of ZnVO_SS. The most intensive peaks at 2Θ—15.4; 18.7; 21.4; 26.5; 27.1; 29.1; 31.0; 34.9; 36.0; 41.1; and 62.9 correspond to Zn3V2O8 as a main product. Moreover, the impurity phases of Zn2V2O7 (JCPDS card no. 00-070-1532) (2Θ—16.7; 25.8; 28.64; 33.6; 40.6; and 42.2) and ZnO (2Θ—31.7) were registered. It should be noted not long synthesis time (11 h) in our case compared to the conventional synthesis in other references [17,18,19]. Based on the XRD data, the following reactions can be assumed:

XRD patterns of Zn3V2O8 synthesized by solid-state synthesis from ZnO and V2O5: *—Zn3V2O8, ●—Zn2V2O7, ■—ZnO
The synthesis of pyrovanadates by SS method usually is long-term and requires high temperatures. Therefore, the authors [11] synthesized vanadates by SS method during 1 week at T = 797 °C (XRD pattern was not shown; also there is no information about the impurities). In [10], synthesis was carried out during 60 h, and authors failed to synthesize pure Zn3V2O8 phase. The XRD patterns of their final product except Zn3V2O8 also have several peaks corresponded to a little amount of impurity phase (the impurity phase was not identified). Kurzawa et al. [20] investigated V2O5–ZnO system and corresponding phase diagram. It was pointed out that the molar ZnO to V2O5 ratio of 3:1 led to the formation of both Zn3V2O8 as Zn2V2O7 phases. It should be noted that the traditional solid-state synthesis of salts from oxides often leads to the formation of the main product with some amount of impurity phases [6, 25]. Comparison of the X-ray diffraction patterns of the product obtained as a result of ultrasonic and solvothermal synthesis allows to make an assumption about the key role of water in the formation of zinc pyrovanadate salt from oxides. And the key product is exactly zinc pyrovanadate crystal hydrate:
The absence of any impurity phases in the X-ray diffraction patterns of both ZnVO_US_100 and ZnVO_US_500 (Fig. 1b, c) also makes it possible to assume the formation of pure Zn3V2O7 (OH)2 phase with the subsequent transformation into pure Zn3V2O8 phase.
SEM
SEM image of ZnVO_US_100 is shown in Fig. 3. Morphology of ZnVO_US_100 has a typical overall view for Zn3V2O7(OH)2·2(H2O) obtained by a conventional co-precipitate method [6, 9, 26]. Uniform size distribution of the particles in the sample is observed. The sample consists of nanoparticles in the form of nanosheets, each of which has a flat size of 0.1–1 µm and a thickness of 10–40 nm. The data of energy-dispersive X-ray spectroscopy (determined at four different points) demonstrate the uniform elements distribution in the sample (Table 1). The data of SEM and EDX are in a good agreement with the XRD data (Fig. 1a) and confirm complete oxides conversion to Zn3V2O7(OH)2·2(H2O).

SEM image of Zn3V2O7(OH)2·2(H2O)
TEM
The TEM images of the initial oxides V2O5 and ZnO used for the preparation of zinc vanadate by both ultrasonic and solid-state methods are presented in Fig. 4a, b, respectively. Typical particle of Zn3V2O7(OH)2·2(H2O) is displayed in TEM image Fig. 5a, b. TEM images of Zn3V2O7(OH)2·2(H2O) don’t contain the particles of the initials oxides (Fig. 4a, b) instead, the particles with sheet-like structure gathered in agglomerates can be observed. The size of the particle plane ranges from several nm to 1 µm. The light color of the Zn3V2O7(OH)2·2(H2O) particles indicate their small thickness. This is in a good agreement with the literature data about the sheet-like particles of Zn3V2O7(OH)2·2(H2O) obtained by a co-precipitation method [6, 11]. In addition, some amount of particles with hexagonal structure is present in TEM images. As known, Zn3V2O7(OH)2·2(H2O) crystallizes in the hexagonal space group P-6 [6, 11, 24].

TEM images of the initial oxides a V2O5 and b ZnO

TEM images of ZnVO_US_100
Specific surface area
The data of nitrogen ad(de)sorption (Fig. 6) show that the sample Zn3V2O7 obtained by a solid-state reaction is characterized by the low specific surface area (SBET within 1 m2g−1) while the sample obtained by an US method have higher specific surface area (SBET ca. 14 m2g−1), and ad(de)sorption isotherm has hysteresis in a wide area of relative nitrogen pressure indicating the presence of mesopores with different sizes.

Nitrogen ad(de)sorption isotherms (77 K) for Zn3V2O8 synthesized by the solid-state method and Zn3V2O8 synthesized by the ultrasonic method
DTA
The thermal stability of Zn3V2O7(OH)2·2(H2O) was studied using DTA in air (Fig. 7). The TG curve has total weight loss of about 12.73% in an interval from 60 to 350 °C. The DTA curve has two endothermic peaks at 164 °C and 267 °C, being agreed well with weight loss on the TG curve. The removal of water of crystallization and coordinated water corresponds to the weight loss of 11.27%: Zn3V2O7(OH)2·2(H2O) = Zn3V2O8 + 3H2O. Other 1.46% of weight loss can be explained by losing water adsorbed in the pore or on the surface. Published TG and DTA data for Zn3V2O7(OH)2·2(H2O) are contradictory. The maximum of the first endothermic peak is located at 163 °C well corresponding to [11]. Next endothermic peak is located at 267 °C being more similar to the data described in [27]. Moreover, DTA curves presented in [11, 27] have exothermic peak at 452 °C and 550 °C, respectively. However, in our case, no exothermic peak in DTA curves was observed as well as in [15]. Such differences in the location of the maxima of endothermic peaks can be explained by the differences in the synthesis or TG and DTA analysis conditions.

TG and DTA analysis of Zn3V2O7(OH)2·2(H2O)
Additionally, TG and DTA of the initial mixture of oxides were carried out, but no thermodynamic effects and weight loss were observed.
4 Conclusions
In summary, a facile, eco-friendly method of synthesis of Zn3V2O7(OH)2·2(H2O) followed by the formation of pure Zn3V2O8 phase was proposed. This approach in contrast to conventional co-precipitation synthesis doesn’t require a lot of water for washing from polluting ions. The advantages of the US synthesis compared to the conventional SS synthesis are lower duration and temperature, as well as the formation of pure Zn3V2O7(OH)2·2(H2O) phase further transformed into pure Zn3V2O8 phase. Moreover, US synthesis allows obtain samples with nanosheet structures and high SBET. Thus, the ultrasonic method is much more environmentally friendly than other traditional methods of synthesis.
References
Putluru SSR, Schill L, Godiksen A, Poreddy R, Mossin S, Jensen AD, Fehrmann R (2016) Promoted V2O5/TiO2 catalysts for selective catalytic reduction of NO with NH3 at low temperatures. Appl Catal B 183:282–290. https://doi.org/10.1016/j.apcatb.2015.10.044
Zazhigalov VA, Diyuk EA, Sidorchuk VV (2014) Development of VPO catalysts supported on mesoporous modified material based on an aerosol gel. Kinet Catal 55(3):399–408. https://doi.org/10.1134/S002315840904017X
Pessoa JC, Etcheverry S (2015) Vanadium compounds in medicine. Coordination Chem Rev 301–302:24–48. https://doi.org/10.1016/j.ccr.2014.12.002
Semiz S (2022) Vanadium as potential therapeutic agent for COVID-19: A focus on its antiviral, antiinflamatory, and antihyperglycemic effects. J Trace Elem Med Biol 69:126887. https://doi.org/10.1016/j.jtemb.2021.126887
Qian T, Fan B, Wang H, Zhu S (2019) Structure and luminescence properties of Zn3V2O8 yellow phosphor for white light emitting diodes. Chem Phys Lett 715:34–39. https://doi.org/10.1016/j.cplett.2018.11.022
Gan L, Deng D, Zhang Y, Li G, Wang X, Jiang L, Wang C (2014) Zn3V2O8 hexagon nanosheets: a high-performance anode material for lithium-ion batteries. J Mater Chem A 2:2461–2466. https://doi.org/10.1039/C3TA14242F
Xia C, Guo J, Lei Y, Liang H, Zhao C, Alshareef H (2017) Rechargeable aqueous Zinc-Ion battery based on porous framework Zinc pyrovanadate intercalation cathode. Adv Mater 1705580. https://doi.org/10.1002/adma.201705580
Khallouk K, Solhy A, Kherbeche A, Dubreucq E, Kouisni L, Barakat A (2020) Effective catalytic delignification and fractionation of lignocellulosic biomass in water over Zn3V2O8 mixed oxide. ACS Omega 5(1):304–316. https://doi.org/10.1021/acsomega.9b02159
Khallouk K, Solhy A, Idrissi N, Flaud V, Kherbeche A, Barakat A (2020) Microwave-assisted selective oxidation of sugars to carboxylic acids derivatives in water over zinc-vanadium mixed oxide. Chem Eng J 385:123914. https://doi.org/10.1016/j.cej.2019.123914
Wang D, Tang J, Zou Z, Ye J (2005) Photophysical and photocatalytic properties of a new series of visible-light-driven photocatalysts M3V2O8 (M = Mg, Ni, Zn). Chem Mater 17:5177–5182. https://doi.org/10.1021/cm051016x
Shi R, Wang Y, Zhoub F, Zhu Y (2011) Zn3V2O7(OH)2(H2O)2 and Zn3V2O8 nanostructures: controlled fabrication and photocatalytic performance. J Mater Chem 21:6313–6320. https://doi.org/10.1039/C0JM04451B
Mondal C, Ganguly M, Sinha AK, Pal J, Sahoo R, Pal T (2013) Robust cubooctahedron Zn3V2O8 in gram quantity: a material for photocatalytic dye degradation in water. Cryst Eng Comm 15:6745–6751. https://doi.org/10.1039/C3CE40852C
Liu F, Guan Y, Sun R, Liang X, Sun P, Liu F, Lu G (2015) Mixed potential type acetone sensor using stabilized zirconia and M3V2O8 (M: Zn, Co and Ni) sensing electrode. Sens Actuat B 221:673–680. https://doi.org/10.1016/j.snb.2015.07.023
Hua K, Cui M, Luo Z, Fang D, Bao R, Qi Q, Yi J, Sun B, Chen C (2019) Fabrication of Zinc pyrovanadate (Zn3(OH)2V2O7·2H2O) nanosheet spheres as an ethanol gas sensor. J Alloy Compd 801:581–588. https://doi.org/10.1016/j.jallcom.2019.06.015
Zhang SY, Xiao X, Lu M, Li ZQ (2013) Zn3V2O7(OH)22H2O and Zn3(VO4)2 3D microspheres as anode materials for lithium-ion batteries. J Mater Sci 48:3679–3685. https://doi.org/10.1007/s10853-013-7164-5
Zhang S, Lei N, Ma W, Zhang Z, Sun Z, Wang Y (2014) Fabrication of ultralong Zn3V2O7(OH)22H2O nanobelts and its application in lithium-ion batteries. Mat Lett 129:91–94. https://doi.org/10.1016/j.matlet.2014.05.047
Gopal R, Calvo C (1971) Crystal structure of a-Zn3(VO4)2. Can J Chem 49:3056–3059. https://doi.org/10.1139/v71-510
Clark GM, Pick AN (1975) DTA study of the reactions of V2O5 with metal (II) oxides. J Therm Anal 7:289–300. https://doi.org/10.1007/BF01911939
Nord AG, Stefanidis T (1985) Crystal chemistry of a—(Zn, M)2V2O7 solid solutions correlation between preference for five-coordination and extension of solid solubility. Mat Res Bull 20:845–851. https://doi.org/10.1016/0025-5408(85)90064-9
Kurzawa M, Rychlowska-Himmel I, Bosacka M, Blonska-Tabero A (2001) Reinvestigation of phase equilibria in the V2O5–ZnO system. J Therm Anal Calorim 64:1113–1119. https://doi.org/10.1023/A:1011524424682
Yang G, Li S, Wu M, Wang C (2016) Zinc pyrovanadate nanosheet of atomic thickness: excellent li-storage properties and investigation of electrochemical mechanism. J Mater Chem A 4:10974–10985. https://doi.org/10.1039/C6TA02782B
Mazloom F, Masjedi-Arani M, Salavati-Niasari M (2017) Rapid and solvent-free solid-state synthesis and characterization of Zn3V2O8 nanostructures and their phenol red aqueous solution photodegradation. Solid State Sci 70:101–109. https://doi.org/10.1016/j.solidstatesciences.2017.06.013
Low WH, Khiew PS, Lim SS, Siong CW, Chia CH, Ezeigwe ER (2019) Facile synthesis of graphene-Zn3V2O8 nanocomposite as a high performance electrode material for symmetric supercapacitor. J Alloy Compd 784:847–858. https://doi.org/10.1016/j.jallcom.2019.01.137
Bayat A, Mahjoub AR, Amini MM (2018) Optical and magnetic properties of zinc vanadates: synthetic design of colloidal Zn3V2O7(OH)2(H2O)2, ZnV2O4 and Zn3V2O8 nanostructures. J Mater Sci: Mater Electron 29:2915–2926. https://doi.org/10.1007/s10854-017-8222-6
Diyuk OA, Zazhigalov VA, Shcherban ND, Permyakov VV, Diyuk NV, Shcherbakov SM, Sachuk1 OV, Dulian P (2021) Kinetics of ZnMoO4·0.8H2O and α-ZnMoO4 formation at ultrasonic treatment of ZnO and MoO3 mixture. In: Fesenko O, Yatsenko L (eds) Nanocomposites, nanostructures, and their applications. Springer Proceedings in Physics book series, vol 263, 87–101. https://doi.org/10.1007/978-3-030-74741-1_6
Luo J, Ning X, Zhan L, Zhou X (2021) Facile construction of a fascinating Z-scheme AgI/Zn3V2O8 photocatalyst for the photocatalytic degradation of tetracycline under visible light irradiation. Sep Purif Technol 255:117691. https://doi.org/10.1016/j.seppur.2020.117691
Hoyos DA, Echavarria A, Saldarriaga C (2001) Synthesis and structure of a porous zinc vanadate, Zn3(VO4)2·3H2O. J Mater Sci 36:5515–5518. https://doi.org/10.1023/A:1012418706071
Acknowledgements
This work was financially supported by NASU Program “New functional substances and materials of chemical production” (project 13-21).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this paper

Cite this paper
Diyuk, O.A. et al. (2023). Ultrasonic Synthesis and Characterization of Zinc Pyrovanadate Nanostructures. In: Fesenko, O., Yatsenko, L. (eds) Nanomaterials and Nanocomposites, Nanostructure Surfaces, and Their Applications . Springer Proceedings in Physics, vol 279. Springer, Cham. https://doi.org/10.1007/978-3-031-18096-5_16
Download citation
DOI: https://doi.org/10.1007/978-3-031-18096-5_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-18095-8
Online ISBN: 978-3-031-18096-5
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)