
Abstract
Type 2 diabetes mellitus is associated with a high risk of heart failure, in part because of its potential to induce impaired cardiac function even in the absence of coronary artery disease and hypertension. The pathogenesis of diabetic cardiomyopathy is not yet fully understood, although it is known that alterations in the cardiac substrate metabolism and energy play a significant role in the development and progression of this condition. Among these abnormalities, increased fatty acid storage and utilization, associated with reduced glucose oxidation and altered mitochondrial oxidative phosphorylation stand out. One of the key connections linking diabetes and the remarkably prevalence of cardiomyopathy is the renin angiotensin system (RAS) hyperactivation. RAS has been described for its crucial involvement in the generation and progression of diabetic cardiovascular complications, as its exacerbated activation supports mechanisms that lead to cardiomyocyte death and myocardial fibrosis. The importance of RAS blockage in the prevention of diabetic cardiomyopathy exhibits the fundamental role that RAS plays in the onset and development of this pathology. Angiotensin-converting enzyme inhibitors and blockers of angiotensin II actions denote the first-line therapy for primary and secondary prevention of cardiovascular disease in type 2 diabetic. Recent studies have revealed new features of RAS and, consequently, new therapeutic potential against diabetic cardiomyopathy. In this chapter, a description of the main mechanisms involved in the correlation between excessive activation of the RAS and type 2 diabetes is presented, with attention on mechanisms associated to pathogenesis and progression of diabetic cardiomyopathy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Diabetic cardiomyopathy
- Renin angiotensin system
- Metabolic dysfunction
- Cardiac remodeling
- Therapeutic actions
Introduction
Diabetes mellitus has emerged as a critical public health concern and represents a significant threat to the global population. It is estimated that by 2040 almost 700 million people will be affected if no effective prevention methods are adopted [1]. Type 2 diabetes is a chronic metabolic disorder represented by insulin resistance and hyperglycemia, also characterizing one of the greatest risks for the development of heart failure [2]. Patients with type 2 diabetes are 2–5 times more likely to develop heart failure than non-diabetic and age-matched patients, independent of other medical conditions. It suggests an intrinsic and specific mechanism that generates pathological cardiac remodeling in this population, currently known as diabetic cardiomyopathy [3].
Diabetic cardiomyopathy is characterized by the impaired cardiac structure and function that happens when other cardiac risk factors, such as coronary artery disease, hypertension, and valvular disease are not present [4]. It was first described more than forty years ago, with hyperglycemia and abnormal insulin signaling in the heart comprising key roles in its genesis and progression [5]. The diabetic heart is mainly characterized by diastolic dysfunction with preserved ejection fraction, changes caused by the pathological cardiac remodeling. Increased perivascular and interstitial fibrosis, in addition to left ventricle hypertrophy, are structural features commonly observed in diabetic hearts [6]. The underlying pathogenic mechanisms of diabetic cardiomyopathy include but are not limited to abnormal extracellular matrix deposition, enhance in inflammation and oxidative stress associated with mitochondrial dysfunction, and alterations in energy production and cardiac metabolic profile [7].
One of the key connections linking diabetes and the remarkably prevalence of cardiomyopathy is the renin angiotensin system (RAS) hyperactivation. RAS has been described for its crucial involvement in the generation and progression of diabetic cardiovascular complications, as its exacerbated activation supports mechanisms that lead to cardiomyocyte death and myocardial fibrosis [6,7,8,9]. In this chapter, a description of the main mechanisms involved in the correlation between excessive activation of the RAS and type 2 diabetes is presented, with attention on mechanisms associated to pathogenesis and progression of diabetic cardiomyopathy.
Diabetes-Induced Activation of the Renin Angiotensin System
Overview of the Renin Angiotensin System
The RAS has a extensive and substantial history that began in 1892 with the discovery of renin [10]. Over the past 130 years, many other elements of this intricate and vital system, in conjunction with angiotensinogen, angiotensin-converting enzyme (ACE) isoforms, angiotensin peptides, and their associated receptors, have been described. Concurrently, pharmacological approaches have also emerged to regulate this system at several different stages. RAS performs key physiological functions in fluid and electrolyte regulation, systemic vascular resistance, blood pressure, and cardiovascular homeostasis [7]. Notwithstanding, its hyperactivation may induce oxidative stress, inflammation, and endothelial dysfunction, leading to many problems such as hypertension, kidney disease, and heart failure. Thus, ACE inhibitors and angiotensin II (Ang II) type 1 (AT1) receptor blockers have been at the forefront of the clinical management of various cardiovascular diseases. Angiotensinogen is the precursor of the various RAS family peptides, which are generated by tightly regulated biochemical processes (Fig. 15.1). Although the liver is the primary systemic source of angiotensinogen, it is also expressed in various other tissues. To date, five main bioactive angiotensin peptides have been studied: Ang II, Angiotensin-(1–7) (Ang-(1–7)), Angiotensin-(1–9) (Ang-(1–9)) and Alamandine [11, 12]. Ang II is the most studied peptide of the RAS family and is considered the effector peptide of the RAS. Besides to its well knows renal and vascular effects, Ang II also has direct actions at the cellular level, affecting cell inflammation, differentiation and survival [13,14,15].

Classical and counter-regulatory renin angiotensin pathways. In the renin angiotensin system, angiotensinogen is processed by renin to form Angiotensin I. Angiotensin I can be cleaved by angiotensin-converting enzyme (ACE) to generate Angiotensin II, which can bind to the Angiotensin II type 1 receptor (AT1R) and Angiotensin II type 2 receptor (AT2R). Angiotensin II can be further processed by aminopeptidase A (APA) to form Angiotensin III, which also binds to the AT1R. In addition, Angiotensin I can be processed by ACE2 and neprilysin (NEP) to respectively produce Angiotensin-(1–9) and Angiotensin-(1–7). AT2R can also be activated by Angiotensin-(1–9), while Angiotensin-(1–7) binds to the proto-oncogene Mas receptor (MasR). Angiotensin-(1–7) can additionally be produced from Angiotensin II cleavage by ACE2 and be further metabolized to Alamandine. Occasionally, Angiotensin II may be cleaved by aspartate decarboxylase (AD) to produce angiotensin A, which can be converted to Alamandine by ACE2. Alamandin binding to the Mas-related G protein-coupled receptor member D (MrgD) promotes similar effects to those reported for Angiotensin-(1–7). Hyperactivation of AT1 receptor signaling are well recognized for their role in inducing cardiac hypertrophy and fibrosis, whereas the activation of AT2, Mas, and MrgD receptors are known primarily for its role in vasodilation and cell survival through their cardioprotective properties. Figure created using Biorender.com
To generate the octapeptide, Ang II, Angiotensinogen is cleaved into the decapeptide Angiotensin I (Ang I) by Renin. Ang I is further processed by ACE, a key dipeptidyl carboxypeptidase, leading to Ang II production. To mediate its physiological functions, Ang II binds to two main G-protein coupled receptors (AT1 and AT2 receptors) (Fig. 15.1). Ang II can be further converted to Angiotensin III (Ang III) through the aminopeptidase A. Ang III binds to the AT1 receptor with a lower affinity than Ang II and to the AT2 receptor with a higher affinity than Ang II [16]. To form the heptapeptide Ang-(1–7), Ang II is cleaved by angiotensin-converting enzyme type 2 (ACE2), an ACE isoform first described in 2000, and then binds to its receptor, the Mas receptor [17]. With less efficiency, ACE2 is also able to cleave Ang I into Ang-(1–9), which can be further cleaved into Ang-(1–7) by ACE or by Neutral Endopeptidase or Neprilysin (NEP) [18]. Furthermore, Ang-(1–7) can be formed directly from Ang I, through the action of the enzymes NEP, Timet Oligopeptidase (TOP) and Prolyl Endopeptidase (PEP). Although effects of Ang-(1–7) are thought to be primarily mediated via the activation of Mas receptor, Ang-(1–7) can also bind with lower affinity to the AT2 receptor and the MrgD receptor (Fig. 15.1) [19]. Recently, Alamandine, a heptapeptide analogous to Ang-(1–7) and formed by its decarboxylation, was shown to bind with and activate MrgD, which led to the inclusion of MrgD receptor the RAS family [20]. Alamandine may also be produced by the decarboxylation of Ang II to Angiotensin A (Ang A), which is later cleaved by ACE2 [20].
ACE is well known for its action in converting Ang I to Ang II, and although Ang II can bind to both AT1 and AT2 receptors, increased ACE activity pointedly conduct to higher activation of AT1 receptor. AT1 receptor hyperactivation and increased ACE levels are well recognized for their vasoconstrictor effects, in addition to cell death and inflammation [21, 22]. Whereas the activation of AT2, Mas, and MrgD receptors are known primarily for its role in vasodilation and cell survival through their anti-inflammatory and antioxidant properties [22, 23]. Despite binding with greater affinity to the Mas receptor, Ang-(1–7) can also bind to AT2 and MrgD receptors. Emerging evidence suggests that the signaling pathways of Mas, MrgD, and AT2 receptors are inter-dependent. The activation of the AT2 receptor has been shown to increase the expression of ACE2, an enzyme primarily involved in Ang-(1–7) formation. Moreover, elimination of AT2 receptor results in a marked reduction in the levels of Mas receptor and ACE2, suggesting a potential role of AT2 receptor signaling in the regulation of ACE2/Ang-(1–7)/MasR signaling [24]. Notably, Mas and AT2 receptors activation can directly antagonize the AT1 receptor through a heterodimer formation, leading to decreased signaling pathway of the primary Ang II receptor [25]. Additionally, ACE2 significantly increases the formation of these heterodimers, facilitating the ACE2/Ang-(1–7) actions. On the contrary, the activation of the AT1 receptor negatively regulates ACE2 levels, impeding the ACE2/Ang-(1–7) actions [18]. The complex nature of these actions substantiates the synergistic interaction between RAS receptors and enzymes to preserve an intricate balance.
Ang-(1–7) has been at the center of various seminal preclinical and clinical research works, primarily due to its cardio and vasculoprotective effects that are mediated by antagonizing the deleterious actions of Ang II on the cardiovascular system. In this regard, various shreds of evidence point to the concept of a RAS formed by two antagonistic axes. (i) a classic axis, where the primary mediator is Ang II, activation of which leads to vasoconstriction, inflammation, oxidative stress, apoptosis, and cell proliferation; (ii) an alternative axis, where the primary mediator is Ang-(1–7), activation of which leads to vasodilation, anti-inflammatory, antioxidant, anti-apoptotic, and angiogenic actions [26,27,28,29].
In addition to its systemic effects, known for key outcomes on blood pressure, fluid and electrolyte balance [30, 31], RAS elements have also been detected in several tissues, including heart, kidney, brain, nerve fibers, reproductive organs, blood vessels, liver, skeletal muscle, and pancreas [3]. The demonstration of components of the RAS in these tissues has established the concept of a tissue or local RAS, whose regulation is believed to be independent of the systemic RAS.
Renin Angiotensin System in Type II Diabetes
Studies of RAS in diabetes have shown a tissue-specific activation of this system mainly in the kidneys, pancreas, heart, vasculature, and adipose tissue [32]. Furthermore, Ang II has been shown to play a key role in the development of insulin resistance at the cellular level, mainly via increased oxidative stress and altered insulin signaling [33, 34]. Physical inactivity is a major contributor to obesity, and together they are known to be crucial pitfalls for the progression of insulin resistance and, consequently, type 2 diabetes. The reduction in insulin sensitivity causes a continuous burden on the impaired pancreatic islet β-cells. At a certain point, insulin secretion is no longer sufficient to support normoglycemia, and glucose levels start to rise to the prediabetic stage, leading to impairment of glucose tolerance and/or changes in fasting glucose [34]. β-cell failure precedes the progress of type 2 diabetes, and the decline of β-cell power, as well as the loss of operational β-cell mass, are the main predictors for the development of this condition [3, 33].
It is noticeable that insulin displays deleterious effects on the functional and structural characteristics of islet cells by generating Ang II-mediated oxidative stress. Via AT1 receptor, Ang-II interferes with the effects of insulin on skeletal muscle and vascular tissue, through disruption of insulin signaling via phosphatidylinositol 3-kinase, and its downstream protein kinase B (Akt) signaling pathway [9, 35]. In animal models, Ang II reduces blood flow to pancreatic islets, leading to decreased insulin deliver from β-cells. Hence, RAS inhibition increases pancreatic blood flow, improving islet’s function [36, 37]. In fact, both in vitro and in vivo assessments indicate that RAS activation leads to inflammation, oxidative stress, fibrosis in pancreatic islets and defective insulin secretion. Meanwhile, RAS repression with AT1 receptor blockers or ACE inhibitors increases systemic glucose tolerance, and improves islets morphology and function [38,39,40]. Despite substantial data obtained from pre-clinical research, there is still a lack of corroboration of these findings in humans subjects. Collectively, the available evidence suggests that the hyperactivated RAS in pancreatic islets may be involved in compromised insulin secretion, and RAS inhibition can be a feasible approach to improve insulin secretion in individuals with a high potential to have type 2 diabetes.
RAS activation from both systemic and cardiac tissue also plays a key direct contribution in the development and progression of diabetic cardiomyopathy. A recent study showed systemic activation of RAS in response to hyperglycemia, which was associated with increased vascular resistance and, consequently, increased blood pressure [32]. The hyperactivation of RAS in diabetes is strongly supported by the effects of AT1 receptor antagonism that results in a more significant decrease in blood pressure in hyperglycemic conditions, when compared to normoglycemic conditions [33]. In fact, Ang II directly stimulates oxidative stress by increasing nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity. Furthermore, RAS hyperactivation activates the mTOR-S6K1 pathway, leading to systemic and cardiac insulin resistance [18]. Concurrently, the hyperactivation of AT1 receptor signaling in the heart reflects in exacerbated proinflammatory immune responses [5]. These changes further lead to the activation of hypertrophic and profibrotic pathways that result in cardiac remodeling associated with cardiac fibrosis, diastolic dysfunction, and ultimately, heart failure.
Metabolic Disturbances in the Diabetic Heart
Normal Cardiac Energetics
Over the course of an average human life, 200 million liters of blood are pumped by the heart in about 2−3 billion heartbeats. This high workload requires a high consumption of adenosine triphosphate (ATP), which can reach up 30 kg of ATP per day [41]. Accordingly, the heart becomes the most energy-demanding organ in the human body, and even small changes in energy availability can quickly lead to changes in cardiac function [42]. The high energy demand also requires an uninterrupted generation of ATP that depends on the continuous supply of substrates and oxygen, in addition to a functional oxidative phosphorylation, since this efficient metabolic pathway produces almost all the ATP used by the heart [43]. Roughly 70% of the ATP produced in the myocardium is used in the cardiac contractions, while the remnant is used for ion pumps, such as sarcoplasmic reticulum Ca2+-ATPase (SERCA). Albeit the heart has the ability to change its substrate preference according to its needs, the two main energy substrates used are fatty acids and glucose [44].
Abnormal Cardiac Fatty Acid Uptake and Utilization in Diabetes
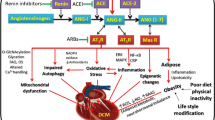
The preference of the heart for fatty acids as energetic substrates is amplified in type 2 diabetes. This is a result of the increased plasma fatty acid levels due to increased lipolysis from insufficient insulin action and triglycerides production in the liver, in addition to inefficient adipocyte triglycerides storage (Fig. 15.2). The intensified delivery of fatty acids then elevates fatty acid uptake and triglycerides stores in cardiomyocytes [45]. As a result, fatty acid abundance in the cardiac muscle activates peroxisome proliferator-activated receptor (PPAR)-α, which heightens the dependence on fatty acids through the upregulation of its uptake, storage, and β-oxidation, while the glucose utilization is suppressed [46]. In addition, PPAR-α acts in the regulation of key factors that lead to fatty acid β-oxidation, and therefore is considered an essential regulator in the homeostatic control of energy balance. Marked PPAR-α activity has been shown in animal models of diabetic cardiomyopathy, and hearts from PPAR-α knockout mice do not change when they become insulin resistant [47]. In humans with type 2 diabetes, increased cardiac fatty acid oxidation has been described to be two times higher when compared with healthy individuals [48], and aberrant fatty acid metabolism has been associated with adverse cardiac damage.

Molecular mechanisms involved in the development of diabetic cardiomyopathy. Resistance to the metabolic actions of insulin, compensatory hyperinsulinemia, and the progression of hyperglycemia promoted by type II diabetes result in several effects at the cardiac level. The mobilization of free fatty acids and activation of the renin angiotensin system can cause lipotoxicity, mitochondrial dysfunction and oxidative stress that result in impaired insulin signaling with changes in calcium metabolism and cardiomyocyte death. These abnormalities further induce fibrosis and myocardial stiffness, and eventually that result in hypertrophy, diastolic dysfunction and diabetic cardiomyopathy. IRS-1: insulin receptor substrate 1; AT1R: Angiotensin II type 1 receptor; mTOR: mechanistic target of rapamycin; S6K1: S6 kinase 1; PI3K: phosphatidylinositol 3-kinase; AKT: protein kinase B; FA: fatty acids; PPAR-α: peroxisome proliferator-activated receptor-α; GLUT4: insulin-regulated glucose transporter type 4. Figure created using Biorender.com
Exaggerated fatty acid retention is highly toxic to cardiac cells, and fatty accumulation in the heart is correlated with cardiac dysfunction in type 2 diabetes. Myocardial triglycerides levels have been shown to be increased by up two-fold in type 2 diabetic patients, with greater triglyceride content associated with impaired left ventricular systolic and diastolic function [48, 49]. The increase in fatty acids within the heart is proposed to also elevate mitochondrial reactive oxygen species by augmenting β-oxidation and electron transport chain activity [50]. Elevated levels of reactive oxygen species are consecutively involved in cardiac hypertrophy and inefficient excitation–contraction coupling due to uncoupled oxidative phosphorylation and mitochondrial dysfunction [51, 52]. Additionally, elevated lipid intermediary metabolites can unbalance signaling, inducing cardiomyocyte apoptosis through cytochrome c pathway. These circumstances determine the lipotoxicity involved in the development of diabetic cardiomyopathy. Considering its greater oxygen demand—approximately 86% higher than glucose—increased fatty acid metabolism eventually leads to cardiac inefficiency [53].
Defective Cardiac Glucose Uptake and Utilization in Diabetes
Although systemic hyperglycemia still leads to suitable glucose uptake in some tissues, myocardial glucose oxidation is decreased by 30 to 40% in type 2 diabetic patients [54]. This mainly refers to fatty acid accumulation in the heart. Lower glycolysis is followed by reduction in the pentose phosphate pathway flux and simultaneous increase in polyol, hexosamine biosynthetic and glycogenic pathways, which may contribute to the cardiac damage [55, 56]. Hyperglycemia plus the resulting end-products of the altered glucose dislocation facilitate several features of diabetic cardiomyopathy (Fig. 15.2). Hyperglycemia increases myocardium glucose levels that may glycate proteins to generate advanced glycation end-products (AGEs) in a non-enzymatic manner. AGEs elevate reactive oxygen species, and can interact and damage macromolecules such as the ryanodine receptor (inducing contractile impairment), SERCA (causing diastolic impairment) and collagen (generating myocardial fibrosis and stiffness) [57, 58]. The elevation in polyol flux and the reduced pentose phosphate pathway activity decreases NADPH levels, enhancing oxidative stress and damage [55, 56]. Increased hexosamine biosynthetic pathway flux elevates the production of glycosaminoglycans, and that can also assist to impaired SERCA activity, matrix remodeling and cardiac fibrosis [59].
Cardiac Mitochondrial Oxidative Phosphorylation Impairment in Diabetes
Cardiac mitochondria show a decrease in rates of oxidative phosphorylation, along with higher uncoupling of respiration and impaired ATP production in type 2 diabetes. This happen even with the confirmation of higher cellular anaplerosis [60] and lower transportation of the Krebs cycle end-products FADH2 and NADH to the electron transport chain. This reveals that elements related to the Krebs cycle narrow oxidative phosphorylation. In fact, preclinical and clinical studies show a coherent depletion in the levels and catalytic activity of hexosamine biosynthetic pathway complexes in type 2 diabetes, and this depletion has been linked to oxidative stress and myocardial impairment [61, 62]. In animal models, reduced oxidative phosphorylation also results from free fatty acid-mediated upregulation of uncoupling proteins that uncouple mitochondria, reductions in mitochondrial quantity, size, and complexity [63, 64]. In type 2 diabetic patients, attenuations in mitochondrial size respond to compromised mitochondrial fusion with reduced mitofusin-2 levels [65].
Mitochondrial dysfunction is related to impaired cardiac structure and function in type 2 diabetes. In animals and humans with type 2 diabetes, depletion in oxidative phosphorylation is evidenced by reduction in myocardial phosphocreatine and ATP content. Pronounced energetic deficiency due to reduced electron transport chain activities impairs diastolic function and exercise capacity [66, 67]. Furthermore, in atrial trabeculae extracted from surgical type 2 diabetic patients, lower oxidative phosphorylation is related with compromised contractility [68]. As mitochondrial dysfunction may lead to systolic and diastolic impairment through its potential to restrict, respectively, ATP supply to myofibrils and SERCA, it also increases reactive oxygen species generation, which is involved in cardiac remodeling [69, 70]. Enhanced reactive oxygen species generation follow the abundance of electrons distributed to the electron transport chain, in response to the higher fatty acid oxidation.
Cardiac Remodeling in Diabetic Cardiomyopathy
The progression of diabetic cardiomyopathy is characterized by a short physiological adaptation to metabolic challenges, that gradually culminates in deteriorating alterations that the myocardium is unable to repair, ultimately leading to irreversible pathological remodeling [71]. Hyperglycemia and hyperlipidemia with progressive increasing of their substrates in the heart cause structural and functional changes by various mechanisms. These changes, which establish themselves slowly, begin with diastolic dysfunction, accompanied by impaired left ventricular systolic function. This heart failure is related with altered myocardial energy metabolism with gradual myocardial hypertrophy and fibrosis, leading to early death if not diagnosed and treated promptly [72].
Cardiac remodeling can be understood as a physiological adaptation to a prolonged stressful stimulus that increases the workload of the myocardium or as a response to reduced myocardial contractility or changes in the composition of myocardial tissue associated with heart disease. Thus, changes in left ventricular mass and/or volume represent only a phenotype common to various pathological processes [73]. Although cardiac remodeling appears to be an adaptive process, many studies conducted in patients with cardiovascular disease have shown a poor association between left ventricular hypertrophy and clinical outcome. For example, in a large prospective study, cardiovascular risk was directly correlated with increasing ventricular mass [74]. In addition, left ventricular hypertrophy was associated with higher mortality in some patient groups, such as the elderly [75], and is considered a step in the progression to dilated cardiomyopathy and, therefore, heart failure [76]. Thus, left ventricular hypertrophy is considered the main predictor of morbidity and mortality in cardiovascular diseases. In obese and diabetic individuals, left ventricular hypertrophy has been systematically and consistently observed [73]. It is important to note that while left ventricular hypertrophy associated with systemic arterial hypertension or myocardial infarction can be attenuated or reversed as a function of the pharmacological treatment of these diseases, there are still no defined therapeutic strategies to treat cardiac remodeling associated with type II diabetes mellitus.
Ang II, in addition to its classic actions, stimulates cell hypertrophy and increases collagen synthesis and deposition by cardiac fibroblasts, leading to cardiac remodeling [77]. In the absence of any intervention that causes illness in mice, selective overexpression of the AT1 receptor in hearts induces left ventricular hypertrophy [78]. This experimental demonstration clarifies the hypertrophic role of Ang II on the heart. Ang II also activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, an enzyme involved in the formation of reactive oxygen species. The oxidative stress generated by the excessive formation of reactive oxygen species, with a decrease in antioxidant defense, brings serious complications to cardiac cells, resulting in structural changes and compromising cardiac contractility [79]. Interestingly, when cardiomyocytes are stimulated in vitro with glucose, mimicking diabetes, there is an increase in intracellular production of Ang II [80]. Therefore, hyperglycemia can induce left ventricular hypertrophy via activation of the local intracardiac RAS.
Renin Angiotensin System Modulation as a Treatment of Diabetic Cardiomyopathy
Since the insulin advent in 1921, refinement in diabetes diagnosis and treatment have remarkably decreased mortality from acute metabolic emergencies. However, chronic complications, including cardiovascular disease, remain as the leading cause of morbidity and mortality among patients with type 2 diabetes. The importance of RAS antagonism in the prevention of diabetic cardiomyopathy illustrated the fundamental role that RAS plays in the onset and development of this pathology. ACE inhibitors and blockers of Ang II actions denote the first line therapy for primary and secondary prevention of cardiovascular disease in diabetic patients. The latest studies have revealed new aspects of RAS and, consequently, new potential therapeutic targets that could be used against diabetic cardiomyopathy in the near future.
AT1 Receptor Antagonists
Treatment with olmesartan, an AT1 receptor antagonist, in an animal model of type II diabetes induced by a high-fat diet, attenuated cardiac inflammation and interstitial and perivascular fibrosis. The mechanism of improved cardiac function by olmesartan was attributed to the inhibition of apoptosis signal regulation kinase-1 (ASK1), an enzyme involved in cell death and hypertrophy, showing the participation of Ang II in the activation of these mechanisms [81]. Another work showed that when candesartan was associated with an oral hypoglycemic agent in an animal model of obesity, db/db mice (harbors mutation in leptin receptor), there was an improvement in fibrosis and cardiac inflammation. These effects of candesartan have been associated with improved cardiac antioxidant defense [82], which is in complete agreement with the literature on the contribution of reactive oxygen species to Ang II actions [79].
A study with increasing doses of irbesartan showed a dose-dependent effect on the reversal of left ventricular hypertrophy in diet-induced type II diabetes rats. Improvements in diastolic and systolic dysfunctions were also observed on echocardiographic measurements and histological analysis of myocardial interstitial fibrosis. In this case, treatment with irbesartan inhibited the protein kinase D pathway, a kinase involved in the regulation of cell survival, differentiation, proliferation, and migration [83]. Finally, losartan was used in a normotensive animal model of diet-induced obesity. Losartan treatment not only reversed left ventricular hypertrophy but also partially improved insulin resistance and lipid and glycemic profiles. The beneficial cardiac effects were associated with reduced activity of several mitogen-activated protein kinases (MAPK) [84]. All studies with AT1 receptor antagonists showed a beneficial effect on left ventricular hypertrophy. However, these effects were not always independent of blood pressure control (since some animals become hypertensive) or improvement in insulin resistance. In addition to its contribution to the induction of cardiac remodeling, Ang II is also intimately involved in the pathophysiology of metabolic alterations.
Angiotensin-Converting Enzyme Inhibitors
As ACE is a significant source of Ang II generation, ACE inhibition consistently leads to a reduction in systemic Ang II levels. Although the use of ACE inhibitors is indicated for the treatment of left ventricular hypertrophy, ACE inhibitors are also used for the clinical management of heart failure, coronary artery disease, and renal failure. Furthermore, it has also been suggested that ACEi are able to reduce the incidence of diabetes [85]. In ob/ob mice (lacking functional leptin signaling), 20 weeks with temocapril reduced cardiac hypertrophy. Moreover, temocapril also reversed the obesity-associated loss of cardiac contractility. Notably, temocapril did not affect the blood glucose or insulin levels, suggesting the direct effect of temocapril on hearts with minimal effects on systemic metabolic abnormalities. Importantly, plasminogen activator inhibitor type 1 (PAI-1), the main fibrinolysis factor, was also involved in the effect of temocapril on cardiac hypertrophy [86]. Cardioprotective effects of ACE inhibition were also validated in another preclinical model of obesity and type II diabetes, the Zucker rats (harbors mutation in the leptin receptor gene). Perindopril treatment of Zucker rates led to a reversal of myocardial hypertrophy and cardiac extracellular matrix remodeling, which was accompanied by a decrease in PAI-1. However, these effects were thought to depend on blood pressure reduction, as the ACE inhibition also reduced markedly increased blood pressure in the Zucker rats [87].
Two separate studies with captopril using ob/ob mice showed that the treatment did not reverse the increase in cardiac weight or changes in myocyte contractility in vitro. However, when dyslipidemia and arterial hypertension were co-induced in obese animals, the reduction in myocyte contractility was more severe, and, in this case, the ACE inhibitor significantly reversed it [88]. Initiation of captopril treatment four weeks before the induction of obesity, captopril showed more potent cardioprotective effects, resulting in reduced cardiac hypertrophy and improved metabolic parameters in ob/ob mice [89]. Various preclinical investigations suggest that the cardioprotective effects of ACE inhibition against diabetic cardiomyopathy are at their peak when the ACE inhibitors are administered early and chronically.
Renin Inhibition
Among the therapeutic agents that act on the RAS, the most recently introduced class is renin inhibitors, of which aliskiren is the only available representative. In addition to being an antihypertensive, aliskiren is also cardioprotective. Aliskiren treatment for 3 months reduced diabetic nephropathy in db/db mice, which was related with decreased urinary albumin excretion, glomerulosclerosis, and suppressed profibrotic and proinflammatory cytokines synthesis [90]. Consistently, aliskiren also prevented cardiac fibrosis, and myocardial inflammation in db/db mice [91]. Cardioprotective effects of aliskiren are considered independent of hemodynamic improvement, as hydralazine, a vasodilator, did not produce similar prevention of left ventricular hypertrophy in type 2 diabetic animals [91].
ACE2/Ang-(1-7)/Mas Axis Activation
ACE2 and Ang-(1–7) are potent negative regulators of the ACE/Ang II/AT1 receptor axis of the RAS. Ang-(1–7) is a biologically active heptapeptide generated by proteolytic cleavage of either Ang I or Ang II by the actions of several endopeptidases and carboxypeptidases, including ACE and ACE2, respectively [92]. Ang-(1–7) antagonizes Ang II/AT1 receptor axis and exhibits vasodilatory, anti-fibrotic, anti-proliferative, and antihypertrophic properties [17, 93]. While the loss of ACE2 exacerbates diabetic cardiomyopathy [94], enhancing ACE2 has a protective effect on Ang II-induced cardiac dysfunction [95] and streptozotocin-induced diabetic cardiomyopathy [96]. These reports suggested the role of the ACE2/Ang-(1–7)/Mas receptor axis as a potential target for the development of novel treatment for diabetic cardiomyopathy. Notably, a critical research report suggests that the majority of cardioprotective effects of ACE2 are mediated via activation of Ang-(1–7)/Mas receptor signaling, signifying the importance of Ang-(1–7) as a potential therapeutic agent in various cardiovascular disease [97, 98].
Overexpression of ACE2 in a rat model of diabetic cardiomyopathy demonstrated markedly reduced myocyte hypertrophy, myocardial fibrosis, and improved left ventricle remodeling and function [96]. The underlying mechanism of protective effects encompassed ACE2 overexpression-mediated inhibition of collagen deposition by converting Ang II to Ang-(1–7) and by increasing MMP-2 activity. Moreover, the administration of A779, a Mas receptor antagonist, in the group injected with the adenoviral ACE2 (Ad-ACE2) overexpression exhibited increased collagen protein expression in response to Mas receptor inhibition, indicating a key role of Ang-(1–7)/Mas receptor signaling in negative regulation of cardiac collagen synthesis. Significantly, ACE2 overexpression exhibited superior cardioprotective potential compared to losartan, an angiotensin receptor blocker (ARB), as evident by reduced left ventricular end-diastolic diameter (LVEDD) and collagen expression along with increased left ventricular ejection fraction (LVEF).
Ang-(1–7) exhibited protective effects against diabetic cardiomyopathy by attenuation of pathological characteristics such as cardiac hypertrophy and lipotoxicity, adipose inflammation, myocardial oxidative stress, and upregulation of adipose triglyceride lipase (Fig. 15.3). In a db/db murine model of diabetic cardiomyopathy, Ang-(1–7) alleviated diastolic dysfunction and proved to be a promising therapeutic target [99]. The right ventricular (RV) fibrosis, a distinctive feature of diabetic cardiomyopathy, was ameliorated by Ang-(1–7) treatment in streptozotocin-induced diabetic cardiomyopathy in rats [100]. In diabetic rats, administration of Ang-(1–7) reduced cardiac fibrosis and dysfunction via a intricate interaction of AT2 receptor and Mas receptor for consequent downregulation of ACE expression and activity along with AT1 receptor expression. This complex interaction facilitated upregulation of ACE2 activity, as well as increased expression of AT2 receptor and SERCA2a. As the high dose of Ang-(1–7) was found to be superior to perindopril in mitigating cardiac remodeling, the study suggested the clinical potential of Ang-(1–7) as a novel treatment for diabetic cardiomyopathy [100]. Along with its prominent vasodilatory, anti-proliferative, and anti-fibrotic properties, Ang-(1–7) treatment resulted in a significant decrease in dyslipidemia, a major predisposing factor of cardiac dysfunction in type 2 diabetes. Ang-(1–7) alleviated left ventricle hypertrophy, cardiac fibrosis, and improved endothelial function, thus attenuating diabetic cardiomyopathy in streptozotocin-induced type 1 diabetic rats with cardiomyopathy [101].

Potential role of Ang 1–7 as a therapeutic agent in diabetic cardiomyopathy. Schematic representation of protective effects mediated by Ang 1–7 shows attenuation of hallmarks of diabetic cardiomyopathy i.e. cardiac fibrosis, inflammation, hypertrophy, and cardiac remodeling. Ang 1–7 exerts beneficial effects via binding to Mas receptor and antagonizes Ang II/AT1R axis of the renin angiotensin system. Under obesity and hyperglycemia-induced pathological conditions, activation of Ang II/AT1R axis potentiates increase in expression of genes (Col1a1, Col131, Mmp2) and proteins (OPN, CTGF, ANP) associated with cardiac fibrosis and hypertrophy. Pathological stress stimuli increase oxidative stress via ROS generation and induce inflammation by proinflammatory cytokines (IL-6, TNF-α, MCP-1). Ang 1–7 suppresses inflammation, oxidative stress, and cardiac remodeling, thus preserving cardiac function. Figure created using Biorender.com
Pharmacological agents alone or in combination with Ang-(1–7) were investigated to identify therapeutic potential in diabetic cardiomyopathy through modulation of ACE2/Ang-(1–7)/Mas receptor axis of RAS. A study using db/db murine model of diabetic cardiomyopathy revealed that majority of cardioprotective effects of azilsartan, an AT1 receptor blocker, were mediated by activation of ACE2/Ang-(1–7)/Mas receptor signaling cascade. Azilsartan significantly abrogated downregulation of ACE2 and Mas receptor in db/db mice and reduced pathological cardiac remodeling, oxidative stress, cardiac fibrosis, thus alleviated diabetic cardiomyopathy in db/db mice by modulating ACE2/Ang-(1–7)/ Mas receptor pathway [102]. In a preclinical study, administration of Ang-(1–7) dose-dependently alleviated left ventricle remodelling and cardiac dysfunction in diabetic rats by reducing cardiac hypertrophy, myocardial fibrosis, and apoptosis, effects which were mediate via activation of both the Mas and AT2 receptors. The underlying mechanism of cardioprotection involved upregulation of ACE2 activity and increased Ang 1–9 levels, resulting in reduced oxidative stress, collagen synthesis and attenuated inflammatory cytokine expression, TGFβ1 expression, and ERK1/2 and p38-MAPK phosphorylation. Combination of Ang-(1–7) and perindopril, an ACE inhibitor, demonstrated superior cardioprotective effect than single therapy, suggesting synergistic effects of blockade of ACE/Ang II/AT1 receptor pathway and activation of Ang-(1–7)/Mas receptor pathway [103]. Hence, various preclinical studies provide strong scientific evidence suggesting that Ang-(1–7) alleviates pathological processes such as cardiac remodeling, inflammation and oxidative stress involved in the onset and progression of diabetic cardiomyopathy; enhancing Ang-(1–7) actions may provide a promising therapeutic approach for the treatment of diabetic cardiomyopathy (Fig. 15.3).
Conclusion
Resistance to the metabolic actions of insulin, compensatory hyperinsulinemia, and the progression of hyperglycemia promoted by type II diabetes activate RAS resulting in several effects at the cardiac level, such as loss of cardiomyocytes by apoptosis, increased proliferation of fibroblasts with fibrosis, and hypertrophy. The resulting heart failure intensifies neurohormonal responses and the activity of RAS, which is related to the progression of diabetic cardiomyopathy, generating a vicious cycle. Diabetes leads to other changes, such as a downregulation of the ACE2 and an increased expression of AT1 receptors in cardiomyocytes, which is associated with a decreased left ventricular systolic pressure and increased in diastolic pressure. It also demonstrates that the ACE/Ang II/AT1 receptor pathway is activated in diabetic cardiomyopathy, while the ACE2/Ang-(1–7)/Mas receptor is not. Therefore, the goal of RAS modulation-based therapies goes beyond the inhibition of Ang II deleterious effects, but also aims to enhance the actions and activity of potentially helpful pathways, by ACE2 replenishing strategies, Ang-(1–7) administration, and Mas receptor agonists.
References
Borghetti G, von Lewinski D, Eaton DM, Sourij H, Houser SR, Wallner M (2018) Diabetic cardiomyopathy: current and future therapies. Beyond Glycemic Control. Front Physiol 9:1514. https://doi.org/10.3389/fphys.2018.01514
Jia G, Hill MA, Sowers JR (2018) Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res 122(4):624–638. https://doi.org/10.1161/CIRCRESAHA.117.311586
Goossens GH, Blaak EE, van Baak MA (2003) Possible involvement of the adipose tissue renin-angiotensin system in the pathophysiology of obesity and obesity-related disorders. Obes Rev 4(1):43–55. https://doi.org/10.1046/j.1467-789x.2003.00091.x
Engeli S, Negrel R, Sharma AM (2000) Physiology and pathophysiology of the adipose tissue renin-angiotensin system. Hypertension 35(6):1270–1277. https://doi.org/10.1161/01.hyp.35.6.1270
Ferrario CM, Strawn WB (2006) Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol 98(1):121–128. https://doi.org/10.1016/j.amjcard.2006.01.059
Kim S, Iwao H (2000) Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev 52(1):11–34
Unger T (2002) The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol 89(2):3–9. https://doi.org/10.1016/s0002-9149(01)02321-9
Henriksen EJ (2007) Improvement of insulin sensitivity by antagonism of the renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 293(3):R974-980. https://doi.org/10.1152/ajpregu.00147.2007
Leung PS (2007) Mechanisms of protective effects induced by blockade of the renin-angiotensin system: novel role of the pancreatic islet angiotensin-generating system in type 2 diabetes. Diabet Med 24(2):110–116. https://doi.org/10.1111/j.1464-5491.2007.02072.x
Tigerstedt R, Bergman PQ (1898) Niere und Kreislauf1. Skandinavisches Archiv Für Physiologie 8(1):223–271. https://doi.org/10.1111/j.1748-1716.1898.tb00272.x
Grobe JL, Xu D, Sigmund CD (2008) An intracellular renin-angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology (Bethesda) 23:187–193. https://doi.org/10.1152/physiol.00002.2008
Bodiga VL, Bodiga S (2013) Renin angiotensin system in cognitive function and dementia. Asian J Neurosci 2013:1–18. https://doi.org/10.1155/2013/102602
Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J (2003) Inflammation and angiotensin II. Int J Biochem Cell Biol 35(6):881–900. https://doi.org/10.1016/s1357-2725(02)00271-6
Paul M, Poyan Mehr A, Kreutz R (2006) Physiology of local renin-angiotensin systems. Physiol Rev 86(3):747–803. https://doi.org/10.1152/physrev.00036.2005
Fouda AY, Artham S, El-Remessy AB, Fagan SC (2016) Renin-angiotensin system as a potential therapeutic target in stroke and retinopathy: experimental and clinical evidence. Clin Sci (Lond) 130(4):221–238. https://doi.org/10.1042/CS20150350
Wright JW, Harding JW (2011) Brain renin-angiotensin–a new look at an old system. Prog Neurobiol 95(1):49–67. https://doi.org/10.1016/j.pneurobio.2011.07.001
Santos RA, Simoes e Silva AC, Maric C, et al (2003) Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA 100(14):8258−8263. https://doi.org/10.1073/pnas.1432869100
Jiang T, Gao L, Lu J, Zhang YD (2013) ACE2-Ang-(1–7)-Mas axis in brain: a potential target for prevention and treatment of ischemic stroke. Curr Neuropharmacol 11(2):209–217. https://doi.org/10.2174/1570159X11311020007
Tetzner A, Gebolys K, Meinert C et al (2016) G-Protein-coupled receptor MrgD Is a receptor for angiotensin-(1–7) involving adenylyl cyclase, cAMP, and phosphokinase A. Hypertension 68(1):185–194. https://doi.org/10.1161/HYPERTENSIONAHA.116.07572
Lautner RQ, Villela DC, Fraga-Silva RA et al (2013) Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res 112(8):1104–1111. https://doi.org/10.1161/CIRCRESAHA.113.301077
Xia H, Lazartigues E (2008) Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem 107(6):1482–1494. https://doi.org/10.1111/j.1471-4159.2008.05723.x
Ahmed HA, Ishrat T, Pillai B et al (2018) Role of angiotensin system modulation on progression of cognitive impairment and brain MRI changes in aged hypertensive animals—a randomized double-blind pre-clinical study. Behav Brain Res 346:29–40. https://doi.org/10.1016/j.bbr.2017.12.007
Labandeira-Garcia JL, Rodriguez-Perez AI, Garrido-Gil P, Rodriguez-Pallares J, Lanciego JL, Guerra MJ (2017) Brain renin-angiotensin system and microglial polarization: implications for aging and neurodegeneration. Front Aging Neurosci 9:129. https://doi.org/10.3389/fnagi.2017.00129
Costa-Besada MA, Valenzuela R, Garrido-Gil P et al (2018) Paracrine and intracrine angiotensin 1–7/mas receptor axis in the substantia nigra of rodents, monkeys, and humans. Mol Neurobiol 55(7):5847–5867. https://doi.org/10.1007/s12035-017-0805-y
Leonhardt J, Villela DC, Teichmann A et al (2017) Evidence for heterodimerization and functional interaction of the angiotensin type 2 receptor and the receptor MAS. Hypertension 69(6):1128–1135. https://doi.org/10.1161/HYPERTENSIONAHA.116.08814
Santos RAS, Campagnole-Santos MJ, SlP A (2000) Angiotensin-(1–7): an update. Regul Pept 91(1–3):45–62. https://doi.org/10.1016/s0167-0115(00)00138-5
Passos-Silva DG, Brandan E, Santos RA (2015) Angiotensins as therapeutic targets beyond heart disease. Trends Pharmacol Sci 36(5):310–320. https://doi.org/10.1016/j.tips.2015.03.001
Passos-Silva DG, Verano-Braga T, Santos RA (2013) Angiotensin-(1–7): beyond the cardio-renal actions. Clin Sci (Lond) 124(7):443–456. https://doi.org/10.1042/CS20120461
Gironacci MM, Cerniello FM, Longo Carbajosa NA, Goldstein J, Cerrato BD (2014) Protective axis of the renin-angiotensin system in the brain. Clin Sci (Lond) 127(5):295–306. https://doi.org/10.1042/CS20130450
Timmermans PB, Wong PC, Chiu AT et al (1993) Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev 45(2):205–251
Peach MJ (1977) Renin-angiotensin system: biochemistry and mechanisms of action. Physiol Rev 57(2):313–370. https://doi.org/10.1152/physrev.1977.57.2.313
Ribeiro-Oliveira AJ, Nogueira AI, Pereira RM, Boas WW, Santos RA, Simões e Silva AC (2008) The renin–angiotensin system and diabetes: an update. Vascular Health and Risk Managem 4(4):787–803
Kahn CR (2008) Medicine. Can we nip obesity in its vascular bud? Science 322(5901):542–543. https://doi.org/10.1126/science.1165667
Kahn SE (2003) The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 46(1):3–19. https://doi.org/10.1007/s00125-002-1009-0
Tikellis C, Cooper ME, Thomas MC (2006) Role of the renin-angiotensin system in the endocrine pancreas: implications for the development of diabetes. Int J Biochem Cell Biol 38(5–6):737–751. https://doi.org/10.1016/j.biocel.2005.08.007
Kampf C, Lau T, Olsson R, Leung PS, Carlsson PO (2005) Angiotensin II type 1 receptor inhibition markedly improves the blood perfusion, oxygen tension and first phase of glucose-stimulated insulin secretion in revascularised syngeneic mouse islet grafts. Diabetologia 48(6):1159–1167. https://doi.org/10.1007/s00125-005-1761-z
Huang Z, Jansson L, Sjoholm A (2006) Pancreatic islet blood flow is selectively enhanced by captopril, irbesartan and pravastatin, and suppressed by palmitate. Biochem Biophys Res Commun 346(1):26–32. https://doi.org/10.1016/j.bbrc.2006.05.144
Chu KY, Lau T, Carlsson PO, Leung PS (2006) Angiotensin II type 1 receptor blockade improves beta-cell function and glucose tolerance in a mouse model of type 2 diabetes. Diabetes 55(2):367–374. https://doi.org/10.2337/diabetes.55.02.06.db05-1022
Shao J, Iwashita N, Ikeda F et al (2006) Beneficial effects of candesartan, an angiotensin II type 1 receptor blocker, on beta-cell function and morphology in db/db mice. Biochem Biophys Res Commun 344(4):1224–1233. https://doi.org/10.1016/j.bbrc.2006.04.011
Yuan L, Li X, Li J, Li HL, Cheng SS (2013) Effects of renin-angiotensin system blockade on the islet morphology and function in rats with long-term high-fat diet. Acta Diabetol 50(4):479–488. https://doi.org/10.1007/s00592-010-0210-8
Ashrafian H, Neubauer S (2009) Metabolomic profiling of cardiac substrate utilization: fanning the flames of systems biology? Circulation 119(13):1700–1702. https://doi.org/10.1161/CIRCULATIONAHA.109.849919
Neubauer S (2007) The failing heart–an engine out of fuel. N Engl J Med 356(11):1140–1151. https://doi.org/10.1056/NEJMra063052
Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M (2004) Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci 1015:202–213. https://doi.org/10.1196/annals.1302.017
Neely JR, Rovetto MJ, Oram JF (1972) Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis 15(3):289–329. https://doi.org/10.1016/0033-0620(72)90029-1
Labbe SM, Grenier-Larouche T, Noll C et al (2012) Increased myocardial uptake of dietary fatty acids linked to cardiac dysfunction in glucose-intolerant humans. Diabetes 61(11):2701–2710. https://doi.org/10.2337/db11-1805
Ferre P (2004) The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes 53(Suppl 1):S43-50. https://doi.org/10.2337/diabetes.53.2007.s43
Cha DR, Han JY, Su DM et al (2007) Peroxisome proliferator-activated receptor-alpha deficiency protects aged mice from insulin resistance induced by high-fat diet. Am J Nephrol 27(5):479–482. https://doi.org/10.1159/000106485
Rijzewijk LJ, van der Meer RW, Lamb HJ et al (2009) Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: studies with cardiac positron emission tomography and magnetic resonance imaging. J Am Coll Cardiol 54(16):1524–1532. https://doi.org/10.1016/j.jacc.2009.04.074
Ng AC, Delgado V, Bertini M et al (2010) Myocardial steatosis and biventricular strain and strain rate imaging in patients with type 2 diabetes mellitus. Circulation 122(24):2538–2544. https://doi.org/10.1161/CIRCULATIONAHA.110.955542
Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M (2006) Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J Clin Invest 116(4):1071–1080. https://doi.org/10.1172/JCI23354
Fillmore N, Mori J, Lopaschuk GD (2014) Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol 171(8):2080–2090. https://doi.org/10.1111/bph.12475
Hafstad AD, Nabeebaccus AA, Shah AM (2013) Novel aspects of ROS signalling in heart failure. Basic Res Cardiol 108(4):359. https://doi.org/10.1007/s00395-013-0359-8
How OJ, Aasum E, Severson DL, Chan WY, Essop MF, Larsen TS (2006) Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes 55(2):466–473. https://doi.org/10.2337/diabetes.55.02.06.db05-1164
Hafstad AD, Solevag GH, Severson DL, Larsen TS, Aasum E (2006) Perfused hearts from type 2 diabetic (db/db) mice show metabolic responsiveness to insulin. Am J Physiol Heart Circ Physiol 290(5):H1763-1769. https://doi.org/10.1152/ajpheart.01063.2005
Chung SS, Ho EC, Lam KS, Chung SK (2003) Contribution of polyol pathway to diabetes-induced oxidative stress. J Am Soc Nephrol 14(8 Suppl 3):S233-236. https://doi.org/10.1097/01.asn.0000077408.15865.06
Sochor M, Gonzalez A-M, McLean P (1984) Regulation of alternative pathways of glucose metabolism in rat heart in alloxan diabetes: changes in the pentose phosphate pathway. Biochem Biophys Res Commun 118(1):110–116. https://doi.org/10.1016/0006-291x(84)91074-x
Petrova R, Yamamoto Y, Muraki K et al (2002) Advanced glycation endproduct-induced calcium handling impairment in mouse cardiac myocytes. J Mol Cell Cardiol 34(10):1425–1431. https://doi.org/10.1006/jmcc.2002.2084
Brownlee M (1995) Advanced protein glycosylation in diabetes and aging. Annu Rev Med 46:223–234. https://doi.org/10.1146/annurev.med.46.1.223
McNulty PH (2007) Hexosamine biosynthetic pathway flux and cardiomyopathy in type 2 diabetes mellitus. Focus on “Impact of type 2 diabetes and aging on cardiomyocyte function and O-linked N-acetylglucosamine levels in the heart”. Am J Physiol Cell Physiol 292(4):C1243–1244. https://doi.org/10.1152/ajpcell.00521.2006
Roche E, Farfari S, Witters LA et al (1998) Long-term exposure of beta-INS cells to high glucose concentrations increases anaplerosis, lipogenesis, and lipogenic gene expression. Diabetes 47(7):1086–1094. https://doi.org/10.2337/diabetes.47.7.1086
Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD (2009) Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol 54(20):1891–1898. https://doi.org/10.1016/j.jacc.2009.07.031
Croston TL, Thapa D, Holden AA et al (2014) Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am J Physiol Heart Circ Physiol 307(1):H54-65. https://doi.org/10.1152/ajpheart.00845.2013
Boudina S, Sena S, Theobald H et al (2007) Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56(10):2457–2466. https://doi.org/10.2337/db07-0481
Giardino I, Edelstein D, Brownlee M (1996) BCL-2 expression or antioxidants prevent hyperglycemia-induced formation of intracellular advanced glycation endproducts in bovine endothelial cells. J Clin Invest 97(6):1422–1428. https://doi.org/10.1172/JCI118563
Zorzano A, Liesa M, Palacin M (2009) Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol 41(10):1846–1854. https://doi.org/10.1016/j.biocel.2009.02.004
Scheuermann-Freestone M, Madsen PL, Manners D et al (2003) Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation 107(24):3040–3046. https://doi.org/10.1161/01.CIR.0000072789.89096.10
Savabi F (1991) Alteration of the phosphocreatine energy shuttle components in diabetic rat heart. J Mol Cell Cardiol 23(11):1323–1333. https://doi.org/10.1016/0022-2828(91)90089-5
Montaigne D, Marechal X, Coisne A et al (2014) Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130(7):554–564. https://doi.org/10.1161/CIRCULATIONAHA.113.008476
Andreyev AY, Kushnareva YE, Starkov AA (2005) Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 70(2):200–214. https://doi.org/10.1007/s10541-005-0102-7
Nishikawa T, Edelstein D, Du XL et al (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404(6779):787–790. https://doi.org/10.1038/35008121
Schannwell CM, Schneppenheim M, Perings S, Plehn G, Strauer BE (2002) Left ventricular diastolic dysfunction as an early manifestation of diabetic cardiomyopathy. Cardiology 98(1–2):33–39. https://doi.org/10.1159/000064682
Pereira L, Ruiz-Hurtado G, Rueda A, Mercadier JJ, Benitah JP, Gomez AM (2014) Calcium signaling in diabetic cardiomyocytes. Cell Calcium 56(5):372–380. https://doi.org/10.1016/j.ceca.2014.08.004
Murarka S, Movahed MR (2010) Diabetic cardiomyopathy. J Card Fail 16(12):971–979. https://doi.org/10.1016/j.cardfail.2010.07.249
Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP (1990) Prognostic implications of echocardiographically determined left ventricular mass in the framingham heart study. N Engl J Med 322(22):1561–1566. https://doi.org/10.1056/NEJM199005313222203
Selmeryd J, Sundstedt M, Nilsson G, Henriksen E, Hedberg P (2014) Impact of left ventricular geometry on long-term survival in elderly men and women. Clin Physiol Funct Imaging 34(6):442–448. https://doi.org/10.1111/cpf.12114
Lieb W, Gona P, Larson MG et al (2014) The natural history of left ventricular geometry in the community: clinical correlates and prognostic significance of change in LV geometric pattern. JACC Cardiovasc Imaging 7(9):870–878. https://doi.org/10.1016/j.jcmg.2014.05.008
Forbes JM, Cooper ME (2013) Mechanisms of diabetic complications. Physiol Rev 93(1):137–188. https://doi.org/10.1152/physrev.00045.2011
Paradis P, Dali-Youcef N, Paradis FW, Thibault G, Nemer M (2000) Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc Natl Acad Sci U S A 97(2):931–936. https://doi.org/10.1073/pnas.97.2.931
Munzel T, Gori T, Keaney JF Jr, Maack C, Daiber A (2015) Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur Heart J 36(38):2555–2564. https://doi.org/10.1093/eurheartj/ehv305
Kumar R, Thomas CM, Yong QC, Chen W, Baker KM (2012) The intracrine renin-angiotensin system. Clin Sci (Lond) 123(5):273−284. https://doi.org/10.1042/CS20120089
Yamamoto E, Dong YF, Kataoka K et al (2008) Olmesartan prevents cardiovascular injury and hepatic steatosis in obesity and diabetes, accompanied by apoptosis signal regulating kinase-1 inhibition. Hypertension 52(3):573–580. https://doi.org/10.1161/HYPERTENSIONAHA.108.112292
Fukuda M, Nakamura T, Kataoka K et al (2010) Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. J Hypertens 28(2):340–352. https://doi.org/10.1097/HJH.0b013e32833366cd
Liu X, Xu Q, Wang X et al (2015) Irbesartan ameliorates diabetic cardiomyopathy by regulating protein kinase D and ER stress activation in a type 2 diabetes rat model. Pharmacol Res 93:43–51. https://doi.org/10.1016/j.phrs.2015.01.001
Oliveira-Junior SA, Martinez PF, Guizoni DM, et al. (2014) AT1 receptor blockade attenuates insulin resistance and myocardial remodeling in rats with diet-induced obesity. PLoS One 9(1):e86447. https://doi.org/10.1371/journal.pone.0086447
Huynh K, Bernardo BC, McMullen JR, Ritchie RH (2014) Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther 142(3):375–415. https://doi.org/10.1016/j.pharmthera.2014.01.003
Zaman AK, Fujii S, Sawa H et al (2001) Angiotensin-converting enzyme inhibition attenuates hypofibrinolysis and reduces cardiac perivascular fibrosis in genetically obese diabetic mice. Circulation 103(25):3123–3128. https://doi.org/10.1161/01.cir.103.25.3123
Toblli JE, Cao G, DeRosa G, Forcada P (2005) Reduced cardiac expression of plasminogen activator inhibitor 1 and transforming growth factor beta1 in obese Zucker rats by perindopril. Heart 91(1):80–86. https://doi.org/10.1136/hrt.2003.022707
Nevelsteen I, Bito V, Van der Mieren G et al (2013) ACE-inhibition, but not weight reduction restores cardiomyocyte response to beta-adrenergic stimulation in the metabolic syndrome. BMC Cardiovasc Disord 13:51. https://doi.org/10.1186/1471-2261-13-51
Tabbi-Anneni I, Buchanan J, Cooksey RC, Abel ED (2008) Captopril normalizes insulin signaling and insulin-regulated substrate metabolism in obese (ob/ob) mouse hearts. Endocrinology 149(8):4043–4050. https://doi.org/10.1210/en.2007-1646
Kang YS, Lee MH, Song HK et al (2011) Aliskiren improves insulin resistance and ameliorates diabetic vascular complications in db/db mice. Nephrol Dial Transplant 26(4):1194–1204. https://doi.org/10.1093/ndt/gfq579
Dong YF, Liu L, Lai ZF et al (2010) Aliskiren enhances protective effects of valsartan against type 2 diabetic nephropathy in mice. J Hypertens 28(7):1554–1565. https://doi.org/10.1097/HJH.0b013e328338bb11
Jiang F, Yang J, Zhang Y et al (2014) Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat Rev Cardiol 11(7):413–426. https://doi.org/10.1038/nrcardio.2014.59
Santos SH, Fernandes LR, Mario EG et al (2008) Mas deficiency in FVB/N mice produces marked changes in lipid and glycemic metabolism. Diabetes 57(2):340–347. https://doi.org/10.2337/db07-0953
Patel VB, Bodiga S, Basu R et al (2012) Loss of angiotensin-converting enzyme-2 exacerbates diabetic cardiovascular complications and leads to systolic and vascular dysfunction: a critical role of the angiotensin II/AT1 receptor axis. Circ Res 110(10):1322–1335. https://doi.org/10.1161/circresaha.112.268029
Zhong J, Basu R, Guo D, et al (2010) Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 122(7):717–728. 718 p following 728. https://doi.org/10.1161/circulationaha.110.955369
Dong B, Yu QT, Dai HY et al (2012) Angiotensin-converting enzyme-2 overexpression improves left ventricular remodeling and function in a rat model of diabetic cardiomyopathy. J Am Coll Cardiol 59(8):739–747. https://doi.org/10.1016/j.jacc.2011.09.071
Patel VB, Takawale A, Ramprasath T et al (2015) Antagonism of angiotensin 1–7 prevents the therapeutic effects of recombinant human ACE2. J Mol Med (Berl) 93(9):1003–1013. https://doi.org/10.1007/s00109-015-1285-z
Patel VB, Zhong JC, Grant MB, Oudit GY (2016) Role of the ACE2/Angiotensin 1–7 axis of the renin-angiotensin system in heart failure. Circ Res 118(8):1313–1326. https://doi.org/10.1161/CIRCRESAHA.116.307708
Mori J, Patel VB, Abo Alrob O et al (2014) Angiotensin 1–7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ Heart Fail 7(2):327–339. https://doi.org/10.1161/CIRCHEARTFAILURE.113.000672
Hao PP, Yang JM, Zhang MX et al (2015) Angiotensin-(1–7) treatment mitigates right ventricular fibrosis as a distinctive feature of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 308(9):H1007-1019. https://doi.org/10.1152/ajpheart.00563.2014
Singh K, Singh T, Sharma PL (2011) Beneficial effects of angiotensin (1–7) in diabetic rats with cardiomyopathy. Ther Adv Cardiovasc Dis 5(3):159–167. https://doi.org/10.1177/1753944711409281
Sukumaran V, Tsuchimochi H, Tatsumi E, Shirai M, Pearson JT (2017) Azilsartan ameliorates diabetic cardiomyopathy in young db/db mice through the modulation of ACE-2/ANG 1–7/Mas receptor cascade. Biochem Pharmacol 144:90–99. https://doi.org/10.1016/j.bcp.2017.07.022
Hao P, Yang J, Liu Y et al (2015) Combination of angiotensin-(1–7) with perindopril is better than single therapy in ameliorating diabetic cardiomyopathy. Sci Rep 5:8794. https://doi.org/10.1038/srep08794
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gomes, K.P., Jadli, A.S., Patel, V.B. (2023). Renin Angiotensin System in the Pathophysiology of Diabetic Cardiomyopathy in Type 2 Diabetes. In: Dhalla, N.S., Bhullar, S.K., Shah, A.K. (eds) The Renin Angiotensin System in Cardiovascular Disease. Advances in Biochemistry in Health and Disease, vol 24. Springer, Cham. https://doi.org/10.1007/978-3-031-14952-8_15
Download citation
DOI: https://doi.org/10.1007/978-3-031-14952-8_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-14951-1
Online ISBN: 978-3-031-14952-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)