
Abstract
The Renin Angiotensin System (RAS) is clearly implicated in the physiopathology of diabetes mellitus (DM). The frequent association of diabetes mellitus (DM) with hypertension, retinopathy, nephropathy, and cardiovascular disease has implicated the RAS in the initiation and progression of these complications of DM. This has been supported by clinical trials in which RAS inhibitors significantly reduced the incidence of vascular complications in DM patients. The main RAS mediator, Angiotensin II (Ang II), exerts several deleterious actions in patients with DM, including increase in insulin resistance, endothelial damage and deterioration of renal function. On the other hand, only few studies have reported the potential protective role of the stimulation of the conter-regulatory RAS axis formed by the enzyme homologue to ACE, ACE2, the heptapeptide Angiotensin-(1-7) [Ang-(1-7)] and its receptor, the proto-oncogene Mas. In this review, we report recent experimental and clinical evidence in relation to ACE2 stimulation and Mas receptor agonists as potential therapeutic targets for DM.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Diabetes mellitus
- Renin angiotensin system
- Angiotensin II
- Angiotensin-(1-7)
- ACE2
- Mas receptor
- Diabetic nephropathy
1 Introduction
The renin-angiotensin system (RAS) has been implicated in complications linked with diabetes mellitus, including insulin resistance, endothelial damage and diabetic nephropathy [1,2,3]. Measurements of the RAS components in diabetic patients have shown conflicting results: some have found elevated levels, others, reduced, and others yet, found no change [4,5,6]. The picture can be further confusing given the activity of local and independently regulated RASs [7, 8]. However, the significant reno and cardioprotection that have been achieved by blockade of the RAS with angiotensin-converting enzyme (ACE ) inhibitors and angiotensin receptor antagonists (ARAs) are strong compelling evidence for the role of the RAS in this disease [2, 9, 10].
Many studies have shown that Angiotensin (Ang) II exerts physiological and biochemical actions that may contribute to cardiovascular and renal damage [11]. The Angiotensin type 1 (AT1) receptor mediates the main actions of Ang II [12]. Over the recent past, our view of Ang II has changed from being a simple vasoconstrictor to that of a complex growth factor mediating effects through diverse signaling pathways [8]. It has also become clear that Ang II is a key player in vascular inflammation . Through increased generation of reactive oxygen species (ROS) and activation of redox-sensitive transcription factors, Ang II promotes expression of cell adhesion molecules and induces synthesis of proinflammatory mediators and growth factors [8, 12]. These processes increased vascular permeability, leukocyte recruitment and fibrosis leading to tissue injury and structural remodeling. Targeting some of these signaling events with novel therapeutic strategies may provide important tissue protection in many forms of cardiovascular, renal and metabolic diseases.
On the other hand, it was originally thought that Ang II mediates all actions of the RAS. Over the past few years, other angiotensin peptides, like Ang III, Ang IV, and especially Ang-(1-7), were shown to selectively mediate different RAS effects [11, 13]. In regard to Ang-(1-7), this heptapetide can be formed from Ang I by neutral-endopepdidase 24.11 or prolyl-endopeptidase or from Ang II via prolyl-endopeptidase, prolylcarboxypeptidase [14] or mainly by ACE2 , an enzyme homologue to ACE [15, 16]. Ang-(1-7) binds to a G-protein coupled receptor, named Mas receptor [17], and, in general, plays a counter-regulatory role in the RAS by opposing the vascular and proliferative effects of Ang II [11, 13]. Currently, RAS is conceived as a system formed by two opposite axes: the first and classical one composed by ACE , Ang II and AT1 receptor and the second and counter-regulatory axis comprising ACE2 , Ang-(1-7) and Mas receptor [11, 13]. Experimental studies clearly support a role for the counter-regulatory RAS axis in diabetes. However, a limited number of studies have evaluated the components of ACE2-Ang-(1-7)-Mas receptor axis in diabetic patients [18,19,20,21,22,23,24,25], and the majority of them investigated only ACE2 [18,19,20, 23,24,25]. In this chapter, we report evidence for the role of ACE2-Ang-(1-7)-Mas receptor axis in diabetes mellitus and its complications , including poor glycemic control, diabetic nephropathy and cardiovascular alterations.
2 Role of ACE2 -Angiotensin-(1-7) -Mas Axis in Glycemic Control
Identification of a local pancreatic RAS has led to a better understanding of the role of the RAS in the physiopathology of diabetes. RAS blockers seem to be able to reverse Ang-II-induced impairment in insulin sensitivity, insulin secretion, and pancreatic β-cell function [26,27,28,29]. However, the role of RAS blockers in this context is an ongoing matter of debate due to studies showing the inefficiency of RAS blockers in controlling hyperglycemic symptoms [30, 31].
On the other hand, ACE2 has received significant attention over the past years, being considered a promising target due to its beneficial role in glycemic control [32]. ACE2 was discovered in 2000 and shares 42% sequence homology with ACE , but cannot be inhibited by ACE inhibitors [15, 16].ACE2 gene is located on the Xchromosome and cleaves various substrates, including Ang II, angiotensin I (Ang I), apelin, neurotensin, and des-Arg bradykinin with the highest catalytic efficiency towards Ang II [15, 16]. Indeed, ACE2 is the main enzyme responsible for the conversion of Ang II into Ang-(1-7) in many organs and tissues [33].
ACE2 overexpression has been shown to reverse the detrimental phenotypes in cardiovascular disease [34, 35], diabetes [36, 37],and its related complications , an effect known to occur by suppressing the overactive Ang II levels [38]. The beneficial effects of ACE2 have been attributed to its capacity to increase Ang-(1-7) levels [15, 16, 33]. It has reported that Ang-(1-7) improves insulin sensitivity and glucose tolerance in experimental animal models, possibly by stimulating the insulin signaling via Mas receptor [39, 40]. Supporting this hypothesis, mice with genetic deletion of Mas receptor exhibit disturbances in glucose and lipid metabolism [41]. Furthermore, the increase in circulating levels of Ang-(1-7) improves glucose tolerance and dyslipidemia [42]. Even under physiological conditions, mice with genetic deletion of ACE2 progressively reduce insulin secretion and glucose tolerance [43]. However, when these knockout animals were under a high-fat high-sucrose diet,the degree of glucose intolerance is higher than in wild type mice [44]. This effect was attributed to the reduced skeletal muscle levels of GLUT4 and myocyte enhancer factor 2A expression [44]. It should be mentioned that the administration of Ang-(1-7) restored glucose tolerance [44]. These results support the importance of Ang-(1-7) signaling in maintaining glucose tolerance and insulin sensitivity.
The importance of the counter-regulatory ACE2 -Ang-(1-7)-Mas axis in maintaining pancreatic β-cell function has been investigated in vitro [45]. The results showed that an upregulation in the expression of ACE2 and of Mas is associated with an increase in insulin secretion at high glucose concentrations [45]. In experimental models of diabetes, ACE2 levels decrease as the disease progresses, leading to an uninhibited rise in the activity of the classical ACE -Ang II-AT1 axis [36, 46].Thus, it can be speculated that, as the Ang II levels increase in hyperglycemic state, ACE2 is also upregulated as a compensatory mechanism and aids in the degradation and reduction of Ang II. Moreover, ACE2 gene therapy and the administration of ACE2 activators, including xanthenone and diminazene aceturate [47], exerted beneficial effects in the face of diabetes [36, 48] and its complications [49, 50].
Various mechanisms have been proposed by which ACE2 elicits opposing effects on Ang II signaling. Oxidative stress has been reported to be one of the predisposing factors of pancreatic β-cell dysfunction during hyperglycemic states [51, 52]. Ang II activates reactive oxygen species (ROS) [53]. ACE2 over expression reduced oxidative stress and corrected Ang II-induced imbalance in the relationship between the expressions of AT1 receptor and of ACE2 [37]. Both mechanisms improved glycemic control [37]. Pharmacological inhibition of ACE2 and of Mas receptor increased ROS formation induced by Ang II [54], further supporting the hypothesis that ACE2 reduces thecapacity of Ang II to form ROSby converting Ang II into Ang-(1-7).
Other mechanisms of impaired glucose homeostasis include endoplasmic reticulum stress, tissue fibrosis and inflammation [55,56,57]. The lack of ACE2 has been reported to exacerbate fibrosis and inflammation in the kidney [58, 59]and in the heart [60], whereas the overexpression of ACE2 decreased fibrosis in the heart [61], lungs [62], and pancreas [63].Moreover, Ang-(1-7) improved insulin sensibility, at least in part, via its anti-inflammatory properties in the liver [64].This effect was also associated with an up regulation in ACE2 expression in the liver. On the other hand, there are scarce information on the role of ACE2-Ang-(1-7)-Mas axis in preventing fibrosis in pancreatic islets [63]. The role of ACE2 in modulating endoplasmic reticulum stress, fibrosis, and inflammation in the islets warrants further investigation.
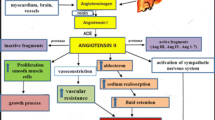
Figure 11.1 summarizes the main actions of the classical and the counter-regulatory RAS axes in the control of the glycemia and of the insulin secretion.

The role of components of the classical and the counter regulatory renin-angiotensisn system axes in the control of glycemia Nd of the insulin secretion
3 Role of ACE2 -Ang-(1-7)-Mas Receptor Axis in Diabetic Nephropathy
Diabetic nephropathy is one of the most common causes of end-stage renal disease and one of the main complications of diabetes, but the factors responsible for the development of diabetic nephropathy have not been fully elucidated [65].
There is growing interest in a possible role of ACE2 in diabetic kidney disease [19, 46, 65]. Activation of the classical RAS axis, ACE -Ang II- AT1 receptor, is widely believed to contribute to kidney injury in diabetes [66]. ACE2 may act as a negative regulator of the classical RAS axis, exerting a renoprotective action [65]. Obese db/db mice (C57BLKS/JLepr) have been used as a model of type 2 diabetes , and their lean littermates (db/m) have served as nondiabetic controls [18, 19, 24, 65]. In renal cortical tubules of db/db mice, the pattern of ACE and ACE2 expression was characterized by low ACE , but increased ACE2 protein [65]. These alterations in ACE2 protein in renal tubules from diabetic mice are accompanied by corresponding changes in enzymatic activity [19]. Ye and co-workers [24]examined the localization of ACE and ACE2 in the glomerulus of control and diabetic mice. The glomerulus is the site of the nephron where the lesions of diabetic nephropathy appear earlier, and an increase in glomerular permeability is an early manifestation of diabetic kidney disease as reflected by the presence of albuminuria. The authors found that in glomeruli from db/db mice, ACE staining was higher than in control mice, while strong ACE2 staining in glomeruli from diabetic mice was less frequently seen than in controls [24]. In addition, the same research group reported that chronic blockade of ACE2 with the enzyme inhibitor, MLN-4760, in control or diabetic mice produced albuminuria and matrix proteins deposition [18]. In this regard, Wong and co-workers [67] examined the effect of deletion of the ACE2 gene on diabetic kidney injury. In this study, ACE2 knockout mice [ACE2−/−]were crossed with Akita mice (Ins2WT/C96Y), a model of type 1 diabetes mellitus, and four groups of mice were studied at 3 months of age: ACE2+/yIns2WT/WT, ACE2−/yIns2WT/WT, ACE2+/yIns2WT/C96Y, and ACE2−/yIns2WT/C96Y. ACE2−/yIns2WT/C96Y mice exhibited increased mesangial matrix scores, glomerular basement membrane thicknesses, glomerular deposit of fibronectin and a twofold augmentation in the urinary albumin excretion rate compared with ACE2+/yIns2WT/C96Y [67]. The treatment with an AT1 receptor blocker, irbesartan, reversed the alterations in renal histology and reduced proteinuria in ACE2−/yIns2WT/C96Y mice [67]. More recently, ACE2 knockout mice with streptozotocin-induced diabetes presented an increase in serum creatinine, urea levels and albuminuria in comparison with wild type diabetic animals [66]. In addition, glomerular and tubulointerstitial injuries and macrophage infiltration were significantly more severe inACE2 knockout mice than in wild type controls. AT1receptor blocked with olmersartan attenuated the effects of ACE2 deficiency, but only partially [66]. Taken together, these studies suggested that ACE2 plays a protective role in the diabetic kidney, and ACE2 is an important determinant of diabetic nephropathy .
Some studies have also suggested a close correlation between albuminuria and ACE2 . The treatment of HK-2 cells with bovine serum albumin has led to significant changes in ACE /ACE2 expression favoring Ang II formation [52]. More recently, Marquez and co-workers [68] showed that insulin increases ACE2 gene, protein expression, and enzymatic activity in cultured podocytes and these increases were maintained over time. In the presence of albumin, the beneficial effect of insulin on ACE2 expression and activity disappeared [68]. Therefore, ACE2 reduction might increase urinary albumin excretion, while albuminuria, in turn, could disrupt the balance of ACE /ACE2 expression [68]. In this regard, Riera and co-workers [69] studied the non-obese diabetic mice model, since these animals develop autoimmune diabetes that resembles human type 1 diabetes. At an early stage of diabetes, diabetic mice exhibited tenfold increase in urinary albumin excretion, glomerular enlargement, increased glomerular filtration rate and higher blood pressure in comparison to controls [69]. At a later stage, diabetic mice had a 20-fold increase in albuminuria, mesangial expansion and reduced podocyte number. Circulating and urine ACE2 activity were markedly increased at early and late stage of diabetes. Insulin administration prevented albuminuria, markedly reduced GFR, blood pressure, and glomerular enlargement at the early stage; and prevented mesangial expansion and the reduced podocyte number at the late stage of diabetes. The increase in serum and urine ACE2 activity was normalized by insulin administration at the early and late stages of diabetes. The authors conclude that diabetic mice develop features of early kidney disease associated with increased activity of ACE2 in both serum and urine and these alterations can be completely prevented by the administration of insulin.
Ang-(1-7) has also a role in experimental models of diabetes. The administration of Ang-(1-7) was able to normalize creatinine clearance and significantly attenuate proteinuria in Zucker diabetic fattyrats, a model of type 2 diabetes and diabetic nephropathy [70]. Diabetic rats treated with Ang-(1-7) displayed markedly reduction in renal fibrosis, presenting levels of extracellular matrix proteins similar to control animals [70]. Levels of TNF-α, IL-6, endothelin-1, and hypoxia inducible factor (HIF)-1α in the kidneys were also decreased to levels similar of those of control animals. The same effect was observed in renal and urinary levels of neutrophil gelatinase-associated lipocalin (NGAL), a marker of kidney damage [70]. Accordingly, chronic infusion of Ang-(1-7) also had significant protective effects in leptin deficient db/db mice, another model of type2 diabetes and diabetic nephropathy [25]. Animals treated with Ang-(1-7) for 28 days normalized urinary albumin excretion and significantly decreased kidney weight and mesangial expansion. Phosphorylation of STAT3 and renal fibrosis were also significantly reduced, as well macrophage infiltration in perirenal adipose tissue [71]. These findings suggest that both elevated levels of Ang II and decreased levels of Ang-(1-7) may contribute to renal damage [68].
In contrast to experimental studies, limited data were obtained in regard to ACE2 -Ang-(1-7)-Mas axis in patients with diabetic nephropathy [22]. Most studies measured urinary levels of ACE2 in patients with type 2 diabetes [18, 20, 23, 24] and few others investigated mRNA and/or protein expression for ACE2 in human renal tissue [21, 25].
Concerning the studies that measured ACE2 in urine, Park and co-wotkers investigated whether urinary ACE2 levels are associated with abnormal glucosehomeostasis and urinary albumin excretion [23]. The authors found that urinary ACE2 levels were an independent predictor of microalbuminuria afteradjusting for other clinical risk factors in patients with type 2 diabetes [23]. In patients with type 2 diabetes and chronic kidney disease, Abe and co-workers showed that the treatment with the AT1 receptor antagonist olmesartansignificantly increases urinary ACE2 levels independently of blood pressure andplasma aldosterone levels and reduces albuminuria, urinary liver-type fatty acidbinding protein, and plasma aldosterone levels [18]. The authors raised the possibility that increased ACE2 contributes to renoprotection elicited by olmesartan [18]. More recently, Liang and co-workers reported that urinary levels of ACE2 are increased in type 2 diabetic patients with various degrees of albuminuria [20]. Furthermore, the treatment with RAS inhibitors reduced urinary ACE2 excretion [20]. The authors concluded that urinary ACE2 measurement mightpotentially function as a marker for monitoring the metabolic status and therapeutic response to RAS inhibitors in diabetes [20]. Only one study investigated ACE2 in patients with type 1 diabetes and found that urinary ACE2 activity and protein expression are increased prior to the onset of clinical complications [19]. None of these studies have investigated the mechanisms that promote the elevation of ACE2 in the urine of diabetic patients. A possible explanation is that the augmentation of urinary ACE2 levels might be a compensatory mechanism in response to kidney injury in diabetic patients.
In regard to the evaluation of Ang-(1-7) and Mas receptor in diabetic patients, Mizuiri and co-workers reported that the proximal tubules from type 2 diabetic patients with nephropathy exhibited higherexpression of ACE and lower expression of ACE2 , Ang-(1-7) and Mas receptor in comparison to healthy controls and to patients with minimal change nephrotic syndrome [21].
Figure 11.2 displays the effects of the classical and the counter-regulatory RAS axes in diabetic nephropathy .

The role of components of the classical and the counter regulatory renin-angiotensisn system axes in diabetic nephropathy
4 Role of ACE2 -Ang-(1-7)-Mas Receptor Axis in Diabetic Cardiovascular Disease
Diabetes mellitus is associated with substantial risk of heart failure and has been described as the leading cause of morbidity and mortality related with cardiovascular diseases (CVD) worldwide. Diabetic CVD includes myocardial infarction , mainly associated with premature atherosclerosis , and diabetic cardiomyopathy, characterized by left ventricular (LV) remodeling and dysfunction, both leading to heart failure [72, 73]. Indeed, diabetes has been considered not only a risk factor for CVD, but also a cardiovascular event equivalent, since diabetic subjects had a risk of cardiovascular complications similar to patients with previous myocardial infarction [74]. Accordingly, diabetic patients with myocardial infarction have worse prognosis than non-diabetic patients with myocardial infarction [75].
The pathophysiological mechanisms underlying diabetic CVD remain poorly elucidated. The discover that RAS key components are also locally expressed in different organs, including the heart, opens the road for the hypothesis that RAS exerted both hemodynamic and non-hemodynamic effects [76]. In fact, RAS components, including renin, angiotensinogen, ACE and Ang II receptors, were up-regulated in the heart after cardiac injury, volume overload, myocardial infarction , and heart failure [77,78,79,80]. In the context of diabetes, over the past decades, clinical and experimental studies have been linked the classical RAS axis to diabetic CVD pathophysiology. For instance, ACE inhibitors, like perindopril, and AT1 receptor blockers improved cardiovascular morbidity and mortality in patients with diabetes [81, 82] and prevented atherosclerosis and myocardial infarction in diabetic apolipoprotein E-deficient mice and in a streptozotocin-induced diabetes model [83,84,85]. There is evidence that Ang II by binding to its AT1 receptors might mediate cardiovascular damage by inducing reactive oxygen species generation, tissue inflammation , fibrosis, and apoptosis [83, 84, 86,87,88,89].
A more modern concept has been supported that diabetic CVD depends on a balance between both RAS axes, the classical (ACE -Ang II-AT1 receptor) and the counter-regulatory (ACE2 -Ang-(1-7)-Mas receptor ) [50, 90, 91].In line with this view, an elegant study demonstrated a significant reduction in cardiac ACE2 expression and activity along with elevated circulating levels of AngII and reduced Ang-(1-7) concentration in the heart in streptozotocin-induced diabetic mice. The changes in RAS components in response to diabetes induction were associated with a significant cardiovascular damage, which included thinning of the LV wall, mild ventricular dilatation, increased cardiomyocyte apoptosis and compensatory heart hypertrophy [90]. Interestingly, the induction of diabetes by streptozotocin in mice genetically deficient for ACE2 did not change Ang II and Ang-(1-7) concentrations; neither led to cardiovascular dysfunction. Moreover, the absence of ACE2 also prevented the accelerated atherosclerosis found in diabetic apolipoprotein E-deficient mice. Altogether, these findings suggest that ACE2 might be a key factor in RAS activation in diabetic CVD, mainly by regulating cardiac levels of Ang II and of Ang-(1-7) [91]. Accordingly, in a model of human diabetes by employing the Akita mice with the loss of ACE2 expression increased plasma and heart tissue levels of Ang II, leading to systolic dysfunction on a background of impaired diastolic function [91]. The cardiovascular systolic alterations were associated with increased oxidative stress , degradation of the extracellular matrix activation of protein kinase C and loss of Akt and endothelial nitric oxide synthase phosphorylation, all of which prevented by the administration of the AT1 receptor blocker, irbesartan [90].Similarly, diabetes induction by streptozotocin in male Wistar rats resulted in diastolic dysfunction, cardiac hypertrophy and fibrosis along withACE2/ACE ratios imbalance, ERK1/2 phosphorylation and changes in the AMP-activated protein kinases, AMPK-α and AMPK-β1 expression. All these changes were prevented by the oral administration of the ACE2 activator XNT, suggesting that increase in ACE2 activity might be a promise therapy for diabetic CVD [50]. This hypothesis was supported by further studies showing that ACE2 over expression induced by a gene therapy with adenovirus was superior to losartan in attenuating diabetic cardiomyopathyas indicated by a decrease in myocyte hypertrophy, myocardial fibrosis, and LV remodeling and an improvement in LV systolic and diastolic function [92]. A protective effect was also found following an oral administration of the ACE2 activator, diminazene aceturate (DIZE), reflected by the improvement in cardiac electrical function in streptozotocin-induced diabetic rats [93].
Emerging evidence have been supported the idea that the beneficial effects of ACE2 is related with its capacity to convert AngII into Ang-(1-7). For instance, increased plasma levels of Ang-(1-7) were independently associated with a protection of left ventricular function in patients with type 2 diabetes mellitus [94]. Moreover, a growing body of experimental studies showed that the administration of Ang-(1-7) or of the Mas receptor oral agonist, AVE0991, significantly protects against diabetes-induced cardiovascular dysfunction [95,96,97,98,99]. Importantly, the opposite effect was observed with the administration of the Mas receptor antagonist, A779 [95, 98]. In this scenario, the elevation of Ang-(1-7) levels might also represent a promise therapeutic strategy for diabetic CVD.
The mechanisms underlying Ang-(1-7) cardiac protection might rely on the inhibition of inflammation and of oxidative stress by decreasing the transcript factor NF-kB activity and the NADPH oxidase activation, by restoring lipid profile alterations, and by reducing collagen and fibronectin-1 production, and TGF-β1 expression [95, 98, 99]. More recent studies, by employing the db/db mice, a well-established model of type 2 diabetic cardiomyopathy, showed that Ang-(1-7) improves myocardial hypertrophy and fibrosis by decreasing the lipotoxicity and the inflammatory response [100, 101]. Similar findings were reported by an in vitro study showing that Ang-(1-7) protects cardiomyocytes against high glucose-induced injuries by inhibiting the activation of the reactive oxygen species-activated leptin -p38 MAPK /ERK1/2 pathways [102].
It has been also reported that the cardioprotective effects of Ang-(1-7) may result from a complex interaction between AT2 and Mas receptors with a subsequent down-regulation of ACE expression and activity and of AT1receptor expression, as well as up-regulation of ACE2 expression and activity [98, 103]. In addition, an increase in AT2 expression was associated with higher apoptosis rate of cardiomyocytes in diabetic rats [104]. In fact, the exogenous Ang-(1-7) significantly increased myocardial ACE2 activity and Ang-(1–9) levels, possibly via its effect on AT2 receptor. The increased activity of ACE2 leads to higher conversion rate of Ang II into Ang-(1-7), thus forming a positive feedback that elevates Ang-(1-7) levels, which, in turn, produce protective effects in diabetes-induced CVD [98].
Figure 11.3 shows the role of the classical and the counter-regulatory RAS axes in heart alterations of diabetes.

The role of components of the classical and the counter regulatory renin-angiotensisn system axes in heart alterations in diabetes
5 Conclusion
Despite available treatments for diabetes, a substantial population is still suffering from renal injury, cardiovascular alterations and other associated comorbidities. The inhibition of the classical RAS axis with angiotensin receptor blockers and/or ACE inhibitors is not effective for all cases and, in such conditions, we may speculate the usefulness of therapies to activate the counter-regulatory RAS axis. Therefore, different ways to activate ACE2 -Ang-(1-7)-Mas receptor axis emerge as a promise therapeutic strategy for diabetes and its co-morbidities.
References
Gallagher H, Suckling RJ (2016) Diabetic nephropathy: where are we on the journey from pathophysiology to treatment? Diabetes Obes Metab 18(7):641–647. doi:10.1111/dom.12630
Johnson SA, Spurney RF (2015) Twenty years after ACEIs and ARBs: emerging treatment strategies for diabetic nephropathy. Am J Physiol Ren Physiol 309(10):F807–F820. doi:10.1152/ajprenal.00266.2015
Ribeiro-Oliveira A Jr, Nogueira AI, Pereira RM, Boas WW, Dos Santos RA, Simoes e Silva AC (2008) The renin-angiotensin system and diabetes: an update. Vasc Health Risk Manag 4(4):787–803
Bojestig M, Nystrom FH, Arnqvist HJ, Ludvigsson J, Karlberg BE (2000) The renin-angiotensin-aldosterone system is suppressed in adults with type 1 diabetes. J Renin-Angiotensin-Aldosterone Syst 1(4):353–356. doi:10.3317/jraas.2000.065
Hollenberg NK, Price DA, Fisher ND, Lansang MC, Perkins B, Gordon MS, Williams GH, Laffel LM (2003) Glomerular hemodynamics and the renin-angiotensin system in patients with type 1 diabetes mellitus. Kidney Int 63(1):172–178. doi:10.1046/j.1523-1755.2003.00701.x
Stevanovic RD, Price DA, Lansang MC, Fisher NDL, Laffel LMB, Hollenberg NK (2005) Renin release in response to renin system blockade: activation of the renin system in type 1 diabetes mellitus. J Renin-Angiotensin-Aldosterone Syst 6(2):78–83. doi:10.3317/jraas.2005.013
Carey RM, Siragy HM (2003) The intrarenal renin-angiotensin system and diabetic nephropathy. Trends Endocrinol Metab: TEM 14(6):274–281
Simoes ESAC, Flynn JT (2012) The renin-angiotensin-aldosterone system in 2011: role in hypertension and chronic kidney disease. Pediatr Nephrol (Berlin, Germany) 27(10):1835–1845. doi:10.1007/s00467-011-2002-y
Bernardi S, Michelli A, Zuolo G, Candido R, Fabris B (2016) Update on RAAS modulation for the treatment of diabetic cardiovascular disease. J Diabetes Res 2016:8917578. doi:10.1155/2016/8917578
Roscioni SS, Heerspink HJ, de Zeeuw D (2014) The effect of RAAS blockade on the progression of diabetic nephropathy. Nat Rev Nephrol 10(2):77–87. doi:10.1038/nrneph.2013.251
Simoes ESAC, Teixeira MM (2016) ACE inhibition, ACE2 and angiotensin-(1-7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol Res 107:154–162. doi:10.1016/j.phrs.2016.03.018
Horiuchi M, Iwanami J, Mogi M (2012) Regulation of angiotensin II receptors beyond the classical pathway. Clin Sci (Lond) 123(4):193–203. doi:10.1042/cs20110677
Prestes TR, Rocha NP, Miranda AS, Teixeira AL, Simoes ESAC (2016) The anti-inflammatory potential of ACE2/angiotensin-(1-7)/mas receptor axis: evidence from basic and clinical research. Curr Drug Targets
Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM (2004) Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J 383(Pt 1):45–51. doi:10.1042/bj20040634
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 87(5):E1–E9
Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ (2000) A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275(43):33238–33243. doi:10.1074/jbc.M002615200
Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SV, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, Walther T (2003) Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A 100(14):8258–8263. doi:10.1073/pnas.1432869100
Abe M, Oikawa O, Okada K, Soma M (2015) Urinary angiotensin-converting enzyme 2 increases in diabetic nephropathy by angiotensin II type 1 receptor blocker olmesartan. J Renin-Angiotensin-Aldosterone Syst: JRAAS 16(1):159–164. doi:10.1177/1470320314551443
Cherney DZI, Xiao F, Zimpelmann J, Har RLH, Lai V, Scholey JW, Reich HN, Burns KD (2014) Urinary ACE2 in healthy adults and patients with uncomplicated type 1 diabetes. Can J Physiol Pharmacol 92(8):703–706. doi:10.1139/cjpp-2014-0065
Liang Y, Deng H, Bi S, Cui ZAL, Zheng D, Wang Y (2015) Urinary angiotensin converting enzyme 2 increases in patients with type 2 diabetic mellitus. Kidney Blood Press Res 40(2):101–110. doi:10.1159/000368486
Mizuiri S, Nishizawa Y, Hamanoue M, Hemmi H, Arita M, Shibuya K, Aoki T, Ohashi Y, Sakai K, Aikawa A (2012) ACE2- Ang 1-7-MAS axis in human diabetic nephropathy. J Nephrol Ther S2 005:6. doi:10.4172/2161-0959.S2-005
Nogueira AI, Souza Santos RA, Simoes ESAC, Cabral AC, Vieira RL, Drumond TC, Machado LJ, Freire CM, Ribeiro-Oliveira A Jr (2007) The pregnancy-induced increase of plasma angiotensin-(1-7) is blunted in gestational diabetes. Regul Pept 141(1–3):55–60. doi:10.1016/j.regpep.2006.12.014
Park SE, Kim WJ, Park SW, Park JW, Lee N, Park CY, Youn BS (2013) High urinary ACE2 concentrations are associated with severity of glucose intolerance and microalbuminuria. Eur J Endocrinol 168(2):203–210. doi:10.1530/eje-12-0782
Soro-Paavonen A, Gordin D, Forsblom C, Rosengard-Barlund M, Waden J, Thorn L, Sandholm N, Thomas MC, Groop PH (2012) Circulating ACE2 activity is increased in patients with type 1 diabetes and vascular complications. J Hypertens 30(2):375–383. doi:10.1097/HJH.0b013e32834f04b6
Wang G, Lai FM, Lai KB, Chow KM, Kwan CH, Li KT, Szeto CC (2009) Discrepancy between intrarenal messenger RNA and protein expression of ACE and ACE2 in human diabetic nephropathy. Am J Nephrol 29(6):524–531. doi:10.1159/000185629
Lau T, Carlsson PO, Leung PS (2004) Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia 47(2):240–248. doi:10.1007/s00125-003-1295-1
Madec AM, Cassel R, Dubois S, Ducreux S, Vial G, Chauvin MA, Mesnier A, Chikh K, Bosco D, Rieusset J, Van Coppenolle F, Thivolet C (2013) Losartan, an angiotensin II type 1 receptor blocker, protects human islets from glucotoxicity through the phospholipase C pathway. FASEB J: Off Publ Fed Am Soc Exp Biol 27(12):5122–5130. doi:10.1096/fj.13-234104
Wang HW, Mizuta M, Saitoh Y, Noma K, Ueno H, Nakazato M (2011) Glucagon-like peptide-1 and candesartan additively improve glucolipotoxicity in pancreatic beta-cells. Metab Clin Exp 60(8):1081–1089. doi:10.1016/j.metabol.2010.11.004
Wei Y, Sowers JR, Clark SE, Li W, Ferrario CM, Stump CS (2008) Angiotensin II-induced skeletal muscle insulin resistance mediated by NF-kappaB activation via NADPH oxidase. Am J Phys Endocrinol Metab 294(2):E345–E351. doi:10.1152/ajpendo.00456.2007
Bokhari S, Israelian Z, Schmidt J, Brinton E, Meyer C (2007) Effects of angiotensin II type 1 receptor blockade on beta-cell function in humans. Diabetes Care 30(1):181. doi:10.2337/dc06-1745
Lindholm LH, Ibsen H, Dahlof B, Devereux RB, Beevers G, de Faire U, Fyhrquist F, Julius S, Kjeldsen SE, Kristiansson K, Lederballe-Pedersen O, Nieminen MS, Omvik P, Oparil S, Wedel H, Aurup P, Edelman J, Snapinn S (2002) Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet (London, England) 359(9311):1004–1010. doi:10.1016/s0140-6736(02)08090-x
Chhabra KH, Chodavarapu H, Lazartigues E (2013) Angiotensin converting enzyme 2: a new important player in the regulation of glycemia. IUBMB Life 65(9):731–738. doi:10.1002/iub.1190
Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P (2002) Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 277(17):14838–14843. doi:10.1074/jbc.M200581200
Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, Ferrario CM, Raizada MK (2005) Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp Physiol 90(5):783–790. doi:10.1113/expphysiol.2005.031096
Zhao YX, Yin HQ, Yu QT, Qiao Y, Dai HY, Zhang MX, Zhang L, Liu YF, Wang LC, Liu DS, Deng BP, Zhang YH, Pan CM, Song HD, Qu X, Jiang H, Liu CX, Lu XT, Liu B, Gao F, Dong B (2010) ACE2 overexpression ameliorates left ventricular remodeling and dysfunction in a rat model of myocardial infarction. Hum Gene Ther 21(11):1545–1554. doi:10.1089/hum.2009.160
Bindom SM, Hans CP, Xia H, Boulares AH, Lazartigues E (2010) Angiotensin I-converting enzyme type 2 (ACE2) gene therapy improves glycemic control in diabetic mice. Diabetes 59(10):2540–2548. doi:10.2337/db09-0782
Chhabra KH, Xia H, Pedersen KB, Speth RC, Lazartigues E (2013) Pancreatic angiotensin-converting enzyme 2 improves glycemia in angiotensin II-infused mice. Am J Phys Endocrinol Metab 304(8):E874–E884. doi:10.1152/ajpendo.00490.2012
Feng Y, Yue X, Xia H, Bindom SM, Hickman PJ, Filipeanu CM, Wu G, Lazartigues E (2008) Angiotensin-converting enzyme 2 overexpression in the subfornical organ prevents the angiotensin II-mediated pressor and drinking responses and is associated with angiotensin II type 1 receptor downregulation. Circ Res 102(6):729–736. doi:10.1161/circresaha.107.169110
Giani JF, Mayer MA, Munoz MC, Silberman EA, Hocht C, Taira CA, Gironacci MM, Turyn D, Dominici FP (2009) Chronic infusion of angiotensin-(1-7) improves insulin resistance and hypertension induced by a high-fructose diet in rats. Am J Phys Endocrinol Metab 296(2):E262–E271. doi:10.1152/ajpendo.90678.2008
Prasannarong M, Santos FR, Henriksen EJ (2012) ANG-(1-7) reduces ANG II-induced insulin resistance by enhancing Akt phosphorylation via a Mas receptor-dependent mechanism in rat skeletal muscle. Biochem Biophys Res Commun 426(3):369–373. doi:10.1016/j.bbrc.2012.08.093
Santos SH, Fernandes LR, Mario EG, Ferreira AV, Porto LC, Alvarez-Leite JI, Botion LM, Bader M, Alenina N, Santos RA (2008) Mas deficiency in FVB/N mice produces marked changes in lipid and glycemic metabolism. Diabetes 57(2):340–347. doi:10.2337/db07-0953
Santos SH, Braga JF, Mario EG, Porto LC, Rodrigues-Machado Mda G, Murari A, Botion LM, Alenina N, Bader M, Santos RA (2010) Improved lipid and glucose metabolism in transgenic rats with increased circulating angiotensin-(1-7). Arterioscler Thromb Vasc Biol 30(5):953–961. doi:10.1161/atvbaha.109.200493
Niu MJ, Yang JK, Lin SS, Ji XJ, Guo LM (2008) Loss of angiotensin-converting enzyme 2 leads to impaired glucose homeostasis in mice. Endocrine 34(1–3):56–61. doi:10.1007/s12020-008-9110-x
Takeda M, Yamamoto K, Takemura Y, Takeshita H, Hongyo K, Kawai T, Hanasaki-Yamamoto H, Oguro R, Takami Y, Tatara Y, Takeya Y, Sugimoto K, Kamide K, Ohishi M, Rakugi H (2013) Loss of ACE2 exaggerates high-calorie diet-induced insulin resistance by reduction of GLUT4 in mice. Diabetes 62(1):223–233. doi:10.2337/db12-0177
Hardtner C, Morke C, Walther R, Wolke C, Lendeckel U (2013) High glucose activates the alternative ACE2/Ang-(1-7)/Mas and APN/Ang IV/IRAP RAS axes in pancreatic beta-cells. Int J Mol Med 32(4):795–804. doi:10.3892/ijmm.2013.1469
Tikellis C, Johnston CI, Forbes JM, Burns WC, Burrell LM, Risvanis J, Cooper ME (2003) Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension (Dallas, Tex: 1979) 41(3):392–397. doi:10.1161/01.hyp.0000060689.38912.cb
Mecca AP, O’Connor TE, Dooies KA, Katovich MJ, Sumners C (2009) Cerebroprotective action of angiotensin 1-7 in a rat model of ischemic stroke. FASEB J 23(1 Supplement):947.941
Bindom SM, Lazartigues E (2009) The sweeter side of ACE2: physiological evidence for a role in diabetes. Mol Cell Endocrinol 302(2):193–202. doi:10.1016/j.mce.2008.09.020
Jarajapu YP, Bhatwadekar AD, Caballero S, Hazra S, Shenoy V, Medina R, Kent D, Stitt AW, Thut C, Finney EM, Raizada MK, Grant MB (2013) Activation of the ACE2/angiotensin-(1-7)/Mas receptor axis enhances the reparative function of dysfunctional diabetic endothelial progenitors. Diabetes 62(4):1258–1269. doi:10.2337/db12-0808
Murca TM, Moraes PL, Capuruco CA, Santos SH, Melo MB, Santos RA, Shenoy V, Katovich MJ, Raizada MK, Ferreira AJ (2012) Oral administration of an angiotensin-converting enzyme 2 activator ameliorates diabetes-induced cardiac dysfunction. Regul Pept 177(1–3):107–115. doi:10.1016/j.regpep.2012.05.093
Harmon JS, Stein R, Robertson RP (2005) Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem 280(12):11107–11113. doi:10.1074/jbc.M410345200
Ihara Y, Toyokuni S, Uchida K, Odaka H, Tanaka T, Ikeda H, Hiai H, Seino Y, Yamada Y (1999) Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes 48(4):927–932
Hitomi H, Kiyomoto H, Nishiyama A (2007) Angiotensin II and oxidative stress. Curr Opin Cardiol 22(4):311–315. doi:10.1097/HCO.0b013e3281532b53
Gwathmey TM, Pendergrass KD, Reid SD, Rose JC, Diz DI, Chappell MC (2010) Angiotensin-(1-7)-angiotensin-converting enzyme 2 attenuates reactive oxygen species formation to angiotensin II within the cell nucleus. Hypertension (Dallas, Tex: 1979) 55(1):166–171. doi:10.1161/hypertensionaha.109.141622
Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A, Ehses JA (2008) Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care 31(Suppl 2):S161–S164. doi:10.2337/dc08-s243
Hayden MR, Sowers JR (2007) Isletopathy in type 2 diabetes mellitus: implications of islet RAS, islet fibrosis, islet amyloid, remodeling, and oxidative stress. Antioxid Redox Signal 9(7):891–910. doi:10.1089/ars.2007.1610
Negi S, Park SH, Jetha A, Aikin R, Tremblay M, Paraskevas S (2012) Evidence of endoplasmic reticulum stress mediating cell death in transplanted human islets. Cell Transplant 21(5):889–900. doi:10.3727/096368911x603639
Liu Z, Huang XR, Chen HY, Penninger JM, Lan HY (2012) Loss of angiotensin-converting enzyme 2 enhances TGF-beta/Smad-mediated renal fibrosis and NF-kappaB-driven renal inflammation in a mouse model of obstructive nephropathy. Lab Invest; J Tech Methods Pathol 92(5):650–661. doi:10.1038/labinvest.2012.2
Zhong J, Guo D, Chen CB, Wang W, Schuster M, Loibner H, Penninger JM, Scholey JW, Kassiri Z, Oudit GY (2011) Prevention of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension (Dallas, Tex: 1979) 57(2):314–322. doi:10.1161/hypertensionaha.110.164244
Alghamri MS, Weir NM, Anstadt MP, Elased KM, Gurley SB, Morris M (2013) Enhanced angiotensin II-induced cardiac and aortic remodeling in ACE2 knockout mice. J Cardiovasc Pharmacol Ther 18(2):138–151. doi:10.1177/1074248412460124
Feng Y, Hans C, McIlwain E, Varner KJ, Lazartigues E (2012) Angiotensin-converting enzyme 2 over-expression in the central nervous system reduces angiotensin-II-mediated cardiac hypertrophy. PLoS One 7(11):e48910. doi:10.1371/journal.pone.0048910
Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Diez-Freire C, Dooies A, Jun JY, Sriramula S, Mariappan N, Pourang D, Venugopal CS, Francis J, Reudelhuber T, Santos RA, Patel JM, Raizada MK, Katovich MJ (2010) The angiotensin-converting enzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med 182(8):1065–1072. doi:10.1164/rccm.200912-1840OC
Chodavarapu H, Chhabra K, Shenoy V, Raizada MK, Yue X, Lazartigues E (2013) ACE2 gene therapy decreases fibrosis in the pancreas of high fat diet-fed mice. FASEB J 27(1 Supplement):1154.1157
Santos SH, Giani JF, Burghi V, Miquet JG, Qadri F, Braga JF, Todiras M, Kotnik K, Alenina N, Dominici FP, Santos RA, Bader M (2014) Oral administration of angiotensin-(1-7) ameliorates type 2 diabetes in rats. J Mol Med (Berlin, Germany) 92(3):255–265. doi:10.1007/s00109-013-1087-0
Pofi R, Di Mario F, Gigante A, Rosato E, Isidori AM, Amoroso A, Cianci R, Barbano B (2016) Diabetic nephropathy: focus on current and future therapeutic strategies. Curr Drug Metab 17(5):497–502
Márquez E, Riera M, Pascual J, Soler MJ (2015) Renin-angiotensin system within the diabetic podocyte. Am J Physiol Ren Physiol 308(1):F1
Wong DW, Oudit GY, Reich H, Kassiri Z, Zhou J, Liu QC, Backx PH, Penninger JM, Herzenberg AM, Scholey JW (2007) Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am J Pathol 171(2):438–451. doi:10.2353/ajpath.2007.060977
Marquez E, Riera M, Pascual J, Soler MJ (2014) Albumin inhibits the insulin-mediated ACE2 increase in cultured podocytes. Am J Physiol Ren Physiol 306(11):F1327–F1334. doi:10.1152/ajprenal.00594.2013
Riera M, Marquez E, Clotet S, Gimeno J, Roca-Ho H, Lloreta J, Juanpere N, Batlle D, Pascual J, Soler MJ (2014) Effect of insulin on ACE2 activity and kidney function in the non-obese diabetic mouse. PLoS One 9(1):e84683. doi:10.1371/journal.pone.0084683
Giani JF, Burghi V, Veiras LC, Tomat A, Munoz MC, Cao G, Turyn D, Toblli JE, Dominici FP (2012) Angiotensin-(1-7) attenuates diabetic nephropathy in Zucker diabetic fatty rats. Am J Physiol Ren Physiol 302(12):F1606–F1615. doi:10.1152/ajprenal.00063.2012
Mori J, Patel VB, Ramprasath T, Alrob OA, Desaulniers J, Scholey JW, Lopaschuk GD, Oudit GY (2014) Angiotensin 1-7 mediates renoprotection against diabetic nephropathy by reducing oxidative stress, inflammation, and lipotoxicity. Am J Physiol Ren Physiol 306(8):F812–F821. doi:10.1152/ajprenal.00655.2013
Ginter E, Simko V (2012) Type 2 diabetes mellitus, pandemic in 21st century. Adv Exp Med Biol 771:42–50
Nichols GA, Joshua-Gotlib S, Parasuraman S (2013) Glycemic control and risk of cardiovascular disease hospitalization and all-cause mortality. J Am Coll Cardiol 62(2):121–127. doi:10.1016/j.jacc.2013.04.031
Schramm TK, Gislason GH, Kober L, Rasmussen S, Rasmussen JN, Abildstrom SZ, Hansen ML, Folke F, Buch P, Madsen M, Vaag A, Torp-Pedersen C (2008) Diabetes patients requiring glucose-lowering therapy and nondiabetics with a prior myocardial infarction carry the same cardiovascular risk: a population study of 3.3 million people. Circulation 117(15):1945–1954. doi:10.1161/circulationaha.107.720847
Miettinen H, Lehto S, Salomaa V, Mahonen M, Niemela M, Haffner SM, Pyorala K, Tuomilehto J (1998) Impact of diabetes on mortality after the first myocardial infarction. The FINMONICA Myocardial Infarction Register Study Group. Diabetes Care 21(1):69–75
Johnston CI (1992) Franz Volhard Lecture. Renin-angiotensin system: a dual tissue and hormonal system for cardiovascular control. J Hypertens Suppl: Off J Int Soc Hypertens 10(7):S13–S26
Hokimoto S, Yasue H, Fujimoto K, Yamamoto H, Nakao K, Kaikita K, Sakata R, Miyamoto E (1996) Expression of angiotensin-converting enzyme in remaining viable myocytes of human ventricles after myocardial infarction. Circulation 94(7):1513–1518
Pieruzzi F, Abassi ZA, Keiser HR (1995) Expression of renin-angiotensin system components in the heart, kidneys, and lungs of rats with experimental heart failure. Circulation 92(10):3105–3112
Schunkert H, Dzau VJ, Tang SS, Hirsch AT, Apstein CS, Lorell BH (1990) Increased rat cardiac angiotensin converting enzyme activity and mRNA expression in pressure overload left ventricular hypertrophy. Effects on coronary resistance, contractility, and relaxation. J Clin Invest 86(6):1913–1920. doi:10.1172/jci114924
Wollert KC, Drexler H (1999) The renin-angiotensin system and experimental heart failure. Cardiovasc Res 43(4):838–849
Daly CA, Fox KM, Remme WJ, Bertrand ME, Ferrari R, Simoons ML (2005) The effect of perindopril on cardiovascular morbidity and mortality in patients with diabetes in the EUROPA study: results from the PERSUADE substudy. Eur Heart J 26(14):1369–1378. doi:10.1093/eurheartj/ehi225
Patel A, MacMahon S, Chalmers J, Neal B, Woodward M, Billot L, Harrap S, Poulter N, Marre M, Cooper M, Glasziou P, Grobbee DE, Hamet P, Heller S, Liu LS, Mancia G, Mogensen CE, Pan CY, Rodgers A, Williams B (2007) Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet (London, England) 370(9590):829–840. doi:10.1016/s0140-6736(07)61303-8
Candido R, Allen TJ, Lassila M, Cao Z, Thallas V, Cooper ME, Jandeleit-Dahm KA (2004) Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation 109(12):1536–1542. doi:10.1161/01.cir.0000124061.78478.94
Candido R, Jandeleit-Dahm KA, Cao Z, Nesteroff SP, Burns WC, Twigg SM, Dilley RJ, Cooper ME, Allen TJ (2002) Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 106(2):246–253
Matsusaka H, Kinugawa S, Ide T, Matsushima S, Shiomi T, Kubota T, Sunagawa K, Tsutsui H (2006) Angiotensin II type 1 receptor blocker attenuates exacerbated left ventricular remodeling and failure in diabetes-associated myocardial infarction. J Cardiovasc Pharmacol 48(3):95–102. doi:10.1097/01.fjc.0000245405.41317.60
Fabris B, Candido R, Bortoletto M, Zentilin L, Sandri M, Fior F, Toffoli B, Stebel M, Bardelli M, Belgrado D, Giacca M, Carretta R (2007) Dose and time-dependent apoptotic effects by angiotensin II infusion on left ventricular cardiomyocytes. J Hypertens 25(7):1481–1490. doi:10.1097/HJH.0b013e328121aae7
Kumar R, Yong QC, Thomas CM, Baker KM (2012) Review: intracardiac intracellular angiotensin system in diabetes. Am J Phys Regul Integr Comp Phys 302(5):R510–R517. doi:10.1152/ajpregu.00512.2011
Schnee JM, Hsueh WA (2000) Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc Res 46(2):264–268
Thomas MC, Pickering RJ, Tsorotes D, Koitka A, Sheehy K, Bernardi S, Toffoli B, Nguyen-Huu TP, Head GA, Fu Y, Chin-Dusting J, Cooper ME, Tikellis C (2010) Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ Res 107(7):888–897. doi:10.1161/circresaha.110.219279
Patel VB, Bodiga S, Basu R, Das SK, Wang W, Wang Z, Lo J, Grant MB, Zhong J, Kassiri Z, Oudit GY (2012) Loss of angiotensin-converting enzyme-2 exacerbates diabetic cardiovascular complications and leads to systolic and vascular dysfunction: a critical role of the angiotensin II/AT1 receptor axis. Circ Res 110(10):1322–1335. doi:10.1161/circresaha.112.268029
Tikellis C, Pickering R, Tsorotes D, Du XJ, Kiriazis H, Nguyen-Huu TP, Head GA, Cooper ME, Thomas MC (2012) Interaction of diabetes and ACE2 in the pathogenesis of cardiovascular disease in experimental diabetes. Clin Sci (Lond) 123(8):519–529. doi:10.1042/cs20110668
Dong B, Yu QT, Dai HY, Gao YY, Zhou ZL, Zhang L, Jiang H, Gao F, Li SY, Zhang YH, Bian HJ, Liu CX, Wang N, Xu H, Pan CM, Song HD, Zhang C, Zhang Y (2012) Angiotensin-converting enzyme-2 overexpression improves left ventricular remodeling and function in a rat model of diabetic cardiomyopathy. J Am Coll Cardiol 59(8):739–747. doi:10.1016/j.jacc.2011.09.071
Coutinho DC, Monnerat-Cahli G, Ferreira AJ, Medei E (2014) Activation of angiotensin-converting enzyme 2 improves cardiac electrical changes in ventricular repolarization in streptozotocin-induced hyperglycaemic rats. Europace: Eur Pacing, Arrhythmias, Card Electrophysiol: J Work Groups Card Pacing, Arrhythmias, Cardiac Cell Electrophysiol Eur Soc Cardiol 16(11):1689–1696. doi:10.1093/europace/euu070
Hao PP, Chen YG, Liu YP, Zhang MX, Yang JM, Gao F, Zhang Y, Zhang C (2013) Association of plasma angiotensin-(1-7) level and left ventricular function in patients with type 2 diabetes mellitus. PLoS One 8(5):e62788. doi:10.1371/journal.pone.0062788
Al-Maghrebi M, Benter IF, Diz DI (2009) Endogenous angiotensin-(1-7) reduces cardiac ischemia-induced dysfunction in diabetic hypertensive rats. Pharmacol Res 59(4):263–268. doi:10.1016/j.phrs.2008.12.008
Benter IF, Yousif MH, Cojocel C, Al-Maghrebi M, Diz DI (2007) Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. Am J Physiol Heart Circ Physiol 292(1):H666–H672. doi:10.1152/ajpheart.00372.2006
Ebermann L, Spillmann F, Sidiropoulos M, Escher F, Heringer-Walther S, Schultheiss HP, Tschope C, Walther T (2008) The angiotensin-(1-7) receptor agonist AVE0991 is cardioprotective in diabetic rats. Eur J Pharmacol 590(1–3):276–280. doi:10.1016/j.ejphar.2008.05.024
Hao P, Yang J, Liu Y, Zhang M, Zhang K, Gao F, Chen Y, Zhang C, Zhang Y (2015) Combination of angiotensin-(1-7) with perindopril is better than single therapy in ameliorating diabetic cardiomyopathy. Sci Rep 5:8794. doi:10.1038/srep08794
Singh K, Singh T, Sharma PL (2011) Beneficial effects of angiotensin (1-7) in diabetic rats with cardiomyopathy. Ther Adv Cardiovasc Dis 5(3):159–167. doi:10.1177/1753944711409281
Mori J, Patel VB, Abo Alrob O, Basu R, Altamimi T, Desaulniers J, Wagg CS, Kassiri Z, Lopaschuk GD, Oudit GY (2014) Angiotensin 1-7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ Heart Fail 7(2):327–339. doi:10.1161/circheartfailure.113.000672
Papinska AM, Mordwinkin NM, Meeks CJ, Jadhav SS, Rodgers KE (2015) Angiotensin-(1-7) administration benefits cardiac, renal and progenitor cell function in db/db mice. Br J Pharmacol. doi:10.1111/bph.13225
Lei Y, Xu Q, Zeng B, Zhang W, Zhen Y, Zhai Y, Cheng F, Mei W, Zheng D, Feng J, Lan J, Chen J (2016) Ang-(1-7) protects cardiomyocytes against HG-induced injuries through inhibiting ROS-activated leptin-p38 MAPK/ERK1/2 pathways but not leptin-JNK pathway in vitro. J Diabetes Investig:n/a-n/a. doi:10.1111/jdi.12603
Hao PP, Yang JM, Zhang MX, Zhang K, Chen YG, Zhang C, Zhang Y (2015) Angiotensin-(1-7) treatment mitigates right ventricular fibrosis as a distinctive feature of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 308(9):H1007–H1019. doi:10.1152/ajpheart.00563.2014
Li C, Cao L, Zeng Q, Liu X, Zhang Y, Dai T, Hu D, Huang K, Wang Y, Wang X, Li D, Chen Z, Zhang J, Li Y, Sharma R (2005) Taurine may prevent diabetic rats from developing cardiomyopathy also by downregulating angiotensin II type2 receptor expression. Cardiovasc Drugs Ther 19(2):105–112. doi:10.1007/s10557-005-0443-x
Acknowledgements
This study was partially supported by CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior), CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil – Grant # 470472/2014-6 and Grant # 460334/2014-0) and FAPEMIG (Fundação deAmparo à Pesquisa do Estado de Minas Gerais, Brazil -Grant # 00555-15). Dr. AC Simões e Silva also received a research productivity grant from CNPq.
Conflict of Interest
None declared
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Simões e Silva, A.C., Ferreira, R.N., Miranda, A.S. (2017). The Renin Angiotensin System and Diabetes. In: Kartha, C., Ramachandran, S., Pillai, R. (eds) Mechanisms of Vascular Defects in Diabetes Mellitus. Advances in Biochemistry in Health and Disease, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-319-60324-7_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-60324-7_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-60323-0
Online ISBN: 978-3-319-60324-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)