
Abstract
The renin–angiotensin–aldosterone system (RAAS) plays a key role in the pathological process of many cardiovascular diseases, including hypertension. Angiotensin II (Ang II) and its two receptors, Ang II type 1 receptor (AT1R) and Ang II type 2 receptor (AT2R), exert many physiological and pathophysiological effects. Lectin-like oxidized low-density lipoprotein (LDL) receptor-1 (LOX-1) is responsile for the uptake and degradation of oxidized low density lipoprotein (ox-LDL). The activation of LOX-1 is involved in various cardiovascular diseases including hypertension. The interplay of LOX-1 and AT1R or AT2R has been recently shown to play important part in hypertension and the related cardiovascular diseases such myocardial infarction and stroke. There is increasing evidence that the immune system is a critical mediator in these physiopathological processes. This chapter reviews the role of the immune system in the crosstalk between RAAS and LOX-1 in the genesis of hypertension and the related pathological consequences.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Renin–angiotensin–aldosterone system
- Hypertension
- Angiotensin II
- Ang II type 1 receptor (AT1R)
- Ang II type 2 receptor (AT2R)
- Lectin-like oxidized low-density lipoprotein (LDL) receptor-1 (LOX-1)
Introduction
Hypertension is one of the most important risk factors for the development of cardiovascular diseases including myocardial infarction (MI), ischemic stroke, chronic heart failure, and atherosclerosis. The RAAS is also a major regulator of blood pressure and salt-water homeostasis [1,2,3,4,5]. Ang II and its receptors are critical for mediating vasoconstrictor responses, excitation of peripheral sympathetic nerve and release of arginine vasopressin. Ang II induces its downstream signaling by activating two of its major G protein coupled receptors, AT1R and AT2R [6,7,8]. The subsequent G protein-mediated signaling induces most of the effects of RAAS. These two receptors have distinct functions in both physiological and pathophysiological regulation of blood pressure. Ang II-AT1R complex induces several molecular and cellular events while the activation of AT2R has the opposite effects.
AT1R is evolutionarily expressed in various species including humans, rats, mice, dogs and pigs. Its relatively high expression has been found in heart, kidney, blood vessels, liver, lung, brain, gastric mucosa, adrenal gland, ovine, placenta and adipose tissues. Among the cell types, AT1R is expressed in cardiomyocytes, endothelial cells, vascular smooth muscle cells (VSMCs), monocytes, fibroblasts, neurons, embryonic stem cells, intestinal epithelial cells, T lymphocytes and podocytes.
AT1R expression has been determined to be affected in many pathological conditions, including coronary atherosclerotic plaque, myocardial infarct and peri-infarct regions, failing heart, hypertensive heart, several tissues in type 2 diabetics as well as in various tumor tissues [9].
The human AT1R gene is located in chromosome 3q21-3q25 and its encoded protein is a 359 amino acids protein with a 41 KD molecular mass. The protein belongs to G protein-coupled receptor (GPCR) protein superfamily with extracellular N-terminus, seven-transmembrane domains and intracellular C-terminus. The serine/threonine and tyrosine-rich residues located in the intracellular C terminus are the phosphorylation sites of protein kinase C (PKC). Similar to other GPCRs, AT1R is also a protein that initiates downstream signaling pathway by interacting with different adaptor G protein, Gq/11, G12/13, and Gi/o. The activated Gq/11 mainly induces the activation PLC-β and hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) on the plasma membrane. The generation of the second messenger inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) are subsequently induced, leading to an increase of cytosolic calcium concentration. The increased intracellular calcium subsequently causes the activation of PKC dependent signaling pathway [10, 11]. In cardiomyocytes, calcium-loaded calmodulin (CaM) induces the activation of Calcineurin (phosphatase-2B), which then enhances the expression of hypertrophic markers such as atrial natriuretic peptide (ANP), β-myosin heavy chain (β-MHC) and α-skeletal muscle (α-SKA) [12, 13]. G12/13 mainly triggers the activation of PLC and Rho kinases. Rho kinases are involved in various cellular functions including cell morphology, polarity, cytoskeletal remodeling, cell–cell adhesion, cell proliferation and cell migration. Rac, a member of the Rho family, is a component of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex which is involved in the production of reactive oxygen species (ROS). ROS, in turn, enhance NAPDH oxidase activity through RhoA and Rac [12, 14] (Fig. 1.1).

Activation of AT1R results in the induction of PKC-PI3K and ROS-MAPKs with subsequently production of cytokines and transcription factors. Activation of AT2R has the opposite effect by promoting the activation of eNOS, MKP-1, SHP-1 and PP2A thus opposing the effects of AT1R-mediated signaling. Ox-LDL and LOX-1 interaction leads to the activation of PKC-PI3K and ROS-MAPKs. All the above signaling pathways facilitate cellular functions including cell proliferation, differentiation, apoptosis, migration, adhesion and inflammation which are critical in the pathogenesis of different cardiovascular diseases such as hypertension, atherosclerosis and heart failure
Similar to the calcium signal, ROS act as potent secondary messengers in G12/13- and Gq/11-induced signaling pathways. Several studies have shown that AT1R-mediated ROS production is essential for its downstream signaling, mitogen-activated protein kinases (MAPKs) such as p38 MAPK, P42/44 MAPK and JNK [15, 16] (Fig. 1.1). MAPKs are important signals in ATR-mediated cell functions such as cell proliferation, differentiation, apoptosis and inflammation [17]. In addition to Ang II-AT1R-G12/13-Rho/Rac-ROS-JNK/p38MAPK pathway, Ang II-AT1R–Gq–Ca2+/PKC-PI3K-Akt pathway is another primary signaling pathway in Ang II-AT1R-mediated cell activities. The activation of PKC induced by Gq/11 activates PI3 Kinase (PI3K) and its downstream effector Akt. This PKC-PI3K-Akt pathway is often involved in many pathological conditions including hypertension, heart failure, ischemic stroke, diabetes and inflammation [18,19,20,21] (Fig. 1.1). Gi/o proteins inhibit adenylyl cyclase and suppress the activation of PKA in some specific tissues.
Besides its primary effect in regulation of systemic blood pressure and salt-water balance, increasing evidence suggests that Ang II-AT1R complex participates in various immune responses. AT1R activation facilitates the release of various inflammatory cytokines such as Interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor-α (TNF-α), and initiates inflammatory response. Expression of matrix-metalloprotease 2 (MMP2) in vascular cells plays a role in the attraction and recruitment of monocytes and T cells.
TNF-α increases MMP2 expression in vascular cells and plays a pivotal role in the recruitment of infiltrating inflammatory cells such as T cells and monocytes. The differentiation process of monocyte-derived macrophages is regulated by Ang II-induced cytokines [22, 23]. Ang II-induced inflammatory responses and vascular remodeling, both contribute to endothelial dysfunction and further promotes the formation of arteriosclerotic plaques and abdominal aortic aneurisms [24,25,26,27].
AT2R is an alternative component of Ang II-mediated downstream physiological functions. The receptor also belongs to seven transmembrane GPCRs. AT2R is generally expressed in low levels in a variety of organs such as heart, brain, kidney, pancreas and skin [28,29,30,31,32,33,34]. Similar to AT1R, AT2R expression is also enhanced in various pathological conditions including myocardial infarction, type 2 diabetes, chronic heart failure and inflammation. AT2R mediates its signal transduction by interacting with Gq/11 and Gi/o. However, the downstream signal cascades are different from those induced by AT1R. So far, three AT2R-mediated signalling pathways have been identified. The first molecular event is the activation of Src homology-2 domain-containing Tyr phosphatase-1 (SHP-1), protein phosphatase 2A (PP2A), and mitogen-activated protein (MAP) kinase phosphatase (MKP)-I with a subsequent inactivation of MAPKs including ERK1/2, JNK and p38 (Fig. 1.1). The inactivation of MAPKs is important in cell growth, proliferation and apoptosis [24]. The second signal transduction cascade is cyclic guanine 3′,5′-monophosphate (cGMP)/nitric oxide (NO) pathway [24,25,26]. Numerous studies have suggested that NO and its downstream effector cGMP are critical for several physiological process such as vasodilation, cell growth, apoptosis, and inflammation. AT2R activation promotes NO generation by facilitating the expression of endothelial nitric oxide synthase (eNOS). The accumulation of NO initiates the activation of cGMP, and induction of vasodilation, cell migration and proliferation. The last signaling pathway is stimulation of phospholipase A2 (PLA2) and release of arachidonic acid (AA). The enhanced PLA2 activity and AA release activate lipid signaling pathways [24, 35].
The role of AT2R in the development of blood pressure is controversial. Generally, AT2R regulates blood pressure by negatively controlling AT1R’s effects. A variety of hypertension models, including renal hypertension, genetic modification hypertension and L-NAME-induced hypertension, have been used to identify the role of AT2R in the regulation of blood pressure [36,37,38,39]. AT2R-mediated blood pressure-lowering effect has been demonstrated in a hypertension model, renal wrap hypertensive rats [40]. Additional observations have been made in the normotensive Sprague–Dawley rats [41]. A long-term study suggested that AT2R ligand CGP42112A alone had only a mild blood pressure-lowering and vasodilatory effect in the conscious rats which could be enhanced by combination with Ang II receptor blockers [41, 42]. Two pharmacologic studies showed that AT2R blocker PD123319 amplified the blood pressure increase in response to Ang II in both brain and uterine arteries [43, 44]. More definitive evidence for the role of AT2R activation in blood pressure was shown in the AT2R-deficient mice. The AT2R-deficient mice had the elevated basal blood pressure, sustained hypertension and sodium excretion. These mice also displayed an enhanced vasopressor response upon Ang II administration [45]. In addition, long-term administration of Ang II to the AT2R overexpressing mice totally eliminated the AT1R-mediated pressor effect [46]. AT2R opposes the pressor actions of Ang II by regulating the generation of cGMP and NO in various tissues including mesenteric, renal, coronary, cerebral and uterine vascular beds.
Dyslipidemia is observed in a large proportion of hypertensive patients that leads to high levels of oxidized low-density lipoprotein (ox-LDL) [47, 48]. LOX-1, is a major single type II transmembrane receptor for binding and uptake of ox-LDL in various tissues and cell types including VSMCs, endothelial cells, monocytes, macrophages, cardiomyocytes and platelets [49,50,51]. The expression of LOX-1 is extremely low under physiological conditions but is upregulated in a series of pathological states, for example, hyperlipidemia, diabetic nephropathy, hypertension, atherosclerosis, myocardial infarction and chronic renal failure [52,53,54,55,56,57,58,59].
LOX-1 belongs to class E scavenger receptor with a C-type lectin like domain and totally contains four functional domains: a short N-terminal cytoplasmic domain, a connecting neck region, a single transmembrane domain and the extracellular C-terminal ligand binding domain which is the functional domain for recognizing its stimuli.
The elevated level of soluble form of LOX-1 (sLOX-1) in blood has been proposed as a biomarker for several cardiovascular diseases including acute coronary syndrome. sLOX-1 is derived from shortening of the full-length LOX-1 through proteolytic cleavage [60, 61]. Increased sLOX-1 levels have been shown in hypertension, and sLOX-1 levels have been thought of as a diagnostic predictor for early endothelial damage in hypertension [62]. sLOX-1 level also acts as a biomarker for plaque instability in patients with acute coronary syndrome and sLOX-1 concentration is higher in the coronary circulation compared to the systemic circulation [63, 64]. sLOX-1 levels are also much higher in early disease onset of systemic lupus erythematosus patients, indicating that sLOX-1 may be a diagnostic marker in SLE patients with high cardiovascular risk. The elevated sLOX-1 levels correlate with the upregulated levels of IL-8, high-density lipoprotein, ox-LDL and high-sensitivity C-reactive protein as well as reduced IFN-γ. Membrane LOX-1 expression is significantly increased in CD14+ and CD16+ monocytes in patients with systemic lupus erythematosus. Ox-LDL enhances the susceptibility of monocytes to DNA-IC stimulation. Monocytes challenged with both ox-LDL and DNA-immune complex (DNA-IC) result in TNF-α, IL-1β and IL-6 release. Polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) are important immune regulators facilitating progression of tumor or different autoimmune diseases [60,61,62]. LOX-1 expression is increased in low density granulocytes (LDGs) from patients with systemic lupus erythematosus. These LDGs have similar phenotype as MDSCs. Ox-LDL-LOX-1 signaling facilitates the generation of neutrophil extracellular traps (NETs) in LDGs, promoting endothelial injury and inflammatory responses [65].
After binding of ox-LDL to LOX-1, the complex internalizes to the cytoplasmic compartments and elicits its intracellular signaling, PKC activation, which further induces the subsequent signals including MAPKs (p38, p42/44, ERK, JNK), nuclear factor-kappaB (NF-κB), p21-activated kinase (PAK), nuclear translocation of the transcription factor (Nrf2) and activating protein-1 (AP-1). The activation of PI3K/Akt, and protein tyrosine kinase (PTK), AT1R, sirtuin-1 (SIRT1) and NADPH oxidase are also observed [66,67,68,69,70,71,72,73,74,75,76,77]. The NF-κB-mediated ROS generation is prompted by the induction of NADPH oxidase 2 (Nox2) and Nox4 which are the main regulators in vessel walls [78]. Ox-LDL increases the activity of epithelial sodium channel (ENaC) in endothelial cells through LOX-1-NADPH oxidase-ROS production. The activation of ENaC can be inhibited by the increased NO. Since LOX-1-PI3K/Akt-mediated decrease of eNOS-NO production was also been determined, blocking the activity of ENaC and increasing NO production may protect the ox-LDL-LOX-1-caused endothelial dysfunction [79]. It has also been shown that rapamycin downregulates phosphorylation of its mechanistic target mammalian target of rapamycin (mTOR) and downstream NF-κB activation, resulting in an attenuated LOX-1 expression and, ox-LDL uptake in endothelial cells. This finding suggests an important role of mTOR in ox-LDL-mediated LOX-1 expression [80]. In addition, the activation of PKC-CD40/CD40L and NADPH oxidase-ROS signaling pathways leads to an upregulated inflammatory response and LOX-1 expression, facilitating a vicious effect in LOX-1 activation [71]. A study confirmed the positive function of ROS formation in the process of atherosclerosis [60]. NF-κB activation is also pivotal for the expression of adhesion molecules and chemokines [75, 76]. Also, induction of PKC, p-p38MAPK, p42/44 MAPK, p-JNK, NF-κB, AP-1and Nrf2 upregulates LOX-1 expression in human VSMCs and macrophages and amplifies LOX-1-mediated cellular responses [70, 72], suggesting a positive feedback loop between above-mentioned signaling activators and LOX-1 expression.
There is evidence that the inhibition of LOX-1 suppresses ox-LDL- or TNF-α-mediated NOD-like receptor pyrin domain containing (NLRP3) inflammasome activation in endothelial cells [81]. In addition, LOX-1 mediates NLRP3 activation through the upregulated ROS generation in SMCs and monocytes treated with xanthine oxidase [33]. Clinical data suggests that electronegative LDL cholesterol (L5-LDL) is significantly increased in patients with ST-segment elevation myocardial infarction. In these studies, L5-LDL induced the activation of caspase-1, NF-κB and IL-1β, which was impaired by the downregulation of NLRP3 or LOX-1 in human macrophages. Furthermore, blocking LOX-1 with a specific antibody suppressed IL-1β generation, indicating the critical role of LOX-1 in the activation of NLRP3 inflammasomes [82].
LOX-1 has also been demonstrated to be involved in numerous physiological and pathological events, including inflammation, cell proliferation, differentiation, apoptosis, autophagy, the enhanced production of inducible nitric oxide synthase (iNOS), MCP-1, MMP1, ROS, adhesion molecules and proinflammatory cytokines, as well as the downregulated secretion of endothelial nitric oxide synthase (eNOS) and NO. LOX-1 activation is also critical in the pathogenesis of the CVD-related diseases such as atherosclerosis, ischemic stroke, acute coronary syndrome, chronic renal failure, myocardial infarction, hypertension, obesity, hyperlipidemia, diabetic nephropathy, and arthritis [59, 66, 70, 83,84,85,86,87,88,89]. In mice with sustained hypertension, loss of LOX-1 dramatically decreased the cardiac fibroblast number and expression of fibronectin and procoallagen-1/collagen in the hearts, indicating the essential role of LOX-1 in cytoskeletal organization and growth of cardiac fibroblasts [90]. Recent studies showed that GATA Binding Protein 4 (GATA4) was essential in LOX-1-mediated proliferation of cardiac fibroblasts. Knockdown of LOX-1 inhibits PI3K/Akt activation, GATA4 expression and cardiac fibroblast proliferation and this effect can be restored by overexpressed LOX-1, indicating the important role in the LOX-1-PI3K/Akt-GATA4-induced proliferation of cardiac fibroblasts which contributes to cardiac remodeling [91].
Innate and Adaptive Immune Responses in Hypertension
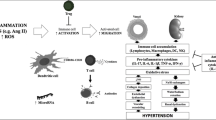
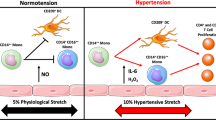
Numerous studies have indicated that the immune system is involved in the development and maintenance of hypertension. Also, different immune cells including adaptive immune cells such as CD8+ T cells, CD4+ T cells, and B cells as well innate immune cells such as macrophages, monocytes, dendritic cells (DCs) also play a critical role in the pathogenesis of hypertension and the hypertension-induced diseases (Fig. 1.2). The immune system is composed of two subtypes, the innate and adaptive immunity. The innate immune system recognizes pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) by the pathogen recognition receptors (PRRs) (Fig. 1.2). Toll-like receptors (TLRs), the most important PRR expressed on various innate immune cells including neutrophils, monocytes, macrophages, and DCs, are activated by DAMPs from the damaged tissues under the stress condition in the context of hypertension. TLRs were identified in human. TLR2, 4 and 5 are expressed on the cell surface while TLR3, 7 and 9 are located in the intracellular compartments, such as endosome, endolysosome and endoplasmic reticulum (ER). The TLRs utilize different adaptor proteins, myeloid differentiation primary response gene 88 (MyD88), TIR-domain–containing adaptor/MyD88 adaptor–like (TIRAP/Mal), TIR-domain–containing adaptor molecule 1/TIR-domain–containing adaptor-inducing interferon β (TICAM1/TRIF), and TRIF-related adaptor molecule (TRAM) to induce two distinct downstream signalings. Upon binding to its stimulator, TLR2,5,7 and 9 induce MyD88 dependent signaling and TLR3 activates TRIF dependent signaling. In addition, TLR4 is the only TLR that can induce both MyD88 and TRIF signaling pathways. Several groups have shown an enhanced TLR4 expression in different tissues and cells including heart and VSMCs from hypertensive animal models [92,93,94,95]. Inhibition of TLR4 by its specific antibody downregulates expression of TLR4 and its downstream MyD88-mediated NF-кB and proinflammatory cytokine signaling pathway, resulting in the decrease of blood pressure in hypertensive animal models [96]. TLR9 can recognize circulating mitochondrial DNA (mtDNA) that is associated with vascular dysfunction in spontaneously hypertensive rats [97]. In the hypertensive environment, the activation of TLRs by diseases-derived molecules significantly trigger the production of pro-inflammatory cytokines, chemokines, and the generation of ROS, leading to a low-grade inflammation [98] (Fig. 1.2). These pathological inflammatory responses including oxidative stress, vascular injury and remodeling as well as multiple organ damages reveal an association between TLRs and the hypertension-related diseases. Circulating monocytes and tissue resident macrophages also play a key role in the pathogenesis of hypertension. The levels of TNFα and IL-1β are elevated in monocytes from hypertensive patients. The release of TNFα, IL-1β and ROS by macrophages increases the blood pressure by suppressing vasculature endothelial and smooth muscle function with consequent vasoconstriction. The hypertension induced by impaired renal sodium excretion also attributes to higher levels of TNFα, IL-1β and ROS-mediated endothelial cell dysfunction [99].

The key role of innate and adaptive immunity in hypertension. Adaptive immune cells including CD8+ T cells, CD4+ T cells, and B cells as well innate immune cells including macrophages, monocytes, dendritic cells (DCs), release cytokines, ROS and antibodies promote the progress of hypertension and hypertension-induced diseases. The NLRP3 inflammasome activation in monocytes and DCs is critical in hypertension
Furthermore, antigen-presenting cells (APC) can uptake these DAMPs and prime T cells by major histocompatibility complex II (MHC II) dependent antigen presentation pathway, initiating the activation of T and B lymphocytes [100, 101]. The accumulation of memory T cells have been observed in hypertensive patients [102]; this phenomenon has also been identified in animal models [103]. In spontaneously hypertensive rats, reduced numbers of T cells, especially non-helper T cells population, were identified in the thymus [104]. Restoring thymic function inhibited the development of hypertension [105,106,107]. In RAG1−/− mice that has deficiency in both T and B cell development, Ang II-induced hypertension is impaired and the vascular injury is also suppressed. In another T and B cell deficiency model, SCID (severe combined immunodeficiency) mouse strain, dramatically decreased heart and kidney injury was determined following Ang II challenge [105]. In hypertensive patients, percentage of pro-inflammatory and cytotoxic CD8+ cells were increased together with upregulated production of perforin, granzyme B, interferon-γ (IFN-γ) as well as TNF-α (Fig. 1.2).
The Interplay of LOX-1 and RAS System in Hypertension
There is increasing evidence that the crosstalk between LOX-1 and AT1R plays a key role in pathogenesis of hypertension and hypertension-related diseases. The positive effect of Ang II in the expression of LOX-1 and consequently uptake of ox-LDL has been determined in several in vitro studies [108,109,110,111,112]. This dose-dependent Ang II-mediated response has been determined to be totally inhibited by the AT1R blocker losartan, but not by the AT2R blocker PD123319 [68, 112]. In in vivo studies, losartan showed its capability of inhibiting the elevated expression of LOX-1 in endothelium and neointima of autologous vein grafts and preventing the development of atherosclerosis in vein grafts [54, 57]. Another clinical investigation showed a similar effect of Ang II in LOX-1 expression under the stress of obstructive sleep apnea [113]. High concentration of LDL-cholesterol in hypercholesterolemic state regulates AT1R expression. Synergistic effect between LDL-cholesterol and Ang II in enhanced AT1R levels was observed in the cultured SMCs derived from rat aortas [113]. This effect was shown to be modulated by ox-LDL rather than native-LDL in the cultured SMCs [114]. Thus, ox-LDL mainly acts as a secondary messenger in the increased AT1R expression of hyperlipidemia. Subsequent in vivo experiments showed that AT1R expression was predominantly increased in smooth muscle layers of aortic intima derived from animal models with hypercholesterolemia. Several-fold upregulation of AT1R was shown in the aortic intima of hypercholesterolemic rabbits [115]. Further, the AT1R expression in a myocardial infarction model was shown to be potentiated by high serum cholesterol and reversed to baseline level by atorvastatin [116].
The transmembrane and cytoplasmic domains of LOX-1 are required for the proximity between LOX-1 on cell-surface membranes. Interaction of ox-LDL to LOX-1 induces AT1-dependent signaling pathway by interactions between the intracellular domain of LOX-1 and AT1R. This signaling event in human vascular endothelial cells is attenuated by either inhibiting the expression of AT1R or use of AT1R blocker, leading to relaxation of vascular rings from mouse thoracic aorta. Ox-LDL significantly increases cytosolic G protein which can be inhibited by AT1R blockade [117]. Azilsartan, a potent AT1R antagonist, was shown to inhibit ox-LDL-induced upregulation of LOX-1, MCP-1, and CXCL-1 as well as the reduced production eNOS and NO. In addition, azilsartan blocked the reduced the expression of occludin. This AT1R inhibitor also reversed the enhanced effect of ox-LDL in endothelial monolayer permeability. Further, azilsartan inhibited ox-LDL-mediated activation of Krüppel-like factor 2 (KLF2) which is also required for the activation of Occluding [118]. This study showed the role of AT1R blockade in repair of damaged tight junction.
Loss of LOX-1 significantly decreases Ang II-induced hypertension in the aged mice. LOX-1 deficiency leads to impaired fibrosis and attenuated fibronectin and collagen-3 expression, as well as ROS production rather than the expression of collagen-1 and collagen-4 in the hearts of aged mice. This effect was further determined in the aged mice with sustained hypertension by long-term Ang II infusion [119]. Pyrogallol-phloroglucinol-6,6-bieckol, a derivative of Ecklonia cava (E. cava), dramatically inhibits the excessive expression of adhesion molecule expression, and VSMCs proliferation and migration, and suppresses the increase of blood pressure, lipoprotein and cholesterol. This compound has also been shown to exert an inhibitory effect on high-fat diet or ox-LDL induced activation of LOX-1-PKC-α signaling pathway (Fig. 1.3). This compound reversed the increased expression of mesenchymal cell markers (α-SMA and vimentin) and reduction of endothelial cell markers (PECAM-1 and vWF) in the aorta or endothelial cells. Moreover, this compound reduced intima-media thickness as well as high fat diet-induced hypertension [120].

The positive feedback loop of interplay between LOX-1 and AT1R is PKC-PI3K and ROS-MAPKs-dependent. The AT2R in this crosstalk between LOX-1 and AT1R inhibits ROS-MAPKs signaling pathways
Stroke-prone spontaneously hypertensive rat is an animal model of severe hypertension and spontaneous stroke. Liang et al. showed that LOX-1 depletion dramatically slowed the development of hypertension-associated cerebral ischemic injury and brain damage, suggesting a potential role of LOX-1 in blood-brain barrier disruption after cerebral ischemia [121]. Grell et al. showed that the expression of LOX-1 to be significantly upregulated in the middle cerebral arteries from spontaneously hypertensive rats compared to wild-type controls, indicating a link between LOX-1 and hypertension-mediated vascular changes and organ injury [122]. LOX-1 deficiency leads to attenuation of angiogenesis in mice infused with Ang II. Lastly, LOX-1 depletion decreases the expression of pro-inflammatory MCP-1 and IL-1β in the hypertensive mice hearts. These observations suggest that LOX-1 is important in Ang II-mediated cardiac angiogenesis in hypertension and its association with inflammation [123].
Several studies have indicated that ROS might be the common downstream mediators in the crosstalk between LOX-1 and AT1R, and ROS generation is NADPH oxidase dependent [124,125,126]. Activation of AT1R regulates the generation of oxidative stress and this process can be inhibited by AT1R antagonist losartan or the NADPH oxidase (NOX) inhibitor DPI [124, 127]. The increase of LOX-1 expression leads to ROS generation and AT1R expression (and activity) via Akt/eNOS and Ca2+ signaling pathways [126]. Ox-LDL and Ang II combination induces capillary tube formation in a ROS dependent manner which is attenuated by NADPH oxidase inhibitor apocynin [38, 190]. The suppression of the NADPH oxidase activity also impedes the expression of AT1R and LOX-1 [128, 129]. Other intracellular molecular factors including angiotensin-converting enzyme 2 (ACE2) and MAPKs, are also involved in the crosstalk between AT1R and LOX-1 (Fig. 1.3). The increased ACE2 expression and activity in HUVECs and in abdominal aorta lead to the downregulated expression of LOX-1 and AT1R simultaneously. The ACE2 via ROS generation plays a regulatory role in the expression of LOX-1 and AT1R [130, 131]. The ROS generation subsequently induces the activation of ROS-PKC-MAPKs signaling pathway which is critical in the crosstalk of LOX-1 and AT1R [16, 66, 124, 132] (Fig. 1.3). The synergistic function of LOX-1 and AT1R has been demonstrated in the pathogenesis and development of different cardiovascular diseases. For example, several investigators have shown that these two receptors coordinate in the formation of foam cells and progression of atherosclerotic lesions. The increased expression and activity of LOX-1 and AT1R serve as key biomarkers of early atherogenesis. In high cholesterol diet fed animal models, simultaneous activation of AT1R and LOX-1 promotes atherogenesis by activation of ROS generation and vascular inflammation [115, 133, 134].
It has been shown that right ventricular systolic pressure is increased in LOX-1 transgenic mice with the hypoxic-pulmonary hypertension. In a rat model of hypoxic-pulmonary hypertension, expression of LOX-1 was noted to be increased which was responsible for pulmonary vascular remodeling. Knocking down or blocking LOX-1 dramatically decreased pulmonary arterial SMCs dedifferentiation via inhibiting ERK1/2 dependent signaling pathway, suggesting a key role of LOX-1 in the maintenance of pulmonary arterial SMCs phenotype [135, 136]. Furthermore, a recent study showed that LOX-1-NOX-ROS pathway plays a critical role in hypoxic-pulmonary hypertension induced right ventricular hypertrophy and cardiac fibrosis in rats [137].
The interplay between LOX-1 and AT2R has not been determined in detail. Recent studies showed that Ang II-LOX1 interplay might be critical in a variety of diseases including hyperlipidemia, atherosclerosis, hypertension, myocardial infarction, ischemic stroke and inflammation. However, Watanabe et al. observed no inter-regulatory effect between LOX-1 and AT2R as the increased LOX-1 expression did not influence AT2R expression [138]. These studies suggest that AT1R inhibition, but not AT2R alteration, can block the increased expression of LOX-1 in the cultured endothelial cells [112]. These data may be attributed to the low expression of AT2R in physiologic states and a relatively modest increase in pathologic states.
On the other hand, LDLR deficiency results in significantly enhanced LOX-1 expression. Hu et al. in our laboratory showed that this effect could be partly reversed by AT2R overexpression utilizing recombinant adeno-associated virus type-2 (AAV) with AT2R cDNA (AAV/AT2R). The AT2R overexpression in the LDLR deficient mice also led to decreased atherogenesis in the aorta [109]. The authors of this study suggested that the increased AT2R expression might play a role in stress states, and play an inhibitory role in the pathogenesis of cardiovascular disease states.
Overall, AT2R may act as an anti-atherosclerotic modulator, while antagonizing LOX-1-mediated pro-atherogenic effects [139]. Thus, the imbalance between LOX-1 and AT2R may determine the severity of atherosclerosis. Of note, different subtypes of collagens have been characterized as key components of atherosclerotic plaques and collagen aggregation has been established as a hallmark of development of atherosclerotic plaques [140,141,142,143]. Dandapat et al. showed that AT2R overexpression may reduce the expression of collagens as well as MMPs in atherosclerotic plaques suggesting a new mechanism of decreased atherogenesis in the aorta [144]. This is important since LOX-1 facilitates collagen aggregation [77, 145]. Loss of LOX-1 suppresses collagen aggregation and increases the expression and activity of MMP2 and MMP9 in atherosclerotic plaques. LOX-1 deficiency also inhibits components of NADPH oxidase expression and ROS production in the LDLR deficient mice. There is also evidence that AT2R activation results in an attenuated ROS generation via enhanced NO levels [146, 147].
The progression of atherosclerosis has been linked to a continuing inflammatory response [148]. Ox-LDL-LOX-1-mediated downstream immune responses have been shown to be essential in the pathogenesis of atherosclerotic lesions [196]. AT2R activation suppresses inflammatory responses during atherogenesis [83, 149]. Thus, the interplay of these two receptors in vascular inflammation might be an important element in the formation of atherosclerotic plaques. LOX-1 and AT2R interaction might be involved in other pathological process including the increased hypertension and fibrosis [150,151,152]. Though significant progress has been obtained in the crosstalk of LOX-1 and AT2R in collagen formation and development of atherosclerotic lesions, further work needs to be done in this realm.
In summary, LOX-1 has been termed as a potent pro-inflammatory mediator in Ang II-mediated hypertension via AT1R and AT2R. Further investigations in immune and inflammatory responses mediated by RAAS system and LOX-1 interaction might provide new insight and potential therapeutic targets in hypertension and its related diseases.
References
Foster GE et al (2010) Intermittent hypoxia increases arterial blood pressure in humans through a Renin-Angiotensin system-dependent mechanism. Hypertension 56(3):369–377
Cruz-Márquez J et al (2011) Effect of altitude training on the renin-angiotensin-aldosterone system and blood pressure in women. Br J Sports Med 45(6):533–533
Saidi S et al (2009) Association between renin–angiotensin–aldosterone system genotypes and haplotypes and risk of ischemic stroke of atherosclerotic etiology. Acta Neurol Scand 119(6):356–363
Sekuri C et al (2005) Renin-angiotensin system gene polymorphisms and premature coronary heart disease. J Renin-Angiotensin-Aldosterone Syst 6(1):38–42
Heffelfinger S (2007) The renin angiotensin system in the regulation of angiogenesis. Curr Pharm Des 13(12):1215–1229
Guo DF et al (2001) The angiotensin II type 1 receptor and receptor-associated proteins. Cell Res 11(3):165–180
Carey RM, Wang Z-Q, Siragy HM (2000) Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 35(1):155–163
Wang C, Jayadev S, Escobedo JA (1995) Identification of a domain in the Angiotensin II Type 1 receptor determining Gq coupling by the use of receptor chimeras (∗). J Biol Chem 270(28):16677–16682
Tanaka N et al (2010) Cis-dichlorodiammineplatinum upregulates angiotensin II type 1 receptors through reactive oxygen species generation and enhances VEGF production in bladder cancer. Mol Cancer Ther 9(11):2982–2992
Cuadra AE et al (2010) A current view of brain renin–angiotensin system: is the (pro) renin receptor the missing link? Pharmacol Ther 125(1):27–38
Yu J et al (2007) Activation of ERK, JNK, Akt, and G-protein coupled signaling by hybrid angiotensin II AT1/bradykinin B2 receptors expressed in HEK-293 cells. J Cell Biochem 101(1):192–204
Nishida M et al (2011) Regulation of angiotensin II receptor signaling by cysteine modification of NF-κB. Nitric Oxide 25(2):112–117
Heineke J, Molkentin JD (2006) Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7(8):589–600
Jin L, Ying Z, Webb RC (2004) Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am J Physiol-Heart Circulatory Physiol 287(4):H1495–H1500
Nishida M et al (2005) Gα12/13-and reactive oxygen species-dependent activation of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase by angiotensin receptor stimulation in rat neonatal cardiomyocytes. J Biol Chem 280(18):18434–18441
Liu B et al (2006) Microarray and phosphokinase screenings leading to studies on ERK and JNK regulation of connective tissue growth factor expression by angiotensin II 1a and bradykinin B2 receptors in Rat1 fibroblasts. J Cell Biochem 97(5):1104–1120
De Gasparo M et al (2000) International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52(3):415–472
Vasudevan KM, Garraway LA (2010) AKT signaling in physiology and disease. In: Phosphoinositide 3-kinase in health and disease, pp 105–133
Brazil DP, Yang Z-Z, Hemmings BA (2004) Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 29(5):233–242
Sbroggio M et al (2011) IQGAP1 regulates ERK1/2 and AKT signalling in the heart and sustains functional remodelling upon pressure overload. Cardiovasc Res 91(3):456–464
Chang Z et al (2010) PKB/Akt signaling in cardiac development and disease. Front Biosci 2:1485–1491
Volpe M et al (2003) Angiotensin II AT2 receptor subtype: an uprising frontier in cardiovascular disease? J Hypertens 21(8):1429–1443
Newby AC (2008) Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol 28(12):2108–2114
Jones ES et al (2008) AT2 receptors: functional relevance in cardiovascular disease. Pharmacol Ther 120(3):292–316
Siragy HM, Carey RM (1996) The subtype-2 (AT2) angiotensin receptor regulates renal cyclic guanosine 3’, 5’-monophosphate and AT1 receptor-mediated prostaglandin E2 production in conscious rats. J Clin Investig 97(8):1978–1982
Siragy HM, Carey RM (1997) The subtype 2 (AT2) angiotensin receptor mediates renal production of nitric oxide in conscious rats. J Clin Investig 100(2):264–269
Walters PE, Gaspari TA, Widdop RE (2005) Angiotensin-(1–7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension 45(5):960–966
Falcón BL et al (2005) Angiotensin II Type 2 receptor-mediated gene expression profiling in human coronary artery endothelial cells. Hypertension 45(4):692–697
Karamyan VT et al (2010) Preliminary biochemical characterization of the novel, non-AT1, non-AT2 angiotensin binding site from the rat brain. Endocrine 37(3):442–448
Naito T et al (2010) Angiotensin type 2 receptor actions contribute to angiotensin type 1 receptor blocker effects on kidney fibrosis. Am J Physiol-Renal Physiol 298(3):F683–F691
Ulmasov B et al (20009) Protective role of angiotensin II type 2 receptor signaling in a mouse model of pancreatic fibrosis. Am J Physiol-Gastrointestinal Liver Physiol 296(2):G284–G294
Zhang X et al (2004) Retinal expression of vascular endothelial growth factor is mediated by angiotensin type 1 and type 2 receptors. Hypertension 43(2):276–281
Dai Y et al (2017) Xanthine oxidase induces foam cell formation through LOX-1 and NLRP3 activation. Cardiovasc Drugs Ther 31(1):19–27
Santos JC et al (2000) Angiotensin-(1–7) increases osmotic water permeability in isolated toad skin. Braz J Med Biol Res 33(9):1099–1104
Lokuta AJ et al (1994) Angiotensin II stimulates the release of phospholipid-derived second messengers through multiple receptor subtypes in heart cells. J Biol Chem 269(7):4832–4838
Jones ES et al (2011) A single β-amino acid substitution to angiotensin II confers AT2 receptor selectivity and vascular function. Hypertension 57(3):570–576
Gao S et al (2013) Angiotensin AT2 receptor agonist stimulates high stretch induced-ANP secretion via PI3K/NO/sGC/PKG/pathway. Peptides 47:36–44
Matavelli LC, Huang J, Siragy HM (2011) Angiotensin AT2 receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension 57(2):308–313
Paulis L et al (2012) Direct Angiotensin II Type 2 receptor stimulation in N ω-Nitro-l-Arginine-Methyl Ester-induced hypertension: the effect on pulse wave velocity and aortic remodeling. Hypertension 59(2):485–492
Siragy HM, Carey RM (1999) Protective role of the angiotensin AT2 receptor in a renal wrap hypertension model. Hypertension 33(5):1237–1242
Carey RM et al (2001) Angiotensin type 2 receptor-mediated hypotension in angiotensin type-1 receptor-blocked rats. Hypertension 38(6):1272–1277
Widdop RE et al (2002) AT2 receptor-mediated relaxation is preserved after long-term AT1 receptor blockade. Hypertension 40(4):516–520
Hannan RE, Davis EA, Widdop RE (2003) Functional role of angiotensin II AT2 receptor in modulation of AT1 receptor-mediated contraction in rat uterine artery: involvement of bradykinin and nitric oxide. Br J Pharmacol 140(5):987–995
Li Z et al (2003) Role of AT2 receptor in the brain in regulation of blood pressure and water intake. Am J Physiol-Heart Circulatory Physiol 284(1):H116–H121
Siragy HM et al (1999) Sustained hypersensitivity to angiotensin II and its mechanism in mice lacking the subtype-2 (AT2) angiotensin receptor. Proc Natl Acad Sci 96(11):6506–6510
Tsutsumi Y et al (1999) Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Investig 104(7):925–935
Mikdashi J et al (2007) Baseline disease activity, hyperlipidemia, and hypertension are predictive factors for ischemic stroke and stroke severity in systemic lupus erythematosus. Stroke 38(2):281–285
Vincent AM et al (2009) Dyslipidemia-induced neuropathy in mice: the role of oxLDL/LOX-1. Diabetes 58(10):2376–2385
Mehta JL et al (2006) Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1): a critical player in the development of atherosclerosis and related disorders. Cardiovasc Res 69(1):36–45
Ishiyama J et al (2010) Palmitic acid enhances lectin-like oxidized LDL receptor (LOX-1) expression and promotes uptake of oxidized LDL in macrophage cells. Atherosclerosis 209(1):118–124
Li D, Mehta JL (2000) Antisense to LOX-1 inhibits oxidized LDL–mediated upregulation of monocyte chemoattractant protein-1 and monocyte adhesion to human coronary artery endothelial cells. Circulation 101(25):2889–2895
Li L, Sawamura T, Renier G (2003) Glucose enhances endothelial LOX-1 expression: role for LOX-1 in glucose-induced human monocyte adhesion to endothelium. Diabetes 52(7):1843–1850
Hayek T et al (2000) Losartan inhibits cellular uptake of oxidized LDL by monocyte-macrophages from hypercholesterolemic patients. Biochem Biophys Res Commun 273(2):417–420
Ge J et al (2004) Upregulation of lectinlike oxidized low-density lipoprotein receptor-1 expression contributes to the vein graft atherosclerosis: modulation by losartan. Atherosclerosis 177(2):263–268
Morawietz H et al (2001) Induction of the oxLDL receptor LOX-1 by endothelin-1 in human endothelial cells. Biochem Biophys Res Commun 284(4):961–965
Kume N et al (1998) Inducible expression of lectin-like oxidized LDL receptor-1 in vascular endothelial cells. Circ Res 83(3):322–327
Chen H et al (2000) Upregulation of LOX-1 expression in aorta of hypercholesterolemic rabbits: modulation by losartan. Biochem Biophys Res Commun 276(3):1100–1104
Mehta JL (2004) The role of LOX-1, a novel lectin-like receptor for oxidized low density lipoprotein, in atherosclerosis. Can J Cardiol 20:32B-36B
Ueno T et al (2003) LOX-1, an oxidized low-density lipoprotein receptor, was upregulated in the kidneys of chronic renal failure rats. Hypertens Res 26(1):117–122
Xu S et al (2013) LOX-1 in atherosclerosis: biological functions and pharmacological modifiers. Cell Mol Life Sci 70(16):2859–2872
Murase T et al (2000) Identification of soluble forms of lectin-like oxidized LDL receptor-1. Arterioscler Thromb Vasc Biol 20(3):715–720
Yavuzer S et al (2015) Endothelial damage in white coat hypertension: role of lectin-like oxidized low-density lipoprotein-1. J Hum Hypertens 29(2):92–98
Hayashida K et al (2005) Serum soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are elevated in acute coronary syndrome: a novel marker for early diagnosis. Circulation 112(6):812–818
Misaka T et al (2014) Significance of soluble lectin-like oxidized LDL receptor-1 levels in systemic and coronary circulation in acute coronary syndrome. In: BioMed research international
Sagar D et al (2020) LOX-1: a potential driver of cardiovascular risk in SLE patients. PLoS ONE 15(3):e0229184
Li D, Mehta JL (2009) Intracellular signaling of LOX-1 in endothelial cell apoptosis. Am Heart Assoc 566–568
Dandapat A et al (2007) Small concentrations of oxLDL induce capillary tube formation from endothelial cells via LOX-1–dependent redox-sensitive pathway. Arterioscler Thromb Vasc Biol 27(11):2435–2442
Mehta JL et al (2003) Pioglitazone inhibits LOX-1 expression in human coronary artery endothelial cells by reducing intracellular superoxide radical generation. Arterioscler Thromb Vasc Biol 23(12):2203–2208
Hyde R et al (2011) PKC-1 acts with the ERK MAPK signaling pathway to regulate Caenorhabditis elegans mechanosensory response. Genes Brain Behav 10(3):286–298
Li L, Sawamura T, Renier G (2004) Glucose enhances human macrophage LOX-1 expression: role for LOX-1 in glucose-induced macrophage foam cell formation. Circ Res 94(7):892–901
Li D et al (2003) LOX-1, an oxidized LDL endothelial receptor, induces CD40/CD40L signaling in human coronary artery endothelial cells. Arterioscler Thromb Vasc Biol 23(5):816–821
Anwar AA et al (2005) Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: role of mitogen-activated protein kinases and Nrf2. Free Radical Biol Med 39(2):227–236
Hu C et al (2008) Modulation of Angiotensin II–mediated hypertension and cardiac remodeling by lectin-like oxidized low-density lipoprotein receptor-1 deletion. Hypertension 52(3):556–562
Stein S et al (2010) SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur Heart J 31(18):2301–2309
Ogura S et al (2009) LOX-1 the multifunctional receptor underlying cardiovascular dysfunction. Circulation J 0909280496–0909280496
Clarke MC et al (2006) Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med 12(9):1075–1080
Hu C et al (2008) LOX-1 deletion decreases collagen accumulation in atherosclerotic plaque in low-density lipoprotein receptor knockout mice fed a high-cholesterol diet. Cardiovasc Res 79(2):287–293
Langbein H et al (2016) NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J 37(22):1753–1761
Liang C et al (2018) Oxidized low-density lipoprotein stimulates epithelial sodium channels in endothelial cells of mouse thoracic aorta. Br J Pharmacol 175(8):1318–1328
Zhou Y-D et al (2016) Rapamycin inhibits oxidized low density lipoprotein uptake in human umbilical vein endothelial cells via mTOR/NF-κB/LOX-1 pathway. PLoS ONE 11(1):e0146777
Wang S et al (2017) Statins attenuate activation of the NLRP3 inflammasome by oxidized LDL or TNFα in vascular endothelial cells through a PXR-dependent mechanism. Mol Pharmacol 92(3):256–264
Yang T-C, Chang P-Y, Lu S-C (2017) L5-LDL from ST-elevation myocardial infarction patients induces IL-1β production via LOX-1 and NLRP3 inflammasome activation in macrophages. Am J Physiol-Heart Circulatory Physiol 312(2):H265–H274
Lee W-J et al (2010) Ellagic acid inhibits oxidized LDL-mediated LOX-1 expression, ROS generation, and inflammation in human endothelial cells. J Vasc Surg 52(5):1290–1300
Sun Y, Chen X (2011) Ox-LDL-induced LOX-1 expression in vascular smooth muscle cells: role of reactive oxygen species. Fundam Clin Pharmacol 25(5):572–579
Vohra RS et al (2006) Atherosclerosis and the lectin-like oxidized low-density lipoprotein scavenger receptor. Trends Cardiovasc Med 16(2):60–64
Chen M, Masaki T, Sawamura T (2002) LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: implications in endothelial dysfunction and atherosclerosis. Pharmacol Ther 95(1):89–100
Hattori H et al (2006) G501C polymorphism of oxidized LDL receptor gene (OLR1) and ischemic stroke. Brain Res 1121(1):246–249
Xu J, Zhu J, Shi M (2010) Value of serum soluble lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) and LOX-1 mRNA in peripheral mononuclear cells in early diagnosis of acute coronary syndrome. Nan fang yi ke da xue xue bao=. J Southern Med Univ 30(12):2749–2751
Yamamoto N et al (2009) Lectin-like oxidized LDL receptor-1 (LOX-1) expression in the tubulointerstitial area likely plays an important role in human diabetic nephropathy. Intern Med 48(4):189–194
Wang X et al (2012) Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) and cardiac fibroblast growth. Hypertension 60(6):1437–1442
Liu B et al (2016) Inhibition of lectin-like oxidized low-density lipoprotein receptor-1 reduces cardiac fibroblast proliferation by suppressing GATA binding protein 4. Biochem Biophys Res Commun 475(4):329–334
Bomfim GF et al (2012) Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci 122(11):535–543
De Batista PR et al (2014) Toll-like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLoS ONE 9(8):e104020
Eißler R et al (2011) Hypertension augments cardiac toll-like receptor 4 expression and activity. Hypertens Res 34(5):551–558
Hernanz R et al (2015) Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br J Pharmacol 172(12):3159–3176
Bomfim G et al (2015) Toll-like receptor 4 inhibition reduces vascular inflammation in spontaneously hypertensive rats. Life Sci 122:1–7
McCarthy CG et al (2015) Circulating mitochondrial DNA and toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res 107(1):119–130
Vallejo JG (2011) Role of toll-like receptors in cardiovascular diseases. Clin Sci 121(1):1–10
Rucker AJ, Crowley SD (2017) The role of macrophages in hypertension and its complications. Pflügers Archiv-Eur J Physiol 469(3–4):419–430
Hevia D et al (2018) Myeloid CD11c+ antigen-presenting cells ablation prevents hypertension in response to angiotensin II plus high-salt diet. Hypertension 71(4):709–718
Kirabo A et al (2014) DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Investig 124(10):4642–4656
Itani HA et al (2016) Activation of human T cells in hypertension: studies of humanized mice and hypertensive humans. Hypertension 68(1):123–132
Itani HA et al (2016) CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res 118(8):1233–1243
Fannon LD, Braylan RC, Phillips MI (1992) Alterations of lymphocyte populations during development in the spontaneously hypertensive rat. J Hypertens 10(7):629–634
Takeichi N et al (1980) Immunological depression in spontaneously hypertensive rats. Clin Exp Immunol 40(1):120
Takeichi N, Suzuki K, Kobayashi H (1981) Characterization of immunological depression in spontaneously hypertensive rats. Eur J Immunol 11(6):483–487
Ba D et al (1982) Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J Immunol 128(3):1211–1216
Morawietz H et al (1999) Angiotensin II induces LOX-1, the human endothelial receptor for oxidized low-density lipoprotein. Circulation 100(9):899–902
Hu C et al (2008) Over-expression of angiotensin II type 2 receptor (agtr2) reduces atherogenesis and modulates LOX-1, endothelial nitric oxide synthase and heme-oxygenase-1 expression. Atherosclerosis 199(2):288–294
Keidar S et al (1995) Angiotensin II stimulates macrophage-mediated oxidation of low density lipoproteins. Atherosclerosis 115(2):201–215
Keidar S, Attias J (1997) Angiotensin II injection into mice increases the uptake of oxidized LDL by their macrophages via a proteoglycan-mediated pathway. Biochem Biophys Res Commun 239(1):63–67
Li D et al (1999) Upregulation of endothelial receptor for oxidized low-density lipoprotein (LOX-1) in cultured human coronary artery endothelial cells by angiotensin II type 1 receptor activation. Circ Res 84(9):1043–1049
Nickenig G, Wassmann S, Böhm M (2000) Regulation of the angiotensin AT1 receptor by hypercholesterolaemia. Diabetes Obes Metab 2(4):223–228
Nickenig G et al (1997) Upregulation of vascular angiotensin II receptor gene expression by low-density lipoprotein in vascular smooth muscle cells. Circulation 95(2):473–478
Yang BC et al (1998) Increased angiotensin II type 1 receptor expression in hypercholesterolemic atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol 18(9):1433–1439
Mączewski M, Mączewska J, Duda M (2008) Hypercholesterolaemia exacerbates ventricular remodelling after myocardial infarction in the rat: role of angiotensin II type 1 receptors. Br J Pharmacol 154(8):1640–1648
Yamamoto K et al (2015) Oxidized LDL (oxLDL) activates the angiotensin II type 1 receptor by binding to the lectin-like oxLDL receptor. FASEB J 29(8):3342–3356
He S et al (2015) Kruppel-like factor 2-mediated suppression of microRNA-155 reduces the proinflammatory activation of macrophages. PLoS ONE 10(9):e0139060
Li X et al (2021) LOX-1 deletion attenuates myocardial fibrosis in the aged mice, particularly those with hypertension. Front Cardiovasc Med 1305
Son M et al (2021) Pyrogallol-phloroglucinol-6 6-bieckol on attenuates high-fat diet-induced hypertension by modulating endothelial-to-mesenchymal transition in the aorta of mice. In: Oxidative medicine and cellular longevity
Liang Y-Q et al (2020) LOX-1 (lectin-like oxidized low-density lipoprotein receptor-1) deletion has protective effects on stroke in the genetic background of stroke-prone spontaneously hypertensive rat. Stroke 51(6):1835–1843
Grell A-S et al (2017) Cerebrovascular gene expression in spontaneously hypertensive rats after transient middle cerebral artery occlusion. Neuroscience 367:219–232
Wang X et al (2014) LOX-1 deletion limits cardiac angiogenesis in mice given angiotensin II. Cardiovasc Drugs Ther 28(5):441–446
Kang B-Y, Mehta JL (2009) Rosuvastatin attenuates Ang II—mediated cardiomyocyte hypertrophy via inhibition of LOX-1. J Cardiovasc Pharmacol Ther 14(4):283–291
Hu C, Dandapat A, Mehta JL (2007) Angiotensin II induces capillary formation from endothelial cells via the LOX-1–dependent redox-sensitive pathway. Hypertension 50(5):952–957
Sakamoto N et al (2009) Role of LOX-1 in monocyte adhesion-triggered redox, Akt/eNOS and Ca2+ signaling pathways in endothelial cells. J Cell Physiol 220(3):706–715
Pendergrass KD et al (2009) The Angiotensin II–AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem Biophys Res Commun 384(2):149–154
Nagase M et al (2001) Redox-sensitive regulation of lox-1 gene expression in vascular endothelium. Biochem Biophys Res Commun 281(3):720–725
Ichiki T et al (2001) Reactive oxygen species-mediated homologous downregulation of angiotensin II Type 1 receptor mRNA by Angiotensin II. Hypertension 37(2):535–540
Zhang C et al (2010) Angiotensin-converting enzyme 2 attenuates atherosclerotic lesions by targeting vascular cells. Proc Natl Acad Sci 107(36):15886–15891
Lu J et al (2011) Oxidative stress and lectin-like ox-LDL-receptor LOX-1 in atherogenesis and tumorigenesis. Antioxid Redox Signal 15(8):2301–2333
Li D et al (1999) Proapoptotic effects of ANG II in human coronary artery endothelial cells: role of AT1receptor and PKC activation. Am J Physiol-Heart Circulatory Physiol 276(3):H786–H792
Takanabe-Mori R et al (2010) Lectin-like oxidized low-density lipoprotein receptor-1 is required for the adipose tissue expression of proinflammatory cytokines in high-fat diet-induced obese mice. Biochem Biophys Res Commun 398(3):576–580
Petnehazy T et al (2006) Role of blood cell-associated AT1 receptors in the microvascular responses to hypercholesterolemia. Arterioscler Thromb Vasc Biol 26(2):313–318
Zhang W et al (2018) LOX-1 mediated phenotypic switching of pulmonary arterial smooth muscle cells contributes to hypoxic pulmonary hypertension. Eur J Pharmacol 818:84–95
Ogura S et al (2013) Oxidative stress augments pulmonary hypertension in chronically hypoxic mice overexpressing the oxidized LDL receptor. Am J Physiol-Heart Circulatory Physiol 305(2):H155–H162
Zhu T-T et al (2017) LOX-1 promotes right ventricular hypertrophy in hypoxia-exposed rats. Life Sci 174:35–42
Watanabe T, Barker TA, Berk BC (2005) Angiotensin II and the endothelium: diverse signals and effects. Hypertension 45(2):163–169
Koitka A et al (2010) Angiotensin II subtype 2 receptor blockade and deficiency attenuate the development of atherosclerosis in an apolipoprotein E-deficient mouse model of diabetes. Diabetologia 53(3):584–592
Rekhter MD (1999) Collagen synthesis in atherosclerosis: too much and not enough. Cardiovasc Res 41(2):376–384
Katsuda S et al (1992) Collagens in human atherosclerosis. Immunohistochemical analysis using collagen type-specific antibodies. Arteriosclerosis Thrombosis: J Vascular Biol 12(4):494–502
Kittelberger R, Davis PF, Stehbens W (1990) Type VI collagen in experimental atherosclerosis. Experientia 46(3):264–267
Velleman SG et al (2001) Collagen characteristics and organization during the progression of cholesterol-induced atherosclerosis in Japanese quail. Exp Biol Med 226(4):328–333
Dandapat A et al (2008) Over-expression of angiotensin II type 2 receptor (agtr2) decreases collagen accumulation in atherosclerotic plaque. Biochem Biophys Res Commun 366(4):871–877
Hu C et al (2008) Regulation of TGFβ1-mediated collagen formation by LOX-1: studies based on forced overexpression of TGFβ1 in wild-type and Lox-1. J Biol Chem 283(16):10226–10231
Yin T et al (2008) Angiotensin II promotes NO production, inhibits apoptosis and enhances adhesion potential of bone marrow-derived endothelial progenitor cells. Cell Res 18(7):792–799
Kitamoto S et al (2000) Chronic inhibition of nitric oxide synthesis in rats increases aortic superoxide anion production via the action of angiotensin II. J Hypertens 18(12):1795–1800
Libby P, Ridker PM, Maseri A (2002) Inflammation and atherosclerosis. Circulation 105(9):1135–1143
Gao L, Zucker IH (2011) AT2 receptor signaling and sympathetic regulation. Curr Opin Pharmacol 11(2):124–130
Hollander W (1976) Role of hypertension in atherosclerosis and cardiovascular disease. Am J Cardiol 38(6):786–800
Savoia C et al (2011) Angiotensin type 2 receptor in hypertensive cardiovascular disease. Curr Opin Nephrol Hypertens 20(2):125–132
Dominguez JH et al (2008) Anti-LOX-1 therapy in rats with diabetes and dyslipidemia: ablation of renal vascular and epithelial manifestations. Am J Physiol-Renal Physiol 294(1):F110–F119
Acknowledgements
Grant support: VA Merit Review, Department of Veterans Affairs Technology Transfer Program and VA Merit Review: Senior Clinician Scientist Award (all to Dr Mehta); Henan University Science and Technology Innovation Team (to Dr Wang).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Cheng, W., Shao, F., Mehta, J.L., Wang, X. (2023). Renin–Angiotensin–Aldosterone System and LOX-1 Interaction in Hypertension with a Focus on Modulation of the Immune System. In: Dhalla, N.S., Bhullar, S.K., Shah, A.K. (eds) The Renin Angiotensin System in Cardiovascular Disease. Advances in Biochemistry in Health and Disease, vol 24. Springer, Cham. https://doi.org/10.1007/978-3-031-14952-8_1
Download citation
DOI: https://doi.org/10.1007/978-3-031-14952-8_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-14951-1
Online ISBN: 978-3-031-14952-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)