
Abstract
Emerging evidence has supported a role of inflammation and immunity in the genesis of hypertension. In humans and experimental models of hypertension, cells of the innate and adaptive immune system enter target tissues, including vessels and the kidney, and release powerful mediators including cytokines, matrix metalloproteinases and reactive oxygen species that cause tissue damage, fibrosis and dysfunction. These events augment the blood pressure elevations in hypertension and promote end-organ damage. Factors that activate immune cells include sympathetic outflow, increased sodium within microenvironments where these cells reside, and signals received from the vasculature. In particular, the activated endothelium releases reactive oxygen species and interleukin (IL)-6 which in turn stimulate transformation of monocytes to become antigen presenting cells and produce cytokines like IL-1β and IL-23, which further affect T cell function to produce IL-17A. Genetic deletion or neutralization of these cytokines ameliorates hypertension and end-organ damage. In this review, we will consider in depth features of the hypertensive milieu that lead to these events and consider new treatment approaches to limit the untoward effects of inflammation in hypertension.
Similar content being viewed by others
Introduction
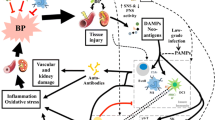
Hypertension is the leading cause of mortality and the major risk factor for myocardial infarction, heart failure, kidney damage, and stroke. The recent American College of Cardiology and American Heart Association recently classified hypertension as a systolic blood pressure >130 mmHg and a diastolic blood pressure of >80 mmHg [1]. These new guidelines classify nearly half of the adult population as hypertensive. Moreover, almost half of patients diagnosed with hypertension are not adequately controlled, despite current therapies. The physiologic mechanisms underlying the development of hypertension are diverse and include renal dysfunction, vascular dysfunction and central neural mechanisms [2]. In addition it has become increasingly evident that inflammation and activation of both innate and adaptive immune cells contribute to this disease [3]. Immunity and inflammation are complex physiological and pathophysiological processes that involve numerous cell types and secreted factors that play a critical role in hypertension [4]. In this review we will focus on the effects of innate immunity on multiple organ systems and the development of systemic hypertension.
Inflammation and hypertension
For more than 50 years, immune cells have been implicated in hypertension. Lymphocytes and macrophages have been observed in the kidneys and blood vessels of hypertensive humans and in animals with experimental hypertension since the 1960s (for review see Norlander et al. [5]). Over the last two decades, there has been expanding interest in the role of immune cells and the cytokines produced by these cells in the development of hypertension. Recombinase activating gene 1 (RAG1) deficient mice, which lack both T and B cells, are protected from angiotensin II (ang II)-induced hypertension [4]. Likewise Crowley and colleagues demonstrated that the SCID (severe combined immunodeficiency) mouse, another model lacking T and B cells, are similarly protected [6]. Moreover, deletion of RAG1 in Dahl salt-sensitive (Dahl SS) rats prevented the increase in systolic blood pressure in response to high salt feeding [7]. Pharmacological blockade or genetic deletion of T cell co-stimulatory receptors markedly inhibited both ang II and Deoxycorticosterone Acetate (DOCA)-hypertension [8]. Likewise, deletion of CD70, important for memory T cell formation, prevents recurrent episodes of experimental hypertension [9]. Caillon et al. demonstrated that genetic or pharmacological deletion of gamma delta T cells ameliorated the hypertensive response and vascular dysfunction caused by ang II infusion [10]. These innate-like T cells usually lack CD4 and CD8 and do not respond to MHC I and MHC II specific antigen presentation. Studies such as these have emphasized the importance of the immune cells in the development of hypertension and its associated end-organ damage.
The mechanisms by which immune cells contribute to hypertension are multifactorial. These cells release powerful mediators, including reactive oxygen species (ROS), matrix metalloproteinases and cytokines that have profound effects on the tissues they invade. Cytokines including interleukin (IL)-17A, IL-6, and IL-1β potentiate the development of hypertension. Madhur et al. showed that serum levels of IL-17A are significantly increased humans with hypertension compared to those without, that the T cell production of IL-17A is increased in ang II infused mice. IL-17A deficient mice were found to be protected from endothelial cell dysfunction and hypertension caused by chronic infusion of ang II [11]. Likewise, immunoclearing of IL-17A but not IL-17F reduced blood pressure in ang II-infused mice [12]. In this study both Th17 and γδT cells were found to produce equal amounts of IL-17A during ang II hypertension [12]. In a subsequent study, Norlander et al. demonstrated that IL-17A stimulates sodium and water retention via acting on several sodium transporters within the kidney [13]. Nquyen et al. showed that IL-17A promotes inhibitory phosphorylation of the endothelial nitric oxide synthase, reducing endothelial production of nitric oxide, and when infused in mice, causes hypertension [14]. In keeping with these findings, IL-17A infusion has been shown to augment the hypertensive response to ang II infusion [15]. Luther et al. demonstrated that ang II infusion for 3 h increased plasma IL-6 levels in humans and this was inhibited by treatment with spironolactone, suggesting a role of aldosterone in this response [16]. Moreover, mice that lack IL-6 develop blunted hypertension in response to chronic infusion of ang II [17]. A potent cytokine produced by myeloid cells that contributes to the development of inflammation associated with hypertension is IL-1β. Alexander et al. performed next generation RNA sequencing of monocytes from normotensive and hypertensive humans and found a significant increase in ~60 genes related to IL-1β in the hypertensive subjects [18]. They demonstrated that lactoferrin, peptidoglycan recognition protein 1, and IL-18 receptor accessory protein (IL18RAP) are elevated in peripheral blood monocytes of hypertensive subjects compared to normotensive individuals [18]. These studies demonstrated the potential importance of immune cell-derived cytokines in augmenting the development of hypertension and its associated inflammation in end-organs.
ROS also seem to activate myeloid cells in hypertension and in turn are produced by these cells to become mediators of tissue damage and dysfunction. Several studies have demonstrated that genetic deletion of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits, p47phox, Nox1 or Nox4 in mice prevent the development of hypertension. Landmesser et al. showed that mice lacking p47phox, a subunit of NADPH oxidase, prevented the development of vascular superoxide production and blunted the development of hypertension in response to ang II infusion [19]. Transgenic overexpression of p22phox, the small membrane bound docking subunit of cytochrome b558 leads to age-related hypertension [20], and augments hypertension in response to ang II [21]. Likewise, overexpression of Nox1 potentiates hypertension and vascular remodeling in experimental hypertension [22]. while deletion of Nox1 protects against these phenomena [23, 24]. There is an interplay between the NADPH oxidase and other sources of ROS, such that ROS produced by the NADPH oxidase seem to activate further ROS production by the mitochondria, uncoupled nitric oxide synthases and xanthine oxide [25].
The predominant product of the NADPH oxidases is superoxide, formed by a one-electron reduction of molecular oxygen. Superoxide is very short-lived and charged and therefore unlikely reaches adjacent cells. In contrast, downstream products of superoxide, including hydrogen peroxide, peroxynitrite and lipid peroxides are longer lived and clearly can diffuse to other cells and have potent biological effects. A particularly important role of ROS in post-translational modification of proteins. Our group demonstrated that hypertension is associated with accumulation isolevuglandins (isoLGs) in antigen presenting cells [26]. IsoLGs are products of lipid peroxidation that covalently react with lysines on self-proteins, and there is substantial evidence that these modified proteins act as neoantigens. IsoLG adducted peptides are presented in the context of major histocompatibility complexes and stimulate subsets of both CD4+ and CD8+ T cells. Unlike wild-type mice, mice lacking the Nox2 subunit of the NADPH oxidase did not develop isoLGs within dendritic cells (DCs). It is also likely that the NADPH oxidase in other cells can modify proteins that are ultimately phagocytosed by professional antigen presenting cells. In keeping with a role of isoLGs and the NADPH oxidase in immune activation overexpression of p22phox significantly increased renal and vascular accumulation of activated CD4+ and CD8+ T cells. In parallel experiments, we found that mice with specific deletion of superoxide dismutase 3 in vascular smooth muscle cells had aortic stiffening by 6 months of age and significantly elevated blood pressure by 9 months of age compared to appropriate controls, indicating that defects ROS scavenging can also contribute to immune activation [20]. Taken together, these studies demonstrate that ROS contributes to the pathogenesis of hypertension and its associated inflammation.
Innate immunity in hypertension
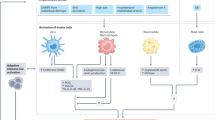
Seminal studies have investigated the role of innate immunity, specifically monocytes and macrophages in antigen presentation to T cells and the production of cytokines in hypertension. Ciuceis et al. demonstrated that Op/Op mice, that lack the macrophage-colony stimulating factor (m-CSF), and therefore exhibit a deficiency of myeloid cells, are protected from the rise in blood pressure, endothelial dysfunction and oxidative stress caused by ang II infusion [27]. In another study, ablation of myeloid cells expressing lysozyme M+ cells in mice prevented the raise in systolic blood pressure in response to ang II-induced hypertension and abrogated both vascular smooth muscle and endothelial dysfunction through a reduction in the production of superoxide [28]. In a subsequent study, Kossmann et al. showed that deletion of monocytes in mice completely prevented the hypertension caused by ang II, attenuated endothelial nitro-oxidative stress and endothelial nitric oxide synthase (eNOS) uncoupling [29]. As discussed above, we discovered that during hypertension, DCs accumulate highly reactive isoLG-adducts to T cells that promote T cell activation, proliferation and end-organ inflammation [26]. We also found that adoptive transfer of DCs from hypertensive mice to naïve recipient mice augmented the raise in blood pressure in response to infusion of a suppressor dose of ang II and this could be prevented if the donor mice were treated with the isoLG scavenger 2-hydroxybenzilamine (2-HOBA). Moreover, DCs from hypertensive mice produce copious amounts of proinflammatory cytokines including IL-1β, TNFα, IL-6, and IL-23 and skew CD8+ T cells to produce IFN-y and IL-17 [26]. This study demonstrated the critical importance of monocyte-derived DCs in the promotion of hypertension and end-organ damage.
In depth phenotyping of myeloid cells using single cell RNA-sequencing or mass cytometry by time of flight (CyTOF) have identified new subsets of these cells that likely contribute. Some macrophages and DCs are derived during in utero development and take residence in peripheral tissues, where they exhibit self-renewal [30]. These tissue resident cells seem to have an anti-inflammatory, tissue patrolling role. In contrast, there are inflammatory macrophages and DCs that are monocyte derived. Traditionally, it was thought that when monocytes enter peripheral tissues via endothelial diapedesis, they transform into either DCs or macrophages [31]. Jakabuzick et al., however, demonstrated that monocytes can enter and remerge from peripheral tissues with minimal differentiation. These experienced monocytes appear to be efficient in presenting antigen and exhibit proinflammatory surface markers.
Our laboratory and others have demonstrated have identified at least 3 mechanisms that are likely involved in the activation of myeloid cells [32,33,34]. First, myeloid cells receive signals including physical interaction, chemokines, and cytokines from an activated endothelium. Second, myeloid cells encounter high sodium microenvironments, such as the renal medulla and papilla. Third, central nervous system activation drives sympathetic outflow to numerous organs including the spleen, kidney, and vasculature.
Activation of innate immune cells
In a seminal study Randolph et al., showed that the endothelium activated by either LPS, Zymosan, or IL-1β can promote conversion of monocytes to DCs [31]. Although the roles of LPS and zymosan in hypertension are not clear, a factor that can activate the endothelium in hypertension is increased mechanical stretch. Our group recently demonstrated that human aortic endothelial cells (HAECs) undergoing uniaxial cyclical hypertensive stretch (10% stretch; 1 Hz) for 48 h released factors that promoted the conversion of human classical monocytes (CD14++/CD16−) to a proinflammatory intermediate monocyte phenotype (CD14+/CD16++) [32]. This conversion was prevented by either co-treatment with catalase or neutralization of IL-6, implicating these mediators in this process. Moreover, monocytes activated by endothelial cells undergoing hypertensive stretch produce copious amounts of the proinflammatory cytokines IL-6, IL-1β, IL-23, and TNF-α and potently drive T cell proliferation from the same human subjects (Fig. 1). Of interest, 10% HAEC stretch promoted acquisition of CD209 (DC-SIGN), which is expressed on DCs and activated macrophages, demonstrating a critical role of monocyte-derived DC activation in a translational in vitro model of hypertension [32]. Of note, we also found that the percent of circulating intermediate monocytes is increased in hypertensive compared to normotensive humans.

Endothelial cells under physiological stretch (5%) release nitric oxide (NO) that inhibits the conversion of classical monocytes (CD14++) to the intermediate monocyte phenotype (CD14+ CD16++). During hypertension, endothelial cells undergoing increased stretch which releases hydrogen peroxide (H2O2) and proinflammatory cytokines including IL-6 that promotes the formation of CD14++ monocytes to the proinflammatory CD14+ CD16++ monocytes and monocytes expressing CD209, a classical dendritic cell (DC) marker. In turn these proinflammatory cells potently drive both CD4+ and CD8+ T cell proliferation.
Another key stimulus in the activation of innate immune cells is exposure to high sodium microenvironments. It has recently been suggested that sodium concentrations in peripheral tissues may reach substantially higher levels than those in plasma. In recent studies it has been demonstrated in a model of chronic renal failure, 5/6th nephrectomy, there is a significant increase in the Na+ and K+ concentration in the skin due to increased renal excretion of water, leading to vasoconstriction of the vasculature and increased blood pressure [35]. In a model of psoriasis, there is vasoconstriction of the dermal microcirculation that occurs to prevent transepidermal water loss that promotes the elevation of arterial blood pressure [36]. Importantly, hypertension promotes the accumulation of myeloid cells, including monocytes, macrophages and DCs in the renal medulla, where interstitial concentrations of sodium normally reach 800–1200 mMol/L. Our group demonstrated that sodium enters DCs via a variant of the epithelial sodium channel (ENaC), composed of alpha and gamma ENaC subunits. Once in the DC, sodium is rapidly exchanged for calcium, and calcium in turn activates the NADPH oxidase, promoting ROS production and isoLG formation in these cells [33]. Modestly elevated levels of extracellular sodium also promoted production of proinflammatory cytokines including IL-6 and IL-1β in DCs. An important step in this process is activation of the serum-glucocorticoid kinase-1 (SGK1), which modulates protein levels of the ENaC. In keeping with this, we recently showed that mice lacking SGK1 in DCs had a marked reduction of the ENaC alpha and gamma subunits and were protected against salt-sensitive hypertension, vascular dysfunction, and renal inflammation, demonstrating a critical role of SGK1 in regulation of antigen presentation [37]. In a related study, we recently demonstrated that human monocytes accumulate isoLG-adducts upon exposure to modestly elevated concentrations of sodium [34]. Exposure to high salt upregulated markers of activation, including co-stimulatory molecules CD83 and CD86, and the production of proinflammatory cytokines IL-1β, TNFα and IL-6 compared to normal salt. We also found that salt feeding of immunodeficient (NOD-scid IL2Ry null; NSG) mice that had been adoptively transferred with human T cells led to proliferation of these cells (a consequence of T cell activation) only if monocytes had also been adoptively transferred [34]. T cell proliferation was not observed in these mice if monocytes were not present, nor if they were fed a low salt diet, indicating the sodium must act via these myeloid cells to promote T cell activation (Fig. 2). When fed a high salt diet, Dahl SS rats have a significant increase in macrophages infiltrating the kidney compared to Sprague Dawley and SSBN13 controls [38]. The majority of these macrophages exhibit a proinflammatory M1-like macrophages phenotype. These studies demonstrate that myeloid cells encountering a high sodium microenvironment drive monocyte, DC, and macrophages activation to promote hypertension and end-organ damage.

Monocytes from microenvironments of high interstitial sodium, including the renal medulla and skin are activated. These activated monocytes convert to dendritic-like antigen presenting cells that accumulate the highly reactive isolevuglandins (isoLGs) and produce copious amounts of IL-1β and potently drive T cell proliferation. Sodium (Na+) enters the monocyte through the epithelial sodium channel (ENaC) which is stabilized by serum glucocorticoid kinase 1 (SGK1) and is exchanged for calcium through the Na+/Ca2+ exchanger (NCX). Intracellular Ca2+ activates PKC and phosphorylates p47phox, which leads to the activation of the NADPH oxidase.
A potential mechanism of the action of high salt diets is via changes in the microbiome. The gut microbiome, comprised of trillions of microbes, regulates both physiological and pathophysiological states [39]. It is now clear that these release metabolites and genetic material that impact systemic responses including blood pressure, immunity, vascular function, and the sympathetic nervous system. Wiick et al. showed that salt feeding of mice depleted the gut microbiome of Lactobacillus murinus and the loss of this species induced TH17 cells. Treatment of mice with Lactobacillus murinus prevented hypertension caused by excess salt intake. Likewise, these investigators showed that salt feeding reduced the human gut microbiome of Lactobacillus species, induced TH17 cells and raised blood pressure. Ferguson et al. showed that humans that consume excess salt have altered gut microbial levels of Firmicutes, Proteobacteria and Prevotella bacteria. These investigators also showed that salt feeding of mice induced intestinal inflammation, increased intestinal Firmicutes and decreased Prevotella species. Fecal transfer from salt fed mice to germ-free mice primed hypertension in response to a usually subpressor dose of ang II in these germ-free recipients [40]. These events were accompanied by accumulation of isoLGs in the gut wall and increased CD8+ T cells within mesenteric lymph nodes. Of interest, Karbach et al. showed that germ-free mice are markedly protected against ang II-induced hypertension [41]. While hypertension was associated with increased vascular superoxide in normal mice, this was markedly reduced in the germ-free mice. Likewise, these animals exhibited a striking attenuation of vascular myelomonocytic infiltration and did not exhibit the reduction of endothelium-dependent vasodilatation and elevation of blood pressure observed in wild-type mice. A mechanism by which the gut microbiota influences physiology of the host is via production of metabolic byproducts including short chain fatty acids (SCFAs), which act on G-protein coupled receptors. Interestingly, Pluznick et al. showed that the olfactory receptor 78 (Olfr78) is present in the kidney and vessels, and controls renin release in response to propionate stimulation [42]. Mice lacking Olfr78 exhibited reduced renin release and a lower blood pressure at baseline as compared to wild type mice. Propionate injection was shown to raise BP in wild-type mice, but to produce hypotension in mice lacking Olfr78. Thus, SCFAs produced by the gut microbiota likely influence peripheral vascular and renal receptors to affects blood pressure. Very recently Nakai et al. analyzed ambulatory blood pressure, fecal microbiome, plasma and fecal metabolites and expression of their receptors in normotensive and hypertensive humans [43]. While they did not observe a difference in microbiome diversity, a significant alteration of microbial gene pathways was observed. One of these pathways involved the production of acetyl-CoA, responsible for production SCFAs. In keeping with this, the investigators observed an increase in circulating levels of acetate and butyrate in hypertensive subjects. A striking finding was that in circulating leukocytes, there was almost a complete absence of mRNA for the SCFA-sensing receptor GPR43 in hypertensive subjects, while this was abundant in normotensives. The authors speculated that this absence of GPR43 might contribute to a proinflammatory state in hypertensive subjects.
In addition, numerous studies have established a critical role of sympathetic tone on activation of immune cells in hypertension. In a recent study, we demonstrated that central sympathetic outflow is increased salt-sensitive hypertension, and this seems to be due to activation of prostaglandin E2 receptor (EP3) in the lamina terminalis of the forebrain [44]. In mice globally deficient in EP3, the elevations of blood pressure, accumulation of isoLG-adducts in splenic DCs was reduced as was renal T cell accumulation and the production of IL-17A and IFN-y during L-NAME/high salt hypertension. In subsequent experiments. silencing of EP3 in these forebrain sites by local injection of shRNA reduced sympathetic outflow in response to high salt and significantly reduced DC activation and accumulation of isoLG-adducts [44]. These studies demonstrated a critical role for increased sympathetic tone, mediated by EP3 receptors in the forebrain, in activation of DCs during the development of salt-sensitive hypertension.
Suppressor cell activity of myeloid cells
As mentioned in the proceeding sections, various myeloid cells, including monocytes, macrophages, and DCs promote a proinflammatory environment driving the development of hypertension and inflammation. However, some myeloid cell subsets orchestrate an anti-inflammatory response, limiting the raise in blood pressure, inflammation, and end-organ damage. Shah et al. demonstrated that myeloid-derived suppressor cells (MDSCs), prevent the increase in blood pressure and end-organ inflammation in response to ang II, L-NG-nitroarginine methyl ester, and high salt feeding [45]. They further demonstrated that depletion of MSDCs led to an increase in inflammatory cells and blood pressure. In keeping with this, Chassion et al., using a cyclosporine-A model of hypertension showed that adoptive transfer of MSDCs prevented both renal and vascular inflammation and improved vascular function [46]. Most recently, Crowley and colleagues showed that expression of A20 in DCs, an ubiquitin-editing protein that plays a role in immune system homeostasis, prevented the elevation in blood pressures and renal accumulation of activated T cells in a model of ang II-induced hypertension [47]. Thus, there appears to be a tolerogenic or immune inhibitory role of myeloid-derived suppressor cells and subsets of DCs in hypertension.
Inflammasome and Il-1β in hypertension
A key signaling mechanism which has garnered attention in the pathogenesis of hypertension is the production of IL-1β and inflammasome signaling. Factors in the hypertensive milieu, including the vasoactive molecules ang II, aldosterone, and endothelin-1 are able to activate the NLRP3 (nucleotide oligomerization domain-like receptor family pyrin domain containing 3) inflammasome [48]. Classical assembly and activation of the NLRP3 inflammasome occur in response to various stimuli including pathogen associated molecular patterns (PAMPs), damage associated molecular patterns (DAMPs), and uric acid crystals [48]. The NLRP3 inflammasome can also be activated by adenosine triphosphate (ATP) induced K+ efflux or the generation of reactive oxygen species [49]. Major products of NLRP3 inflammasome are IL-1β and IL-18 [49], which in turn exert potent inflammatory effects on both immune and non-immune cells alike. Clinically, polymorphisms in the NLRP3 gene are associated with elevations in blood pressures, and humans with hypertension have elevated plasma IL-1β levels compared to normotensive human subjects [50, 51]. Krishnan et al. recently demonstrated that the selective NLRP3 inhibitor, MCC950, prevented the increase in systolic blood pressure, renal inflammation, and renal fibrosis in salt-sensitive hypertension [52]. Furthermore, mice with NLRP3 gene deletion were protected against the vascular inflammation and prevented vascular remodeling in response to ang II-infusion [53]. Another study showed leukocytes from human subjects with hypertension have higher protein levels of NLRP3, caspase-1, and mature IL-1β compared to healthy control subjects [54]. A recent secondary analysis of the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial showed that while canakinumab did not lower blood pressure, that there was a trend for the greatest reduction in major cardiovascular events, death and myocardial infarction among those in the highest quartile of blood pressure [55]. In patients with a systolic blood pressure of ≥130 mm Hg at baseline, canakinumab reduced absolute risk by 3.4%. This reduction in events was approximately half this (1.8%) in patients with a baseline systolic blood pressure <130 mm Hg. Although this interaction between Canakinumab benefit and blood pressure levels was not significant and could have occurred by chance, this outcome is compatible with the benefit observed with IL-1β blockade and NLRP inhibition in experimental studies.
Clinical perspectives
The realization that inflammation and activation of immune cells of both the innate and adaptive immune system contribute to hypertension has provided insight into mechanisms of end-organ damage that occurs in this disease. It is conceivable that in depth analysis of circulating immune cells, perhaps using single cell sequencing could reveal subsets of patients at greater risk for end-organ damage. In this regard, Siedlinski et al. recently demonstrated a positive association between quintiles of lymphocytes, neutrophils, and monocytes and systolic blood pressure, diastolic blood pressure, and calculated pulse pressure [56]. Specifically, the authors found that the strongest correlations were between neutrophils and monocytes and all three-blood pressure indices measured in the study suggesting a possible additional stratification criterion for hypertensive patients. As demonstrated by Nakai et al. [43], the ability to leukocytes to sense signals from the gut might be altered in hypertension and promote inflammatory end-organ damage. Further in-depth analyses of these cells, their products and signaling pathways will almost certainly provide additional insight.
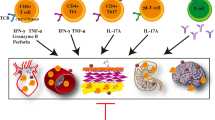
Moreover, the use of anti-inflammatory and immunomodulatory therapies has been demonstrated to provide blood pressure benefits in numerous autoinflammatory disease states. For instance, Yoshida et al. treated 16 rheumatoid arthritis patients with the tumor necrosis factor-alpha (TNF-α) inhibitor, Infliximab, and found a significant reduction in 24-h ambulatory blood pressure when compared to before treatment [57]. Herrera et al. investigated the role of mycophenolate mofetil (MMF) on blood pressure in 140 human subjects with psoriasis or rheumatoid arthritis. MMF reduced both systolic (152.3 mmHg to 136.6 mmHg) and diastolic (91.7 mmHg to 82.5 mmHg) during MMF treatment compared to baseline. Moreover, they found a trend for a reduction in TNF-α, RANTES, and malondialdehyde (MDA), implicating a role for renal inflammation and oxidative stress in the development of hypertension [58]. Lastly, monoclonal antibodies against IL-17, IL-6, and TNF-α are undergoing clinical trials for several autoimmune pathologies including systemic lupus nephritis, psoriasis, and rheumatoid arthritis (Fig. 3). While these current anti-inflammatory or immune suppressive therapy will probably not be used in routine cases of hypertension, targeted treatment that addresses some of the pathways described in this review might prove helpful, at least in high-risk individuals. and might in the future prove effective reducing systemic complications of hypertension. Patient populations with resistant hypertension, who require 3 or more anti-hypertensive therapies, could possibly benefit from such therapeutic options as they are at increased risk for chronic kidney disease, stroke, and coronary artery disease, and all-cause mortality. Furthermore, as evident from the blood pressure lowering effects of MMF and TNF-α blockade, patients in the highest quartile of blood pressure could possibly benefit from additional anti-inflammatory therapies reducing hypertension-mediate organ disease. As an example, small molecules have been developed that effectively scavenge isoLGs, preventing adduct formation. Such agents have been used in humans [59], and might in the future prove effective reducing systemic complications of hypertension.

Numerous hypertensive stimuli (aldosterone, ang II, endothelial cell stretch, and NaCl) activate the innate immune system including monocytes, dendritic cells, and macrophages. These activated cells produce copious amounts of proinflammatory (IL-1β, TNF-α, etc.) cytokines and present antigen that potently drive T cell proliferation and activation. The precise orchestration of these inflammatory events drives immune cell infiltration into target organs including the vasculature and kidneys which promote the formation of hypertension. Immunomodulatory therapeutics including Mycophenolate Mofetil, and monoclonal antibodies that inhibit specific cytokine and/or chemokine signals could be potential therapies for human subjects with uncontrolled hypertension on anti-hypertensive therapies.
Another caveat regarding translation of animal model studies to treatment of humans is worth discussion. Virtually all animal models involve relatively young mice or rats, and interventions are usually imposed during the onset of disease. In contrast, we treat humans who are middle aged or older and who have often had hypertension for many years. Inflammation during the early phases of hypertension likely engenders irreversible alterations of vascular and renal structure that make hypertension permanent and not amenable to later treatment with anti-inflammatory approaches. As an example, we have observed vascular fibrosis occurs after a short period of hypertension in mice, and that this is not reversed after even after prolonged removal of the hypertensive stimulus [60]. Likewise, renal damage and fibrosis can occur as a result of inflammation and is likely irreversible once established. Thus, early detection and treatment of hypertension, and use of immune modulatory agents during initial phases of the disease might be necessary to observe a reduction in blood pressure. Such interventions might continue to prevent ongoing end-organ damage even in the absence of blood pressure lowering.
Summary table
What is known on this topic?
-
Cells of the innate and adaptive immune system have been observed in the kidneys and vasculature of experimental animals with hypertension and in humans with hypertension for one-half of a century.
-
Recent studies have delineated a role of these cells, and the cytokines they produce in increasing the blood pressure elevation in experimental models.
-
In addition to cytokines, these cells also release reactive oxygen species, matrix metalloproteinases and other factors that promote tissue remodeling and end-organ damage in hypertension.
What this review adds?
-
We provide a synthesis of existing literature on this topic and suggest future directions for therapeutic interventions in hypertension.
References
Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease. Circulation. 2019;140:e596–e646.
Harrison DG, Coffman TM, Wilcox CS. Pathophysiology of hypertension. Circ Res. 2021;128:847–863.
Madhur MS, Elijovich F, Alexander MR, Pitzer A, Ishimwe J, Beusecum JPV, et al. Hypertension: do inflammation and immunity hold the key to solving this epidemic? Circ Res. 2021;128:908–933.
Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II–induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460.
Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med. 2018;215:21–33.
Crowley SD, Song Y-S, Lin EE, Griffiths R, Kim H-S, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–R1097.
Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. 2013;304:R407–R414.
Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–2537.
Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, et al. CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118:1233–1243.
Caillon A, Mian MOR, Fraulob-Aquino JC, Huo K-G, Barhoumi T, Ouerd S, et al. γ/δ T cells mediate angiotensin II-induced hypertension and vascular injury. Circulation. 2017;135:2155–2162.
Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II–induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507.
Saleh MA, Norlander AE, Madhur MS. Inhibition of interleukin-17A, but not interleukin-17F, signaling lowers blood pressure, and reduces end-organ inflammation in angiotensin II–induced hypertension. Jacc Basic Transl Sci. 2016;1:606–616.
Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, et al. Interleukin-17A regulates renal sodium transporters and renal injury in angiotensin II–induced hypertension. Hypertension. 2018;68:167–174.
Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97:696–704.
Itani HA, Dikalova AE, McMaster WG, Nazarewicz RR, Bikineyeva AT, Harrison DG, et al. Mitochondrial cyclophilin D in vascular oxidative stress and hypertension. Hypertension. 2016;67:1218–1227.
Luther JM, Gainer JV, Murphey LJ, Yu C, Vaughan DE, Morrow JD, et al. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor–dependent mechanism. Hypertension. 2006;48:1050–1057.
Lee DL, Sturgis LC, Labazi H, Osborne JB, Fleming C, Pollock JS, et al. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ. 2006;290:H935–H940.
Alexander MR, Norlander AE, Elijovich F, Atreya RV, Gaye A, Gnecco JS, et al. Human monocyte transcriptional profiling identifies IL‐18 receptor accessory protein and lactoferrin as novel immune targets in hypertension. Brit J Pharm. 2019;176:2015–2027.
Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, et al. Role of p47phox in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515.
Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KRC, Xiao L, et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2016;126:50–67.
Weber DS, Rocic P, Mellis AM, Laude K, Lyle AN, Harrison DG, et al. Angiotensin II-induced hypertrophy is potentiated in mice overexpressing p22phox in vascular smooth muscle. Am J Physiol-Heart Circ. 2005;288:37–42.
Dikalova A, Clempus R, Lassègue B, Cheng G, McCoy J, Dikalov S, et al. Nox1 overexpression potentiates angiotensin ii-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676.
Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, et al. Nox1 is involved in angiotensin II–mediated hypertension. Circulation. 2005;112:2677–2685.
Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, et al. Decreased blood pressure in NOX1‐deficient mice. Febs Lett. 2006;580:497–504.
Mueller CF, Laude K, McNally JS, Harrison DG. ATVB in focus: redox mechanisms in blood vessels. Arteriosclerosis Thrombosis Vasc Biol. 2005;25:274–278.
Kirabo A, Fontana V, Faria APC, de, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124:4642–4656.
Ciuceis CD, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II–infused macrophage colony-stimulating factor–deficient mice. Arteriosclerosis Thrombosis Vasc Biol. 2005;25:2106–2113.
Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, et al. Lysozyme M–positive monocytes mediate angiotensin II–induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381.
Kossmann S, Hu H, Steven S, Schönfelder T, Fraccarollo D, Mikhed Y, et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem. 2014;289:27540–27550.
Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804.
Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, Muller WA. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–483.
Loperena R, Beusecum JPV, Itani HA, Engel N, Laroumanie F, Xiao L, et al. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. 2018;114:1547–1563.
Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21:1009–1020.
Barbaro NR, Beusecum JV, Xiao L, Carmo LD, Pitzer A, Loperena R. et al. Sodium activates human monocytes via the NADPH oxidase and isolevuglandin formation. Cardiovasc Res. 2021;117:1358–1371.
Kovarik JJ, Morisawa N, Wild J, Marton A, Takase‐Minegishi K, Minegishi S, et al. Adaptive physiological water conservation explains hypertension and muscle catabolism in experimental chronic renal failure. Acta Physiol. 2021;232:e13629.
Wild J, Jung R, Knopp T, Efentakis P, Benaki D, Grill A, et al. Aestivation motifs explain hypertension and muscle mass loss in mice with psoriatic skin barrier defect. Acta Physiol. 2021;232:e13628.
Beusecum JPV, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK, et al. High salt activates CD11c+ antigen-presenting cells via SGK (serum glucocorticoid kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertension. 2019;74:555–563.
Fehrenbach DJ, Abais-Battad JM, Dasinger JH, Lund H, Mattson DL. Salt-sensitive increase in macrophages in the kidneys of Dahl SS rats. Am J Physiol Ren. 2019;317:F361–F374.
Tang WHW, Kitai T, Hazen SL. Gut microbiota in cardiovascular health and disease. Circ Res. 2017;120:1183–1196.
Ferguson JF, Aden LA, Barbaro NR, Beusecum JPV, Xiao L, Simmons AJ, et al. High dietary salt-induced dendritic cell activation underlies microbial dysbiosis-associated hypertension. JCI Insight. 2019; 4. https://doi.org/10.1172/jci.insight.126241.
Karbach SH, Schönfelder T, Brandão I, Wilms E, Hörmann N, Jäckel S, et al. Gut microbiota promote angiotensin ii–induced arterial hypertension and vascular dysfunction. J Am Heart Assoc. 2016; 5. https://doi.org/10.1161/jaha.116.003698.
Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA. 2013;110:4410–4415.
Nakai M, Ribeiro RV, Stevens BR, Gill P, Muralitharan RR, Yiallourou S, et al. Essential hypertension is associated with changes in gut microbial metabolic pathways: a multisite analysis of ambulatory blood pressure. Hypertension. 2021;78:804–815.
Xiao L, Itani HA, Carmo LS, do, Carver LS, Breyer RM, Harrison DG. Central EP3 (E Prostanoid 3) receptors mediate salt-sensitive hypertension and immune activation. Hypertension. 2019;74:1507–1515.
Shah KH, Shi P, Giani JF, Janjulia T, Bernstein EA, Li Y, et al. Myeloid suppressor cells accumulate and regulate blood pressure in hypertension. Circ Res. 2015;117:858–869.
Chiasson VL, Bounds KR, Chatterjee P, Manandhar L, Pakanati AR, Hernandez M, et al. Myeloid-derived suppressor cells ameliorate cyclosporine A–induced hypertension in mice. Hypertension. 2018;71:199–207.
Lu X, Rudemiller NP, Wen Y, Ren J, Hammer GE, Griffiths R, et al. A20 in myeloid cells protects against hypertension by inhibiting dendritic cell-mediated t-cell activation. Circ Res. 2019;125:1055–1066.
Shao B-Z, Xu Z-Q, Han B-Z, Su D-F, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharm. 2015;6:262.
Krishnan SM, Sobey CG, Latz E, Mansell A, Drummond GRIL‐1β. and IL‐18: inflammatory markers or mediators of hypertension? Brit J Pharm. 2014;171:5589–5602.
Dorffel Y, Latsch C, Stuhlmuller B, Schreiber S, Scholze S, Burmester GR, et al. Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension. 1999;34:113–117.
Omi T, Kumada M, Kamesaki T, Okuda H, Munkhtulga L, Yanagisawa Y, et al. An intronic variable number of tandem repeat polymorphisms of the cold-induced autoinflammatory syndrome 1 (CIAS1) gene modifies gene expression and is associated with essential hypertension. Eur J Hum Genet. 2006;14:1295–1305.
Krishnan SM, Ling YH, Huuskes BM, Ferens DM, Saini N, Chan CT, et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc Res. 2018;115:776–787.
Ren X-S, Tong Y, Ling L, Chen D, Sun H-J, Zhou H, et al. NLRP3 gene deletion attenuates angiotensin II-induced phenotypic transformation of vascular smooth muscle cells and vascular remodeling. Cell Physiol Biochem. 2018;44:2269–2280.
Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho F, Pequeno IO, Olivon VC, et al. NLRP3 inflammasome mediates aldosterone-induced vascular damage. Circulation. 2016;134:1866–1880.
Rothman AM, MacFadyen J, Thuren T, Webb A, Harrison DG, Guzik TJ, et al. Effects of interleukin-1β inhibition on blood pressure, incident hypertension, and residual inflammatory risk. Hypertension. 2019;75:477–482.
Siedlinski M, Jozefczuk E, Xu X, Teumer A, Evangelou E, Schnabel RB, et al. White blood cells and blood pressure: a Mendelian randomization study. Circulation. 2020;141:1307–1317.
Yoshida S, Takeuchi T, Kotani T, Yamamoto N, Hata K, Nagai K, et al. Infliximab, a TNF-α inhibitor, reduces 24-h ambulatory blood pressure in rheumatoid arthritis patients. J Hum Hypertens. 2014;28:165–169.
Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol. 2006;17:S218–S225.
Pitchford LM, Driver PM, Fuller JC, Akers WS, Abumrad NN, Amarnath V, et al. Safety, tolerability, and pharmacokinetics of repeated oral doses of 2-hydroxybenzylamine acetate in healthy volunteers: a double-blind, randomized, placebo-controlled clinical trial. Bmc Pharm Toxicol. 2020;21:3.
Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, et al. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res. 2014;114:616–625.
Author information
Authors and Affiliations
Contributions
JVB, HM and DGH composed and edited this review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Van Beusecum, J.P., Moreno, H. & Harrison, D.G. Innate immunity and clinical hypertension. J Hum Hypertens 36, 503–509 (2022). https://doi.org/10.1038/s41371-021-00627-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41371-021-00627-z
- Springer Nature Limited
This article is cited by
-
Gut bacteria-derived extracellular vesicles and hypertension
Hypertension Research (2024)
-
Life is really simple, but we insist on making it complicated. (Confucius)
Hypertension Research (2023)
-
Lifestyle Medicine as a Treatment for Resistant Hypertension
Current Hypertension Reports (2023)
-
Sodium Homeostasis and Hypertension
Current Cardiology Reports (2023)
-
Treatment of male and female spontaneously hypertensive rats with TNF-α inhibitor etanercept increases markers of renal injury independent of an effect on blood pressure
Biology of Sex Differences (2022)