
Abstract
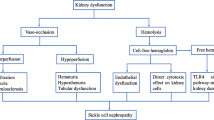
Early in life, patients with sickle cell anemia are at high risk for the development of hyposthenuria, enuresis, and hyperfiltration. Importantly, sickle cell patients are at risk for the development of chronic kidney disease (CKD) and end-stage kidney disease, often starting in the first or second decade. Patients with CKD have poor outcomes, and access to kidney transplant remains a barrier. Patients require annual monitoring for CKD by screening for albuminuria starting no later than 10 years of age. In addition to glomerular disease, patients with sickle cell anemia are at risk for the development of renal papillary necrosis and tubular dysfunction, which leads to a variety of complications, including polyuria, nocturnal enuresis and impaired excretion of acid and potassium.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Sickle cell anemia
- Chronic kidney disease
- Hyposthenuria
- Renal papillary necrosis
- Hyperfiltration
- Hypertension
- Acute kidney injury
- End-stage kidney disease
Sickle cell disease (SCD) is the most common inherited red blood cell disorder in the United States, impacting approximately 100,000 Americans and 1 in 365 African American births [1]. SCD is most prevalent in sub-Saharan Africa due to its protective inheritance against malaria. About 1000 children are born in Africa each day with SCD [2]. Some Hispanic and Indian populations have also been identified with up to 40% of residents having at least one sickle gene mutation.
The sickle cell mutation causes a hydrophobic valine to replace a hydrophilic glutamic acid in the sixth amino acid position of the β-globin protein. This mutation allows polymerization of hemoglobin S in the deoxyhemoglobin state. Two inherited β-globin sickle cell mutations result in the diagnosis of hemoglobin SS (HbSS). Different mutations in one of the β-globin subunits cause other forms of SCD. These include mutations that lead to no beta globin synthesis (β0 thalassemia) or minimal globin synthesis (β+ thalassemia) as well as other mutations, including hemoglobin C (HbC). The most prevalent, severe forms of SCD are HbSS and hemoglobin Sβ0 (HbSβ0) thalassemia; these genotypes are referred to as sickle cell anemia (SCA) and occur in about 70% of SCD patients. Other genotypes usually have less severe disease, and include hemoglobin Sβ+ (HbSβ+) thalassemia and hemoglobin SC (HbSC).
Sickle cell trait, which occurs in 1 in 13 African Americans, has been studied for its association with progressive kidney disease in adults, hematuria, renal papillary necrosis, and pyelonephritis during pregnancy. Sickle cell trait is associated with the rare cancer, renal medullary carcinoma. Patients with sickle cell trait do not have a significant pediatric clinical disease course due to the protective effect of one normal β-globin gene. Therefore, patients with sickle cell trait do not have SCD and, aside from counseling, do not receive follow-up care by a pediatric hematologist.
Hyposthenuria, Renal Papillary Necrosis, and Nocturnal Enuresis
Hyposthenuria
Pathophysiology: The development of hyposthenuria and renal papillary necrosis begin with sickling of the red blood cells in the vasa recta, leading to marked vascular changes. Necropsy studies have demonstrated almost complete destruction of the vasa recta and medullary capillaries in patients with SCA as compared to a reduced number of vasa recta in patients with sickle cell trait and HbSC disease [3]. These findings may be related to the impact of the hyperosmolar environment of the kidney on red cell rheology. In vitro models have demonstrated that sickle red blood cells exposed to even mildly hypertonic environments (sodium levels of 141 mEq/L) experience a delay in transit time, an increase in red cell rigidity and an increase in red cell adherence to vascular walls [4]. The renal medulla is an extremely hypertonic, hyperosmotic environment (800–1200 mOsm/kg) relative to the milder in vitro environments studied, which likely contributes to the more pronounced pathophysiologic changes seen in vivo. In addition, the renal medulla is a relatively hypoxic environment. Sickle red blood cells are stable in normoxia, but hemoglobin S begins to polymerize during the hypoxic state. Finally, acidic environments promote sickling of RBCs. Therefore, the combination of hypertonicity, hypoxia, and the acidotic environment in the renal medulla causes sickling of red blood cells within the vasa recta, with subsequent ischemic and reperfusion injury.
Epidemiology: A diminished urine concentrating ability is a well-established complication in patients with SCD [5,6,7,8]. The prevalence of hyposthenuria increases with disease severity (SCA > HbSC disease > sickle cell trait) [9, 10]. Urine concentration defects are first noted in infants and toddlers with SCA. In the baseline analysis of the BABY HUG study of children with SCA, only 30% of infants concentrated their urine >500 mOsm/kg, and only 13% of infants concentrated their urine >2 times their serum osmolality [11]. Of note, infants whose urine osmolality was >500 mOsm/kg after fluid deprivation had higher mean fetal hemoglobin concentrations, and the urine osmolality correlated with glomerular filtration rate (GFR).
Treatment: The BABY HUG study treated infants with 24 months of hydroxyurea, which increases fetal hemoglobin, versus placebo. While treatment with hydroxyurea for 24 months did not affect GFR, it did result in a significantly higher mean urine osmolality (495 mOsm/kg hydroxyurea vs. 452 mOsm/kg placebo) and a higher percentage of infants with a urine osmolality >500 mOsm/kg after fluid deprivation [12]. Murine models also suggest that a higher percentage of fetal hemoglobin and higher hemoglobin levels are associated with less severe concentrating defects [13]. Thus, hydroxyurea may decrease the urine concentrating defect early in life in SCA patients. Chronic transfusion therapy may also improve hyposthenuria [5, 6, 14]. However, preventing hyposthenuria is not an indication for initiating chronic transfusion; it is restricted to patients with increased risk of a poor central nervous system outcome or a very severe clinical course.
Interventions to prevent hyposthenuria should be initiated early in life. Red blood cell transfusions before the age of 10 years are effective in reversing hyposthenuria, but this reversibility was lost when initiated after the first decade [3, 5]. In a study of HbSC patients with a mean age of 11 years, hydroxyurea treatment for 12 months did not improve urinary concentrating ability [15]. This contrasts with the BABY HUG study and suggests loss of reversibility in older children.
Renal Papillary Necrosis
Another complication resulting from destruction of the renal vasculature is renal papillary necrosis. The pathophysiology is likely due to the renal medullary environment that promotes a higher level of sickling of red blood cells. It is believed that occlusion of the blood supply causes ischemia-induced necrosis of the renal medulla and papillae. This necrosis initiates subclinical and clinical hematuria. Pathology studies in SCD patients identified papillary necrosis in about one-third, most often located in the tips of the papillae [16]. Hematuria is secondary to changes in the permeability of the vasculature that allows red cell leakage into the collecting system.
Epidemiology: In radiologic studies, 30–75% of SCD patients have evidence of papillary necrosis [17,18,19]. Studies in Africa suggest 2% of patients will develop symptomatic renal papillary necrosis [20]. Risk factors for renal papillary necrosis are female sex, older age, more severe anemia and hypertension. Renal papillary necrosis is more common in the left kidney.
Diagnosis: While renal papillary necrosis is a common etiology of hematuria in pediatric patients with SCD, a standard diagnostic workup for hematuria should be performed on initial presentation. Since hemoglobinuria may cause a positive urine dipstick result, microscopic evaluation of the urine is necessary to confirm hematuria. SCD patients should also have a complete blood count (CBC) and creatinine to identify acute changes in hemoglobin and kidney function. Patients should be queried regarding other acute symptoms of SCD, prior history of hematuria, and medication use.
Several diagnostic imaging techniques can be considered in the initial evaluation of hematuria in a SCD patient. Ultrasound, which does not require contrast and is readily available, will identify hydronephrosis, and may provide evidence for a kidney stone as a cause of hematuria. A renal mass suggesting renal medullary carcinoma is more often identified in patients with sickle cell trait than SCA [21]. Findings on ultrasound suggestive of renal papillary necrosis include filling defects and necrosed papillae in cavities [22]. However, ultrasound is not the optimal imaging for identifying renal papillary necrosis. Most patients require a contrast evaluation, including intravenous urography, retrograde pyelography, or CT with contrast [23]. Intravenous urography and retrograde pyelography have less radiation exposure than CT [23]. Findings on a contrast study will often demonstrate filling defects of the renal calyx, including deformities of the renal papillae (hooks, spurs) and a blunted calyx [22].
Treatment: Some cases of hematuria due to papillary necrosis will be mild, painless, and self-resolve; other cases may require therapy. There are no guidelines addressing the efficacy of supportive care or when patients should be admitted to the hospital for care. Treatment may include intravenous (IV) fluids, analgesia, bedrest, alkalization, and low-dose aminocaproic acid [24, 25]. Patients who develop severe anemia require transfusion. Surgical interventions such as papillary tamponade, shunt placement, or nephrectomy are rarely indicated unless a pediatric patient is experiencing severe, persistent, life-threatening hemorrhage [16, 26].
The goal of fluid therapy is to ensure adequate hydration and to maintain high urine output. For mild cases, aggressive oral hydration as an outpatient is sufficient. For more severe cases, IV fluids can be prescribed in either outpatient day hospital settings or inpatient units. Patients can be administered fluids at maintenance to 1.5 times maintenance with or without a loop diuretic to further ensure adequate urinary output [27]. Since acidosis promotes sickling, adding base to the IV fluids to create a more alkaline environment to reduce sickling is theoretically appealing; however, there are no trials demonstrating benefit in SCD patients with papillary necrosis [28]. Bedrest may reduce the risk for dislodging of clots. Clinicians should closely monitor for the development of respiratory symptoms or fluid overload in patients receiving higher rates of IVF and/or suggested bedrest. Analgesia is used in patients with painful hematuria. If patients presenting with painless hematuria progress to painful hematuria, additional imaging may be required to evaluate new obstructive disease due to blood clots. Low dose aminocaproic acid at 20–50 mg/kg IV or po every 8 or 12 h has been used in severe cases as well as lower maintenance oral dosing [24, 25, 29]. SCD is a hypercoagulable state so close monitoring for new clot formation or ureteral obstruction should occur when using aminocaproic acid. Transfusion therapy can improve the anemia and reduce the concentration of sickle red blood cells, which should reduce sickling. In very severe cases, exchange transfusion may be used to significantly reduce the concentration of sickle cells, often to a sickle cell concentration of less than 30%. Reducing the sickle cell concentration may not treat the acute complication, but may allow more rapid recovery and prevent early recurrence of another renal injury.
Nocturnal Enuresis
Epidemiology: Primary nocturnal enuresis (PNE) is bedwetting that occurs in an individual who has never been dry at night, and is usually not associated with daytime wetting symptoms. PNE affects approximately 15% of children aged 5 years in the general population, and spontaneously remits at a rate of 15% per year [30]. The prevalence of PNE in SCD is much higher; children with SCD aged 14–17 years old were five times more likely to have PNE than controls [31,32,33,34,35]. Similar to the general population, PNE is more common in males, younger children and children with a positive family history of PNE [31,32,33,34,35]. Children with SCA and PNE are more likely to have sleep-disordered breathing [36]. In a study of 8-year-old Jamaican children, the prevalence of enuresis was 52% for boys and 38% for girls with HbSS disease, and 10% for boys and 20% for girls with HbSC disease. The prevalence of PNE in HbSS disease was significantly more common than HbSC disease or controls, but there was no significant difference between HbSC disease and controls. There was no significant difference by sex [37].
Risk Factors: PNE in HbSS disease has been attributed to hyposthenuria and polyuria. However, in a study comparing HbSS patients with and without enuresis, there was no difference in urinary concentrating ability or overnight urine volume [38]. However, the bladder capacity corrected to body surface area was lower, and the ratio of overnight urine volume divided by bladder capacity was higher in enuretic compared to non-enuretic children. A high prevalence of daytime symptoms of overactive bladder was also present in those with nocturnal enuresis and SCA [34]. These results suggest that hyposthenuria-induced polyuria may not be the primary cause of PNE in HbSS disease. In addition to urinary tract pathology, an association has been identified between sleep disordered breathing and PNE [36].
Therapy: Several pharmacological and behavioral therapies have been identified to treat patients with PNE in the general populations [39]. However, there are no controlled trials demonstrating benefit in children with SCD. One prospective study of 10 patients with SCD treated with desmopressin reported improvement in six patients [32]. Hence, the evidence for pharmacologic treatment is inadequate, and thus the focus of therapy is watchful waiting and bed-wetting alarms in some patients [40].
Albuminuria
Epidemiology: Fifty to seventy percent of adults with SCA have albuminuria in large cross-sectional studies [41,42,43]. Children with SCD develop albuminuria around 5–10 years of age and the prevalence increases throughout adolescence. The reported prevalence of albuminuria in cross-sectional pediatric SCA studies is 20–40% [44,45,46]. There is a need for longitudinal data on the natural history of the progression of kidney disease and development of end-stage kidney disease (ESKD) among pediatric patients with albuminuria as they transition into adulthood [47].
Pathology: Patients with SCA develop albuminuria due to either direct glomerular injury or impaired tubular reabsorption of albumin. In adults with SCD, glomerular complications include glomerular hypertrophy, focal segmental glomerulosclerosis, and membranoproliferative glomerulonephritis [48]. Kidney pathologic data was reported from 36 pediatric patients that underwent renal biopsy for proteinuria or low estimated GFR (eGFR) [49]. The majority had glomerular hypertrophy. In addition, the majority of the biopsies had mesangial hypercellularity and/or increased mesangial matrix. All patients in this cohort with mesangial hypercellularity, had proteinuria, including 88% with nephrotic range proteinuria. Eleven of the 36 patients had focal segmental glomerulosclerosis, six of the 11 also had global sclerosis. Five patients had membranoproliferative glomerulonephritis. On electron microscopy, more than 50% of biopsies had podocyte effacement, and a quarter had mesangial deposits. This study supports the importance of nephrology evaluation of children with SCD and significant proteinuria or decreased GFR.
Impaired tubular reabsorption of albumin may also contribute to albuminuria independent of glomerular disease in patients with SCA. Some albumin is normally filtered at the glomerulus and then reabsorbed by receptor-mediated endocytosis by megalin and cubilin in the proximal tubule [50]. However, internalization of albumin is subject to competition from many other proteins. Importantly in SCD, hemoglobin dimers can also be reabsorbed in the proximal tubule. In preclinical models, the addition of oxyhemoglobin to proximal tubule cells significantly reduced albumin uptake [51]. As SCD patients have daily variations in the amount of hemolysis consequent hemoglobinuria, it is plausible that albuminuria may vary depending on the amount of hemolysis. Studies have demonstrated that a single urine sample with albuminuria may not represent persistent albuminuria [47, 52]. While free hemoglobin or heme reuptake may lead to non-glomerular albuminuria, the uptake of free heme by the proximal tubule may induce pathologic changes and kidney disease progression, as described in the section on acute kidney injury.
Risk factors: Older age is a risk factor for albuminuria in SCD [43, 47]. Inheritance of two apolipoprotein L1 (APOL1) risk alleles, which are common in people of African descent, is associated with increased risk of albuminuria and CKD in SCD [53, 54]. Albuminuria in SCD patients with two APOL1 risk alleles begins in the first decade of life [44]. Serum hemoglobin is inversely related to risk of albuminuria [45, 55,56,57]. Patients with severe anemia in their second year of life are more likely to develop albuminuria earlier in life [45]. There is inconsistent data on the association of albuminuria with leukocytosis, hemolytic markers (lactate dehydrogenase), and blood pressure [46, 58,59,60,61].
Similar to diabetes, hyperfiltration is a risk factor for albuminuria in SCD. In cross-sectional studies, there are conflicting results on the association of eGFR with albuminuria [55, 62, 63]. A large, prospective, pediatric cohort evaluated the impact of hyperfiltration on progression of albuminuria; patients with hyperfiltration in the first decade of life were more likely to develop albuminuria at an earlier age [64]. Patients that developed albuminuria had a significant increase in eGFR prior to developing albuminuria while patients without albuminuria did not experience a significant rise in eGFR during the first decade of life. Adult data also suggests that hyperfiltration is associated with albuminuria [65].
Diagnosis: The National Heart, Lung, and Blood Institute (NHLBI) guidelines recommend screening for albuminuria by age 10 years, with annual screening thereafter [66]. Patients with a positive screening result should have a first morning test for albuminuria (albumin/creatinine ratio [ACR] >30 mg/g), with referral to a kidney specialist if positive. If a first morning void is not available or feasible, scheduling patients for an early morning appointment to obtain a second morning void may be of benefit. This second urine measurement is important as patients with SCD experience intermittent albuminuria, but may not have persistent albuminuria. About 25–50% of SCA patients with ACR <100 mg/g do not have persistent albuminuria; patients with ACR >100 mg/g are more likely to have persistent albuminuria [47, 67].
Albuminuria can begin prior to 10 years of age; therefore, some centers may begin screening for albuminuria earlier than 10 years of age [44, 64]. This is especially relevant in patients with two APOL1 risk alleles since almost 25% of these patients with SCD develop albuminuria prior to age 10 years.
The presence of severe albuminuria (ACR >300 mg/g) in SCD is less likely in pediatric patients than adults. As severe albuminuria is a rare complication in children with SCA, patients presenting with severe albuminuria require a complete diagnostic workup for proteinuria.
Treatment: The therapeutic approach in patients with SCD and albuminuria focuses on traditional modifiers of SCD and interventions to reduce hyperfiltration.
SCD Modifying Therapies: All patients with SCA should be offered hydroxyurea, a daily oral therapy, starting at 9 months of age regardless of clinical complications [66]. Therefore, patients that have progressed to albuminuria should be encouraged to begin hydroxyurea or improve adherence if nonadherence is present. Hydroxyurea induces fetal hemoglobin, raises hemoglobin, and reduces hemolysis and inflammation. The dose of hydroxyurea may be increased to 35 mg/kg in children to allow for maximal effect without reducing the absolute neutrophil count below 1000–2000/μL [68]. Patients on hydroxyurea have reduced hospitalizations, pain events, and episodes of acute chest syndrome; this may provide downstream renoprotective effect [69]. Pediatric patients on hydroxyurea with higher fetal hemoglobin levels have a lower prevalence of albuminuria [70]. As hydroxyurea improves anemia and reduces hemolysis, biologic plausibility suggests that early use of hydroxyurea should decrease glomerular hyperperfusion and reduce free hemoglobin exposure to proximal tubule cells; however, randomized controlled data demonstrating this benefit is lacking. Longitudinal adult studies have demonstrated some benefit of hydroxyurea when started prior to the development of severe albuminuria [71].
Chronic transfusion therapy is a proven therapy to reduce progressive SCD and treat acute SCD complications. By performing chronic monthly transfusion therapy, patients are maintained with a very low sickle cell concentration, often to a lower percentage of sickle cells than patients with sickle cell trait. Chronic transfusion protocols have demonstrated a clear benefit to prevent a first or second stroke in patients at risk. The use of transfusion therapy would likely benefit pediatric patients with albuminuria or other renal complications, but using chronic transfusion for this indication has not been systematically studied for benefit or cost-effectiveness in large studies [72]. One complication of chronic transfusion is the development of iron overload. After 10–12 transfusions, many patients will be started on iron chelation, which can have nephrotoxic effects [73].
A few SCD modifying therapies have been recently approved by the Food and Drug Administration (FDA), but these approvals were not based on a renal indication. Crizanlizumab is a P-selectin inhibitor administered intravenously monthly that can prolong the time to the next pain event in patients with 2–10 pain events annually [74]. It is currently being studied for renoprotective effects (NCT04053764). Voxelotor is a daily oral medication that increases the oxygen affinity of sickle red blood cells. This increased oxygen affinity reduces sickling of red blood cells and was FDA approved for its ability to raise hemoglobin [75]. As a medication that reduces hemoglobinuria and likely reduces heme exposure in the kidneys, this therapy may have renoprotective effects and is being studied in adults with high risk for CKD (NCT04335721). Finally, L-glutamine received FDA approval for the reduction in painful events among pediatric and adult patients with two or more painful events in the last year [76]. L-glutamine may protect SCD patients by reducing oxidative stress through an increase in the availability of glutathione. Future studies will need to be performed to determine the effect of glutamine on renal endpoints.
Angiotensin Blockade: Based on the benefit of angiotensin converting enzyme inhibitors (ACEI) and angiotensin receptor blockers (ARBs) in patients with diabetes to reduce proteinuria and lower intraglomerular pressure, SCD patients with albuminuria may be started on these medications. The American Society of Hematology (ASH) Guidelines for the management of SCD includes an evidence-based review of ACEI/ARBs in patients with albuminuria [77]. Based on a low certainty in the evidence, the guideline panel decided to “suggest” the use of ACEI/ARBs for albuminuria rather than “recommend” it for albuminuria. One randomized controlled trial and several observational studies that suggested benefit were evaluated [78]. The methodologic experts and guideline panel members determined from these studies that 63% of patients who received ACEI showed improvement in albuminuria; the panel also identified that 100% of 30 patients who received ARBs had improved albuminuria at some point during the treatment [77,78,79,80,81,82]. Patients started on ACEI/ARBs should be reassessed in one week and then serially for changes in GFR and serum potassium. In addition, these medications should be held during acute illness, planned IV contrast administration, or prior to surgery or other procedures.
Endothelin Receptor Antagonist: Endothelin receptor A (ETA) is responsible for vasoconstriction, inflammation, and adhesion. Endothelin B receptor mediates nitric oxide (NO) production and vasodilation. It is likely that specific blockade of the ETA receptor could improve renal and SCD outcomes as compared to dual blockade. Murine sickle cell data showed a benefit after 10 weeks of ETA receptor blockade, as compared to a combined ETA/B antagonist, in reducing plasma ET-1 levels, urinary protein and albumin excretion and maintaining a stable GFR [83]. In adults with SCA, a phase I double blind study of ETA antagonist ambrisentan has been completed [84]. The data suggests a potential benefit for albuminuria and improved microvascular function.
Glomerular Filtration Rate
Epidemiology: Patients with SCD experience hyperfiltration early in life. Infants enrolled in a randomized trial underwent measured GFR (mGFR) at around one year of age and again at study exit 2 years later. At one year of age, participants had a normal mGFR; 2 years later, the mGFR had increased by a mean of 20 mL/min/1.73 m2 to 146 mL/min/1.73 m2 [69]. This increase in GFR occurs throughout early life and cross-sectional studies have identified that around 40–80% of SCD patients will develop hyperfiltration [12, 46, 58, 85]. After this early rise in GFR, pediatric patients likely experience a plateau in GFR around 6–12 years of age before they begin a decline in GFR [58, 64]. Some pediatric patients will progress to a GFR <90 mL/min/1.73 m2; these patients should be evaluated to investigate non-SCD related causes of CKD [62, 86].
Risk Factors: Many cross-sectional studies have identified potential associations with GFR. Data is conflicting for the role of anemia, leukocytosis, blood pressure, male sex, and LDH [12, 58, 85, 87]. A few studies have explored the association of uric acid and GFR in SCD; these studies suggest that uric acid may be an early marker of risk for GFR decline [88,89,90]. As discussed in the albuminuria section, several studies have identified a link between hyperfiltration (elevated GFR) and albuminuria.
Treatment: Treatment approaches to prevent hyperfiltration or glomerular injury have not been fully explored. Treatment of infants with hydroxyurea for 2 years did not prevent the increase in eGFR or reduce the number of participants who developed hyperfiltration [12]. In contrast, some older participants started on hydroxyurea experience a decrease in eGFR that was associated with change in fetal hemoglobin and LDH [91]. The studies of angiotensin targeted therapies often focused on albuminuria as the primary outcome; however, some of these studies have also reported the impact of treatment on change in GFR. In a well-designed study of enalapril in adults, treatment for 2 weeks did not change the mean GFR, effective renal plasma flow, or filtration fraction [80]. Two studies using losartan therapy for 6 months or 1 year also did not identify a change in GFR while on treatment [92, 93].
One concern in clinical care and conducting research to evaluate eGFR or change in eGFR in patients with SCA is that the equations used to estimate GFR in children and adults have low precision and accuracy [12, 94,95,96]. In addition to the concern for the bias in a single eGFR measurement, individual patients do not demonstrate concordance between eGFR and mGFR in longitudinal studies [96]. Therefore, clinicians and researchers need to evaluate GFR changes at several time points to reduce these inaccuracies. Formulas using either cystatin C alone or cystatin C and creatinine may have the best precision and accuracy among the current formulas. A current study is attempting to develop novel eGFR equations for children and adult participants with SCA (NCT 04380610). A second concern is that after a period of hyperfiltration in early childhood, a decline in eGFR occurs. Several studies have evaluated the change in eGFR after an intervention or in the evaluation of risk factors. A rapid decline in eGFR in adults is associated with higher morbidity and mortality [97,98,99]. However, shorter-term longitudinal pediatric studies of patients with baseline hyperfiltration struggle to determine whether a decline in eGFR during the study is related to an improvement in renal function back to baseline, the beginning of a progressive decline in renal function to CKD, or regression to the mean among participants with higher eGFR at one time point. Long-term pediatric research is vital to determine whether a change in eGFR represents a benefit or risk.
Progressive CKD, End-Stage Kidney Disease, and Mortality
It is important to monitor pediatric patients with SCA for progression from glomerular hypertrophy with elevated GFR to CKD with decreased GFR due to sclerosis and fibrosis [80]. CKD in SCA is an independent risk factor for early death [100]. The management of CKD in SCD is similar to other patients with CKD, including control of hypertension and proteinuria with an ACEI or ARB. Currently, there are no FDA-approved medications for the specific treatment of sickle cell nephropathy.
Renal Replacement Therapy: The average age of initiation of renal replacement therapy (RRT) in SCD patients is 40–45 years [101,102,103]. Patients with SCD are currently a small minority of U.S. dialysis patients, but this population is expected to grow as overall life expectancy improves [104]. A longitudinal cohort study of SCA patients published in 1991 reported that the median survival of patients requiring dialysis was a mere 4 years [105]. In a more contemporary study (2005–2009), the hazard ratio for mortality among SCD patients with ESKD was 2.8 (95% CI 2.31–3.38) compared to those without SCD as the primary cause of renal failure, and 26.3% of incident SCD ESKD patients died within the first year of dialysis [101].
SCD patients are less likely than other ESKD patients to have a functioning arteriovenous fistula at the time of hemodialysis initiation, an important quality metric tied to improved RRT survival [103]. SCD patients on RRT experience greater rates of bacteremia and sepsis, atrial flutter and fibrillation, congestive heart failure exacerbations, and major hemorrhage than other ESKD patients [103]. SCD patients on RRT have more RBC transfusions than other RRT patients. SCD patients not receiving erythropoietin stimulating agents (ESAs) have the highest transfusion burden, while those treated with ESAs and hydroxyurea have the lowest transfusion burden [103]. The 2019 ASH guidelines recommend that hydroxyurea and ESAs be used in combination to promote fetal hemoglobin production. Clinicians should use a lower hemoglobin threshold (<10 g/dL) when prescribing this combination therapy as higher hemoglobin levels, especially with higher HbS percentage, may be associated with increased SCD complications [77].
Renal Transplantation: For those with advanced CKD, the ASH evidenced-based guidelines suggests referral for renal transplant. As SCD is associated with chronic inflammation, the guidelines suggest judicious use of corticosteroids in post-transplant protocols due to the risk for vaso-occlusive pain with the increase in WBCs that accompanies steroid use [77]. Referral rates for transplantation for SCA patients are lower than other ESKD patients, even after adjusting for covariates [102]. One potential explanation for this disparity may be related to historical data reporting high rates of complication and poor graft survival in SCD patients. An analysis of the U.S. Renal Data System data from 1984–1996 showed that SCA patients and other renal transplant recipients have similar 1-year cadaveric graft survival [106]. Recipients had higher rates of 3-year graft loss (RR 1.60) and a significantly higher adjusted mortality rate at 1 year (RR 2.95) and 3 years (RR 2.82) compared to non-SCA transplant recipients [106]. However, more recent data (2000–2011 compared to 1988–1999) showed that 6-year survival among SCA recipients improved in the more recent era compared to the early era (78% versus 55.7%, p < 0.001) [107]. While the 6-year patient survival was still significantly lower than non-SCA recipients (HR for mortality 2.03, 95% CI 1.31–3.16), it was equal to that of black diabetic transplant recipients [107].
An additional concern that may hinder referral for renal transplantation is the perceived risk of alloimmunization if blood transfusion is required in anemic SCD transplant recipients. It is not surprising, given the lifetime exposure to RBC transfusions, that a greater proportion of SCA renal transplant recipients had allosensitization, with panel reactive antibodies >20% [107]. One retrospective multicenter study compared the proportion of de novo donor specific antibodies (DSAs) and graft survival among SCD renal transplant recipients who received regular automated exchange blood transfusions (EBT) pre or post renal transplant versus those who did not receive regular EBT [108]. Goals for EBT were to maintain hemoglobin >9 g/dL, to reduce HbS to <30%, and to reduce sickling-related complications. The median number of red blood cell units transfused per year was 37 and 8 in the EBT and non-EBT group, respectively. Overall, patient survival, graft survival, and graft function were superior in those who were on EBT, and the proportion of patients who developed de novo DSAs was not different (20% and 21%) between the groups. In addition, the incidence of rejection was lower in those on EBT (28% vs 54%). These data, while limited by sample size, indicate that blood transfusions peri-transplant are effective, safe and lead to improved outcomes in the SCA renal transplant population.
SCA patients with advanced CKD should therefore be counseled on the shortened graft survival and increased complication rates expected after transplantation, but should not be restricted from renal transplant access. Moreover, they should receive blood transfusions as needed pre or post transplantation.
Acute Kidney Injury
Patients with SCD may be at increased risk for acute kidney injury (AKI) due to high use of nephrotoxic medications and hemolysis causing proximal tubule injury from free heme and hemoglobin exposure [51, 109]. Acute pain events are a leading cause of hospitalization in children with SCD and repeated acute pain events can progress to chronic pain. Aggressive and early pain management is important, and many centers utilize individualized pain plans for home and during hospitalizations [110]. These pain plans often include non-steroidal anti-inflammatory medications (NSAIDs) during home pain events and inpatient IV ketorolac as adjunctive therapy to opioids [111,112,113]. The 2014 NIH guidelines provide a moderate recommendation for the use of NSAIDs for mild to moderate pain in the absence of contraindications [110]. The 2019 ASH guidelines suggest a short course of NSAIDs in addition to opioids for acute pain management based on a low certainty of evidence [113]. The ASH guidelines remark that patients with known risk for renal toxicity should be identified as the mild potential benefit to NSAIDs for pain may not outweigh the risks associated with NSAID use. One concern with these guidelines is that pediatric patients may utilize a significant amount NSAIDs without appropriate monitoring of kidney function or fluid intake. Rarely, a single dose of ketorolac in the absence of volume depletion may precipitate irreversible renal failure [114].
In addition to acute pain events, infections may lead to use of nephrotoxic medications in SCD patients, who are increased risk for infection with pneumococcus and other encapsulated bacteria [115,116,117]. Patients may receive vancomycin or other nephrotoxic antibiotics for acute chest syndrome, fever, sepsis, or skin infections. It is important to consider the pros and cons of nephrotoxic medications given the potential long-term exposure and underlying risk of developing sickle cell nephropathy; monitoring of kidney function is needed when patients with SCD are at risk of AKI from nephrotoxins, volume depletion or infection.
When pediatric SCD patients are admitted for pain events or acute chest syndrome, AKI, when defined as an increase in creatinine occurs in 10–20% [118,119,120]. Risk factors for AKI in these studies include ketorolac exposure and an acute drop in hemoglobin, which probably reflects increased hemolysis and tubular exposure to free heme and hemoglobin. In another study using coding data, 1.4% of hospitalized SCD patients develop AKI, with risk factors including HbSS genotype, older age and greater number of total hospitalizations [121].
Adult studies of SCD patients demonstrate a higher incidence of AKI during hospitalization than described in children [122]. In a prospective study of adult SCD patients observed for a median of 5.5 years, 46% developed AKI [122]. Patients with AKI were older, had lower hemoglobin levels, higher white blood cell counts, and higher use of vancomycin. Moreover, genetic variants of heme catabolism (HMOX1) were independently associated with the development of AKI. Finally, adults with AKI were likely to develop CKD sooner, with the highest risk associated with more severe AKI.
Tubular Abnormalities and Acidification Defects
Along with defects in urinary concentrating ability (see above), SCD patients have well-described defects in other tubular functions, including urinary acidification and potassium excretion. SCA patients have an incomplete distal renal tubular acidosis as evidenced by decreased urine acidification in response to a systemic acid load [10, 123]. In one study, 42% of adult SCA patients had a metabolic acidosis [124]. Defects in acid excretion are associated with poor outcomes. In one adult study, the lowest tertile of urinary ammonia excretion increased the risk of ESKD [125]. Acidification of the urine was impaired in 52% of adults SCD patients, and was associated with older age, higher serum uric acid, increased hemolysis, lower eGFR, and lower serum bicarbonate [121]. Poor urinary ammonium excretion, as a measure of acid excretion, is associated with poor urinary concentrating ability [121].
SCA patients also have impaired potassium excretion [126]. In this study, patients had a normal renin-aldosterone axis and normal GFR, but had impaired potassium excretion, urine acidification and urinary concentrating ability, indicating that a severe distal tubular dysfunction is present in SCA patients despite preservation of glomerular function.
In one study of 24 children without decreased GFR, 75% had hyperphosphatemia, but serum calcium was normal [127]. Seventy-nine percent of participants had elevated FGF-23 levels, which is the expected physiologic response to hyperphosphatemia and would be expected to increase renal phosphate excretion. However, patients had evidence of impaired phosphate excretion, suggesting t that SCA patients have a proximal tubular resistance to FGF-23 before evidence of GFR decline.
Hypertension
A meta-analysis conducted for the 2014 NHLBI guidelines demonstrated that patients with HbSS genotype have lower diastolic and systolic blood pressures than healthy children. A large cohort study developed 90th percentile curves for children with SCD [128]. Hence, it may be appropriate to use SCD-specific blood pressure (BP) tables when evaluating BP in children with SCD.
There is an association in SCD patients between elevated BP and higher hemoglobin values [128, 129]. Hypertension in SCD patients has been associated with increased risk of acute stroke, although causality has not been established [128, 130]. Finally, data suggests that there is a direct correlation between higher blood pressure and increased mortality [128].
SCD patients have a high prevalence of masked hypertension and “white coat” hypertension in studies using 24-h ambulatory blood pressure monitoring (ABPM). Patients with SCA have only moderate concordance between in-clinic blood pressure and ABPM, with 25–33% having masked hypertension and 60% having white coat hypertension [61, 90, 131,132,133]. This may explain why some studies of in-clinic BP did not find a correlation between albuminuria and hypertension while there was an association when using ABPM [90, 132, 133]. Hence, since ABPM is the gold standard for measurement of BP, it is especially important to follow the American Academy of Pediatrics Guidelines and perform ABPM prior to initiation of anti-hypertensive treatment in SCD patients. Screening for masked hypertension may be appropriate in children with SCD and evidence of kidney disease. Patients identified with nocturnal hypertension or abnormal nocturnal dipping may have an increased risk for more rapid annual decline in GFR, albuminuria, and silent cerebral infarcts [61, 90, 131,132,133].
The ASH evidence-based guidelines for the management of SCD patients recommends a lower BP goal (<130/80) than the usual goal in adults without additional co-morbidities (<140/90) [77]. This strong recommendation was based on a moderate certainty in the evidence. The guidelines did not address BP management in children, and thus there are no recommendations to have a lower BP goal in SCD patients than in healthy children. Hence, BP management in children with SCD should follow the guidelines for healthy children.
References
Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38:S512–21.
Piel FB, Patil AP, Howes RE, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010;1:104.
Statius van Eps LW, Pinedo-Veels C, de Vries GH, et al. Nature of concentrating defect in sickle-cell nephropathy. Microradioangiographic studies. Lancet. 1970;1:450–2.
Carden MA, Fay ME, Lu X, et al. Extracellular fluid tonicity impacts sickle red blood cell deformability and adhesion. Blood. 2017;130:2654–63.
Itano HA, Keitel HG, Thompson D. Hyposthenuria in sickle cell anemia: a reversible renal defect. J Clin Invest. 1956;35:998–1007.
Cochran RT Jr. Hyposthenuria in sickle cell states. Arch Intern Med. 1963;112:222–5.
Perillie PE, Epstein FH. Sickling phenomenon produced by hypertonic solutions: a possible explanation for the hyposthenuria of sicklemia. J Clin Invest. 1963;42:570–80.
Kunz HW, Pratt EL, Mellin GW, et al. Impairment of urinary concentration in sickle cell anemia. Pediatrics. 1954;13:352–6.
Schlitt L, Keitel HG. Pathogenesis of hyposthenuria in persons with sickle cell anemia or the sickle cell trait. Pediatrics. 1960;26:249–54.
Badr M, El Koumi MA, Ali YF, et al. Renal tubular dysfunction in children with sickle cell haemoglobinopathy. Nephrology (Carlton). 2013;18:299–303.
Miller ST, Wang WC, Iyer R, et al. Urine concentrating ability in infants with sickle cell disease: baseline data from the phase III trial of hydroxyurea (BABY HUG). Pediatr Blood Cancer. 2010;54:265–8.
Alvarez O, Miller ST, Wang WC, et al. Effect of hydroxyurea treatment on renal function parameters: results from the multi-center placebo-controlled BABY HUG clinical trial for infants with sickle cell anemia. Pediatr Blood Cancer. 2012;59:668–74.
Lebensburger JD, Pestina TI, Ware RE, et al. Hydroxyurea therapy requires HbF induction for clinical benefit in a sickle cell mouse model. Haematologica. 2010;95:1599–603.
Statius van Eps LW, Schouten H, La Porte-Wijsman LW, et al. The influence of red blood cell transfusions on the hyposthenuria and renal hemodynamics of sickle cell anemia. Clin Chim Acta. 1967;17:449–61.
Iyer R, Baliga R, Nagel RL, et al. Maximum urine concentrating ability in children with Hb SC disease: effects of hydroxyurea. Am J Hematol. 2000;64:47–52.
Mostofi FK, Vorder Bruegge CF, Diggs LW. Lesions in kidneys removed for unilateral hematuria in sickle-cell disease. AMA Arch Pathol. 1957;63:336–51.
Pandya KK, Koshy M, Brown N, et al. Renal papillary necrosis in sickle cell hemoglobinopathies. J Urol. 1976;115:497–501.
Odita JC, Ugbodaga CI, Okafor LA, et al. Urographic changes in homozygous sickle cell disease. Diagn Imaging. 1983;52:259–63.
Eckert DE, Jonutis AJ, Davidson AJ. The incidence and manifestations of urographic papillary abnormalities in patients with S hemoglobinopathies. Radiology. 1974;113:59–63.
Madu AJ, Okoye AE, Ajuba IC, et al. Prevalence and associations of symptomatic renal papillary necrosis in sickle cell anemia patients in south-eastern Nigeria. Niger J Clin Pract. 2016;19:471–4.
Alvarez O, Rodriguez MM, Jordan L, et al. Renal medullary carcinoma and sickle cell trait: a systematic review. Pediatr Blood Cancer. 2015;62:1694–9.
Sutariya HC, Pandya VK. Renal papillary necrosis: role of radiology. J Clin Diagn Res. 2016;10:TD10–2.
Henderickx M, Brits T, De Baets K, et al. Renal papillary necrosis in patients with sickle cell disease: how to recognize this ‘forgotten’ diagnosis. J Pediatr Urol. 2017;13:250–6.
Gabrovsky A, Aderinto A, Spevak M, et al. Low dose, oral epsilon aminocaproic acid for renal papillary necrosis and massive hemorrhage in hemoglobin SC disease. Pediatr Blood Cancer. 2010;54:148–50.
Black WD, Hatch FE, Acchiardo S. Aminocaproic acid in prolonged hematuria of patients with sicklemia. Arch Intern Med. 1976;136:678–81.
Kiryluk K, Jadoon A, Gupta M, et al. Sickle cell trait and gross hematuria. Kidney Int. 2007;71:706–10.
Zadeii G, Lohr JW. Renal papillary necrosis in a patient with sickle cell trait. J Am Soc Nephrol. 1997;8:1034–9.
Meyersfield SA, Morganstern SL, Seery W, et al. Medical management of refractory hematuria in sickle-cell trait. Urology. 1976;8:112–3.
Baldree LA, Ault BH, Chesney CM, et al. Intravenous desmopressin acetate in children with sickle trait and persistent macroscopic hematuria. Pediatrics. 1990;86:238–43.
Arda E, Cakiroglu B, Thomas DT. Primary nocturnal enuresis: a review. Nephrourol Mon. 2016;8:e35809.
Esezobor CI, Akintan P, Nwaogazie U, et al. Enuresis in children and adolescents with sickle cell anaemia is more frequent and substantially different from the general population. PLoS One. 2018;13:e0201860.
Figueroa TE, Benaim E, Griggs ST, et al. Enuresis in sickle cell disease. J Urol. 1995;153:1987–9.
Barakat LP, Smith-Whitley K, Schulman S, et al. Nocturnal enuresis in pediatric sickle cell disease. J Dev Behav Pediatr. 2001;22:300–5.
Portocarrero ML, Portocarrero ML, Sobral MM, et al. Prevalence of enuresis and daytime urinary incontinence in children and adolescents with sickle cell disease. J Urol. 2012;187:1037–40.
Ekinci O, Celik T, Unal S, et al. Nocturnal enuresis in sickle cell disease and thalassemia major: associated factors in a clinical sample. Int J Hematol. 2013;98:430–6.
Lehmann GC, Bell TR, Kirkham FJ, et al. Enuresis associated with sleep disordered breathing in children with sickle cell anemia. J Urol. 2012;188:1572–6.
Readett DR, Morris JS, Serjeant GR. Nocturnal enuresis in sickle cell haemoglobinopathies. Arch Dis Child. 1990;65:290–3.
Readett DR, Morris J, Serjeant GR. Determinants of nocturnal enuresis in homozygous sickle cell disease. Arch Dis Child. 1990;65:615–8.
Neveus T, Eggert P, Evans J, et al. Evaluation of and treatment for monosymptomatic enuresis: a standardization document from the International Children’s Continence Society. J Urol. 2010;183:441–7.
Wolf RB, Kassim AA, Goodpaster RL, et al. Nocturnal enuresis in sickle cell disease. Expert Rev Hematol. 2014;7:245–54.
Drawz P, Ayyappan S, Nouraie M, et al. Kidney disease among patients with sickle cell disease, hemoglobin SS and SC. Clin J Am Soc Nephrol. 2016;11:207–15.
Asnani MR, Reid ME. Renal function in adult Jamaicans with homozygous sickle cell disease. Hematology. 2015;20:422–8.
Guasch A, Navarrete J, Nass K, et al. Glomerular involvement in adults with sickle cell hemoglobinopathies: prevalence and clinical correlates of progressive renal failure. J Am Soc Nephrol. 2006;17:2228–35.
Zahr RS, Rampersaud E, Kang G, et al. Children with sickle cell anemia and APOL1 genetic variants develop albuminuria early in life. Haematologica. 2019;104:e385–7.
Aban I, Baddam S, Hilliard LM, et al. Severe anemia early in life as a risk factor for sickle-cell kidney disease. Blood. 2017;129:385–7.
King L, MooSang M, Miller M, et al. Prevalence and predictors of microalbuminuria in Jamaican children with sickle cell disease. Arch Dis Child. 2011;96:1135–9.
Niss O, Lane A, Asnani MR, et al. Progression of albuminuria in patients with sickle cell anemia: a multicenter, longitudinal study. Blood Adv. 2020;4:1501–11.
Maigne G, Ferlicot S, Galacteros F, et al. Glomerular lesions in patients with sickle cell disease. Medicine (Baltimore). 2010;89:18–27.
Zahr RS, Yee ME, Weaver J, et al. Kidney biopsy findings in children with sickle cell disease: a Midwest Pediatric Nephrology Consortium study. Pediatr Nephrol. 2019;34:1435–45.
Dickson LE, Wagner MC, Sandoval RM, et al. The proximal tubule and albuminuria: really! J Am Soc Nephrol. 2014;25:443–53.
Eshbach ML, Kaur A, Rbaibi Y, et al. Hemoglobin inhibits albumin uptake by proximal tubule cells: implications for sickle cell disease. Am J Physiol Cell Physiol. 2017;312:C733–40.
Lebensburger JD, Miller ST, Howard TH, et al. Influence of severity of anemia on clinical findings in infants with sickle cell anemia: analyses from the BABY HUG study. Pediatr Blood Cancer. 2012;59:675–8.
Saraf SL, Shah BN, Zhang X, et al. APOL1, alpha-thalassemia, and BCL11A variants as a genetic risk profile for progression of chronic kidney disease in sickle cell anemia. Haematologica. 2017;102:e1–6.
Ashley-Koch AE, Okocha EC, Garrett ME, et al. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. Br J Haematol. 2011;155:386–94.
Hamideh D, Raj V, Harrington T, et al. Albuminuria correlates with hemolysis and NAG and KIM-1 in patients with sickle cell anemia. Pediatr Nephrol. 2014;29:1997–2003.
Becton LJ, Kalpatthi RV, Rackoff E, et al. Prevalence and clinical correlates of microalbuminuria in children with sickle cell disease. Pediatr Nephrol. 2010;25:1505–11.
McBurney PG, Hanevold CD, Hernandez CM, et al. Risk factors for microalbuminuria in children with sickle cell anemia. J Pediatr Hematol Oncol. 2002;24:473–7.
Aygun B, Mortier NA, Smeltzer MP, et al. Glomerular hyperfiltration and albuminuria in children with sickle cell anemia. Pediatr Nephrol. 2011;26:1285–90.
McPherson ME, Hutcherson D, Olson E, et al. Safety and efficacy of targeted busulfan therapy in children undergoing myeloablative matched sibling donor BMT for sickle cell disease. Bone Marrow Transplant. 2011;46:27–33.
Youssry I, Makar S, Fawzy R, et al. Novel marker for the detection of sickle cell nephropathy: soluble FMS-like tyrosine kinase-1 (sFLT-1). Pediatr Nephrol. 2015;30:2163–8.
Becker AM, Goldberg JH, Henson M, et al. Blood pressure abnormalities in children with sickle cell anemia. Pediatr Blood Cancer. 2014;61:518–22.
McPherson Yee M, Jabbar SF, Osunkwo I, et al. Chronic kidney disease and albuminuria in children with sickle cell disease. Clin J Am Soc Nephrol. 2011;6:2628–33.
Gurkan S, Scarponi KJ, Hotchkiss H, et al. Lactate dehydrogenase as a predictor of kidney involvement in patients with sickle cell anemia. Pediatr Nephrol. 2010;25:2123–7.
Lebensburger JD, Aban I, Pernell B, et al. Hyperfiltration during early childhood precedes albuminuria in pediatric sickle cell nephropathy. Am J Hematol. 2019;94:417–23.
Vazquez B, Shah B, Zhang X, et al. Hyperfiltration is associated with the development of microalbuminuria in patients with sickle cell anemia. Am J Hematol. 2014;89:1156–7.
Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312:1033–48.
Zahr RS, Hankins JS, Kang G, et al. Hydroxyurea prevents onset and progression of albuminuria in children with sickle cell anemia. Am J Hematol. 2019;94:E27–9.
Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010;115:5300–11.
Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377:1663–72.
Lebensburger J, Johnson SM, Askenazi DJ, et al. Protective role of hemoglobin and fetal hemoglobin in early kidney disease for children with sickle cell anemia. Am J Hematol. 2011;86:430–2.
Bartolucci P, Habibi A, Stehle T, et al. Six months of hydroxyurea reduces albuminuria in patients with sickle cell disease. J Am Soc Nephrol. 2016;27:1847–53.
Alvarez O, Montane B, Lopez G, et al. Early blood transfusions protect against microalbuminuria in children with sickle cell disease. Pediatr Blood Cancer. 2006;47:71–6.
Coates TD, Wood JC. How we manage iron overload in sickle cell patients. Br J Haematol. 2017;177:703–16.
Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376:429–39.
Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of Voxelotor in sickle cell disease. N Engl J Med. 2019;381:509–19.
Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018;379:226–35.
Liem RI, Lanzkron S, Coates TD, et al. American Society of Hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease. Blood Adv. 2019;3:3867–97.
Foucan L, Bourhis V, Bangou J, et al. A randomized trial of captopril for microalbuminuria in normotensive adults with sickle cell anemia. Am J Med. 1998;104:339–42.
Aoki RY, Saad ST. Enalapril reduces the albuminuria of patients with sickle cell disease. Am J Med. 1995;98:432–5.
Falk RJ, Scheinman J, Phillips G, et al. Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin-converting enzyme. N Engl J Med. 1992;326:910–5.
Fitzhugh CD, Wigfall DR, Ware RE. Enalapril and hydroxyurea therapy for children with sickle nephropathy. Pediatr Blood Cancer. 2005;45:982–5.
Haymann JP, Hammoudi N, Stankovic Stojanovic K, et al. Renin-angiotensin system blockade promotes a cardio-renal protection in albuminuric homozygous sickle cell patients. Br J Haematol. 2017;179:820–8.
Kasztan M, Fox BM, Speed JS, et al. Long-term endothelin-a receptor antagonism provides robust renal protection in humanized sickle cell disease mice. J Am Soc Nephrol. 2017;28:2443–58.
Kutlar A, Pollock J, Meiler SE, et al. Phase-I study of ETA receptor antagonist Ambrisentan in sickle cell disease. Blood. 2019;134:617.
Aloni MN, Ngiyulu RM, Ekulu PM, et al. Glomerular hyperfiltration is strongly correlated with age in Congolese children with sickle cell anaemia. Acta Paediatr. 2017;106:819–24.
Bodas P, Huang A, O'Riordan MA, et al. The prevalence of hypertension and abnormal kidney function in children with sickle cell disease -a cross sectional review. BMC Nephrol. 2013;14:237.
Ephraim RK, Osakunor DN, Cudjoe O, et al. Chronic kidney disease is common in sickle cell disease: a cross-sectional study in the Tema metropolis, Ghana. BMC Nephrol. 2015;16:75.
Kaspar CDW, Beach I, Newlin J, et al. Hyperuricemia is associated with a lower glomerular filtration rate in pediatric sickle cell disease patients. Pediatr Nephrol. 2020;35:883–9.
Lebensburger JD, Aban I, Hilliard LM, et al. Hyperuricemia and abnormal nocturnal dipping impact glomerular filtration rate in patients with sickle cell anemia. Am J Hematol. 2021;96(5):E143–6.
Lebensburger JD, Cutter GR, Howard TH, et al. Evaluating risk factors for chronic kidney disease in pediatric patients with sickle cell anemia. Pediatr Nephrol. 2017;32:1565–73.
Aygun B, Mortier NA, Smeltzer MP, et al. Hydroxyurea treatment decreases glomerular hyperfiltration in children with sickle cell anemia. Am J Hematol. 2013;88:116–9.
Quinn CT, Saraf SL, Gordeuk VR, et al. Losartan for the nephropathy of sickle cell anemia: a phase-2, multicenter trial. Am J Hematol. 2017;92:E520–8.
Yee ME, Lane PA, Archer DR, et al. Losartan therapy decreases albuminuria with stable glomerular filtration and permselectivity in sickle cell anemia. Blood Cells Mol Dis. 2018;69:65–70.
Yee MEM, Lane PA, Archer DR, et al. Estimation of glomerular filtration rate using serum cystatin C and creatinine in adults with sickle cell anemia. Am J Hematol. 2017;92:E598–9.
Arlet JB, Ribeil JA, Chatellier G, et al. Determination of the best method to estimate glomerular filtration rate from serum creatinine in adult patients with sickle cell disease: a prospective observational cohort study. BMC Nephrol. 2012;13:83.
Lebensburger JD, Gossett J, Zahr R, et al. High bias and low precision for estimated versus measured glomerular filtration rate in pediatric sickle cell anemia. Haematologica. 2021;106(1):295–8.
Xu JZ, Garrett ME, Soldano KL, et al. Clinical and metabolomic risk factors associated with rapid renal function decline in sickle cell disease. Am J Hematol. 2018;93:1451–60.
Derebail VK, Ciccone EJ, Zhou Q, et al. Progressive decline in estimated GFR in patients with sickle cell disease: an observational cohort study. Am J Kidney Dis. 2019;74:47–55.
Derebail VK, Zhou Q, Ciccone EJ, et al. Rapid decline in estimated glomerular filtration rate is common in adults with sickle cell disease and associated with increased mortality. Br J Haematol. 2019;186:900–7.
Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–44.
Abbott KC, Hypolite IO, Agodoa LY. Sickle cell nephropathy at end-stage renal disease in the United States: patient characteristics and survival. Clin Nephrol. 2002;58:9–15.
Viner M, Zhou J, Allison D, et al. The morbidity and mortality of end stage renal disease in sickle cell disease. Am J Hematol. 2019;94:E138–41.
McClellan AC, Luthi JC, Lynch JR, et al. High one year mortality in adults with sickle cell disease and end-stage renal disease. Br J Haematol. 2012;159:360–7.
Boyle SM, Jacobs B, Sayani FA, et al. Management of the dialysis patient with sickle cell disease. Semin Dial. 2016;29:62–70.
Powars DR, Elliott-Mills DD, Chan L, et al. Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Ann Intern Med. 1991;115:614–20.
Ojo AO, Govaerts TC, Schmouder RL, et al. Renal transplantation in end-stage sickle cell nephropathy. Transplantation. 1999;67:291–5.
Huang E, Parke C, Mehrnia A, et al. Improved survival among sickle cell kidney transplant recipients in the recent era. Nephrol Dial Transplant. 2013;28:1039–46.
Willis JC, Awogbade M, Howard J, et al. Outcomes following kidney transplantation in patients with sickle cell disease: the impact of automated exchange blood transfusion. PLoS One. 2020;15:e0236998.
Ofori-Acquah SF, Hazra R, Orikogbo OO, et al. Hemopexin deficiency promotes acute kidney injury in sickle cell disease. Blood. 2020;135:1044–8.
Yawn BP, Buchanan G, Hassell K. Management of patients with sickle cell disease—reply. JAMA. 2015;313:91–2.
Han J, Saraf SL, Lash JP, et al. Use of anti-inflammatory analgesics in sickle-cell disease. J Clin Pharm Ther. 2017;42:656–60.
Cacciotti C, Vaiselbuh S, Romanos-Sirakis E. Pain management for sickle cell disease in the pediatric emergency department: medications and hospitalization trends. Clin Pediatr (Phila). 2017;56:1109–14.
Brandow AM, Carroll CP, Creary S, et al. American Society of Hematology 2020 guidelines for sickle cell disease: management of acute and chronic pain. Blood Adv. 2020;4:2656–701.
Simckes AM, Chen SS, Osorio AV, et al. Ketorolac-induced irreversible renal failure in sickle cell disease: a case report. Pediatr Nephrol. 1999;13:63–7.
Bala N, Chao J, John D, et al. Prevalence of bacteremia in febrile patients with sickle cell disease: meta-analysis of observational studies. Pediatr Emerg Care. 2021;37(12):e1695–700.
Baskin MN, Goh XL, Heeney MM, et al. Bacteremia risk and outpatient management of febrile patients with sickle cell disease. Pediatrics. 2013;131:1035–41.
Sirigaddi K, Aban I, Jantz A, et al. Outcomes of febrile events in pediatric patients with sickle cell anemia. Pediatr Blood Cancer. 2018;65:e27379.
Baddam S, Aban I, Hilliard L, et al. Acute kidney injury during a pediatric sickle cell vaso-occlusive pain crisis. Pediatr Nephrol. 2017;32:1451–6.
Lebensburger JD, Palabindela P, Howard TH, et al. Prevalence of acute kidney injury during pediatric admissions for acute chest syndrome. Pediatr Nephrol. 2016;31:1363–8.
Oakley J, Zahr R, Aban I, et al. Acute kidney injury during parvovirus B19-induced transient aplastic crisis in sickle cell disease. Am J Hematol. 2018.
Cazenave M, Audard V, Bertocchio JP, et al. Tubular acidification defect in adults with sickle cell disease. Clin J Am Soc Nephrol. 2020;15:16–24.
Saraf SL, Viner M, Rischall A, et al. HMOX1 and acute kidney injury in sickle cell anemia. Blood. 2018;132:1621–5.
Silva Junior GB, Liborio AB, Vieira AP, et al. Evaluation of renal function in sickle cell disease patients in Brazil. Braz J Med Biol Res. 2012;45:652–5.
Maurel S, Stankovic Stojanovic K, Avellino V, et al. Prevalence and correlates of metabolic acidosis among patients with homozygous sickle cell disease. Clin J Am Soc Nephrol. 2014;9:648–53.
Vallet M, Metzger M, Haymann JP, et al. Urinary ammonia and long-term outcomes in chronic kidney disease. Kidney Int. 2015;88:137–45.
DeFronzo RA, Taufield PA, Black H, et al. Impaired renal tubular potassium secretion in sickle cell disease. Ann Intern Med. 1979;90:310–6.
Raj VM, Freundlich M, Hamideh D, et al. Abnormalities in renal tubular phosphate handling in children with sickle cell disease. Pediatr Blood Cancer. 2014;61:2267–70.
Pegelow CH, Colangelo L, Steinberg M, et al. Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am J Med. 1997;102:171–7.
Wolf RB, Saville BR, Roberts DO, et al. Factors associated with growth and blood pressure patterns in children with sickle cell anemia: silent cerebral infarct multi-center clinical trial cohort. Am J Hematol. 2015;90:2–7.
DeBaun MR, Sarnaik SA, Rodeghier MJ, et al. Associated risk factors for silent cerebral infarcts in sickle cell anemia: low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood. 2012;119:3684–90.
Strumph K, Hafeman M, Ranabothu S, et al. Nocturnal hypertension associated with stroke and silent cerebral infarcts in children with sickle cell disease. Pediatr Blood Cancer. 2021;68:e28883.
Ranabothu S, Hafeman M, Manwani D, et al. Ambulatory hypertension in pediatric patients with sickle cell disease and its association with end-organ damage. Cureus. 2020;12:e11707.
Shatat IF, Jakson SM, Blue AE, et al. Masked hypertension is prevalent in children with sickle cell disease: a Midwest Pediatric Nephrology Consortium study. Pediatr Nephrol. 2013;28:115–20.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lebensburger, J., Kaspar, C. (2023). The Kidney in Sickle Cell Disease. In: Schaefer, F., Greenbaum, L.A. (eds) Pediatric Kidney Disease. Springer, Cham. https://doi.org/10.1007/978-3-031-11665-0_31
Download citation
DOI: https://doi.org/10.1007/978-3-031-11665-0_31
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-11664-3
Online ISBN: 978-3-031-11665-0
eBook Packages: MedicineMedicine (R0)