
Abstract
Traditional drug discovery process has made a major contribution to pharmacotherapy, but also confer big challenges: it is time-consuming, expensive, and laborious. In recent years, the computational drug repositioning methods are used to address such challenges and bring up new opportunities. The drug-disease association prediction is a crucial task towards computational drug repositioning. In this paper, we propose a new computational method termed CFDDA which employs graph-based neural collaborative filtering to effectively predict the potential indications of existing drugs. 10-fold cross validation on benchmark dataset shows that the proposed model achieves a promising performance in predicting drug-disease association compared with other state-of-the-art methods. The obtained AUPR of 0.539 absolutely outperforms the baselines and the AUC of 0.9103 is comparative to the best model. Moreover, the predicted drug indications are validated by published literature to confirm the effectiveness of our method in practical application.
X. Xiong and Q. Yuan---These authors contributed equally to this work.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Traditional drug development is a time-consuming, expensive and high-risk process. According to reports, the total development time of a new drug is at least 10–15 years, with an average cost of $2.6 billion [1]. In view of the difficulties and challenges of traditional drug development, it is worth the effort to identify new therapeutic uses for existing drugs. This kind of drug development methods is known as drug repositioning. Computational drug repositioning helps rapidly identify new indications for market drugs using computation-based methods, such as network inference, machine learning and deep learning.
To date, the computational methods used to predict the undetected drug-disease association can be classified into three categories: network-based propagation, machine learning-based methods and deep learning-based methods. Network-based propagation methods generally spread information iteratively to neighboring nodes through edges in the network [2]. Traditional machine learning techniques have been widely used to build biological entity association prediction model [3]. However, feature engineering in machine learning is a laborious and time-consuming task which requires a decent amount of domain knowledge. Instead, deep learning is thriving owing to its capacity of learning distributed feature representation automatically through multi-level nonlinear transformation, and is widely used to complete complex downstream tasks. For instance, an increasing number of deep learning-based recommendation algorithms are used to predict potential drug-disease association [4]. The extensively used techniques in CF recommendation include matrix factorization (MF), neural collaborative filtering (NCF) and graph neural network (GNN), etc. [5,6,7]. Recently, neural graph collaborative filtering (NGCF) model has attracted more and more attention [7, 8]. Inspired by such studies, graph neural collaborative filtering (GNCF) has been used in biological entity prediction tasks such as drug-disease associations prediction and polypharmacy side effects prediction [9, 10].
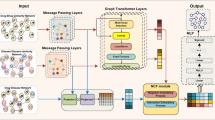
Inspired by the succeed of CF methods in recommendations, this paper focuses on collaborative filtering models for drug-disease associations (DDA) extraction, especially on NGCF model. Here we propose a new end-to-end framework, termed CFDDA, using collaborative filtering methods for DDA prediction. First, we conduct a comparative study on various collaborative filtering algorithms and investigate their advantages in our task. Next, we combine the advantages of several typical CF models to explore diverse perspectives of node representation for purpose of enhanced prediction performance. Further, considering the cold start problem which is most common in CF recommendation system, we add the auxiliary information from drug/disease similarity measures to our framework. Finally, CFDDA is applied to predict the potential drug-disease associations accurately and effectively. The overall schematic framework of CFDDA is shown in Fig. 1.

The overall schematic framework of CFDDA model
2 Materials and Methods
2.1 Dataset
Known Drug-Disease Associations
Two published datasets, i.e. PREDICT [11] and CDataset [12], were employed in our experiment to validate the proposed model. There are 1933 known drug-disease associations involving 593 drugs and 313 diseases in the PREDICT, and 2532 known drug-disease associations including 663 drugs and 409 diseases in the CDataset. For each dataset, drugs were derived from DrugBank [13], while diseases were acquired from Online Mendelian Inheritance in Man database (OMIM) [14]. The known associations can be viewed as a bipartite network between drugs and diseases. For convenience, the bipartite network is presented as a binary matrix \(Y\in {R}^{m\times n}\), where m and n denote the dimension of the matrix same as the number of drugs and diseases. The entry \({y}_{ij}\) in the matrix is set as 1 if drug \({r}_{i}\) has a known association with disease \({d}_{j}\), or \({y}_{ij}=0\) conversely. It is worth to mention that verified interactions between drugs and diseases are very sparse, and the percentage of unverified associations is above 98%.
Similarity Matrices
The molecular structure of drugs is denoted by the notation of Simplified Molecular Input Line Entry Specification (SMILES) [15]. Base on the SMILES, PubChem fingerprint descriptors are calculated via the Chemical Development Kit (CDK) [16]. Then, pairwise drug similarity based on their feature profiles, i.e. fingerprint, is measured by the Tanimoto score. The calculated drug similarities are denoted by a \(m\times m\) matrix \({s}_{r}\), where the entry \({s}_{r}\)(i, j) is the similarity between drug \({r}_{i}\) and drug \({r}_{j}\). The similarities between diseases are obtained from MimMiner [17], which used text mining approach to compute similarity between two diseases from the OMIM database. The disease similarities are represented by a \(n\times n\) matrix \({s}_{d}\), where the entry \({s}_{d}\)(i, j) is the similarity between disease \({d}_{i}\) and disease \({d}_{j}\).
2.2 GCN-Based CF Module
Here we descript the procedure of node encoding using graph convolutional network (GCN) framework. The common GCNs contain three operations: 1) feature transformation, 2) non-linear activation, and 3) neighborhood aggregation. Specifically, given an undirected graph with nodes \(X\) and adjacency matrix \(A\), a multi-layer neural network is constructed on the graph with the following layer-wise propagation rule:
where \({H}^{(l+1)}\) is the eigenvector matrix of drugs and diseases obtained after l step embedding propagation. \({H}^{(0)}\) is assigned as X that can be randomly initialized. \(\tilde{A }=I+A\) denotes the adjacency matrix with self-connection added and \(D\) is a degree matrix such that \({D}_{ii}=\sum_{i}{\tilde{A }}_{ij}\), thus entry \({d}_{ii}\) denotes the number of non-zero elements of the \(i\)-th row vector in the adjacency matrix \(A\). The Laplacian matrix is defined as \(\tilde{L }={\tilde{D }}^{-\frac{1}{2}}\tilde{A }{\tilde{D }}^{-\frac{1}{2}}\), \(\tilde{D }=I+D\), and \(\mathrm{I}\) denotes the identity matrix. \({W}^{(l)}\) is a layer-specific trainable transformation matrix.
In our experiment, we find that the three-operations in GCN not always are necessary to strengthen a model. So we build two types of GCN models with different components, which are termed as GCN1 and GCN2 respectively. GCN1 combines all the three operations of feature transformation, nonlinear activation and neighborhood aggregation but GCN2 only contains neighborhood aggregation.
GCN1 Formulation
Given the drug-disease interaction matrix \(R\), we get the adjacent matrix \(A\) and its degree matrix \(D\), shown in formula (2). The Laplacian matrix \(L\) is calculated as follows:
According to the propagation rules, the network embeddings of nodes are calculated as follows:
where \({H}^{(l)}\) is the node embeddings obtained by \(l\) iterations of information propagation; \(\tilde{L }=L+I\), and \(\odot \) denotes the element-wise product. \(LeakyReLU\) is the activation function. The other symbols’ definition is similar to formula (1). Finally, we concatenate the outputs of all convolutional layers to obtain the embedding of drug \({r}_{i}\) as formula (4):
where \({h}_{{r}_{i}}^{\left(l\right)}\) and \({h}_{{d}_{j}}^{\left(l\right)}\) denote the \(l\)-th layer embeddings of drug \({r}_{i}\) and disease \({d}_{j}\) respectively; || means concatenation. Similar as \({e}_{{r}_{i}}^{GCN}\), we can obtain \({e}_{{d}_{i}}^{GCN}\) as the GCN embedding of disease \({d}_{i}\). Then the interaction feature of drug-disease pair (\({r}_{i}, {d}_{j}\)) are calculated by a elements-wise product \(\odot \) in formula (5):
GCN2 Formulation
Compared to GCN1, GCN2 only use the convolutional operation as formula (6):
Considering that different convolutional layer has different contribution to the final node embeddings. Instead of equally concatenating the embeddings from different layers as formula (4) did, we apply a weighted sum method to aggregate the embeddings as follows:
where \({\alpha }_{i}\) is a trainable weight for the \(l\)-th layer embedding \({h}^{(l)}\). Similar as \({e}_{{r}_{i}}^{GCN}\), we can obtain \({e}_{{d}_{i}}^{GCN}\) as the GCN embedding of disease \({d}_{i}\). Then the two node embeddings are integrated by a elements-wise product \(\odot \):
2.3 Neural CF Module
The Neural CF module is borrowed from NCF framework which was originally proposed for recommendation [6]. It consists of two parts: MF layer and multiple linear perceptron (MLP) layers.
MF Layer
We use \({e}_{{r}_{i}}^{MF}\) and \({e}_{{d}_{j}}^{MF}\) to respectively denote the embeddings of drug \({r}_{i}\) and disease \({d}_{j}\) that are learned from the input of their one-hot vector pair. MF layer leverages a linear operation \(\odot \) to get the interaction characteristics of the pair (\({r}_{i}\), \({d}_{j}\)):
MLP Layers
First, we learn the node representations \({e}_{{r}_{i}}^{MLP}\) and \({e}_{{d}_{j}}^{MLP}\) from embedding layer for drug \({r}_{i}\) and disease \({d}_{j}.\) Then they are concatenated to get the simple interaction feature \({e}^{MLP(0)}\). Next, an \(l\)-layers MLP uses the non-linear function \(ReLU\) to explore the deep interaction features:
where \(\oplus \) is the concatenation operation; \({W}^{(l)}\) and \({b}^{(l)}\) are the trainable weight and bias value of the \(l\)-th MLP layer respectively.
2.4 Learning Similarity Features by Weighted Random Walk
We view the similarity as a weighted network, then the weighted random walk algorithm (WRW) derived from Deepwalk [18] is employed to learn comprehensive features of drugs and diseases. First, given a start node in the network, the random walk algorithm is used to extract node sequence on the walk path, and then the SkipGram [19] model is applied to learn node embeddings. In this way, we obtain drug embedding \({e}_{{r}_{i}}^{WRW}\) and disease \({e}_{{d}_{j}}^{WRW}\) respectively. Finally, we take \({e}_{{r}_{i}}^{WRW}\) and \({e}_{{d}_{j}}^{WRW}\) as the input to learn further feature representation by MLP. The procedure is similar to formula (11), so we don’t repeat it. The outputs of MLP are interacted by a elements-wise product:
2.5 CFDDA Model
Model Formulation
We model the drug-disease relationships prediction process as a binary classification problem. The output of the model is a probability \({\widehat{y}}_{ij}\), which is used to predict whether there is a potential treatment relationship between sample pair:
where \({e}_{ij}^{MF}\), \({e}_{ij}^{MLP}\), \({e}_{ij}^{GCN}\) and \({e}_{ij}^{WRW}\) have been mentioned above; \(\sigma ,\) \(h\) and \(b\) denote the activation function, weight and bias value of the output layer respectively. Here, we choose sigmoid as the activation function and update the \(h\) value through back propagation in the training stage.
Loss Function and Training Process
The loss between the predicted value and the target value is defined as binary cross-entropy loss:
where \({y}_{i,j}\) denotes whether there is an observed association between the drug \({r}_{i}\) and the disease \({d}_{j}\) (0 or 1), \({\widehat{y}}_{i,j}\) denotes the predicted drug-disease association score, \((i,j)\) denotes a training sample. \(\theta \) denotes the model parameters, \({\uplambda }\) denotes the regularization parameter. \(L\left(\theta \right)\) is the loss function to be minimized, and we use Adam optimizer to minimize the loss function.
Parameter Settings
The training epochs is 80. The learning rate is 0.001. The batch training size is 64. The number of negative samples is 5. The embedding size of MF part is 16. The embedding size of the MLP part is 64, and the dimension of the subsequent hidden layer is [64, 32, 16]. The embedding size of GCN part is 16, and the number of convolution layers is 3. The embedding size of WRW part is 16. Researchers can adjust regularization coefficient by themselves.
3 Results and Discussion
3.1 Evaluation Metrics
We conducted a 10-fold cross-validation using golden standard datasets PREDICT and CDataset to evaluate the performance of CFDDA. In the 10-fold cross-validation, all known drug-disease associations in the dataset are randomly divided into 10 subsets. Each subset in turn serves as the test set, and the other 9 subsets serve as the training set. The cross-validation process is repeated 10 times and the averaged result is taken as the final performance report. In each fold, CFDDA model is trained on the training set, and then used to predict the associations in the test set. According to the prediction scores, the area under curve (AUC) and the area under precision-recall curve (AUPR) are selected as metrics to evaluate the prediction model. Since recent studies have shown that AUPR can provide more informative assessments than AUC on highly unbalanced datasets [20], we attach more importance to AUPR in the following experiments. Besides, we also use the metric of hit ratio (HR)@n that is usually adopted in recommendation system to measure the accuracy of top-n recommendations in the ranking list.
3.2 Performance Comparison of Various CF Models
Each CF model has its distinct advantage in feature representation so as to have different recommendation performance. Here we conducted experiments to explore the properties of several typical CF models, such as MF, NCF, GCN. It is worth mentioning that we developed two different GCN-based models: GCN1 and GCN2. The parameters involved in these CF methods are set through grid search, and the default values will refer to the original papers that proposed these methods. Table 1 shows the AUC, AUPR and HR@10 obtained by four prediction methods on PREDICT.
As the result shown in Table 1, two GCN models achieve better performance than MF and NCF in all metrics, except the AUPR of GCN2. Moreover, GCN1 outperforms GCN2 with the AUPR of 0.5234 and HR@10 of 0.6912 respectively. Specifically, the AUPR of GCN1 is 13% higher than GCN2 method.
MF is a linear model that only uses simple and fixed inner product to estimate complex drug-disease associations in low-dimensional potential space. NCF framework integrates MF and MLP methods. Since MLP uses nonlinear function to learn deep interaction features of drugs and diseases, it greatly enhances NCF performance. However, MF and NCF cannot encode topological information. Comparatively, GCN-base methods perform information propagation on the bipartite graph from drug-disease associations to model the high-order connectivity of the graph structure. Specifically, GCN2 only retains neighborhood aggregation to reduce the model complexity and speeds up the training progress. Accordingly, it achieves a slightly lower performance than GCN1.
3.3 Performance of Integrated CF Models
In light of the results of CF model comparison, we focus on the GCN model as the main framework. To include the ability of various CF model in capturing features of drugs and disease, we try several combinations and compare their contribution to prediction model. The results are reported in Table 2.
From the results, we can see that the addition of NCF to GCN1 almost has no positive effect on the prediction performance. In contrast, the combination of NCF and GCN2 make a significant improvement on AUC, AUPR, and HR@10 which reach to 0.9047, 0.536 and 0.7289 respectively. In particular, the AUPR is 14.3% higher than simple GNC2. Since GCN2 and NCF pay attention to distinct aspect information of drug-disease associations, their combination helps to obtain comprehensive feature representation.
We analyze that GCN1’s complex network structure is enough to model high-order connectivity from bipartite graphs, so adding NCF will not significantly improve performance. Here, we add auxiliary information of drugs and disease from knowledge database to alleviate the cold start problem in CF model. The experiment results demonstrate that the auxiliary information has positive contribution to CFADD as expected, shown in Table 3. Finally, considering the best performance we take NCF + GCN2 + SIM as our standard model, referred to as CFDDA.
3.4 Comparisons with the State-of-the-Arts Models
In this section, we compare CFDDA with four recent methods: NIMCGCN [21], LAGCN [9], DisDrugPred [22], MBiRW [12]. We adopt the published results or open source code in the following experiments. The comparison between baselines and CFDDA is shown in Table 4.
As the results shown in Table 4, we can see that CFDDA has the absolute advantage over all baseline methods with respect to the value of AUPR which reach to 0.539. It is crucial to the model with sparse data. As we above-mentioned, for highly unbalanced datasets, AUPR can provide a more significant evaluation than AUC. AUPR comprehensively considers both real positive rate and false positive rate, so the biased dataset has little effect on it. From Table 5 we can see that AUC of CFDDA is 0.9103 which is a bit lower than the best model MBiRW. The reason may be that our model uses fewer prior auxiliary information by comparison to MBiRW.
3.5 Evaluation on Extra Dataset
In order to make the prediction more convincing, we repeated the above experiment on another dataset. The comparison results are shown in Table 5.
Clearly, we can see that CFDDA has the absolute advantage over all baseline methods with respect to value of AUPR which reaches to 0.5917. It shows that the performance of CFDDA on different datasets is certainly stable.
3.6 Prediction of Novel Drug-Disease Associations
To further validate the prediction ability of CFDDA, we investigate the prediction results of the model and cite the evidence for the discoveries. The prediction scores at top 10 are considered, shown in Table 6. We find that eight out of ten predicted associations to be proved in published literature. After retrieval in PubMed, we find that Trihexyphenidyl has been proved to be one of the most useful drugs to treat Parkinson’s disease [23]. In addition, some publications confirmed that Tiagabine is a novel antiepileptic drug that was designed to block gamma-aminobutyric acid uptake by presynaptic neurons and glial cells [24].
4 Conclusion
In this paper, we formalized the DDA prediction problem into a CF recommendation task aiming to computational drug repositioning. For the purpose of ranking the association probabilities of drug-disease pairs, we investigated several typical collaborative filtering recommendation methods, such as Matrix Factorization (MF), Neural Collaborative Filtering (NCF), Graph Convolutional Network (GCN). The comparison experiment shows that these CF methods provide unique contribution to the overview prediction performance. Understandably, MFs’ linearity limits the ability of feature representation. Both NCF and GNN-based CF leverage the neural network framework to encode the features of drugs and diseases, but GNN-based models focus more on topological neighbors. Correspondingly, we combined the two CF framework aiming to derive comprehensive feature representation. In addition, the addition of auxiliary information contributes to the improvement of system performance through relieving the cold start problem. The final experimental results show that CFDDA has considerable competitiveness compared with other state-of-the-art methods. Moreover, the predicted recommendation list of drug indications is verified by publicly available literatures.
References
Chan, H.C.S., Shan, H., et al.: Advancing drug discovery via artificial intelligence. Trends Pharmacol Sci. 40(8), 592–604 (2019)
Cowen, L., Ideker, T., et al.: Network propagation: a universal amplifier of genetic associations. Nat. Rev. Genet. 18(9), 551–562 (2017)
Wei, X., Zhang, Y., et al.: Predicting drug–disease associations by network embedding and biomedical data integration. Data Technol. Appl. 53(2), 217–229 (2019)
Yuan, Q., Wei, X., et al.: A hybrid neural collaborative filtering model for drug repositioning. In: 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), pp. 515–518 (2020)
He, X., Zhang, H., et al.: Fast matrix factorization for online recommendation with implicit feedback. In: Proceedings of the 39th International ACM SIGIR Conference on Research and Development in Information Retrieval, pp. 549–558 (2016)
He, X., Liao, L., et al.: Neural collaborative filtering. In: Proceedings of the 26th International Conference on World Wide Web, pp. 173–182 (2017)
Wang, X., He, X., et al.: Neural graph collaborative filtering. In: Proceedings of the 42nd International ACM SIGIR Conference on Research and Development in Information Retrieval, pp. 165–174 (2019)
He, X., Deng, K., et al.: LightGCN: simplifying and powering graph convolution network for recommendation. In: Proceedings of the 43rd International ACM SIGIR Conference on Research and Development in Information Retrieval, pp. 639–648, (2020)
Yu, Z., Huang, F., et al.: Predicting drug-disease associations through layer attention graph convolutional network. Brief Bioinform. 22(4) (2021)
Zitnik, M., Agrawal, M., et al.: Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 34(13), i457–i466 (2018)
Gottlieb, A., Stein, G.Y., et al.: PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 7, 496 (2011)
Luo, H., Wang, J., et al.: Drug repositioning based on comprehensive similarity measures and bi-random walk algorithm. Bioinformatics 32(17), 2664–2671 (2016)
Law, V., Knox, C., et al.: DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 42, Database issue, D1091–7 (2014)
Hamosh, A., Scott, A.F., et al.: Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, Database issue, D514–7 (2005)
Weininger, D.: SMILES, a chemical language and information system. 1. introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28(1), 31–36 (1988)
Steinbeck, C., Hoppe, C., et al.: Recent developments of the chemistry development kit (CDK) - an open-source java library for chemo- and bioinformatics. Curr. Pharm. Des. 12(17), 2111–2120 (2006)
van Driel, M.A., Bruggeman, J., et al.: A text-mining analysis of the human phenome. Eur. J. Hum. Genet. 14(5), 535–542 (2006)
Perozzi, B., Al-Rfou, R., et al.: DeepWalk: online learning of social representations. In: Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp. 701–710: Association for Computing Machinery, New York, New York, USA (2014)
Mikolov, T., Sutskever, I., et al.: Distributed representations of words and phrases and their compositionality. In: Advances in Neural Information Processing Systems, vol. 26, pp. 3111–3119, Lake Tahoe, NV(US) (2013)
Saito, T., Rehmsmeier, M.: The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS ONE 10(3), e0118432 (2015)
Li, J., Zhang, S., et al.: Neural inductive matrix completion with graph convolutional networks for miRNA-disease association prediction. Bioinformatics 36(8), 2538–2546 (2020)
Xuan, P., Cao, Y., et al.: Drug repositioning through integration of prior knowledge and projections of drugs and diseases. Bioinformatics 35(20), 4108–4119 (2019)
Schwab, R.S., Doshay, L.J.: Slow-release trihexyphenidyl in Parkinson’s disease. JAMA 180(2), 159–161 (1962)
Shinnar, S.: Tiagabine. In: Seminars in Pediatric Neurology, vol. 4, no. 1, pp. 24–33 (1997)
van Charldorp, K.J., de Jonge, A., et al.: Subclassification of muscarinic receptors in the heart, urinary bladder and sympathetic ganglia in the pithed rat. Naunyn Schmiedebergs Arch. Pharmacol. 331(4), 301–306 (1985)
Gajek, A., Poczta, A., et al.: Chemical modification of melphalan as a key to improving treatment of haematological malignancies. Sci. Rep. 10(1), 4479 (2020)
Faderl, S., Gandhi, V., et al.: The role of clofarabine in hematologic and solid malignancies—development of a next-generation nucleoside analog. Cancer 103(10), 1985–1995 (2005)
Kostelnik, A., Cegan, A., et al.: Anti-parkinson drug biperiden inhibits enzyme acetylcholinesterase. Biomed. Res. Int. 2017, 2532764 (2017)
Jacob, A.: A case of torsion dystonia treated with orphenadrine. Scott. Med. J. 7(3), 139–140 (1962)
DA agonists - Ergot derivaties: Bromocriptine. Mov. Disord. 17, S4, S53–S67 (2002)
Acknowledgments
This work is supported by the Fundamental Research Funds for the Central Universities (No: 2662020XXPY08).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this paper

Cite this paper
Xiong, X., Yuan, Q., Zhou, M., Wei, X. (2022). Graph-Based Neural Collaborative Filtering Model for Drug-Disease Associations Prediction. In: Memmi, G., Yang, B., Kong, L., Zhang, T., Qiu, M. (eds) Knowledge Science, Engineering and Management. KSEM 2022. Lecture Notes in Computer Science(), vol 13368. Springer, Cham. https://doi.org/10.1007/978-3-031-10983-6_43
Download citation
DOI: https://doi.org/10.1007/978-3-031-10983-6_43
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-10982-9
Online ISBN: 978-3-031-10983-6
eBook Packages: Computer ScienceComputer Science (R0)