
Abstract
Voltage-gated calcium channels are important mediators of signal transduction in the primary afferent pain pathway. In rodents, T-type calcium channels regulate the excitability of afferent fibers and spinal cord interneurons, and contribute to synaptic release in dorsal horn synapses. N-type calcium channels are the primary driver of neurotransmission in afferent fiber terminals in the superficial dorsal horn. N-type calcium channel activity in chronic pain states is splice isoform dependent and is regulated by ancillary Cavα2δ subunits. During chronic pain, T-type calcium channel activity can be dysregulated by post-translational modification such as glycosylation and ubiquitination. Both N-type and T-type calcium channels are potential drug targets for treating pain, with the former also being a target of G-protein-coupled receptors such as opioid receptors. R-type calcium channels may also play a possible role in afferent nociceptive signaling.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Nociception is a key physiological process that has a critical protective function across many different species (Dubin & Patapoutian, 2010). The same neuroanatomical pathways that support nociceptive processing can become dysregulated from an adaptive to a maladaptive state and result in chronic hypersensitivity that serves no useful biological purpose (Kuner & Flor, 2017). Chronic pain is a debilitating condition that affects more than 20% of adult North Americans, and treatment of chronic pain represents one of the most serious health challenges facing society (Schopflocher et al., 2011; Reitsma et al., 2012). It is a major cause of lost productivity, and leads to a substantially reduced quality of life in both adults and children, along with staggering health care and economic costs (Gaskin & Richard, 2012; Mayer et al., 2019).
Painful stimuli are first detected by a set of diverse and specialized nociceptors that have their nerve endings in the skin or internal organs, and their cell bodies in either the dorsal root ganglia (DRG) or trigeminal ganglia (Fig. 1). In response to noxious stimuli such as heat, mechanical pressure, or chemicals, action potentials are initiated in the distal nerve endings, which then propagate along the afferent axons to nerve terminals located in the superficial layers of the spinal dorsal horn and brain stem (for cephalic neurons of the trigeminal ganglia) (Dubin & Patapoutian, 2010; Basbaum et al., 2009). Synaptic transmission then leads to the initiation of action potentials in second-order neurons that project to various regions in the brain, where they are processed to ultimately give rise to the perception of an unpleasant sensation. The brain extends descending projections to the spinal cord, which modulate ascending nociceptive inputs through activation of excitatory and inhibitory spinal interneurons. Afferent sensory fibers are heterogeneous and do not just convey nociceptive inputs, but also other input modalities such as touch, proprioception, and itch, and ascending spinal dorsal columns are similarly organized along modalities (Niu et al., 2013). Nociceptive fibers are classified into (1) non-myelinated, slowly conducting peptidergic and non-peptidergic C-fibers, and (2) myelinated, more rapidly conducting A-fibers (Basbaum et al., 2009). These can be further subdivided based on their diameter and more precisely, based on a number of different markers that in many cases show overlapping expression patterns (e.g., tropomyosin receptor kinase A [TRKA], Ret, calcitonin gene-related peptide [CGRP], substance P, islocetin B4, transient receptor potential cation channel subfamily V member 1 [TRPV1], Mas-related G-protein-coupled receptor [MrgpR] among others). Furthermore, different subtypes of afferent fibers terminate in different laminae in the spinal cord (Molliver et al., 1995; Li et al., 2011). A more detailed description of afferent fiber types is beyond the scope of this chapter but can be found in recent transcriptomic analyses and comprehensive reviews (Finnerup et al., 2021; Sharma et al., 2020; Kupari et al., 2021). Equally complex is the organization of the spinal dorsal horn and its role in processing nociceptive information (Peirs et al., 2015; Gangadharan & Kuner, 2015; Harding et al., 2020). Suffice it to say that dorsal horn neurons are responsible for the integration of nociceptive peripheral inputs, and the activity of a heterogeneous set of spinal interneurons, and their inputs from descending pathways.

Schematic representation of the afferent pain pathway, illustrating the roles of Cav3.2 and Cav2.2 calcium channels
Voltage-gated calcium channels play important roles in these processes (Zamponi, 2016). Cav3.2 (T-type) calcium channels regulate the firing of certain types of afferent sensory neurons and contribute to synaptic transmission in dorsal horn synapses. They also appear to be involved in shaping the firing properties of spinal interneurons. Cav2.2 (N-type) channels are the major calcium channel subtype that is involved in synaptic transmission between nociceptive fibers and second-order neurons in the spinal cord (Fig. 1). In this chapter, we focus primarily on the roles of these two calcium channel isoforms in afferent nociceptive signaling.
N-Type Calcium Channels in the Afferent Pain Pathway
N-type (Cav2.2) channels exhibit a mainly neuronal distribution and they are enriched at presynaptic terminals where they coordinate the synchronous release of neurotransmitters (Nowycky et al., 1985; Westenbroek et al., 1992; Wheeler et al., 1994). In terminals of primary afferent neurons, calcium entry via Cav2.2 channels triggers the release of neurotransmitters such as glutamate, substance P, and CGRP (Gruner & Silva, 1994; Holz et al., 1988; Maggi et al., 1990; Santicioli et al., 1992). Specific Cav2.2 channel blockers such as ω-conotoxin GVIA and MVIIA (Olivera et al., 1994) have been used to demonstrate the involvement of this channel type in the afferent pain pathway (basal thermal and mechanical nociception), and they have also highlighted the potential of N-type blockers as potent analgesics (Chaplan et al., 1994; Malmberg & Yaksh, 1994; Bowersox et al., 1996; Diaz & Dickenson, 1997; Sluka, 1998). The participation of Cav2.2 channels in the pain pathway was later confirmed by the phenotype of Cav2.2 channel knockout mice. Indeed, these mice are hyposensitive to pain (Saegusa et al., 2001; Kim et al., 2001; Hatakeyama et al., 2001), and they only present a relatively mild central nervous system (CNS) phenotype that includes reduced anxiety levels and reduced alcohol withdrawal symptoms (Saegusa et al., 2001; Newton et al., 2004).
The CACNA1B gene, which encodes the Cav2.2α1 subunit, is subject to extensive alternative splicing (Gray et al., 2007). The chapter by T.W. Soong (chapter “Splicing and Editing to Fine-Tune Activity of High Voltage-Activated Calcium Channels”) deals in detail with this regulatory mechanism for voltage-gated calcium channels. In this section, we focus on splice variants that are relevant to the afferent pain pathway. Exon 37 encodes a short section (14 amino acids) of the cytoplasmic carboxy-terminal tail of the channel and can generate two mutually exclusive variants: E37a and E37b (Lipscombe et al., 2002; Bell et al., 2004). In heterologous expression systems, Cav2.2 E37a produces larger calcium currents than its counterpart Cav2.2 E37b, which is probably due to the fact that Cav2.2 E37a is both more resistant to degradation by the proteasome and more efficiently trafficked to the plasma membrane (Marangoudakis et al., 2012; Macabuag & Dolphin, 2015). It is noteworthy that Cav2.2 E37a is selectively expressed in peripheral nociceptive neurons that are positive for both TRPV1 and Nav1.8 channels, and its expression is critical for pain signaling (Bell et al., 2004; Altier et al., 2007). Indeed, using an small interfering RNA (siRNA) knockdown strategy, it was shown that Cav2.2 channels containing E37a mediate basal nociception and inflammatory pain (Altier et al., 2007).
In a chronic pain context, an upregulation of Cav2.2 channel expression has been shown in dorsal root ganglia and in the dorsal horn of the spinal cord (Cizkova et al., 2002; Yokoyama et al., 2003). However, it is still unclear whether the increase of Cav2.2 channels in the dorsal horn results from an increase of channels in central neurons or from an accumulation of channels in primary afferent terminals or a combination of both. Interestingly, while several studies have investigated the transcription level of Cav2.2 channels in chronic pain models, none has shown a correlation between level of Cav2.2 channel proteins and level of Cav2.2 channel mRNAs (messenger RNA), suggesting that chronic pain-induced changes in Cav2.2 channel expression occur at a translational and/or post-translational level (Altier et al., 2007; Umeda et al., 2006). Cav2.2-channel-deficient mice subjected to inflammatory and neuropathic insults exhibit reduced pain hypersensitivity (Hatakeyama et al., 2001; Saegusa et al., 2001; Kim et al., 2001). Moreover, whereas thermal hyperalgesia induced by sciatic nerve ligation relies mainly on the expression of E37a, tactile allodynia depends on both E37a and E37b splice variants (Altier et al., 2007). E37a was also shown to play a critical role in morphine-dependent analgesia (Andrade et al., 2010). Finally, in a model of chronic inflammatory pain, the expression of a Cav2.2 channel splice variant that lacks exon 18a was shown to be increased in DRG neurons (Asadi et al., 2009). Exon 18 encodes a region of the channel that corresponds to the II–III linker, which plays a critical role in presynaptic targeting (Zamponi et al., 2015). Although the physiological relevance of this variant switch is still unclear, it has been suggested that it could constitute a regulatory mechanism to attenuate excitability and reduce synaptic transmission (Asadi et al., 2009).
In the central nervous system, excitatory synaptic transmission usually depends on the dual activity of Cav2.2 and Cav2.1 channels. However, using a multi-electrode array recording system, a study has shown that in the anterior cingulate cortex (ACC), Cav2.2 channels are the only voltage-gated calcium channels involved in glutamatergic excitatory synaptic transmission (Kang et al., 2013). Interestingly, the ACC plays important roles in pain perception and chronic pain (Xu et al., 2008; Li et al., 2010).
Interaction Between CRMP2 and Cav2.2: A Therapeutic Target for Chronic Pain
For more than a decade, the interaction between Cav2.2 channels and collapsin response mediator protein 2 (CRMP2) has attracted increasing interest (Brittain et al., 2009). Initially identified as a protein involved in axonal growth, CRMP2 was then shown to directly interact with Cav2.2 channels and to promote their forward trafficking to the plasma membrane (Brittain et al., 2009). The CRMP2/Cav2.2 interaction was shown to modulate vesicular release in hippocampal neurons (Brittain et al., 2009; Chew & Khanna, 2018). In DRG neurons, Cav2.2-dependent secretion of CGRP was enhanced by the over-expression of CRMP2, which pointed to its involvement in the pain pathway (Chi et al., 2009). These initial studies led to the identification of interaction domains between the two proteins and the development of a short 14 amino-acid peptide (CBD3) derived from CRMP2 that can interfere with the binding of CRMP2 to Cav2.2 (Brittain et al., 2011). Conjugated to the HIV1 TAT protein and injected in vivo, CBD3 peptide is able to suppress inflammatory, neuropathic, postoperative, diabetic, migraine, and HIV-related pain (Brittain et al., 2011; Wilson et al., 2011; François-Moutal et al., 2015; Xie et al., 2016; Piekarz et al., 2012; Ripsch et al., 2012).
CRMP2/Cav2.2 interactions can be modulated by several mechanisms (Moutal et al., 2019). Phosphorylation of CRMP2 by cyclin-dependent kinase 5 (Cdk5) has been shown to reinforce its interaction with Cav2.2 channels (Brittain et al., 2012) and thus potentially enhance calcium currents. Interestingly, activation of Cdk5 was increased in animal models of neuropathic pain and inhibiting Cdk5 activity attenuated mechanical allodynia (Li et al., 2014; Yang et al., 2014; Brittain et al., 2012; Moutal et al., 2016a, b). Cdk5 has multiple cellular targets, and thus inhibiting its phosphorylation activity likely disrupts other pathways that (in addition to reducing CRMP2/Cav2.2 interaction) can account for the improvement of the pain phenotype (Yang et al., 2014).
SUMOylation, the post-translational addition of small ubiquitin-related modifier (SUMO) peptide, can modulate CRMP2 function (Dustrude et al., 2013). A putative SUMO-interaction motif in CRMP2 has been linked to its role in regulating calcium influx via voltage-gated calcium channels (Ju et al., 2013). However, there is still no evidence showing involvement of CRMP2 SUMOylation and Cav2.2 channels in pain pathways.
Finally, the tumor suppressor protein neurofibromin has been shown to interact with CRMP2 via its C-terminal domain, inhibiting its function on Cav2.2 channels (Moutal et al., 2017a, b, 2019). Neurofibromatosis type 1 patients present mutations in the NF1 gene, which encodes neurofibromin. These mutations often result in truncated proteins and afflicted patients suffer from idiopathic chronic pain (Gutmann et al., 2017; Esposito et al., 2015). In mutant mice heterozygous for Nf1 (Nf1+/−), N-type current densities are increased in DRG neurons and vesicular release is enhanced compared with wild-type neurons (Duan et al., 2014). Of note, the use of the CBD3 peptide to treat these Nf1+/− mice restores calcium currents and vesicular release to control levels indicating that neurofibromin contributes to the same regulatory pathway as CRMP2–Cav2.2 (Wilson et al., 2012). Moreover, a clustered regularly (CRISPR)/Cas9 strategy was used to truncate the C-terminal domain of neurofibromin in rats and the effect on calcium channel function was examined (Moutal et al., 2017b). This latter study showed an increase in voltage-gated calcium currents in DRG neurons and the animals developed thermal hyperalgesia. Further investigation of the interaction between neurofibromin and CRMP2 led to the design of a 15 amino-acid peptide (CNRP1) derived from CRMP2 C-terminal sequence, which, when coupled to a TAT peptide, was able to mimic the negative regulation of neurofibromin on CRMP2–Cav2.2 interaction in vivo (Moutal et al., 2017a). Indeed, intrathecal injection of TAT-CNRP1 alleviated nociceptive responses in animal models of inflammatory, post-surgical, and neuropathic pain (Moutal et al., 2017a). Interestingly, CNRP1 was also used in a proteomic analysis of a synaptic membrane library to reveal a novel interaction between CRMP2 and the presynaptic protein syntaxin 1A (Moutal et al., 2017a). Syntaxin 1A, which interacts with the synprint (Synaptic Protein Interaction) domain of Cav2.2 and Cav2.1, plays an important role in trafficking the channels to presynaptic terminals and promotes synaptic vesicle docking (Sheng et al., 1994; Gandini & Zamponi, 2021; Bennett et al., 1992). Altogether, these data reveal the importance of CRMP2 as a presynaptic interaction hub regulating Cav2.2 trafficking and highlight its potential as therapeutic target for pain treatment (Khanna et al., 2020).
Cavα2δ Subunits and Neuropathic Pain
Cavα2δ are critical auxiliary subunits that are required for Cav1.X and Cav2.X channel complexes to be trafficked to the plasma membrane, and for Cav2.X to be targeted to presynaptic terminals (Dolphin, 2016; Ferron et al., 2021). A detailed account on the structure and functions of Cavα2δs is provided in the chapter by A.C. Dolphin (chapter “Regulation of Calcium Channels and Synaptic Function by Auxiliary α2δ Subunits”). In this section, we will focus on Cavα2δ subunits, and particularly Cavα2δ-1, in a context of chronic pain. Cavα2δ-1 mRNA expression is enhanced in DRG neurons in animal models of chronic pain (Newton et al., 2001; Wang et al., 2002; Bauer et al., 2009; Lana et al., 2014). This transcriptional effect is associated with an increase in protein expression in the soma of DRG neurons and also along the axons and in the terminals within the spinal cord (Bauer et al., 2009; Luo et al., 2001). Interestingly, in vivo over-expression of Cavα2δ-1 is sufficient to induce tactile allodynia and hyperalgesia (Li et al., 2006). In addition, mice lacking the Cacna2d1 gene that encodes Cavα2δ-1 have a reduced sensitivity to mechanical and cold stimuli, and delayed mechanical hypersensitivity following sciatic nerve ligation (Patel et al., 2013). Altogether, these data support a key role of Cavα2δ-1 in controlling DRG neuron excitability and its involvement in the development of chronic pain.
Cavα2δ-1 is the target of gabapentinoids (gabapentin and pregabalin), a class of drugs prescribed to treat chronic pain in humans (Rosenberg et al., 1997; Field et al., 2006, 2007; Taylor et al., 2007). Chronic application of gabapentinoids to DRG neurons in culture inhibits synaptic transmission by reducing the trafficking of Cavα2δ-1, and consequently Cav2.2, to the plasma membrane (Hendrich et al., 2008, 2012; Cassidy et al., 2014). In animal models of neuropathic pain, chronic treatment with pregabalin partially prevents the increase of presynaptic Cavα2δ-1 in the dorsal horn of the spinal cord and alleviates allodynia (Bauer et al., 2009).
Besides their role as auxiliary subunits for Cav channels, Cavα2δs have been involved in direct interactions with other ion channels and have been linked to other biological functions. Indeed, the N-terminal domain of KCa1.1 (BK) potassium channels competes with Cav2.2 channels for the binding of Cavα2δ-1 and reduces Cav2.2 surface expression and calcium current density (Zhang et al., 2018). Importantly, over-expressing a membrane-bound BK channel N-terminus peptide has an analgesic effect in mouse models of inflammatory and neuropathic pain. In addition, Cavα2δ-1 has been shown to interact with N-methyl-D-aspartate (NMDA) receptors and to increase their delivery at dorsal horn synapses during neuropathic pain development (Chen et al., 2018). Finally, Cavα2δs have been shown to play a role in synaptogenesis via extracellular matrix protein thrombospondins and thrombospondin-4 has been associated with the development of neuropathic pain (Eroglu et al., 2009; Yu et al., 2018; Kim et al., 2012). Altogether, these studies reinforce the interest of Cavα2δ-1 as a therapeutic target for the treatment of chronic pain, and they also highlight the fact that the benefits of gabapentinoid treatments in vivo are not solely attributable to the normalization of Cav2.2-dependent synaptic transmission in the spinal cord.
N-Type Channel Blockers as Pain Therapeutics
In addition to the gabapentinoids discussed in the preceding section, a number of blockers of Cav2.2 channels have been shown to mediate analgesia in preclinical models, and some in a clinical setting. The chapter by Lewis and Adams (chapter “Pharmacology and Structure-Function of Venom Peptide Inhibitors of N-Type (Cav2.2) Calcium Channels”) discusses in detail the inhibition of voltage-gated calcium channels by peptide toxins. Briefly, let us recall that ω-conotoxins are among the most selective inhibitors of N-type calcium channels. Both ω-conotoxins GVIA (Conus geographus) and MVIIA (Conus magus) potently inhibit native and transiently expressed Cav2.2 channels in an almost irreversible manner by occluding the pore of the channel (Olivera et al., 1984; Reynolds et al., 1986; Feng et al., 2001). The selective action of MVIIA, the analgesic effects in preclinical models (Chaplan et al., 1994; Bowersox & Luther, 1998), and the fact that it can be synthesized in vitro ultimately led this molecule to being developed as a potential therapeutic for pain. Under the commercial name Prialt, MVIIA passed phase III clinical trials and was ultimately approved for use in patients with severe cancer pain (Atanassoff et al., 2000; Miljanich, 2004). A major limitation of Prialt is that it does not cross the blood-brain barrier and therefore has to be delivered intrathecally via an implanted pump. Another is the fact that the therapeutic window is relatively narrow, and surprising side effects such as memory problems, hypotension, and vision problems have been described (Penn & Paice, 2000; Staats et al., 2004). Several additional conotoxins derived from other species such as Conus fulmen and Conus catus have been explored as possible analgesics (Adams et al., 2003; Lee et al., 2010; Sadeghi et al., 2013), with ω-conotoxin CVID having been tested in clinical trials, but ultimately not pursued further (Schroeder et al., 2006). Despite the limitations of MVIIA as a human pain therapeutic, it did serve to validate N-type calcium channels as a possible therapeutic target from pain.
Small organic blockers of N-type calcium channels that can penetrate the blood-brain barrier would be able to circumvent the need for intrathecal delivery. These include several distinct classes of compounds, such as amino-acid derivatives, N-triazoles, piperazines, piperidines, and even some dihydropyridines, that are normally thought to act on L-type channels (for review, see Schroeder et al., 2006). Many of these derivatives are not strictly specific for Cav2.2 channels, such as TROX-1, and N-triazole derivative that acts on all Cav2 channel isoforms but mediates analgesia in rodent models of osteoarthritis pain (Rahman et al., 2015; Abbadie et al., 2010; Swensen et al., 2012). L-cysteine (Seko et al., 2002) and amino-piperidine derivatives (Teodori et al., 2004) are N-type channel-blocking molecules with analgesic properties. The mixed L-/N-type channel-blocking dihydropyridine cilnidipine (Uneyama et al., 1997; Koganei et al., 2009; Yamamoto et al., 2016) has shown promise as an analgesic in preclinical models. A new N-type channel-blocking chemical scaffold was developed based on the structure of piperazine-based antipsychotics (Tytgat et al., 1991) with N-type channel-blocking activity (Zamponi et al., 2009; Pajouhesh et al., 2010). This includes a lead compound termed Z160 that was remarkably efficacious in a number of rodent pain models, but unfortunately failed phase II human trials due to lack of efficacy. The discrepancy between the preclinical and clinical data remains unresolved. To our knowledge, there are no small organic direct blockers of N-type calcium channels approved for use in humans.
Opioid and GABA-B Receptor Regulation of N-Type Calcium Channels
N-type calcium channels are subject to powerful modulation by a wide array of G-protein-coupled receptors (GPCRs) (for review, see Tedford & Zamponi, 2006). A detailed description of GPCR modulation of voltage-gated calcium channels is provided in the chapter by Herlitze et al. (chapter “Modulation of VGCCs by G-protein Coupled Receptors and Their Second Messengers”). In a nutshell, N-type calcium channels are modulated in a voltage-dependent manner through the direct interaction of G-protein βγ subunits with binding sites in the N-terminus and domain I–II linker regions of the Cav2.2 α1 subunit (Herlitze et al., 1996; Zamponi et al., 1997; Agler et al., 2005), leading to an inhibition of voltage-dependent activation of the channels that can be reversed by strong membrane depolarizations. In addition, there are also voltage-independent pathways that involve the activation of classical messenger cascades such as protein kinases (Luebke & Dunlap, 1994). In the context of nociceptive signaling, N-type calcium channel modulation by the family of opioid receptors (ORs) is particularly important. ORs comprise a family of four different receptor subtypes (μ-, δ-, κ-opioid receptors [MOR, DOR, KOR], and opioid receptor like receptors [NOP]; Waldhoer et al., 2004; McDonald & Lambert, 2005), that can undergo alternative splicing to create additional diversity (Gavériaux-Ruff et al., 1997; Pasternak, 2018; Piltonen et al., 2019). The different opioid receptor family members share a common ability to inhibit N-type calcium channels, and can heteromerize to form complexes with altered signaling properties (Evans et al., 2010). Different types of ORs are expressed in different types of afferent fibers, with MORs and DORs modulating different pain modalities (Scherrer et al., 2009). Activation of these receptors by their ligands leads to an activation of G-protein-coupled inwardly rectifying potassium channels (Blanchet & Lüscher, 2002; Marker et al., 2004), and direct voltage-dependent inhibition of presynaptic N-type calcium channels. In the afferent pain pathway, this leads to reduced neuronal excitability and reduced synaptic transmission in the spinal dorsal horn, leading to analgesia (Kondo et al., 2005; Beaudry et al., 2011). There appears to be tonic opioid control over nociceptive inputs such that DOR null mice show tactile allodynia (Nozaki et al., 2012). MORs are the pharmacological target of clinically active opioids such as morphine and fentanyl, whereas the pentazocine is a clinically approved KOR agonist (Goldstein, 1985). Action of opioids on MOR and DOR activation can lead to respiratory depression (Field et al., 1999), and repeated opioid use can lead to addiction (Darcq & Kieffer, 2018). Interestingly, KORs do not affect respiration, which would be an advantage in the development of OR agonists for pain. Several DOR agonists have been explored in clinical trials (Spahn & Stein, 2017), and bivalent ligands of OR subtypes are being explored as possibly less addictive pain medications (Vardanyan et al., 2017).
MOR modulation of Cav2.2 channels expressed in tA-201 cells has been shown to occur in an OR splice isoform-dependent manner (Gandini et al., 2019). Furthermore, MORs couple differentially to Cav2.2 channels with alternatively spliced exon 37 sequences (Gandini et al., 2019; Raingo et al., 2007). This highlights an immense potential for functional diversity in OR coupling to N-type calcium channels, and thus fine tuning of synaptic activity. Further complexity is generated by the formation of signaling complexes between NOP receptors and Cav2.2 channels that leads to (1) agonist-independent modulation of Cav2.2 channel activity, and (2) receptor-mediated regulation of forward trafficking and internalization of the channels (Beedle et al., 2004; Altier et al., 2006). Furthermore, co-expression with NOP can recruit other OR subtypes into Cav2.2 complexes (Evans et al., 2010). How these signaling complexes affect pain modulation remains to be determined.
GABA-B receptors have also been shown to modulate pain transmission via actions on N-type channels in rodents (Terrence et al., 1985), but the clinical utility of agonists such as baclofen has not been borne out due to CNS side effects (Bortolato et al., 2010). The α-conotoxin Vc1.1, which has GABA-B receptor agonist activity (Callaghan et al., 2008), has been explored as a possible pain therapeutic, but despite promising preclinical results (Castro et al., 2017, 2018), has failed clinical development (Carstens et al., 2011). This molecule mediates voltage-independent modulation rather than the classical Gβγ-mediated voltage-dependent inhibition of Cav2.2 activity (Callaghan et al., 2008). In addition to OR and GABA-B receptors, many other types of GPCRs are known to modulate pain transmission (for review, see Pan et al., 2008), including cannabinoid, and adrenergic and muscarinic receptors. Although these receptors all modulate N-type channels, it is unclear if and how their analgesic actions can be directly attributed to the modulation of these channels in the afferent pathway and/or brain.
Other Types of Cav2 Calcium Channels
P/Q-type (Cav2.1) calcium channels are critical mediators of certain types of congenital migraines. Their role in headache is described in chapter “Voltage-Gated Calcium Channels and Migraine” by Dr. Pietrobon and will not be discussed here further. R-type (Cav2.3) calcium channels have emerged as potential players in afferent pain signaling. Due to their hyperpolarized activation range, Cav2.3 channels are well suited toward supporting neuronal firing (e.g., Park et al., 2010; Zaman et al., 2011; Gutzmann et al., 2019), and furthermore, they have been shown to contribute to neurotransmitter release at a subset of CNS synapses (Myoga & Regehr, 2011; Dietrich et al., 2003). They are expressed in peripheral and colonic sensory neurons (Yusaf et al., 2001; Fang et al., 2007; Qian et al., 2013). Mice lacking Cav2.3 channels show resistance to chemically induced seizures (in agreement with the role of these channels in neuronal excitability; Weiergräber et al., 2007; Dibué-Adjei et al., 2017), and partial resistance to inflammatory pain (Saegusa et al., 2000, 2002). Cav2.3 channels can be inhibited by ORs (Berecki et al., 2016), and direct inhibitors of Cav2.3 channels have been shown to mediate antinociception (Murakami et al., 2004, 2007), possibly by acting on Cav2.3 channels in the spinal dorsal horn. A potential role of Cav2.3 channels in afferent pain signaling is also supported by the notion that natural mixed blockers of Cav2.3 and Cav2.2 channels are able to reverse neuropathic pain in rodents (Shan et al., 2019). Given the paucity of selective small organic Cav2.3 channel blockers, the precise role of Cav2.3 in pain signaling is not yet fully understood. In this context we note that gain-of-function mutations in Cav2.3 channels have been linked to seizure disorders in children, without any associated pain hypersensitivity (Helbig et al., 2018). On the other hand, the R-type channel blocker topiramate has been shown to mediate analgesia in patients with polyneuropathy (Nazarbaghi et al., 2017; but see Wiffen et al., 2013). Hence, the jury is out on whether Cav2.3 is a suitable target for treating pain.
Role of T-Type Calcium Channels in Afferent Pain Signaling
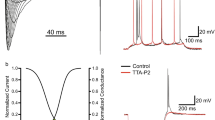
T-type calcium channels are uniquely suited toward regulating cellular excitability (Fig. 2). They activate at hyperpolarized membrane potentials and display a hyperpolarized half inactivation potential (Nowycky et al., 1985; Perez-Reyes, 2003). The overlap between steady-state activation and inactivation curves supports a large window current that allows these channels to be active near typical neuronal resting potentials (Iftinca & Zamponi, 2009; Fig. 2a). At rest, a large fraction of T-type calcium channels is tonically inactivated. Upon membrane hyperpolarization, as occurs, for example, during a GABAergic synaptic input, T-type channels recover from inactivation, giving rise to a greatly increased T-type current upon a subsequent membrane depolarization. This in turn triggers the opening of voltage-gated sodium channels and supports a rapid activation discharge, termed a rebound burst (Coulter et al., 1989; Huguenard & Prince, 1992; Fig. 2b). These collective biophysical properties allow T-type calcium channels to regulate the excitability of primary afferent nociceptive fibers, as well as the firing of spinal interneurons (Candelas et al., 2019). T-type calcium channels also associate with the synaptic vesicle release protein syntaxin 1A to support low-threshold exocytosis (Weiss et al., 2012). This is relevant for synaptic transmission in the dorsal horn of the spinal cord (Jacus et al., 2012; García-Caballero et al., 2014). A role of T-type calcium channels in afferent pain signaling in rodents was originally supported by two lines of evidence. First, delivery of known blockers of T-type calcium channels such as ethosuximide mediated analgesic effects in rodent pain models (Matthews & Dickenson, 2001; Dogrul et al., 2003; Flatters & Bennett, 2004). Second, in an elegant study, Bourinet and coworkers delivered siRNA against the three known T-type calcium channel isoforms, and found that either pan knockdown of Cav3 or specific knockdown of the Cav3.2 isoform resulted in potent analgesia in rat neuropathic pain models, thus identifying a single specific Cav3 channel isoform as a key mediator of afferent pain signaling (Bourinet et al., 2005). Along these lines, similar protection was observed upon knockdown of Cav3.2 in a model of visceral pain induced by intracolonic delivery of butyric acid and in streptozotocin-induced diabetic neuropathy in rats (Messinger et al., 2009). Subsequent experiments in Cav3.2 null mice revealed a more complex picture, with mice lacking Cav3.2 showing reduced sensitivity to intraplantar formalin, but relatively normal response to intraplantar injection of Complete Freund’s Adjuvant (CFA) and in neuropathic pain models (Choi et al., 2007; García-Caballero et al., 2014). This is most likely due to developmental compensatory mechanisms.

(a) Schematic representation of the steady-state activation and inactivation curves for T-type calcium channels, highlighting the large window current. (Adapted with permission from Iftinca & Zamponi, 2009). (b) Effects of ICaT in a model neuron on rebound depolarizations. Neuronal output (top traces) and corresponding inward ICaT current trajectories (lower traces) are shown for the three Cav3 isoforms. (Fernandez et al., 2021)
In rodents, Cav3.2 channels are expressed in specific subtypes of primary afferent fibers including fibers positive for peripherin and vesicular glutamate transporter 3 (vGlut3), which mark a subset of nociceptive C-fibers, and neurofilament protein 200 (NF200) and tropomyosin receptor kinase B (TRKB), which are found in D-hair mechanoreceptors (François et al., 2015). In the spinal cord, Cav3.2 has been shown to overlap with the markers for neuronal nuclear protein (NeuN), Tlx3, Pax2, calretinin, protein kinase C gamma (PKCγ), calbindin, nNos, and parvalbumin, indicating expression in both glutamatergic and GABAergic interneurons (Candelas et al., 2019). The precise function of Cav3.2 channel in spinal cord neurons needs to be explored further, but there is clear evidence that these channels are able to shape the firing properties of lamina II cells (Candelas et al., 2019).
There is also emerging evidence of Cav3.2 channel mutations that are linked to chronic pain. Two heterozygous mutations (P769L and A1059S) in different alleles were identified in a pediatric chronic pain patient (Souza et al., 2019). When introduced and expressed in tsA-201 cells, the mutations mediated slight gain-of-function effects; they were however dependent on the particular recording conditions used for electrophysiological measurements. A number of Cav3.2 channel mutations were also identified in patients with trigeminal neuralgia (Dong et al., 2020); however, to date, they have not been functionally characterized.
Dysregulation of Cav3.2 Channel Expression and Function in Chronic Pain States
An important role of Cav3.2 channels is further underscored by the observation that these channels are upregulated in sensory neurons and/or spinal cord in a number of chronic pain conditions in rodents. This includes peripheral nerve injury (Jagodic et al., 2008; Wen et al., 2010; Feng et al., 2019; Tomita et al., 2020), spinal cord injury (Lauzadis et al., 2020), chronic diabetic conditions (Jagodic et al., 2007), visceral and CFA-induced inflammation (Marger et al., 2011; García-Caballero et al., 2014), osteoarthritis (Shin et al., 2020), certain types of chemotherapy-induced neuropathies (Li et al., 2017; Tomita et al., 2019), and models of post-surgical pain (Joksimovic et al., 2018). Two different molecular mechanisms that may contribute to this enhanced Cav3.2 activity have been identified. First, N-linked glycosylation of a set of four different asparagine residues has been shown to lead to an increase in Cav3.2 channel surface expression (Weiss et al., 2013; Orestes et al., 2013). Importantly, exposing cells to high glucose levels promoted an increase in Cav3.2 surface expression, whereas neuraminidase inhibited Cav3.2 channel membrane expression (Orestes et al., 2013). Thus, glycosylation may at least in part contribute to the upregulation of T-type channel activity during diabetic pain states. Second, Cdk5- and PKC-mediated phosphorylation has been associated with an increase in Cav3.2 channel activity in different models of neuropathic pain (Gomez et al., 2020; Gaifullina et al., 2019). Third, Cav3.2 channels are regulated by ubiquitination by the ubiquitin ligase WWP1 and the deubiquitinase USP5 (García-Caballero et al., 2014). The intracellular linker region between domains III and IV of Cav3.2 contains a ubiquitin ligase consensus motif as well as two lysine residues that are ubiquitinated. Importantly, this region also associates with USP5, an enzyme that is upregulated in the dorsal root ganglia and spinal cord in several mouse models of chronic pain (Gadotti et al., 2015b). This includes models of inflammatory pain induced by intraplantar delivery of CFA, neuropathic pain induced by sciatic nerve injury, visceral pain, chronic diabetes (Gadotti et al., 2015b), and surgery (Joksimovic et al., 2018). Importantly, depletion of USP5 or preventing the association of USP5 with Cav3.2 by delivery of decoy peptides mediated analgesia in these conditions in both male and female mice (García-Caballero et al., 2014, 2016; Gadotti & Zamponi, 2018). Small organic molecules targeting the USP5–Cav3.2 interaction also mediated analgesia (Gadotti et al., 2015b), overall suggesting this molecular interaction as a potential drug target for treating pain. Interestingly, USP5 upregulation could also be observed in response to transcutaneous non-invasive optogenetic activation of C-fibers (Stemkowski et al., 2016). This led to a transient sensitization of the stimulated paw that subsided after 24 h along with a decrease in USP5 levels back to baseline. This then suggests that activity-dependent upregulation of USP5 and an associated increase in Cav3.2 channel activity may have originally evolved as an adaptive (i.e., protective) response that can become maladaptive under certain circumstances. Altogether, there are at least two molecular mechanisms that may contribute to the aberrant upregulation of Cav3.2 channels in chronic pain states, and it is possible that there are others. This could include the Cavα2δ subunit, which has been shown to promote cell surface expression of Cav3.2 channels in expression system even though this does not appear to occur via a physical interaction (Dubel et al., 2004).
We note that the trigeminal system appears to be different from other peripheral afferents in that trigeminal neuropathic pain has been associated with an upregulation of Cav3.3 rather than Cav3.2, and inhibiting these channels mediates analgesia (Montera et al., 2021) suggesting that trigeminal neuralgia may involve different T-type calcium channel signaling than peripheral nerve neuropathy. Finally, T-type calcium channels do not only contribute to nociceptive signaling in the afferent pain pathway, but also there is evidence that they do so in the brain. Cav3.1 null mice have been reported to show increased visceral pain sensitivity, and this was attributed to alterations in function of ventroposterolateral thalamic neurons (Kim et al., 2003). Along these lines, mice lacking Cav3.1 showed a reduction in trigeminal neuropathic pain, due to alterations in thalamic signaling (Choi et al., 2016). Direct delivery of the T-type channel inhibitor NCC-55-0396 into the anterior cingulate cortex was shown to inhibit neuropathic pain in rats (Shen et al., 2015). From a therapeutic point of view, it is thus important to consider all types of Cav3 channels, both peripherally and in the CNS.
T-Type Calcium Channel Blockers as Possible Pain Therapeutics
There is considerable evidence from preclinical models that inhibitors of T-type calcium channels mediate analgesia in a number of different chronic pain models, including diabetic, visceral, inflammatory, and neuropathic pain arising from physical nerve injury. Several detailed reviews of different classes of T-type calcium channel blockers with analgesic properties in rodent pain models were recently published (Snutch & Zamponi, 2018; Weiss & Zamponi, 2019a, b) and we refer the reader to these publications for additional detail. It has been known for some time that certain dihydropyridines can effectively block T-type calcium channels. For example, nimodipine blocks Cav3 channels in the micromolar range, although it should be noted that nimodipine has a much higher affinity for L-type calcium channels, especially at depolarized membrane potentials. Nonetheless, hexahydroquinoline–dihydropyridine derivatives have been shown to block T-type calcium channel preferentially over L-types (Bladen et al., 2014), with several derivatives of this compound series mediating analgesia in mouse models of both neuropathic and inflammatory pain (Bladen et al., 2015a; Gadotti et al., 2015a). Another class of T-type channel inhibitors with analgesic properties is derived from cannabinoids and endocannabinoids. The cannabinoid receptor ligands anandamide (Chemin et al., 2001), as well as N-arachnidonylyglycine (Barbara et al., 2009; Ross et al., 2009) were both shown to potently block Cav3 calcium channels. Importantly, the analgesic effects of these endocannabinoids were abolished in Cav3.2 null mice (Barbara et al., 2009), indicating that they inhibit pain via this calcium channel subtype. Subsequently, a number of compounds related to cannabinoid receptor agonists were developed and shown to not only inhibit Cav3.2 calcium channels but also mediate analgesia by blocking this channel subtype (You et al., 2011; Berger et al., 2014). Derivatives of this compound series that do not act on cannabinoid receptors were subsequently developed, and shown to mediate potent T-type channel inhibition along with analgesic effects (Bladen et al., 2015b), as well as the ability to inhibit acute itch in mice (Gadotti et al., 2020). Piperidine- and piperazine-based compounds such as penfluridol and flunarizine have been known to block Cav3-type calcium channels for some time (Santi et al., 2002). A number of related compounds have since been developed, including TTA-P2 and Z944, and shown to inhibit pain (Choe et al., 2011; Harding et al., 2021). Of note, Z944 has also been shown to mediate analgesic CNS effects by affecting thalamocortical connectivity in rats with neuropathic pain (LeBlanc et al., 2016).
Only a limited number of clinical pain studies with T-type calcium channels have been reported. ABT-639, a potent T-type calcium channel inhibitor, has failed multiple phase II clinical trials (Wallace et al., 2016), including for diabetic pain (Serra et al., 2015; Ziegler et al., 2015). Recently, ethosuximide was tested as a potential pain drug, but failed a randomized controlled trial for neuropathic pain (Kerckhove et al., 2018). Z944 on the other hand successfully completed a phase 1 trial; however, to date no data on efficacy in phase II are available.
The availability of a cryo-electron microscopy (EM) structure for Cav3.1 channels (Zhao et al., 2019) in complex with Z944 has opened new avenues for rational design of T-type calcium channel blockers. Homology models for Cav3.2 and Cav3.3 based on the Cav3.1 structure will aid the development of compounds with specificity for individual T-type calcium channel isoforms, opening new therapeutic options for pain as well as new means for probing the physiological roles of T-type calcium channels in a broader context.
Conclusions
In summary, Cav2.2 and Cav3.2 calcium channels are the predominant calcium channel isoforms involved in the afferent pain pathways, with a possible contribution from Cav2.3. The only clinically approved calcium channel inhibitors for treating chronic pain are the gabapentinoids, which target the ancillary Cavα2δ subunit of Cav2.2 channels; Prialt, which specifically target Cav2.2; and opioids, which inhibit Cav2.2 channel activity via activation of MORs. There remains a paucity of direct small organic inhibitors of Cav2.2 and Cav3.2 channels for clinical use in chronic pain.
Abbreviations
- Cdk5:
-
cyclin-dependent kinase 5
- CFA:
-
Complete Freund’s Adjuvant
- CGRP:
-
calcitonin gene-related peptide
- CRMP2:
-
collapsin response mediator protein 2
- DRG:
-
Dorsal root ganglia
- GPCR:
-
G-protein-coupled receptor
- MrgpR:
-
Mas-related G-protein-coupled receptor
- NeuN:
-
neuronal nuclear protein
- NF200:
-
neurofilament protein 200
- NMDA:
-
N-methyl-D-aspartate
- OR:
-
opioid receptor
- PKC:
-
protein kinase C
- TRKA:
-
tropomyosin receptor kinase A
- TRKB:
-
tropomyosin receptor kinase B
- TRPV1:
-
transient receptor potential cation channel subfamily V member 1
- vGLut3:
-
vesicular glutamate transporter 3
References
Abbadie, C., McManus, O. B., Sun, S. Y., Bugianesi, R. M., Dai, G., Haedo, R. J., et al. (2010). Analgesic effects of a substituted N-triazole oxindole (TROX-1), a state-dependent, voltage-gated calcium channel 2 blocker. The Journal of Pharmacology and Experimental Therapeutics, 334(2), 545–555. https://doi.org/10.1124/jpet.110.166363
Adams, D. J., Smith, A. B., Schroeder, C. I., Yasuda, T., & Lewis, R. J. (2003). Omega-conotoxin CVID inhibits a pharmacologically distinct voltage-sensitive calcium channel associated with transmitter release from preganglionic nerve terminals. The Journal of Biological Chemistry, 278(6), 4057–4062. https://doi.org/10.1074/jbc.M209969200
Agler, H. L., Evans, J., Tay, L. H., Anderson, M. J., Colecraft, H. M., & Yue, D. T. (2005). G protein-gated inhibitory module of N-type (ca(v)2.2) ca2+ channels. Neuron, 46(6), 891–904. https://doi.org/10.1016/j.neuron.2005.05.011
Altier, C., Khosravani, H., Evans, R. M., Hameed, S., Peloquin, J. B., Vartian, B. A., et al. (2006). ORL1 receptor-mediated internalization of N-type calcium channels. Nature Neuroscience, 9(1), 31–40. https://doi.org/10.1038/nn1605
Altier, C., Dale, C. S., Kisilevsky, A. E., Chapman, K., Castiglioni, A. J., Matthews, E. A., et al. (2007). Differential role of N-type calcium channel splice isoforms in pain. The Journal of Neuroscience, 27(24), 6363–6373. https://doi.org/10.1523/JNEUROSCI.0307-07.2007
Andrade, A., Denome, S., Jiang, Y. Q., Marangoudakis, S., & Lipscombe, D. (2010). Opioid inhibition of N-type Ca2+ channels and spinal analgesia couple to alternative splicing. Nature Neuroscience, 13(10), 1249–1256. https://doi.org/10.1038/nn.2643
Asadi, S., Javan, M., Ahmadiani, A., & Sanati, M. H. (2009). Alternative splicing in the synaptic protein interaction site of rat Ca(v)2.2 (alpha (1B)) calcium channels: Changes induced by chronic inflammatory pain. Journal of Molecular Neuroscience, 39(1–2), 40–48. https://doi.org/10.1007/s12031-008-9159-2
Atanassoff, P. G., Hartmannsgruber, M. W., Thrasher, J., Wermeling, D., Longton, W., Gaeta, R., et al. (2000). Ziconotide, a new N-type calcium channel blocker, administered intrathecally for acute postoperative pain. Regional Anesthesia and Pain Medicine, 25(3), 274–278. https://doi.org/10.1016/s1098-7339(00)90010-5
Barbara, G., Alloui, A., Nargeot, J., Lory, P., Eschalier, A., Bourinet, E., et al. (2009). T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. The Journal of Neuroscience, 29(42), 13106–13114. https://doi.org/10.1523/JNEUROSCI.2919-09.2009
Basbaum, A. I., Bautista, D. M., Scherrer, G., & Julius, D. (2009). Cellular and molecular mechanisms of pain. Cell, 139(2), 267–284. https://doi.org/10.1016/j.cell.2009.09.028
Bauer, C. S., Nieto-Rostro, M., Rahman, W., Tran-Van-Minh, A., Ferron, L., Douglas, L., et al. (2009). The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. The Journal of Neuroscience, 29(13), 4076–4088. https://doi.org/10.1523/JNEUROSCI.0356-09.2009
Beaudry, H., Dubois, D., & Gendron, L. (2011). Activation of spinal mu- and delta-opioid receptors potently inhibits substance P release induced by peripheral noxious stimuli. The Journal of Neuroscience, 31(37), 13068–13077. https://doi.org/10.1523/JNEUROSCI.1817-11.2011
Beedle, A. M., McRory, J. E., Poirot, O., Doering, C. J., Altier, C., Barrere, C., et al. (2004). Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nature Neuroscience, 7(2), 118–125. https://doi.org/10.1038/nn1180
Bell, T. J., Thaler, C., Castiglioni, A. J., Helton, T. D., & Lipscombe, D. (2004). Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron, 41(1), 127–138. https://doi.org/10.1016/s0896-6273(03)00801-8
Bennett, M. K., Calakos, N., & Scheller, R. H. (1992). Syntaxin: A synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science, 257(5067), 255–259. https://doi.org/10.1126/science.1321498
Berecki, G., Motin, L., & Adams, D. J. (2016). Voltage-gated R-type calcium channel inhibition via human μ-, δ-, and κ-opioid receptors is voltage-independently mediated by Gβγ protein subunits. Molecular Pharmacology, 89(1), 187–196. https://doi.org/10.1124/mol.115.101154
Berger, N. D., Gadotti, V. M., Petrov, R. R., Chapman, K., Diaz, P., & Zamponi, G. W. (2014). NMP-7 inhibits chronic inflammatory and neuropathic pain via block of Cav3.2 T-type calcium channels and activation of CB2 receptors. Molecular Pain, 10, 77. https://doi.org/10.1186/1744-8069-10-77
Bladen, C., Gündüz, M. G., Şimşek, R., Şafak, C., & Zamponi, G. W. (2014). Synthesis and evaluation of 1,4-dihydropyridine derivatives with calcium channel blocking activity. Pflügers Archiv, 466(7), 1355–1363. https://doi.org/10.1007/s00424-013-1376-z
Bladen, C., Gadotti, V. M., Gündüz, M. G., Berger, N. D., Şimşek, R., Şafak, C., et al. (2015a). 1,4-Dihydropyridine derivatives with T-type calcium channel blocking activity attenuate inflammatory and neuropathic pain. Pflügers Archiv, 467(6), 1237–1247. https://doi.org/10.1007/s00424-014-1566-3
Bladen, C., McDaniel, S. W., Gadotti, V. M., Petrov, R. R., Berger, N. D., Diaz, P., et al. (2015b). Characterization of novel cannabinoid based T-type calcium channel blockers with analgesic effects. ACS Chemical Neuroscience, 6(2), 277–287. https://doi.org/10.1021/cn500206a
Blanchet, C., & Lüscher, C. (2002). Desensitization of mu-opioid receptor-evoked potassium currents: Initiation at the receptor, expression at the effector. Proceedings of the National Academy of Sciences of the United States of America, 99(7), 4674–4679. https://doi.org/10.1073/pnas.072075399
Bortolato, M., Frau, R., Orrù, M., Fà, M., Dessì, C., Puligheddu, M., et al. (2010). GABAB receptor activation exacerbates spontaneous spike-and-wave discharges in DBA/2J mice. Seizure, 19(4), 226–231. https://doi.org/10.1016/j.seizure.2010.02.007
Bourinet, E., Alloui, A., Monteil, A., Barrère, C., Couette, B., Poirot, O., et al. (2005). Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. The EMBO Journal, 24(2), 315–324. https://doi.org/10.1038/sj.emboj.7600515
Bowersox, S. S., & Luther, R. (1998). Pharmacotherapeutic potential of omega-conotoxin MVIIA (SNX-111), an N-type neuronal calcium channel blocker found in the venom of Conus magus. Toxicon, 36(11), 1651–1658. https://doi.org/10.1016/s0041-0101(98)00158-5
Bowersox, S. S., Gadbois, T., Singh, T., Pettus, M., Wang, Y. X., & Luther, R. R. (1996). Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. The Journal of Pharmacology and Experimental Therapeutics, 279(3), 1243–1249.
Brittain, J. M., Piekarz, A. D., Wang, Y., Kondo, T., Cummins, T. R., & Khanna, R. (2009). An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. The Journal of Biological Chemistry, 284(45), 31375–31390. https://doi.org/10.1074/jbc.M109.009951
Brittain, J. M., Duarte, D. B., Wilson, S. M., Zhu, W., Ballard, C., Johnson, P. L., et al. (2011). Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nature Medicine, 17(7), 822–829. https://doi.org/10.1038/nm.2345
Brittain, J. M., Wang, Y., Eruvwetere, O., & Khanna, R. (2012). Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Letters, 586(21), 3813–3818. https://doi.org/10.1016/j.febslet.2012.09.022
Callaghan, B., Haythornthwaite, A., Berecki, G., Clark, R. J., Craik, D. J., & Adams, D. J. (2008). Analgesic alpha-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. The Journal of Neuroscience, 28(43), 10943–10951. https://doi.org/10.1523/JNEUROSCI.3594-08.2008
Candelas, M., Reynders, A., Arango-Lievano, M., Neumayer, C., Fruquière, A., Demes, E., et al. (2019). Cav3.2 T-type calcium channels shape electrical firing in mouse Lamina II neurons. Scientific Reports, 9(1), 3112. https://doi.org/10.1038/s41598-019-39703-3
Carstens, B. B., Clark, R. J., Daly, N. L., Harvey, P. J., Kaas, Q., & Craik, D. J. (2011). Engineering of conotoxins for the treatment of pain. Current Pharmaceutical Design, 17(38), 4242–4253. https://doi.org/10.2174/138161211798999401
Cassidy, J. S., Ferron, L., Kadurin, I., Pratt, W. S., & Dolphin, A. C. (2014). Functional exofacially tagged N-type calcium channels elucidate the interaction with auxiliary α2δ-1 subunits. Proceedings of the National Academy of Sciences of the United States of America, 111(24), 8979–8984. https://doi.org/10.1073/pnas.1403731111
Castro, A., Raver, C., Li, Y., Uddin, O., Rubin, D., et al. (2017). Cortical regulation of nociception of the trigeminal nucleus caudalis. The Journal of Neuroscience, 37(47), 11431–11440. https://doi.org/10.1523/JNEUROSCI.3897-16.2017
Castro, J., Grundy, L., Deiteren, A., Harrington, A. M., O’Donnell, T., et al. (2018). Cyclic analogues of α-conotoxin Vc1.1 inhibit colonic nociceptors and provide analgesia in a mouse model of chronic abdominal pain. British Journal of Pharmacology, 175(12), 2384–2398. https://doi.org/10.1111/bph.14115
Chaplan, S. R., Pogrel, J. W., & Yaksh, T. L. (1994). Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. The Journal of Pharmacology and Experimental Therapeutics, 269(3), 1117–1123.
Chemin, J., Monteil, A., Perez-Reyes, E., Nargeot, J., & Lory, P. (2001). Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. The EMBO Journal, 20(24), 7033–7040. https://doi.org/10.1093/emboj/20.24.7033
Chen, J., Li, L., Chen, S. R., Chen, H., Xie, J. D., Sirrieh, R. E., et al. (2018). The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Reports, 22(9), 2307–2321. https://doi.org/10.1016/j.celrep.2018.02.021
Chew, L. A., & Khanna, R. (2018). CRMP2 and voltage-gated ion channels: Potential roles in neuropathic pain. Neuro-Signals, 2(1), NS20170220. https://doi.org/10.1042/NS20170220
Chi, X. X., Schmutzler, B. S., Brittain, J. M., Wang, Y., Hingtgen, C. M., Nicol, G. D., et al. (2009). Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. Journal of Cell Science, 122(Pt 23), 4351–4362. https://doi.org/10.1242/jcs.053280
Choe, W., Messinger, R. B., Leach, E., Eckle, V. S., Obradovic, A., Salajegheh, R., et al. (2011). TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Molecular Pharmacology, 80(5), 900–910. https://doi.org/10.1124/mol.111.073205
Choi, S., Na, H. S., Kim, J., Lee, J., Lee, S., Kim, D., et al. (2007). Attenuated pain responses in mice lacking Ca(V)3.2 T-type channels. Genes, Brain, and Behavior, 6(5), 425–431. https://doi.org/10.1111/j.1601-183X.2006.00268.x
Choi, S., Yu, E., Hwang, E., & Llinás, R. R. (2016). Pathophysiological implication of CaV3.1 T-type Ca2+ channels in trigeminal neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America, 113(8), 2270–2275. https://doi.org/10.1073/pnas.1600418113
Cizkova, D., Marsala, J., Lukacova, N., Marsala, M., Jergova, S., Orendacova, J., et al. (2002). Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Experimental Brain Research, 147(4), 456–463. https://doi.org/10.1007/s00221-002-1217-3
Coulter, D. A., Huguenard, J. R., & Prince, D. A. (1989). Calcium currents in rat thalamocortical relay neurones: Kinetic properties of the transient, low-threshold current. The Journal of Physiology, 414, 587–604. https://doi.org/10.1113/jphysiol.1989.sp017705
Darcq, E., & Kieffer, B. L. (2018). Opioid receptors: Drivers to addiction? Nature Reviews. Neuroscience, 19(8), 499–514. https://doi.org/10.1038/s41583-018-0028-x
Diaz, A., & Dickenson, A. H. (1997). Blockade of spinal N- and P-type, but not L-type, calcium channels inhibits the excitability of rat dorsal horn neurones produced by subcutaneous formalin inflammation. Pain, 69(1–2), 93–100. https://doi.org/10.1016/s0304-3959(96)03271-x
Dibué-Adjei, M., Kamp, M. A., Alpdogan, S., Tevoufouet, E. E., Neiss, W. F., Hescheler, J., et al. (2017). Cav2.3 (R-type) calcium channels are critical for mediating anticonvulsive and neuroprotective properties of lamotrigine in vivo. Cellular Physiology and Biochemistry, 44(3), 935–947. https://doi.org/10.1159/000485361
Dietrich, D., Kirschstein, T., Kukley, M., Pereverzev, A., von der Brelie, C., Schneider, T., et al. (2003). Functional specialization of presynaptic Cav2.3 Ca2+ channels. Neuron, 39(3), 483–496. https://doi.org/10.1016/s0896-6273(03)00430-6
Dogrul, A., Gardell, L. R., Ossipov, M. H., Tulunay, F. C., Lai, J., & Porreca, F. (2003). Reversal of experimental neuropathic pain by T-type calcium channel blockers. Pain, 105(1–2), 159–168. https://doi.org/10.1016/s0304-3959(03)00177-5
Dolphin, A. C. (2016). Voltage-gated calcium channels and their auxiliary subunits: Physiology and pathophysiology and pharmacology. The Journal of Physiology, 594(19), 5369–5390. https://doi.org/10.1113/JP272262
Dong, W., Jin, S. C., Allocco, A., Zeng, X., Sheth, A. H., Panchagnula, S., et al. (2020). Exome sequencing implicates impaired GABA signaling and neuronal ion transport in trigeminal neuralgia. iScience, 23(10), 101552. https://doi.org/10.1016/j.isci.2020.101552
Duan, J. H., Hodgdon, K. E., Hingtgen, C. M., & Nicol, G. D. (2014). N-type calcium current, Cav2.2, is enhanced in small-diameter sensory neurons isolated from Nf1+/− mice. Neuroscience, 270, 192–202. https://doi.org/10.1016/j.neuroscience.2014.04.021
Dubel, S. J., Altier, C., Chaumont, S., Lory, P., Bourinet, E., & Nargeot, J. (2004). Plasma membrane expression of T-type calcium channel alpha(1) subunits is modulated by high voltage-activated auxiliary subunits. The Journal of Biological Chemistry, 279(28), 29263–29269. https://doi.org/10.1074/jbc.M313450200
Dubin, A. E., & Patapoutian, A. (2010). Nociceptors: The sensors of the pain pathway. The Journal of Clinical Investigation, 120(11), 3760–3772. https://doi.org/10.1172/JCI42843
Dustrude, E. T., Wilson, S. M., Ju, W., Xiao, Y., & Khanna, R. (2013). CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. The Journal of Biological Chemistry, 288(34), 24316–24331. https://doi.org/10.1074/jbc.M113.474924
Eroglu, C., Allen, N. J., Susman, M. W., O’Rourke, N. A., Park, C. Y., Ozkan, E., et al. (2009). Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell, 139(2), 380–392. https://doi.org/10.1016/j.cell.2009.09.025
Esposito, T., Piluso, G., Saracino, D., Uccello, R., Schettino, C., Dato, C., et al. (2015). A novel diagnostic method to detect truncated neurofibromin in neurofibromatosis 1. Journal of Neurochemistry, 135(6), 1123–1128. https://doi.org/10.1111/jnc.13396
Evans, R. M., You, H., Hameed, S., Altier, C., Mezghrani, A., Bourinet, E., et al. (2010). Heterodimerization of ORL1 and opioid receptors and its consequences for N-type calcium channel regulation. The Journal of Biological Chemistry, 285(2), 1032–1040. https://doi.org/10.1074/jbc.M109.040634
Fang, Z., Park, C. K., Li, H. Y., Kim, H. Y., Park, S. H., Jung, S. J., et al. (2007). Molecular basis of Ca(v)2.3 calcium channels in rat nociceptive neurons. The Journal of Biological Chemistry, 282(7), 4757–4764. https://doi.org/10.1074/jbc.M605248200
Feng, Z. P., Hamid, J., Doering, C., Bosey, G. M., Snutch, T. P., & Zamponi, G. W. (2001). Residue Gly1326 of the N-type calcium channel alpha 1B subunit controls reversibility of omega-conotoxin GVIA and MVIIA block. The Journal of Biological Chemistry, 276(19), 15728–15735. https://doi.org/10.1074/jbc.M100406200
Feng, J. X., Ma, L. X., Jiao, C., Kuang, H. X., Zeng, F., et al. (2019). Nerve injury elevates functional Cav3.2 channels in superficial spinal dorsal horn. Molecular Pain, 15, 1744806919836569. https://doi.org/10.1177/1744806919836569
Fernandez, F. R., Iftinca, M. C., Zamponi, G. W., & Turner, R. W. (2021). Modeling temperature- and Cav3 subtype-dependent alterations in T-type calcium mediated burst firing. Molecular Brain, 14, 115.
Ferron, L., Koshti, S., & Zamponi, G. W. (2021). The life cycle of voltage-gated Ca2+ channels in neurons: An update on the trafficking of neuronal calcium channels. Neuro-Signals, 5(1), NS20200095. https://doi.org/10.1042/NS20200095
Field, M. J., Carnell, A. J., Gonzalez, M. I., McCleary, S., Oles, R. J., Smith, R., et al. (1999). Enadoline, a selective kappa-opioid receptor agonist shows potent antihyperalgesic and antiallodynic actions in a rat model of surgical pain. Pain, 80(1–2), 383–389. https://doi.org/10.1016/s0304-3959(98)00237-1
Field, M. J., Cox, P. J., Stott, E., Melrose, H., Offord, J., Su, T. Z., et al. (2006). Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proceedings of the National Academy of Sciences of the United States of America, 103(46), 17537–17542. https://doi.org/10.1073/pnas.0409066103
Field, M. J., Li, Z., & Schwarz, J. B. (2007). Ca2+ channel alpha2-delta ligands for the treatment of neuropathic pain. Journal of Medicinal Chemistry, 50(11), 2569–2575. https://doi.org/10.1021/jm060650z
Finnerup, N. B., Kuner, R., & Jensen, T. S. (2021). Neuropathic pain: From mechanisms to treatment. Physiological Reviews, 101(1), 259–301. https://doi.org/10.1152/physrev.00045.2019
Flatters, S. J., & Bennett, G. J. (2004). Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain, 109(1–2), 150–161. https://doi.org/10.1016/j.pain.2004.01.029
François, A., Schüetter, N., Laffray, S., Sanguesa, J., Pizzoccaro, A., Dubel, S., et al. (2015). The low-threshold calcium channel Cav3.2 determines low-threshold mechanoreceptor function. Cell Reports, 10(3), 370–382. https://doi.org/10.1016/j.celrep.2014.12.042
François-Moutal, L., Wang, Y., Moutal, A., Cottier, K. E., Melemedjian, O. K., Yang, X., et al. (2015). A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain, 156(7), 1247–1264. https://doi.org/10.1097/j.pain.0000000000000147
Gadotti, V. M., & Zamponi, G. W. (2018). Disrupting USP5/Cav3.2 interactions protects female mice from mechanical hypersensitivity during peripheral inflammation. Molecular Brain, 11(1), 60. https://doi.org/10.1186/s13041-018-0405-4
Gadotti, V. M., Bladen, C., Zhang, F. X., Chen, L., Gündüz, M. G., Şimşek, R., et al. (2015a). Analgesic effect of a broad-spectrum dihydropyridine inhibitor of voltage-gated calcium channels. Pflügers Archiv, 467(12), 2485–2493. https://doi.org/10.1007/s00424-015-1725-1
Gadotti, V. M., Caballero, A. G., Berger, N. D., Gladding, C. M., Chen, L., Pfeifer, T. A., et al. (2015b). Small organic molecule disruptors of Cav3.2 – USP5 interactions reverse inflammatory and neuropathic pain. Molecular Pain, 11, 12. https://doi.org/10.1186/s12990-015-0011-8
Gadotti, V. M., Kreitinger, J. M., Wageling, N. B., Budke, D., Diaz, P., & Zamponi, G. W. (2020). Cav3.2 T-type calcium channels control acute itch in mice. Molecular Brain, 13(1), 119. https://doi.org/10.1186/s13041-020-00663-9
Gaifullina, A. S., Lazniewska, J., Gerasimova, E. V., Burkhanova, G. F., Rzhepetskyy, Y., Tomin, A., et al. (2019). A potential role for T-type calcium channels in homocysteinemia-induced peripheral neuropathy. Pain, 160(12), 2798–2810. https://doi.org/10.1097/j.pain.0000000000001669
Gandini, M. A., & Zamponi, G. W. (2021). Voltage-gated calcium channel nanodomains: Molecular composition and function. The FEBS Journal, 289(3), 614–633. https://doi.org/10.1111/febs.15759
Gandini, M. A., Souza, I. A., Raval, D., Xu, J., Pan, Y. X., & Zamponi, G. W. (2019). Differential regulation of Cav2.2 channel exon 37 variants by alternatively spliced μ-opioid receptors. Molecular Brain, 12(1), 98. https://doi.org/10.1186/s13041-019-0524-6
Gangadharan, V., & Kuner, R. (2015). Unravelling spinal circuits of pain and mechanical allodynia. Neuron, 87(4), 673–675. https://doi.org/10.1016/j.neuron.2015.08.013
García-Caballero, A., Gadotti, V. M., Stemkowski, P., Weiss, N., Souza, I. A., Hodgkinson, V., et al. (2014). The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron, 83(5), 1144–1158. https://doi.org/10.1016/j.neuron.2014.07.036
Garcia-Caballero, A., Gadotti, V. M., Chen, L., & Zamponi, G. W. (2016). A cell-permeant peptide corresponding to the cUBP domain of USP5 reverses inflammatory and neuropathic pain. Molecular Pain, 12, 1744806916642444. https://doi.org/10.1177/1744806916642444
Gaskin, D. J., & Richard, P. (2012). The economic costs of pain in the United States. The Journal of Pain, 13(8), 715–724. https://doi.org/10.1016/j.jpain.2012.03.009
Gavériaux-Ruff, C., Peluso, J., Befort, K., Simonin, F., Zilliox, C., & Kieffer, B. L. (1997). Detection of opioid receptor mRNA by RT-PCR reveals alternative splicing for the delta- and kappa-opioid receptors. Brain Research. Molecular Brain Research, 48(2), 298–304. https://doi.org/10.1016/s0169-328x(97)00109-5
Goldstein, G. (1985). Pentazocine. Drug and Alcohol Dependence, 14(3–4), 313–323. https://doi.org/10.1016/0376-8716(85)90064-x
Gomez, K., Vallecillo, T. G. M., Moutal, A., Perez-Miller, S., Delgado-Lezama, R., Felix, R., et al. (2020). The role of cyclin-dependent kinase 5 in neuropathic pain. Pain, 161(12), 2674–2689. https://doi.org/10.1097/j.pain.0000000000002027
Gray, A. C., Raingo, J., & Lipscombe, D. (2007). Neuronal calcium channels: Splicing for optimal performance. Cell Calcium, 42(4–5), 409–417. https://doi.org/10.1016/j.ceca.2007.04.003
Gruner, W., & Silva, L. R. (1994). Omega-conotoxin sensitivity and presynaptic inhibition of glutamatergic sensory neurotransmission in vitro. The Journal of Neuroscience, 14(5 Pt 1), 2800–2808.
Gutmann, D. H., Ferner, R. E., Listernick, R. H., Korf, B. R., Wolters, P. L., & Johnson, K. J. (2017). Neurofibromatosis type 1. Nature Reviews. Disease Primers, 3, 17004. https://doi.org/10.1038/nrdp.2017.4
Gutzmann, J. J., Lin, L., & Hoffman, D. A. (2019). Functional coupling of Cav2.3 and BK potassium channels regulates action potential repolarization and short-term plasticity in the mouse hippocampus. Frontiers in Cellular Neuroscience, 13, 27. https://doi.org/10.3389/fncel.2019.00027
Harding, E. K., Boivin, B., & Salter, M. W. (2020). Intracellular calcium responses encode action potential firing in spinal cord lamina I neurons. The Journal of Neuroscience, 40(23), 4439–4456. https://doi.org/10.1523/JNEUROSCI.0206-20.2020
Harding, E. K., Dedek, A., Bonin, R. P., Salter, M. W., Snutch, T. P., & Hildebrand, M. E. (2021). The T-type calcium channel antagonist, Z944, reduces spinal excitability and pain hypersensitivity. British Journal of Pharmacology, 178(17), 3517–3532. https://doi.org/10.1111/bph.15498
Hatakeyama, S., Wakamori, M., Ino, M., Miyamoto, N., Takahashi, E., Yoshinaga, T., et al. (2001). Differential nociceptive responses in mice lacking the alpha(1B) subunit of N-type Ca(2+) channels. Neuroreport, 12(11), 2423–2427. https://doi.org/10.1097/00001756-200108080-00027
Helbig, K. L., Lauerer, R. J., Bahr, J. C., Souza, I. A., Myers, C. T., Uysal, B., et al. (2018). De novo pathogenic variants in CACNA1E cause developmental and epileptic encephalopathy with contractures, macrocephaly, and dyskinesias. American Journal of Human Genetics, 103(5), 666–678. https://doi.org/10.1016/j.ajhg.2018.09.006
Hendrich, J., Van Minh, A. T., Heblich, F., Nieto-Rostro, M., Watschinger, K., Striessnig, J., et al. (2008). Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proceedings of the National Academy of Sciences of the United States of America, 105(9), 3628–3633. https://doi.org/10.1073/pnas.0708930105
Hendrich, J., Bauer, C. S., & Dolphin, A. C. (2012). Chronic pregabalin inhibits synaptic transmission between rat dorsal root ganglion and dorsal horn neurons in culture. Channels (Austin, Tex.), 6(2), 124–132. https://doi.org/10.4161/chan.19805
Herlitze, S., Garcia, D. E., Mackie, K., Hille, B., Scheuer, T., & Catterall, W. A. (1996). Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature, 380(6571), 258–262. https://doi.org/10.1038/380258a0
Holz, G. G., Dunlap, K., & Kream, R. M. (1988). Characterization of the electrically evoked release of substance P from dorsal root ganglion neurons: Methods and dihydropyridine sensitivity. The Journal of Neuroscience, 8(2), 463–471.
Huguenard, J. R., & Prince, D. A. (1992). A novel T-type current underlies prolonged Ca(2+)-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. The Journal of Neuroscience, 12(10), 3804–3817.
Iftinca, M. C., & Zamponi, G. W. (2009). Regulation of neuronal T-type calcium channels. Trends in Pharmacological Sciences, 30(1), 32–40. https://doi.org/10.1016/j.tips.2008.10.004
Jacus, M. O., Uebele, V. N., Renger, J. J., & Todorovic, S. M. (2012). Presynaptic Cav3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. The Journal of Neuroscience, 32(27), 9374–9382. https://doi.org/10.1523/JNEUROSCI.0068-12.2012
Jagodic, M. M., Pathirathna, S., Nelson, M. T., Mancuso, S., Joksovic, P. M., Rosenberg, E. R., et al. (2007). Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. The Journal of Neuroscience, 27(12), 3305–3316. https://doi.org/10.1523/JNEUROSCI.4866-06.2007
Jagodic, M. M., Pathirathna, S., Joksovic, P. M., Lee, W., Nelson, M. T., Naik, A. K., et al. (2008). Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. Journal of Neurophysiology, 99(6), 3151–3156. https://doi.org/10.1152/jn.01031.2007
Joksimovic, S. L., Joksimovic, S. M., Tesic, V., García-Caballero, A., Feseha, S., Zamponi, G. W., et al. (2018). Selective inhibition of CaV3.2 channels reverses hyperexcitability of peripheral nociceptors and alleviates postsurgical pain. Science Signaling, 11(545), eaao4425. https://doi.org/10.1126/scisignal.aao4425
Ju, W., Li, Q., Wilson, S. M., Brittain, J. M., Meroueh, L., & Khanna, R. (2013). SUMOylation alters CRMP2 regulation of calcium influx in sensory neurons. Channels (Austin, Tex.), 7(3), 153–159. https://doi.org/10.4161/chan.24224
Kang, S. J., Liu, M. G., Shi, T. Y., Zhao, M. G., Kaang, B. K., & Zhuo, M. (2013). N-type voltage gated calcium channels mediate excitatory synaptic transmission in the anterior cingulate cortex of adult mice. Molecular Pain, 9, 58. https://doi.org/10.1186/1744-8069-9-58
Kerckhove, N., Pereira, B., Soriot-Thomas, S., Alchaar, H., Deleens, R., Hieng, V. S., et al. (2018). Efficacy and safety of a T-type calcium channel blocker in patients with neuropathic pain: A proof-of-concept, randomized, double-blind and controlled trial. European Journal of Pain, 22(7), 1321–1330. https://doi.org/10.1002/ejp.1221
Khanna, R., Moutal, A., Perez-Miller, S., Chefdeville, A., Boinon, L., & Patek, M. (2020). Druggability of CRMP2 for neurodegenerative diseases. ACS Chemical Neuroscience, 11(17), 2492–2505. https://doi.org/10.1021/acschemneuro.0c00307
Kim, C., Jun, K., Lee, T., Kim, S. S., McEnery, M. W., et al. (2001). Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage-dependent calcium channel. Molecular and Cellular Neurosciences, 18(2), 235–245. https://doi.org/10.1006/mcne.2001.1013
Kim, D., Park, D., Choi, S., Lee, S., Sun, M., et al. (2003). Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science, 302(5642), 117–119. https://doi.org/10.1126/science.1088886
Kim, S. D., Li, K. W., Boroujerdi, A., Peter Yu, Y., Zhou, C. Y., et al. (2012). Thrombospondin-4 contributes to spinal sensitization and neuropathic pain states. The Journal of Neuroscience, 32(26), 8977–8987. https://doi.org/10.1523/JNEUROSCI.6494-11.2012
Koganei, H., Shoji, M., & Iwata, S. (2009). Suppression of formalin-induced nociception by cilnidipine, a voltage-dependent calcium channel blocker. Biological & Pharmaceutical Bulletin, 32(10), 1695–1700. https://doi.org/10.1248/bpb.32.1695
Kondo, I., Marvizon, J. C., Song, B., Salgado, F., Codeluppi, S., Hua, X. Y., et al. (2005). Inhibition by spinal mu- and delta-opioid agonists of afferent-evoked substance P release. The Journal of Neuroscience, 25(14), 3651–3660. https://doi.org/10.1523/JNEUROSCI.0252-05.2005
Kuner, R., & Flor, H. (2017). Structural plasticity and reorganisation in chronic pain. Nature Reviews. Neuroscience, 18(2), 113. https://doi.org/10.1038/nrn.2017.5
Kupari, J., Usoskin, D., Parisien, M., Lou, D., Hu, Y., Fatt, M., et al. (2021). Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain. Nature Communications, 12(1), 1510. https://doi.org/10.1038/s41467-021-21725-z
Lana, B., Schlick, B., Martin, S., Pratt, W. S., Page, K. M., Goncalves, L., et al. (2014). Differential upregulation in DRG neurons of an α2δ-1 splice variant with a lower affinity for gabapentin after peripheral sensory nerve injury. Pain, 155(3), 522–533. https://doi.org/10.1016/j.pain.2013.12.001
Lauzadis, J., Liu, H., Lu, Y., Rebecchi, M. J., Kaczocha, M., & Puopolo, M. (2020). Contribution of T-type calcium channels to spinal cord injury-induced hyperexcitability of nociceptors. The Journal of Neuroscience, 40(38), 7229–7240. https://doi.org/10.1523/JNEUROSCI.0517-20.2020
LeBlanc, B. W., Lii, T. R., Huang, J. J., Chao, Y. C., Bowary, P. M., Cross, B. S., et al. (2016). T-type calcium channel blocker Z944 restores cortical synchrony and thalamocortical connectivity in a rat model of neuropathic pain. Pain, 157(1), 255–263. https://doi.org/10.1097/j.pain.0000000000000362
Lee, S., Kim, Y., Back, S. K., Choi, H. W., Lee, J. Y., Jung, H. H., et al. (2010). Analgesic effect of highly reversible ω-conotoxin FVIA on N type Ca2+ channels. Molecular Pain, 6, 97. https://doi.org/10.1186/1744-8069-6-97
Li, Y. C., Zhang, X. L., Matthews, E. A., Li, K. W., Kurwa, A., et al. (2006). Calcium channel alpha2delta1 subunit mediates spinal hyperexcitability in pain modulation. Pain, 125(1–2), 20–34. https://doi.org/10.1016/j.pain.2006.04.022
Li, Y. X., Ko, H. G., Chen, T., Descalzi, G., Koga, K., et al. (2010). Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science, 330(6009), 1400–1404. https://doi.org/10.1126/science.1191792
Li, L., Rutlin, M., Abraira, V. E., Cassidy, C., Kus, L., et al. (2011). The functional organization of cutaneous low-threshold mechanosensory neurons. Cell, 147(7), 1615–1627. https://doi.org/10.1016/j.cell.2011.11.027
Li, K., Zhao, G. Q., Li, L. Y., Wu, G. Z., & Cui, S. S. (2014). Epigenetic upregulation of Cdk5 in the dorsal horn contributes to neuropathic pain in rats. Neuroreport, 25(14), 1116–1121. https://doi.org/10.1097/WNR.0000000000000237
Li, Y., Tatsui, C. E., Rhines, L. D., North, R. Y., Harrison, D. S., et al. (2017). Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain, 158(3), 417–429. https://doi.org/10.1097/j.pain.0000000000000774
Lipscombe, D., Pan, J. Q., & Gray, A. C. (2002). Functional diversity in neuronal voltage-gated calcium channels by alternative splicing of Ca(v)alpha1. Molecular Neurobiology, 26(1), 21–44. https://doi.org/10.1385/MN:26:1:021
Luebke, J. I., & Dunlap, K. (1994). Sensory neuron N-type calcium currents are inhibited by both voltage-dependent and -independent mechanisms. Pflügers Archiv, 428(5–6), 499–507. https://doi.org/10.1007/BF00374571
Luo, Z. D., Chaplan, S. R., Higuera, E. S., Sorkin, L. S., Stauderman, K. A., Williams, M. E., et al. (2001). Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. The Journal of Neuroscience, 21(6), 1868–1875.
Macabuag, N., & Dolphin, A. C. (2015). Alternative splicing in Ca(V)2.2 regulates neuronal trafficking via adaptor protein complex-1 adaptor protein motifs. The Journal of Neuroscience, 35(43), 14636–14652. https://doi.org/10.1523/JNEUROSCI.3034-15.2015
Maggi, C. A., Tramontana, M., Cecconi, R., & Santicioli, P. (1990). Neurochemical evidence for the involvement of N-type calcium channels in transmitter secretion from peripheral endings of sensory nerves in guinea pigs. Neuroscience Letters, 114(2), 203–206. https://doi.org/10.1016/0304-3940(90)90072-h
Malmberg, A. B., & Yaksh, T. L. (1994). Voltage-sensitive calcium channels in spinal nociceptive processing: Blockade of N- and P-type channels inhibits formalin-induced nociception. The Journal of Neuroscience, 14(8), 4882–4890.
Marangoudakis, S., Andrade, A., Helton, T. D., Denome, S., Castiglioni, A. J., & Lipscombe, D. (2012). Differential ubiquitination and proteasome regulation of Ca(V)2.2 N-type channel splice isoforms. The Journal of Neuroscience, 32(30), 10365–10369. https://doi.org/10.1523/JNEUROSCI.0851-11.2012
Marger, F., Gelot, A., Alloui, A., Matricon, J., Ferrer, J. F., Barrère, C., et al. (2011). T-type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proceedings of the National Academy of Sciences of the United States of America, 108(27), 11268–11273. https://doi.org/10.1073/pnas.1100869108
Marker, C. L., Stoffel, M., & Wickman, K. (2004). Spinal G-protein-gated K+ channels formed by GIRK1 and GIRK2 subunits modulate thermal nociception and contribute to morphine analgesia. The Journal of Neuroscience, 24(11), 2806–2812. https://doi.org/10.1523/JNEUROSCI.5251-03.2004
Matthews, E. A., & Dickenson, A. H. (2001). Effects of ethosuximide, a T-type Ca(2+) channel blocker, on dorsal horn neuronal responses in rats. European Journal of Pharmacology, 415(2–3), 141–149. https://doi.org/10.1016/s0014-2999(01)00812-3
Matthews, E. A., Bee, L. A., Stephens, G. J., & Dickenson, A. H. (2007). The Cav2.3 calcium channel antagonist SNX-482 reduces dorsal horn neuronal responses in a rat model of chronic neuropathic pain. The European Journal of Neuroscience, 25(12), 3561–3569. https://doi.org/10.1111/j.1460-9568.2007.05605.x
Mayer, S., Spickschen, J., Stein, K. V., Crevenna, R., Dorner, T. E., & Simon, J. (2019). The societal costs of chronic pain and its determinants: The case of Austria. PLoS One, 14(3), e0213889. https://doi.org/10.1371/journal.pone.0213889
McDonald, J., & Lambert, D. G. (2005). Opioid receptors. Continuing Education in Anaesthesia, Critical Care & Pain, 5(1), 22–25. https://doi.org/10.1093/bjaceaccp/mki004
Messinger, R. B., Naik, A. K., Jagodic, M. M., Nelson, M. T., Lee, W. Y., Choe, W. J., et al. (2009). In vivo silencing of the Ca(V)3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain, 145(1–2), 184–195. https://doi.org/10.1016/j.pain.2009.06.012
Miljanich, G. P. (2004). Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Current Medicinal Chemistry, 11(23), 3029–3040. https://doi.org/10.2174/0929867043363884
Molliver, D. C., Radeke, M. J., Feinstein, S. C., & Snider, W. D. (1995). Presence or absence of TrkA protein distinguishes subsets of small sensory neurons with unique cytochemical characteristics and dorsal horn projections. The Journal of Comparative Neurology, 361(3), 404–416. https://doi.org/10.1002/cne.903610305
Montera, M., Goins, A., Cmarko, L., Weiss, N., Westlund, K. N., & Alles, S. R. A. (2021). Trigeminal neuropathic pain is alleviated by inhibition of Cav3.3 T-type calcium channels in mice. Channels (Austin, Tex.), 15(1), 31–37. https://doi.org/10.1080/19336950.2020.1859248
Moutal, A., Chew, L. A., Yang, X., Wang, Y., Yeon, S. K., Telemi, E., et al. (2016a). (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain, 157(7), 1448–1463. https://doi.org/10.1097/j.pain.0000000000000555
Moutal, A., François-Moutal, L., Perez-Miller, S., Cottier, K., Chew, L. A., Yeon, S. K., et al. (2016b). (S)-lacosamide binding to collapsin response mediator protein 2 (CRMP2) regulates CaV2.2 activity by subverting its phosphorylation by Cdk5. Molecular Neurobiology, 53(3), 1959–1976. https://doi.org/10.1007/s12035-015-9141-2
Moutal, A., Wang, Y., Yang, X., Ji, Y., Luo, S., Dorame, A., et al. (2017a). Dissecting the role of the CRMP2-neurofibromin complex on pain behaviors. Pain, 158(11), 2203–2221. https://doi.org/10.1097/j.pain.0000000000001026
Moutal, A., Yang, X., Li, W., Gilbraith, K. B., Luo, S., Cai, S., et al. (2017b). CRISPR/Cas9 editing of Nf1 gene identifies CRMP2 as a therapeutic target in neurofibromatosis type 1-related pain that is reversed by (S)-Lacosamide. Pain, 158(12), 2301–2319. https://doi.org/10.1097/j.pain.0000000000001002
Moutal, A., White, K. A., Chefdeville, A., Laufmann, R. N., Vitiello, P. F., Feinstein, D., et al. (2019). Dysregulation of CRMP2 post-translational modifications drive its pathological functions. Molecular Neurobiology, 56(10), 6736–6755. https://doi.org/10.1007/s12035-019-1568-4
Murakami, M., Nakagawasai, O., Suzuki, T., Mobarakeh, I. I., Sakurada, Y., Murata, A., et al. (2004). Antinociceptive effect of different types of calcium channel inhibitors and the distribution of various calcium channel alpha 1 subunits in the dorsal horn of spinal cord in mice. Brain Research, 1024(1–2), 122–129. https://doi.org/10.1016/j.brainres.2004.07.066
Myoga, M. H., & Regehr, W. G. (2011). Calcium microdomains near R-type calcium channels control the induction of presynaptic long-term potentiation at parallel fiber to purkinje cell synapses. The Journal of Neuroscience, 31(14), 5235–5243. https://doi.org/10.1523/JNEUROSCI.5252-10.2011
Nazarbaghi, S., Amiri-Nikpour, M. R., Eghbal, A. F., & Valizadeh, R. (2017). Comparison of the effect of topiramate versus gabapentin on neuropathic pain in patients with polyneuropathy: A randomized clinical trial. Electronic Physician, 9(10), 5617–5622. https://doi.org/10.19082/5617
Newton, A. R., Bingham, S., Case, P. C., Sanger, G. J., & Lawson, S. N. (2001). Dorsal root ganglion neurons show increased expression of the calcium channel alpha2delta-1 subunit following partial sciatic nerve injury. Brain Research. Molecular Brain Research, 95(1–2), 1–8. https://doi.org/10.1016/s0169-328x(01)00188-7
Newton, M. P., Orr, C. J., Wallace, M. J., Kim, C., Shin, H. S., et al. (2004). Deletion of N-type calcium channels alters ethanol reward and reduces ethanol consumption in mice. The Journal of Neuroscience, 24(44), 9862–9869. https://doi.org/10.1523/JNEUROSCI.3446-04.2004
Niu, J., Ding, L., Li, J. J., Kim, H., Liu, J., Li, H., et al. (2013). Modality-based organization of ascending somatosensory axons in the direct dorsal column pathway. The Journal of Neuroscience, 33(45), 17691–17709. https://doi.org/10.1523/JNEUROSCI.3429-13.2013
Nowycky, M. C., Fox, A. P., & Tsien, R. W. (1985). Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature, 316(6027), 440–443. https://doi.org/10.1038/316440a0
Nozaki, C., Le Bourdonnec, B., Reiss, D., Windh, R. T., Little, P. J., Dolle, R. E., et al. (2012). δ-Opioid mechanisms for ADL5747 and ADL5859 effects in mice: Analgesia, locomotion, and receptor internalization. The Journal of Pharmacology and Experimental Therapeutics, 342(3), 799–807. https://doi.org/10.1124/jpet.111.188987
Olivera, B. M., McIntosh, J. M., Cruz, L. J., Luque, F. A., & Gray, W. R. (1984). Purification and sequence of a presynaptic peptide toxin from Conus geographus venom. Biochemistry, 23(22), 5087–5090. https://doi.org/10.1021/bi00317a001
Olivera, B. M., Miljanich, G. P., Ramachandran, J., & Adams, M. E. (1994). Calcium channel diversity and neurotransmitter release: The omega-conotoxins and omega-agatoxins. Annual Review of Biochemistry, 63, 823–867. https://doi.org/10.1146/annurev.bi.63.070194.004135
Orestes, P., Osuru, H. P., McIntire, W. E., Jacus, M. O., Salajegheh, R., Jagodic, M. M., et al. (2013). Reversal of neuropathic pain in diabetes by targeting glycosylation of Ca(V)3.2 T-type calcium channels. Diabetes, 62(11), 3828–3838. https://doi.org/10.2337/db13-0813
Pajouhesh, H., Feng, Z. P., Ding, Y., Zhang, L., Morrison, J. L., Belardetti, F., et al. (2010). Structure-activity relationships of diphenylpiperazine N-type calcium channel inhibitors. Bioorganic & Medicinal Chemistry Letters, 20(4), 1378–1383. https://doi.org/10.1016/j.bmcl.2010.01.008
Pan, H. L., Wu, Z. Z., Zhou, H. Y., Chen, S. R., Zhang, H. M., & Li, D. P. (2008). Modulation of pain transmission by G-protein-coupled receptors. Pharmacology & Therapeutics, 117(1), 141–161. https://doi.org/10.1016/j.pharmthera.2007.09.003
Park, J. Y., Remy, S., Varela, J., Cooper, D. C., Chung, S., Kang, H. W., et al. (2010). A post-burst after depolarization is mediated by group i metabotropic glutamate receptor-dependent upregulation of Ca(v)2.3 R-type calcium channels in CA1 pyramidal neurons. PLoS Biology, 8(11), e1000534. https://doi.org/10.1371/journal.pbio.1000534
Pasternak, G. W. (2018). Mu opioid pharmacology: 40 years to the promised land. Advances in Pharmacology, 82, 261–291. https://doi.org/10.1016/bs.apha.2017.09.006
Patel, R., Bauer, C. S., Nieto-Rostro, M., Margas, W., Ferron, L., Chaggar, K., et al. (2013). α2δ-1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. The Journal of Neuroscience, 33(42), 16412–16426. https://doi.org/10.1523/JNEUROSCI.1026-13.2013
Peirs, C., Williams, S. P., Zhao, X., Walsh, C. E., Gedeon, J. Y., Cagle, N. E., et al. (2015). Dorsal horn circuits for persistent mechanical pain. Neuron, 87(4), 797–812. https://doi.org/10.1016/j.neuron.2015.07.029
Penn, R. D., & Paice, J. A. (2000). Adverse effects associated with the intrathecal administration of ziconotide. Pain, 85(1–2), 291–296. https://doi.org/10.1016/s0304-3959(99)00254-7
Perez-Reyes, E. (2003). Molecular physiology of low-voltage-activated t-type calcium channels. Physiological Reviews, 83(1), 117–161. https://doi.org/10.1152/physrev.00018.2002
Piekarz, A. D., Due, M. R., Khanna, M., Wang, B., Ripsch, M. S., Wang, R., et al. (2012). CRMP-2 peptide mediated decrease of high and low voltage-activated calcium channels, attenuation of nociceptor excitability, and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Molecular Pain, 8, 54. https://doi.org/10.1186/1744-8069-8-54
Piltonen, M., Parisien, M., Grégoire, S., Chabot-Doré, A. J., Jafarnejad, S. M., Bérubé, P., et al. (2019). Alternative splicing of the delta-opioid receptor gene suggests existence of new functional isoforms. Molecular Neurobiology, 56(4), 2855–2869. https://doi.org/10.1007/s12035-018-1253-z
Qian, A., Song, D., Li, Y., Liu, X., Tang, D., Yao, W., et al. (2013). Role of voltage gated Ca2+ channels in rat visceral hypersensitivity change induced by 2,4,6-trinitrobenzene sulfonic acid. Molecular Pain, 9, 15. https://doi.org/10.1186/1744-8069-9-15
Rahman, W., Patel, R., & Dickenson, A. H. (2015). Electrophysiological evidence for voltage-gated calcium channel 2 (Cav2) modulation of mechano- and thermosensitive spinal neuronal responses in a rat model of osteoarthritis. Neuroscience, 305, 76–85. https://doi.org/10.1016/j.neuroscience.2015.07.073
Raingo, J., Castiglioni, A. J., & Lipscombe, D. (2007). Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nature Neuroscience, 10(3), 285–292. https://doi.org/10.1038/nn1848
Reitsma, M. I., Tranmer, J. E., Buchanan, D. M., & VanDenKerhof, E. G. (2012). The epidemiology of chronic pain in Canadian men and women between 1994 and 2007: Results from the longitudinal component of the National Population Health Survey. Pain Research & Management, 17(3), 166–172.
Reynolds, I. J., Wagner, J. A., Snyder, S. H., Thayer, S. A., Olivera, B. M., & Miller, R. J. (1986). Brain voltage-sensitive calcium channel subtypes differentiated by omega-conotoxin fraction GVIA. Proceedings of the National Academy of Sciences of the United States of America, 83(22), 8804–8807. https://doi.org/10.1073/pnas.83.22.8804
Ripsch, M. S., Ballard, C. J., Khanna, M., Hurley, J. H., White, F. A., & Khanna, R. (2012). A peptide uncoupling CRMP-2 from the presynaptic Ca2+ channel complex demonstrates efficacity in animal models of migraine and AIDS therapy-induced neuropathy. Translational Neuroscience, 3(1), 1–8. https://doi.org/10.2478/s13380-012-0002-4
Rosenberg, J. M., Harrell, C., Ristic, H., Werner, R. A., & de Rosayro, A. M. (1997). The effect of gabapentin on neuropathic pain. The Clinical Journal of Pain, 13(3), 251–255. https://doi.org/10.1097/00002508-199709000-00011
Ross, H. R., Gilmore, A. J., & Connor, M. (2009). Inhibition of human recombinant T-type calcium channels by the endocannabinoid N-arachidonoyl dopamine. British Journal of Pharmacology, 156(5), 740–750. https://doi.org/10.1111/j.1476-5381.2008.00072.x
Sadeghi, M., Murali, S. S., Lewis, R. J., Alewood, P. F., Mohammadi, S., & Christie, M. J. (2013). Novel ω-conotoxins from C. catus reverse signs of mouse inflammatory pain after systemic administration. Molecular Pain, 9, 51. https://doi.org/10.1186/1744-8069-9-51
Saegusa, H., Kurihara, T., Zong, S., Minowa, O., Kazuno, A., Han, W., et al. (2000). Altered pain responses in mice lacking alpha 1E subunit of the voltage-dependent Ca2+ channel. Proceedings of the National Academy of Sciences of the United States of America, 97(11), 6132–6137. https://doi.org/10.1073/pnas.100124197
Saegusa, H., Kurihara, T., Zong, S., Kazuno, A., Matsuda, Y., Nonaka, T., et al. (2001). Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. The EMBO Journal, 20(10), 2349–2356. https://doi.org/10.1093/emboj/20.10.2349
Saegusa, H., Matsuda, Y., & Tanabe, T. (2002). Effects of ablation of N- and R-type Ca(2+) channels on pain transmission. Neuroscience Research, 43(1), 1–7. https://doi.org/10.1016/s0168-0102(02)00017-2
Santi, C. M., Cayabyab, F. S., Sutton, K. G., McRory, J. E., Mezeyova, J., Hamming, K. S., et al. (2002). Differential inhibition of T-type calcium channels by neuroleptics. The Journal of Neuroscience, 22(2), 396–403.
Santicioli, P., Del Bianco, E., Tramontana, M., Geppetti, P., & Maggi, C. A. (1992). Release of calcitonin gene-related peptide like-immunoreactivity induced by electrical field stimulation from rat spinal afferents is mediated by conotoxin-sensitive calcium channels. Neuroscience Letters, 136(2), 161–164. https://doi.org/10.1016/0304-3940(92)90039-a
Scherrer, G., Imamachi, N., Cao, Y. Q., Contet, C., Mennicken, F., O’Donnell, D., et al. (2009). Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell, 137(6), 1148–1159. https://doi.org/10.1016/j.cell.2009.04.019
Schopflocher, D., Taenzer, P., & Jovey, R. (2011). The prevalence of chronic pain in Canada. Pain Research & Management, 16(6), 445–450. https://doi.org/10.1155/2011/876306
Schroeder, C. I., Doering, C. J., Zamponi, G. W., & Lewis, R. J. (2006). N-type calcium channel blockers: Novel therapeutics for the treatment of pain. Medicinal Chemistry, 2(5), 535–543. https://doi.org/10.2174/157340606778250216
Seko, T., Kato, M., Kohno, H., Ono, S., Hashimura, K., Takenobu, Y., et al. (2002). L-cysteine based N-type calcium channel blockers: Structure-activity relationships of the C-terminal lipophilic moiety, and oral analgesic efficacy in rat pain models. Bioorganic & Medicinal Chemistry Letters, 12(17), 2267–2269. https://doi.org/10.1016/s0960-894x(02)00456-0
Serra, J., Duan, W. R., Locke, C., Solà, R., Liu, W., & Nothaft, W. (2015). Effects of a T-type calcium channel blocker, ABT-639, on spontaneous activity in C-nociceptors in patients with painful diabetic neuropathy: A randomized controlled trial. Pain, 156(11), 2175–2183. https://doi.org/10.1097/j.pain.0000000000000249
Shan, Z., Cai, S., Yu, J., Zhang, Z., Vallecillo, T. G. M., Serafini, M. J., et al. (2019). Reversal of peripheral neuropathic pain by the small-molecule natural product physalin F via block of CaV2.3 (R-type) and CaV2.2 (N-type) voltage-gated calcium channels. ACS Chemical Neuroscience, 10(6), 2939–2955. https://doi.org/10.1021/acschemneuro.9b00166
Sharma, N., Flaherty, K., Lezgiyeva, K., Wagner, D. E., Klein, A. M., & Ginty, D. D. (2020). The emergence of transcriptional identity in somatosensory neurons. Nature, 577(7790), 392–398. https://doi.org/10.1038/s41586-019-1900-1
Shen, F. Y., Chen, Z. Y., Zhong, W., Ma, L. Q., Chen, C., Yang, Z. J., et al. (2015). Alleviation of neuropathic pain by regulating T-type calcium channels in rat anterior cingulate cortex. Molecular Pain, 11, 7. https://doi.org/10.1186/s12990-015-0008-3
Sheng, Z. H., Rettig, J., Takahashi, M., & Catterall, W. A. (1994). Identification of a syntaxin-binding site on N-type calcium channels. Neuron, 13(6), 1303–1313. https://doi.org/10.1016/0896-6273(94)90417-0
Shin, S. M., Cai, Y., Itson-Zoske, B., Qiu, C., Hao, X., Xiang, H., et al. (2020). Enhanced T-type calcium channel 3.2 activity in sensory neurons contributes to neuropathic-like pain of monosodium iodoacetate-induced knee osteoarthritis. Molecular Pain, 16, 1744806920963807. https://doi.org/10.1177/1744806920963807
Sluka, K. A. (1998). Blockade of N- and P/Q-type calcium channels reduces the secondary heat hyperalgesia induced by acute inflammation. The Journal of Pharmacology and Experimental Therapeutics, 287(1), 232–237.
Snutch, T. P., & Zamponi, G. W. (2018). Recent advances in the development of T-type calcium channel blockers for pain intervention. British Journal of Pharmacology, 175(12), 2375–2383. https://doi.org/10.1111/bph.13906
Souza, I. A., Gandini, M. A., Zhang, F. X., Mitchell, W. G., Matsumoto, J., Lerner, J., et al. (2019). Pathogenic Cav3.2 channel mutation in a child with primary generalized epilepsy. Molecular Brain, 12(1), 86. https://doi.org/10.1186/s13041-019-0509-5
Spahn, V., & Stein, C. (2017). Targeting delta opioid receptors for pain treatment: Drugs in phase I and II clinical development. Expert Opinion on Investigational Drugs, 26(2), 155–160. https://doi.org/10.1080/13543784.2017.1275562
Staats, P. S., Yearwood, T., Charapata, S. G., Presley, R. W., Wallace, M. S., Byas-Smith, M., et al. (2004). Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: A randomized controlled trial. JAMA, 291(1), 63–70. https://doi.org/10.1001/jama.291.1.63
Stemkowski, P., García-Caballero, A., Gadotti, V. M., M’Dahoma, S., Huang, S., Black, S. A. G., et al. (2016). TRPV1 nociceptor activity initiates USP5/T-type channel-mediated plasticity. Cell Reports, 17(11), 2901–2912. https://doi.org/10.1016/j.celrep.2016.11.047
Swensen, A. M., Herrington, J., Bugianesi, R. M., Dai, G., Haedo, R. J., Ratliff, K. S., et al. (2012). Characterization of the substituted N-triazole oxindole TROX-1, a small-molecule, state-dependent inhibitor of Ca(V)2 calcium channels. Molecular Pharmacology, 81(3), 488–497. https://doi.org/10.1124/mol.111.075226
Taylor, C. P., Angelotti, T., & Fauman, E. (2007). Pharmacology and mechanism of action of pregabalin: The calcium channel alpha2-delta (alpha2-delta) subunit as a target for antiepileptic drug discovery. Epilepsy Research, 73(2), 137–150. https://doi.org/10.1016/j.eplepsyres.2006.09.008
Tedford, H. W., & Zamponi, G. W. (2006). Direct G protein modulation of Cav2 calcium channels. Pharmacological Reviews, 58(4), 837–862. https://doi.org/10.1124/pr.58.4.11
Teodori, E., Baldi, E., Dei, S., Gualtieri, F., Romanelli, M. N., Scapecchi, S., et al. (2004). Design, synthesis, and preliminary pharmacological evaluation of 4-aminopiperidine derivatives as N-type calcium channel blockers active on pain and neuropathic pain. Journal of Medicinal Chemistry, 47(24), 6070–6081. https://doi.org/10.1021/jm049923l
Terrence, C. F., Fromm, G. H., & Tenicela, R. (1985). Baclofen as an analgesic in chronic peripheral nerve disease. European Neurology, 24(6), 380–385. https://doi.org/10.1159/000115830
Tomita, S., Sekiguchi, F., Deguchi, T., Miyazaki, T., Ikeda, Y., Tsubota, M., et al. (2019). Critical role of Cav3.2 T-type calcium channels in the peripheral neuropathy induced by bortezomib, a proteasome-inhibiting chemotherapeutic agent, in mice. Toxicology, 413, 33–39. https://doi.org/10.1016/j.tox.2018.12.003
Tomita, S., Sekiguchi, F., Kasanami, Y., Naoe, K., Tsubota, M., Wake, H., et al. (2020). Cav3.2 overexpression in L4 dorsal root ganglion neurons after L5 spinal nerve cutting involves Egr-1, USP5 and HMGB1 in rats: An emerging signaling pathway for neuropathic pain. European Journal of Pharmacology, 888, 173587. https://doi.org/10.1016/j.ejphar.2020.173587
Tytgat, J., Pauwels, P. J., Vereecke, J., & Carmeliet, E. (1991). Flunarizine inhibits a high-threshold inactivating calcium channel (N-type) in isolated hippocampal neurons. Brain Research, 549(1), 112–117. https://doi.org/10.1016/0006-8993(91)90606-v
Umeda, M., Ohkubo, T., Ono, J., Fukuizumi, T., & Kitamura, K. (2006). Molecular and immunohistochemical studies in expression of voltage-dependent Ca2+ channels in dorsal root ganglia from streptozotocin-induced diabetic mice. Life Sciences, 79(21), 1995–2000. https://doi.org/10.1016/j.lfs.2006.06.039
Uneyama, H., Takahara, A., Dohmoto, H., Yoshimoto, R., Inoue, K., & Akaike, N. (1997). Blockade of N-type Ca2+ current by cilnidipine (FRC-8653) in acutely dissociated rat sympathetic neurones. British Journal of Pharmacology, 122(1), 37–42. https://doi.org/10.1038/sj.bjp.0701342
Vardanyan, R. S., Cain, J. P., Haghighi, S. M., Kumirov, V. K., McIntosh, M. I., Sandweiss, A. J., et al. (2017). Synthesis and investigation of mixed μ-opioid and δ-opioid agonists as possible bivalent ligands for treatment of pain. Journal of Heterocyclic Chemistry, 54(2), 1228–1235. https://doi.org/10.1002/jhet.2696
Waldhoer, M., Bartlett, S. E., & Whistler, J. L. (2004). Opioid receptors. Annual Review of Biochemistry, 73, 953–990. https://doi.org/10.1146/annurev.biochem.73.011303.073940
Wallace, M., Duan, R., Liu, W., Locke, C., & Nothaft, W. (2016). A randomized, double-blind, placebo-controlled, crossover study of the T-type calcium channel blocker ABT-639 in an intradermal capsaicin experimental pain model in healthy adults. Pain Medicine, 17(3), 551–560. https://doi.org/10.1093/pm/pnv068
Wang, H., Sun, H., Della Penna, K., Benz, R. J., Xu, J., Gerhold, D. L., et al. (2002). Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience, 114(3), 529–546. https://doi.org/10.1016/s0306-4522(02)00341-x
Weiergräber, M., Henry, M., Radhakrishnan, K., Hescheler, J., & Schneider, T. (2007). Hippocampal seizure resistance and reduced neuronal excitotoxicity in mice lacking the Cav2.3 E/R-type voltage-gated calcium channel. Journal of Neurophysiology, 97(5), 3660–3669. https://doi.org/10.1152/jn.01193.2006
Weiss, N., & Zamponi, G. W. (2019a). T-type calcium channels: From molecule to therapeutic opportunities. The International Journal of Biochemistry & Cell Biology, 108, 34–39. https://doi.org/10.1016/j.biocel.2019.01.008
Weiss, N., & Zamponi, G. W. (2019b). T-type channel druggability at a crossroads. ACS Chemical Neuroscience, 10(3), 1124–1126. https://doi.org/10.1021/acschemneuro.9b00031
Weiss, N., Hameed, S., Fernández-Fernández, J. M., Fablet, K., Karmazinova, M., Poillot, C., et al. (2012). A Ca(v)3.2/syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. The Journal of Biological Chemistry, 287(4), 2810–2818. https://doi.org/10.1074/jbc.M111.290882
Weiss, N., Black, S. A., Bladen, C., Chen, L., & Zamponi, G. W. (2013). Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflügers Archiv, 465(8), 1159–1170. https://doi.org/10.1007/s00424-013-1259-3
Wen, X. J., Xu, S. Y., Chen, Z. X., Yang, C. X., Liang, H., & Li, H. (2010). The roles of T-type calcium channel in the development of neuropathic pain following chronic compression of rat dorsal root ganglia. Pharmacology, 85(5), 295–300. https://doi.org/10.1159/000276981
Westenbroek, R. E., Hell, J. W., Warner, C., Dubel, S. J., Snutch, T. P., & Catterall, W. A. (1992). Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron, 9(6), 1099–1115. https://doi.org/10.1016/0896-6273(92)90069-p
Wheeler, D. B., Randall, A., & Tsien, R. W. (1994). Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science, 264(5155), 107–111. https://doi.org/10.1126/science.7832825
Wiffen, P. J., Derry, S., Lunn, M. P., & Moore, R. A. (2013). Topiramate for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews, 8, CD008314. https://doi.org/10.1002/14651858.CD008314.pub3
Wilson, S. M., Brittain, J. M., Piekarz, A. D., Ballard, C. J., Ripsch, M. S., Cummins, T. R., et al. (2011). Further insights into the antinociceptive potential of a peptide disrupting the N-type calcium channel-CRMP-2 signaling complex. Channels (Austin, Tex.), 5(5), 449–456. https://doi.org/10.4161/chan.5.5.17363
Wilson, S. M., Schmutzler, B. S., Brittain, J. M., Dustrude, E. T., Ripsch, M. S., Pellman, J. J., et al. (2012). Inhibition of transmitter release and attenuation of anti-retroviral-associated and tibial nerve injury-related painful peripheral neuropathy by novel synthetic Ca2+ channel peptides. The Journal of Biological Chemistry, 287(42), 35065–35077. https://doi.org/10.1074/jbc.M112.378695
Xie, J. Y., Chew, L. A., Yang, X., Wang, Y., Qu, C., Federici, L. M., et al. (2016). Sustained relief of ongoing experimental neuropathic pain by a CRMP2 peptide aptamer with low abuse potential. Pain, 157(9), 2124–2140. https://doi.org/10.1097/j.pain.0000000000000628
Xu, H., Wu, L. J., Wang, H., Zhang, X., Vadakkan, K. I., Kim, S. S., et al. (2008). Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. The Journal of Neuroscience, 28(29), 7445–7453. https://doi.org/10.1523/JNEUROSCI.1812-08.2008
Yamamoto, S., Suzuki, Y., Ono, H., Kume, K., & Ohsawa, M. (2016). N- and L-type calcium channels blocker cilnidipine ameliorates neuropathic pain. European Journal of Pharmacology, 793, 66–75. https://doi.org/10.1016/j.ejphar.2016.11.001
Yang, L., Gu, X., Zhang, W., Zhang, J., & Ma, Z. (2014). Cdk5 inhibitor roscovitine alleviates neuropathic pain in the dorsal root ganglia by downregulating N-methyl-D-aspartate receptor subunit 2A. Neurological Sciences, 35(9), 1365–1371. https://doi.org/10.1007/s10072-014-1713-9
Yokoyama, K., Kurihara, T., Makita, K., & Tanabe, T. (2003). Plastic change of N-type Ca channel expression after preconditioning is responsible for prostaglandin E2-induced long-lasting allodynia. Anesthesiology, 99(6), 1364–1370. https://doi.org/10.1097/00000542-200312000-00019
You, H., Gadotti, V. M., Petrov, R. R., Zamponi, G. W., & Diaz, P. (2011). Functional characterization and analgesic effects of mixed cannabinoid receptor/T-type channel ligands. Molecular Pain, 7, 89. https://doi.org/10.1186/1744-8069-7-89
Yu, P. Y., Gong, N., Kweon, T. D., Vo, B., & Luo, Z. D. (2018). Gabapentin prevents synaptogenesis between sensory and spinal cord neurons induced by thrombospondin-4 acting on pre-synaptic Cavα2δ1 subunits and involving T-type Ca2+ channels. British Journal of Pharmacology, 175(12), 2348–2361. https://doi.org/10.1111/bph.14149
Yusaf, S. P., Goodman, J., Pinnock, R. D., Dixon, A. K., & Lee, K. (2001). Expression of voltage-gated calcium channel subunits in rat dorsal root ganglion neurons. Neuroscience Letters, 311(2), 137–141. https://doi.org/10.1016/s0304-3940(01)02038-9
Zaman, T., Lee, K., Park, C., Paydar, A., Choi, J. H., Cheong, E., et al. (2011). Cav2.3 channels are critical for oscillatory burst discharges in the reticular thalamus and absence epilepsy. Neuron, 70(1), 95–108. https://doi.org/10.1016/j.neuron.2011.02.042
Zamponi, G. W. (2016). Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nature Reviews. Drug Discovery, 15(1), 19–34. https://doi.org/10.1038/nrd.2015.5
Zamponi, G. W., Bourinet, E., Nelson, D., Nargeot, J., & Snutch, T. P. (1997). Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature, 385(6615), 442–446. https://doi.org/10.1038/385442a0
Zamponi, G. W., Feng, Z. P., Zhang, L., Pajouhesh, H., Ding, Y., Belardetti, F., et al. (2009). Scaffold-based design and synthesis of potent N-type calcium channel blockers. Bioorganic & Medicinal Chemistry Letters, 19(22), 6467–6472. https://doi.org/10.1016/j.bmcl.2009.09.008
Zamponi, G. W., Striessnig, J., Koschak, A., & Dolphin, A. C. (2015). The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacological Reviews, 67(4), 821–870. https://doi.org/10.1124/pr.114.009654
Zhang, F. X., Gadotti, V. M., Souza, I. A., Chen, L., & Zamponi, G. W. (2018). BK potassium channels suppress Cavα2δ subunit function to reduce inflammatory and neuropathic pain. Cell Reports, 22(8), 1956–1964. https://doi.org/10.1016/j.celrep.2018.01.073
Zhao, Y., Huang, G., Wu, Q., Wu, K., Li, R., Lei, J., et al. (2019). Cryo-EM structures of apo and antagonist-bound human Cav3.1. Nature, 576(7787), 492–497. https://doi.org/10.1038/s41586-019-1801-3
Ziegler, D., Duan, W. R., An, G., Thomas, J. W., & Nothaft, W. (2015). A randomized double-blind, placebo-, and active-controlled study of T-type calcium channel blocker ABT-639 in patients with diabetic peripheral neuropathic pain. Pain, 156(10), 2013–2020. https://doi.org/10.1097/j.pain.0000000000000263
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Ferron, L., Zamponi, G.W. (2022). Voltage-Gated Calcium Channels in the Afferent Pain Pathway. In: Zamponi, G.W., Weiss, N. (eds) Voltage-Gated Calcium Channels . Springer, Cham. https://doi.org/10.1007/978-3-031-08881-0_18
Download citation
DOI: https://doi.org/10.1007/978-3-031-08881-0_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-08880-3
Online ISBN: 978-3-031-08881-0
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)