
Abstract
MicroRNAs (miRNAs) provide a fundamental layer of regulation in cells. miRNAs act posttranscriptionally through complementary base-pairing with the 3′-UTR of a target mRNA, leading to mRNA degradation and translation arrest. The likelihood of forming a valid miRNA-target duplex within cells was computationally predicted and experimentally monitored. In human cells, the miRNA profiles determine their identity and physiology. Therefore, alterations in the composition of miRNAs signify many cancer types and chronic diseases. In this chapter, we introduce online functional tools and resources to facilitate miRNA research. We start by introducing currently available miRNA catalogs and miRNA-gateway portals for navigating among different miRNA-centric online resources. We then sketch several realistic challenges that may occur while investigating miRNA regulation in living cells. As a showcase, we demonstrate the utility of miRNAs and mRNAs expression databases that cover diverse human cells and tissues, including resources that report on genetic alterations affecting miRNA expression levels and alteration in binding capacity. Introducing tools linking miRNAs with transcription factor (TF) networks reveals miRNA regulation complexity within living cells. Finally, we concentrate on online resources that analyze miRNAs in human diseases and specifically in cancer. Altogether, we introduce contemporary, selected resources and online tools for studying miRNA regulation in cells and tissues and their utility in health and disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Human miRNA Regulation
Molecular View
In multicellular organisms, microRNAs (miRNAs) play a role in driving cell differentiation, identity, and physiology (Wienholds and Plasterk 2005). A miRNA prototype is a single-stranded RNA molecule of approximately 22-nucleotide length that hybridizes to the 3′-UTR of its target transcript. In humans, the ~1900 miRNA genes account for ~2600 mature miRNAs (Djuranovic et al. 2011). The RNA-induced silencing complex (RISC) protects the miRNA from degradation while stabilizing the miRNA–mRNA duplex. Consequently, the paired RISC–miRNA–mRNA shifts the protein translation efficiency by interfering with initiation, elongation, or termination steps. Occasionally, the miRNA–mRNA duplex also activates protein degradation processes. The bound transcript itself may undergo deadenylation, decapping, and further processing (e.g., degradation) (Cai et al. 2009).
The 5′ sequence of mature miRNAs includes the seed region (6–8 nt) that anchors the miRNA to the miRNA-binding sites (MBS) by a perfect base-pairing (Peterson et al. 2014) (Fig. 5.1). However, other sequence-based and structural features govern the actual binding specificity and efficiency. The fraction of the human transcriptome that is subjected to miRNA regulation is unknown. It is estimated that ~60% of the transcripts are regulated and have at least one conserved miRNA-binding site (MBS) at the 3′-UTR. Approximately 50% of all human protein-coding genes harbor alternative polyadenylation sites, resulting in transcripts with different 3′-UTR lengths (Müller et al. 2014). Naturally, alternative transcripts that differ in their 3′-UTR occur at different cell types. Consequently, the actual network of miRNAs and their targets is cell dependent. Whereas some mRNAs lack MBS, others may contain tens of them. From the miRNA perspective, while some miRNAs may pair with a limited number of targets, other miRNAs can pair with 100s of different mRNAs. Moreover, among the ~2600 reported human miRNAs, many have low expression levels and were only identified in specific cellular contexts by NGS (next-generation sequencing) data with an increased depth. Many of these candidate miRNAs lack experimental validation.

A schematic view of stem-loop structure of the primary miRNA, pre-miRNA, and the mature miRNA products. The miRNA names are indicative of the source of the sequence from the stem-loop. For example, hsa-miR-142-5p and miR-142-3p are from the 5′ and 3′ arm, respectively. The dashed sign indicates non-perfect base-pairing in the stem-loop. The seed is 6–8 nt at the 5′ end of the mature miRNA. Often only one arm of the pre-miRNA is selected (guide strand) and loaded onto the RISC to form the miRISC
Cellular View
For most cell systems, a detailed description of the transcriptome (i.e., mRNAs and miRNAs) allows determining each cell type and its origin (Gebert and MacRae 2019). However, knowledge regarding the cell state and its physiology remains untraceable (Fu et al. 2013). Furthermore, regulating transcription factors (TFs) by miRNAs raises the need for assessing the direct and indirect effects within cells and tissues. In steady-state, miRNAs act as molecular agents for attenuating transcriptional noise. Upon changes in external conditions, miRNAs exploit cooperative and competitive modes that are difficult to model. For yielding an accurate model of cell homeostasis, evaluating the robustness of each cellular system to miRNA perturbations is needed (Mahlab-Aviv et al. 2019). Concretely, the molecular interactions of a miRNA with its targets can lead to abrupt changes in cell fate due to alterations in the levels of TFs, protein production, signaling cascades, and cell energetics (Alvarez-Garcia and Miska 2005).
Within living cells, sequestering of miRNAs by pairing with long noncoding RNAs (lncRNAs) leads to an apparent depletion in free miRNAs (sponge-like function). This implies an additional layer of complex regulation driven by the miRNA interactome (Militello et al. 2017). As the competition among miRNAs governs cell physiology, quantifying the amounts and the stoichiometry of miRNAs and mRNAs within cells is critical. The stochastic nature of miRNA-target interaction argues for using a probabilistic framework to describe the dynamics and the steady state of miRNAs and transcripts in living cells (Mahlab-Aviv et al. 2019). Capturing the bound miRNA–mRNA pairs yields a comprehensive and quantitative view of miRNA competition within living cells. Currently, most available miRNA tools fail to address the complexity of miRNA–mRNA pairing within cells. The contribution of miRNAs to the communication among neighboring cells was reported for neurons and glial cells (Morel et al. 2013). The generality of the miRNA-dependent signaling between cells awaits further studies. Merging cell studies (e.g., clinical tissues, cell lines, and single cells) with computational and experimental resources and tools is fundamental to empowering miRNA research.
5.2 The Scope and Organization of the Chapter
Plenty of resources and web tools were developed over the last 18 years (since 2003) for supporting miRNA research (Gomes et al. 2013). Studying miRNA regulation had been expanded along with the maturation of deep sequencing and diverse cross-linking immunoprecipitation (CLIP)-seq technologies. Many of the early developed tools aimed to predict miRNA–mRNA pairs. In reality, the many miRNA–mRNA prediction tools suffer from low consistency between them. Notably, results from computational prediction tools and experimental results show a high degree of inconsistency.
In recent years, numerous review articles have covered the collection of miRNA databases and tools (Shukla et al. 2017; Lukasik and Zielenkiewicz 2019; Akhtar et al. 2016; Shaker et al. 2020). Other publications concentrate on computational miRNA–mRNA prediction tools and their underlying algorithms (Mendes et al. 2009; Schmitz and Wolkenhauer 2013; Riffo-Campos et al. 2016; Monga and Kumar 2019). A recent survey of the literature revisited ~100 review articles that covered ~1000 tools related to the broad field of miRNA (Chen et al. 2019). Unfortunately, many of the original tools and resources are discontinued or unstable. We focus on online tools and webservers and will not discuss stand-alone tools.
We aim to present a contemporary list of selected resources and online tools for studying miRNA regulation in health and disease. To allow an entry point to human-centric research, we will briefly mention tools in the context of the competition of miRNA and other noncoding RNAs (ncRNA) such as pseudogenes and circular RNAs (circRNAs). We will not discuss miRNA-related tools that focus on comparative genomics and evolution conservation (e.g., miROrtho (Gerlach et al. 2009) and CoGemiR (Maselli et al. 2008)).
The chapter starts by introducing a gateway to a human-centric collection of miRNA resources and online tools. We limit ourselves to those developed or updated in the last decade (from 2012) and are fully functional. We highlight tools that associate miRNAs with their targets according to computational and experimental approaches. We focus on tools that apply method integration, including major validated benchmarks. We then discuss resources that highlight dysregulation of miRNA in human diseases, specifically in cancer. In discussing the online tools, we consider the most updated version, as reported by primary publications. Finally, we will briefly review useful databases and online tools that are valuable in solving real-life experimental tasks regarding miRNA regulation in cellular systems.
5.3 Repositories for miRNA: Catalogs and Genome Browsers
MiRNAs are processed from hairpin-containing primary transcripts of ~200-nt (pri-miRNA) that are further processed to a ~70-nt stem-loop structure (pre-miRNA). These transcriptional events occur in the nucleus. All mature miRNAs undergo these biogenesis maturation steps. The pre-miRNA is then transported into the cytoplasm where a set of sequential cleavage events result in a functional miRNA that is bound to the RISC complex (Fig. 5.1). However, for many observed short RNAs that were identified from large-scale deep sequencing experiments, the definition of miRNAs is less definitive, and often indirect evidence from sequence conservation and independent experimental identification remains the sole support.
Several repositories for human miRNAs have been developed over the past 18 years. In the early days, the microRNA Registry, a branch of Rfam (Griffiths-Jones 2004; Kalvari et al. 2018) was compiled to facilitate the development of computational approaches for miRNA-target prediction. This registry was the basis for the current miRBase catalog (Kozomara et al. 2019). At present, miRNA notations and nomenclature are unified and adopted by the research community as presented in miRBase (Fromm et al. 2015).
5.3.1 miRNA Gene Catalogs
miRBase (release 22.1; 10/2018) is an exhaustive and inclusive miRNA catalog that aims to reach completeness. The miRNA collection was initially developed in 2006 and was regularly updated (Kozomara and Griffiths-Jones 2011). Presently, it includes over 1900 human miRNA genes and the notations for >2600 mature miRNA as observed from experimentally sequenced miRNAs. With the expansion of deep sequencing data, evidence from experiments is reported as normalized values (reads per million, RPM). Each miRNA is labeled on the pre-miRNA (stem-loop structure) and the mature processed version is depicted. A graphical viewer aligns the clustered sequences on the pre-miRNA and uses a unified nomenclature for the 3p and 5p arms (Fig. 5.1). Each miRNA in miRBase is associated with relevant publications and a detailed sequence of the precursor. Moreover, miRNAs are assigned to their families. The family members are miRNAs that derive from a common ancestor and have similar physiological functions (but are not necessarily conserved in sequence or structure) (Kamanu et al. 2013). Besides, miRBase provides a predicted secondary structure for miRNA hairpin loop precursors based on the RNAfold software. A confidence comment was added to allow the community to indicate the subjective confidence for the validity of a candidate miRNA. miRBase reports also on neighboring miRNAs by their chromosomal locations (i.e., miRNA clusters).
miRBase search engine allows extracting all cell or tissue-specific experiments. It is a useful entry point for miRNAs that were originally reported in RNAcentral (The RNAcentral Consortium et al. 2017). miRBase Tracker allows to keep track of historical and current miRNA annotations (Van Peer et al. 2014). miRBase FTP downloads allow the user to select data from any organism of choice (total 270).
MirGeneDB (Ver 2.0, 1/2020) is a robust platform for experimental results on small RNA (sRNA). While miRBase provides an exhaustive list of miRNA candidates, it suffers from a high level of false-positive entries. MirGeneDB aims to increase miRNA identification reliability by testing similarity among 45 metazoan species. The challenge for MirGeneDB is to provide an accurate assignment of miRNAs among expressed smRNA fragments derived from other genes (e.g., tRNAs, small nuclear (snRNAs), and small nucleolar RNAs (snoRNAs), piRNAs). The input starts with ~400 publicly available smRNA sequencing datasets extracted from smRNAbench (originally called miRanalyzer) (Aparicio-Puerta et al. 2019), and processed by miRTrace. A uniform annotation for each species relies on MirMiner that uses data derived from experiments of manipulated miRNA biogenesis genes (Fromm et al. 2020). Therefore, MirGeneDB also considers miRNA variants derived from noncanonical biogenesis. The current version compiled 556 annotated human miRNA genes that can be browsed, searched, and downloaded.
miRCarta (Ver 1.1 7/2018) is a database that features miRNA and precursor data from miRBase but complements the list from deep sequencing NGS from miRMaster and selected publications (Backes et al. 2018). The goal of miRCarta is to compile a consistent collection of novel miRNA candidates and augment the information reported by miRBase. The database also includes an integrated information on predicted and experimentally validated targets extracted from miRTarBase (Huang et al. 2020), microT-CDS (Paraskevopoulou et al. 2013), and TargetScan (Agarwal et al. 2015). The functional links of miRCarta include the pathway dictionary of miRPathDB (Backes et al. 2016), and several miRNA-disease association databases (miR2Disease (Jiang et al. 2009), HMDD (Huang et al. 2019)). Besides, miRCarta provides a comprehensive collection of human miRNAs and miRNA candidates. It covers ~40 k miRNAs and precursors which are compressed to 2.9 k genomic clusters and 590 miRNA families.
5.3.2 miRNAs in Genome Browsers
An easily accessible entry point for the collection of miRNAs is supported by the major human genome browsers (e.g., UCSC and Ensembl). Figure 5.2 displays an overview of the main sources of primary data used in miRNA research. The primary data derived from NGS (next-generation sequencing) for genomics and transcriptomics (e.g., RNA-seq and smRNA-seq). The main genomic browsers provide the researcher with a comprehensive and up-to-date repository of genetic variants, transcripts, cross-species information, and a multidimensional rich data on cell regulation (Fig. 5.2).

An overview about sources of primary data for miRNA research combined with generic catalogs. The primary data is driven by recent advances in NGS (next generation sequencing) for genomics and transcriptomics (RNA-seq and smRNA-seq). NGS is applied to diverse biological samples. Specific methodologies for isolated miRNA and their targets include high-throughput (HTP) CLIP experiments (e.g., HITS-CLIP, iCLIP, and PAR-CLIP), CLASH based on miRNA-mRNA ligation protocol, ChIP-seq with TF and more. Rich experimental protocols of low throughput (LTP) include miRNA overexpression (OX), antimiR settings for downregulation of miRNA expression, mass spectrometry (MS), immunoprecipitation (IP) by RISC proteins, and numerous molecular manipulations with designed reporter (e.g., luciferase) for quantifying miRNA-dependent in vivo gene regulation (Thomson et al. 2011). Generic catalogs include the annotation of genes, alternative splicing (AS), Refseq transcripts, catalogs of cancerous cell lines (e.g., CCLE (Ghandi et al. 2019)), and human-related pathways (e.g., KEGG and BioCarta)
The UCSC browser (5/2018) provides a collection of miRNAs as part of an annotation track for snoRNAs and miRNAs (Fujita et al. 2010). The data is matched to genome coordinates from miRBase, with only perfect matches (100% identity) are annotated. A list of miRNAs and genome coordinates are cross-referenced to miRBase. Together with the snoRNAs, it reports on 2230 gene name entries.
The Ensembl browser (Release 102, 11/2020) provides a collection of miRNAs that are annotated as part of the ncRNAs (Aken et al. 2016). Details are extracted from miRBase, with ~1900 entries for gene annotations. Each miRNA is considered by its functional arm of the pre-miRNA (3p or 5p; e.g., hsa-miR-155-3p, Fig. 5.1). When information is available, the relevant miRNA transcript is reported with evidence extracted from TarBase v8 (Karagkouni et al. 2018). The download of miRNA gene information of transcripts is supported by the Ensembl BioMart retrieval system.
5.4 Gateways for miRNAs: Integrative Platforms
The overwhelming number of tools and resources for miRNA analysis calls for easier access along with high-quality assessment. Elixir bio.tools registry (Ison et al. 2019) is such an entry point that provides a comprehensive registry of software and databases in all life science domains. Within the miRNA research domain, there are ~160 listed resources with comparable information and easy access. To ease the classification of the available web-based database, a meta-database was presented (miRandb) covering ~180 miRNA-centric databases (Aghaee-Bakhtiari et al. 2018). An entry point for human miRNAs is designed to specifically answer common miRNA-related research tasks. HumiR is an integrated website that compiled several human-centric databases and online tools for facilitating the selection of an appropriate tool (Solomon et al. 2020).
miRToolsGallery (9/2017) is a portal that provides an effective shortcut to main hubs for bioinformatics tools developed for miRNA research. The tool addresses the need for navigating among 100s of tools and the demand to meet the correct set of tools for any specific application. miRToolsGallery facilitates this process by curating ~950 miRNA analysis tools and resources. The portal provides a querying system for prioritizing the preferred tool for the needed application. The user can refine the search according to different criteria and requirements. Several features make this platform valuable as an entry point for miRNA research. One such feature allows flexible tagging of tools that belong to multiple categories. Moreover, it ranks results according to their popularity in citation and visibility (Chen et al. 2018a).
mirDIP 4.1 (1/2018) compiles a large number of computational miRNA-target prediction tools. It integrates >150 M human miRNA-target predictions across 30 different resources. Altogether, there are >48 M unique interactions, comprising ~2600 unique miRNAs and 28 k human genes. The database presents a statistically-based integrative score for each miRNA-target interaction. Users can search for miRNA-target pairs according to the level of consistency between given resources, and apply several options as their desirable confidence score (Tokar et al. 2018).
Tools4miRs Ver 1.1 (3/2021) is a manually curated platform gathering all available tools that are miRNA related. Currently, there are 205 such tools (based on 170 methods), with the vast majority providing data on humans. The tools and database collections are categorized and further partitioned to more defined themes (e.g., isomiR identification and target functional analysis). Filtering by organisms, availability (e.g., online and downloading) facilitates the search for tools and databases that meet the user’s needs. For example, the target prediction allows the user to define the target MBS positioning to 5′-UTR, coding, or 3′-UTR (Lukasik et al. 2016). The searched tools are presented in a simple or advanced mode. Tools4miRs also provide a meta-server for target prediction in which the user selects the designated methods to be included. It provides an option for reporting the miRNA-target prediction results via unification, intersection, or consensus method. A summary for each of the 205 tools is available along with a publication (Lukasik et al. 2016).
5.5 miRNA Gene Regulation: TFs and Cellular Context
The following set of resources are compiled from large-scale sequencing analysis with complementary information regarding gene expression and regulation. A unique feature unifying all these resources is the use of information from the cross-talk of miRNAs and cell-specific TFs. Many key resources benefit from cross-referenced complementary tools and algorithms and will briefly be mentioned. The ChIPBase database is a comprehensive annotation database from ChIP-Seq data mapping the transcriptional regulation of miRNAs (Yang et al. 2013). Other publicly available databases that address the problem of miRNA gene transcription regulation are TFmiR (Hamed et al. 2015), the tissue-specific miRNAs (TSmiR (Guo et al. 2014)), and more (Fig. 5.3).

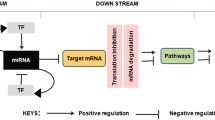
An overview of main components in miRNA research: thematic categories, experimental methods, and the online tools, database, and resources. Experiments are based on miRNA-specific (e.g., CLASH) and generic methodologies (e.g., RNA-seq and MS proteomics). Cell perturbations for investigating miRNAs in cellular systems are based on overexpression (OX), downregulation (CRISPR, AntimiR), and designed reporters (e.g., luciferase) for assessing 3′-UTR regulation of target genes. Databases that were developed to facilitate miRNA research often rely on publicly available genomics, transcriptomics, and proteomics results from large-scale experiments (e.g., TCGA)
DIANA-miRGen v4 (1/2021) is an updated version of experimentally supported functional relationships of miRNA-regulating genes (Alexiou et al. 2010). miRGen’s goal is to provide an exhaustive resource for miRNA transcription start sites (TSS) extracted from a cap-based expression analysis (CAGE) of gene expression as reported by FANTOM (Abugessaisa et al. 2021). The TSS analysis covers most miRNA genes (1534 pre-miRNAs annotated in miRBase) across 135 different cellular contexts of diverse tissues, primary cells, and cell lines. Information on miRNA TSS is combined with the ENCODE ChIP-Seq results for TF binding sites (TFBS > 280) available from (Davis et al. 2018). miRGen provides detailed information on the genomic context of miRNAs, TF regulation (with multiple lines of experimental evidence), and cell-specific gene expression. It compiles a rich resource for miRNAs through cell-specific promoters and transcription regulation (Perdikopanis et al. 2021).
TransmiR v2.0 (1/2019) is a database that provides comprehensive information on TF-miRNA regulation based on surveying the literature and manual curation of >1300 publications. In addition, 1.7 M tissue-specific TF-miRNA regulations from ChIP-seq data were included. Querying the predicted TF-miRNA regulations in humans is based on information of the TF binding motifs. Additional capacities of TransmiR allow presenting the TF-miRNA interaction through a network or disease-centric views. Querying the system with a set of miRNAs allows the identification of TFs which most likely regulate this set of miRNAs. TransmiR provides a rich resource for investigating the regulation of miRNAs (Tong et al. 2019).
CircutesDB (1/2014) is a database of regulatory circuits composed of miRNAs and TFs. This resource integrates transcriptional and posttranscriptional regulatory networks. The basic notion is the existence of several circuit motifs such as feed-forward loops (FFLs) in which a TF regulates a miRNA, and together the expression level of a joint target is determined. A unique feature allows browsing among the catalog of regulatory motifs. Examples are intronic miRNA-mediated self-loops (iMSLs) (Friard et al. 2010).
5.6 miRNA-Target Prediction: Experiments and Validations
Most computational efforts and available tools for miRNA research address the pressing problem of target prediction. Namely, the task of accurately mapping miRNAs to their designated targets in a cellular context (Fig. 5.3). The many-to-many relationships make this problem challenging from a computational and experimental perspective. We will not elaborate on any of the algorithms behind target prediction tools that were thoroughly discussed (Schmitz and Wolkenhauer 2013; Riffo-Campos et al. 2016; Monga and Kumar 2019).
In a nutshell, the main features that are used by almost any of the tens of available miRNA-target prediction tools consider the degree of base-pairing with the seed region at the 5′ sequence with the mRNA (Fig. 5.1) (Biggar and Storey 2015). Additional features include the thermodynamic stability determined via the predicted minimum free energy of a putative miRNA-target duplex (Yue et al. 2009), and the estimated energy for removing the secondary structure of the target mRNA for exposing hidden MBS (i.e., MBS accessibility). Sequence conservation and genomic information across different branches of the taxonomy tree are utilized for removing false annotations, assigning reliability measures for the miRNA-target pairing (Peterson et al. 2014). Many more subtle features are considered for assessing the likelihood of the miRNA-target pairing. In some of the routinely used tools, a machine learning approach (e.g., support vector machine—SVM) was applied along the miRNA-prediction process. Most tools use scores to internally rank their predictions. Unfortunately, tool-specific scores are not easily generalized and rarely used in an integrative approach (Friedman et al. 2014). Recently, an application based on a semi-supervised ML method (RPmirDIP) confirmed the benefit of using prediction scores (Kyrollos et al. 2020).
5.6.1 miRNA-Target Prediction Tools and Resources
Selected tools for target prediction and validation, developed and updated since 2013, are listed in Table 5.1. We indicate the class of the algorithms and features that led to the development of the listed tools. Notably, many of these tools use machine learning (ML) that includes 100s of features and experimental results, allowing the algorithms to optimize the weights for each input data (Zou et al. 2015). As training by deep-learning efforts benefits from an increase in datasets, tools that rely on ML often give a superior prediction.
starBase v2.0 (1/2014) database reports on miRNA interactions as extracted from 108 CLIP-Seq experiments (PAR-CLIP, HITS-CLIP, iCLIP, CLASH). It covers 9 k miRNA–circRNA, 16 k miRNA–pseudogene, and 285 k protein–RNA regulatory relationships. starBase v2.0 provides a comprehensive pairing list for miRNA–mRNA and miRNA-lncRNA interactions based on CLIP-Seq data. A unique feature of starBase v2.0 is the miRFunction and ceRNAFunction servers that allow predicting the function of miRNAs and ncRNAs in the context of their regulatory networks and their coordinated function (Li et al. 2014).
miRDB v6.0 (1/2020) is an online database for miRNA target prediction and functional annotations (Chen and Wang 2020). The comprehensiveness of the training data is evident in the RNA-seq dataset that consists of ~1.5 billion reads. Additionally, features extracted from CLASH (CLIP-ligation protocol) are considered as a validated set for the miRISC-MBS pairs. It covers the expression profiles of >1000 human cell lines and presents target prediction tailored for specific cell models. The underlying algorithm is MirTarget that was developed for analyzing NGS experiments. The predictions are based on a support vector machine (SVM) model. The prediction scores range from 0 to 100 where transcripts with scores of ≥50 are presented as predicted targets. For humans, miRDB covers ~2600 miRNA for 29 k genes leading to 1.6 M pairs. On average, there are ~600 gene targets per miRNA in humans. The miRDB is supplemented with a rich querying system with functional annotations and expression profiles from 100s of cell lines. All can be analyzed for GO annotation functional enrichment.
TargetScan v7.2 (3/2018) is designed to predict miRNA-targets by searching for the presence of conserved sequence features (e.g., seed and its extended variants) that match MBS in the 3′-UTR of mRNA transcripts. It combined a rich set of sequence-based features including the location of MBS relative to the stop codon. TargetScan also sorts miRNAs by their family relation according to the degree of taxonomical conservation (e.g., only mammals). The user can select to activate the analyses while choosing the level of miRNA family conservation (e.g., highly conserved in mammals, broadly conserved, or poorly conserved among mammals), including MBS with mismatches within the seed region. In mammals, the internal scoring reflects the predicted efficacy of targeting. The other scoring system shows the prediction ranking by the probability of conserved targeting (Agarwal et al. 2015).
miRTar2GO (4/2017) is a model trained on the accepted rules of miRNA–target pairing, including experimentally validated interactions from CLIP-seq data. miRTar2GO allows the prediction of cell-type-specific targets. The model provides biological insights through GO enrichment of miRNA-targets (Ahadi et al. 2017). miRTar2GO ranks the interactions predicted for a miRNA based on its distance to the verified interactions of that miRNA. A unique feature is the option to activate the model as highly specific or highly sensitive (Ahadi et al. 2017).
miRGate (4/2015) is a curated database of miRNA–mRNA targets that compare several established miRNA-related experimental databases and integrate major miRNA-target prediction tools (microTar, RNAHybrid, miRanda, TargetScan) (Andres-Leon et al. 2015).
miRror-Suite (6/2014) is an integrative set of tools dealing with miRNA regulation in the context of living cells. Specifically, it allows a query on a set of differentially expressed miRNAs that list the most likely targets that are affected by such a set. It allows the user to reanalyze the data by redefining the statistical significance thresholds. It is based on miRror v2.0 that also provides the opposite view. Namely, from a set of differentially expressed mRNAs as input, find the most likely miRNAs set that plays a role in the regulation. The miRror-Suite miRtegrate algorithm designates statistical criteria that were uniformly applied to a dozen miRNA-target prediction databases. The user can refine the analysis by selecting the desired tissues, cell lines, differential expression, and internal predicting scores (Friedman et al. 2014).
miRGator 3.0. (1/2013) serves as a miRNA portal that relies on NGS data for miRNA diversity, expression, and target relationships. It is based on 73 NGS datasets from major gene expression resources (GEO, SRA, and TCGA) that include 2.5B aligned reads. The database provides expression data by anatomical description and assigned the miRNA data to 38 human diseases with summary statistics (Cho et al. 2013). A unique feature is the availability of tools to facilitate the exploration of massive raw NGS reads for finding miRNAs. By using the miRDeep2 algorithm novel miRNAs, isomiRs, and edited miRNA versions are sought. Moreover, the portal allows comparing gene sets from different studies and extracting biological insights by functional enrichment scheme and gene set analysis.
The utilization of publicly available NGS data such as transcriptomic data for successful use by the miRNA community is challenging. Many databases (e.g., microRNA.org, deepBase, and miRBase) quantify the results from smRNA-seq data for presenting normalized expression values. Many computational and bioinformatics tools combined HTP experimental data with normalized and processed raw data. Still, a set of tools were developed to assist researchers in using miRNA-specific NGS-based pipelines.
miRMine (5/2017) compiled ~350 miRNA-seq datasets from NCBI-SRA. miRMine reported on ~2500 mature RNAs and their RPM (reads per million) expression level for 16 human tissues including body fluids and 24 cell lines (Panwar et al. 2017).
smRNAtoolbox (5/2015) provides a collection of useful tools for NGS experiment analyses (smRNAbench), complemented with several miRNA analysis tools. While it is not a miRNA dedicated platform, it contains seven independent tools that create a workflow for miRNA analyses. The tools are designed to meet a realistic flow for NGS miRNA-seq experiments. Integration of tools allows the user to benefit from a set of established smRNA bioinformatic tools for read mapping, expression profiles, differential expression, genome browser visualization, enrichment of functional annotations, pathway viewer, and cross-species miRNA target prediction (Rueda et al. 2015).
5.6.2 miRNA-Target Prediction Validation Databases
Results from experimental CLIP-seq studies, CLASH, and the advances in NGS led to a wave of datasets that are the basis for updating many miRNA-target prediction tools. Such an effect led to high-quality miRNA-target validated resources as benchmarks in the miRNA field.
DIANA-TarBase v8 (1/2018) database reports on experimentally supported miRNA targets. This resource is considered a benchmark for several prediction methods. The current TarBase reports on ~670 k unique miRNA-target pairs. The database compiles information from a large set of experimental methodologies, conditions, and cellular contexts, covering about 600 cell types and tissues. The database provides an interactive querying system and rich filtering options in addition to the browsing capacity. Retrieval of miRNA-target pairs is activated according to a combined selection of species, supporting methodology, and the selected cell type. TarBase v8 presents a ranking system that is based on the empirical reliability of the method used as evidence (Karagkouni et al. 2018).
miRTarBase 2020 (1/2020) is a comprehensive resource of experimentally validated miRNA-target interactions (denoted MTIs). miRTarBase is a rich experimentally validated MTI database with comprehensively annotated information (Huang et al. 2020). The database covers >380 k validated MTIs for humans. Such MTIs are based on ~2600 miRNAs and >15 k targets with supporting evidence from 7.2 k manually curated publications. A scoring system based on text mining ranks any miRNA-target interaction pair. Evidence from direct assays (e.g., Western blot, qPCR, and reporter gene) are scored higher than those from large-scale methodologies (e.g., CLIP-Seq). Also, a large number of databases were integrated to provide rich information on the number of MTIs within a regulatory network (based on KEGG pathways). The database also provides the current list of validated miRNA-targets according to CLIP-seq technology (Huang et al. 2020).
miRecords (4/2013) consists of experimentally validated miRNA-targets as revealed from literature curation. In addition, it provides a synthesis of many target prediction programs (e.g., DIANA-microT, miTarget, PITA, and TargetScan). miRecords hosts over 2700 records of miRNA-target pairs, with information from direct testing of interaction. It covers about 650 miRNAs and 1900 target genes (Xiao et al. 2009).
5.7 miRNA-Target Databases: Networks and Pathways
On the basis of the predicted and experimentally validated miRNA-target interactions, several databases were developed to address complex regulatory networks in the context of cellular pathways. Assignment of miRNAs to pathways according to individual prediction tools (Table 5.1) suffers from an uncontrolled number of false-positive predictions and poor level of agreement. Relying on the consistency of miRNA-target prediction tools and predetermined topology of human pathways showed that miRNAs are crucial in most pathways from KEGG and the pathway interaction database (PID) (Naamati et al. 2012).
mirDIP v4.1 (1/2018) provides nearly 152 M miRNA-target predictions collected from 30 different resources. The underlying algorithm NAViGaTOR (Shirdel et al. 2011) produces miRNA-target networks from the literature and pathways databases (e.g., KEGG and Reactome). The signaling pathway networks that are signified by miRNA involvement are listed and scored (Tokar et al. 2018).
miRWalk v3.0 (10/2018) is a platform providing an intuitive interface that generates predicted and validated MBS. miRWalk uses a random-forest approach implemented in the TarPmiR algorithm to search for MBS across the entire transcript length (i.e., MBS is not limited to the 3′-UTR). The current version of miRWalk stores predicted data including experimentally verified miRNA-target interactions (Sticht et al. 2018). The human version covers the entire miRNA set (2656 miRNAs according to miRBase Ver 22). The pairing is with regard to the ~20 k RefSeq coding genes and 42 k transcripts. miRWalk provides a cross-reference to other major miRNA-target predictions (e.g., TargetScan and miRDB). The pairwise miRNA–mRNA 100 M reported interactions include the ~0.9 M validated pairs from miRTarBase database (Sticht et al. 2018).
miRPathDB v2.0 (1/2020) is a dictionary of miRNAs and pathways. It covers an exhaustive collection of candidate miRNAs from miRBase v.22.1 and miRCarta (v.1.1), 28 k human targets and ~17 k pathways for Homo sapiens. It uses the validated MTIs from miRTarBase and activates target prediction by using TargetScan (v.7.1) and miRanda. A querying system allows determining the maximal number of miRNAs to be presented based on a reference pathway (e.g., from KEGG, Recatome, and WikiPathways). In addition, it provides new functionality by allowing users to determine a threshold for the reliability of the results. The miRPathDB presents similarity maps for miRNAs and pathways by the statistical significance of overlapping in their targets and pathways. The visualization tools and the downstream analysis are designed to determine a minimal set of candidate regulators that are sufficient to target a gene list (Kehl et al. 2020).
5.8 miRNA Sponging: ceRNA and lncRNA Interactions
An indirect regulatory level of miRNA function is formulated by the concept of competing endogenous RNAs (ceRNAs). In cells, miRNAs may be sequestered by RNA molecules that contain MBS but are not genuine mRNA targets. These RNAs act as miRNA sponges and are often pseudogenes, long noncoding RNAs (lncRNA), or circRNAs. Cell physiological and pathological processes are often regulated by ceRNAs. To fully appreciate the in vivo steady-state in cells, the quantitative aspects of miRNAs in the cellular context and the subtleties of cellular states and molecular events such as miRNA partition between nucleus and cytoplasm, exosome signaling, and miRNA editing may impact the in vivo miRNA-target interaction landscape.
miRSponge (9/2015) is a manually curated database that provides experimentally supported resources for miRNA sponge interactions. miRSponge reports on 298 miRNA–ceRNA interactions in humans that occur between miRNA, pseudogenes, lncRNAs, circRNAs, and human viruses. The database covers 11 species with ~600 miRNA–ceRNA interactions that are supported by ~1200 published articles. miRSponge is a webtool with browsing, retrieval, and downloading capacities. A unique feature is a submission page that allows researchers to enrich the resource by adding validated miRNA sponge data (Wang et al. 2015).
DIANA-LncBase v.2 (1/2016) is a database of experimentally supported and in silico predicted MBS in lncRNAs. DIANA-LncBase is an extensive collection of miRNA–lncRNA interactions with ~70 k experimentally supported interactions derived from publications and the analysis of ~150 AGO CLIP-Seq libraries. In addition to the experimentally validated set, DIANA-LncBase lists in silico predictions based on the DIANA-microT algorithm. A unique feature is the association of the prediction results with information regarding 66 different cell types from 36 tissues. The database includes an exhaustive analysis of six billion RNA-Seq reads for obtaining accurate cell-specific lncRNA expression information (Paraskevopoulou et al. 2016).
5.9 Genomic miRNA Databases: Variations and isomiRs
The following resources address the impact of human genome variations on miRNA regulation via changes in the identity and specificity of MBSs and miRNAs. Also, the immense NGS repository becomes fundamental for identifying isomiRs and other miRNA candidates (Glogovitis et al. 2020).
PolymiRTS v3.0 (1/2014) is a platform for analyzing the impact of genetic polymorphisms in miRNA seed regions and miRNA target sites for humans and mice. The resource provides a comprehensive list of naturally occurring genetic variations in seed regions of miRNAs and the MBS on targets. SNVs and indels in miRNAs and their MBS have the potential to alter miRNA-mediated regulation. This database is a useful resource for genotype and phenotype association studies. The data is based on CLASH experimental results of miRNA–mRNA interactions. Unique features include the use of polymorphic sites of TargetScan scores. Interpretation for the SNVs occurring at the 3′-UTR of the human transcripts are presented by searching the downstream effects on gene expression and pathways in the context of genome wide association studies (GWAS) for human traits and diseases (Bhattacharya et al. 2014).
miRdSNP (1/2012) aims to present the impact of SNVs with regard to diseases. SNVs could lead to gene dysregulation by modifying the efficiency of miRNA binding to the 3′-UTR of the target. miRdSNP is based on a manually curated literature survey with ~800 disease associations SNVs and ~200 disease types, an extended list of experimentally validated miRNA–mRNAs pairs, and sites predicted by key predicting tools (e.g., TargetScan). The tool allows searching for the distance of the identified MBS and SNVs associated with human diseases. It also provides a viewer through the UCSC Genome browser (Bruno et al. 2012).
MIRPIPE (8/2016) is a pipeline for the quantification of miRNA based on smRNA sequencing reads. MIRPIPE allows an automatic trimming of sequencing adapters from raw RNA-seq reads and clustering of isomiRs. MIRPIPE does not rely on the generic reference genome to identify miRNAs. A unique feature is its flexible model for miRNA quantification. Any uploaded database can be considered a reference for the homology search engine (Kuenne et al. 2014).
SomamiR v 2.0 (1.2016) is a database of cancer somatic mutations that potentially affect miRNAs and their targets. It addresses the impact of genetic alterations on ceRNA, including their effect on circRNAs and lncRNAs. SomamiR provides an integrated platform for functional analysis of somatic mutations. To this end, miR2GO is used to analyze the functional consequences of somatic mutations in the seed region of miRNAs. Besides, experimental data (CLASH, CLIP-seq) are analyzed given the somatic mutations. The database highlights mutations in miRNAs and their target sites that change cancer risk as reported in GWAS and various experimental evidence (Bhattacharya and Cui 2016).
Enriching the miRNA variant landscape from external data collections led to the development of dedicated pipelines. An example is miRVar which is based on LOVD v.2.0 (Build 22) (Bhartiya et al. 2011). A machine learning approach using SVM predicts the processing sites of pre-miRNA and the guide strand selection (Fig. 5.1). The possible effect of variations in miRNAs was assessed based on the expected penetrance in the human population.
5.10 miRNA Dysregulation: Diseases, Cancer, and Signaling
The regulation by miRNAs on their intended target only represents a snapshot of a dynamic circuit (Re et al. 2017). miRNAs take active parts in many pathologies and altered signaling pathways. The regulatory networks are produced from small network motifs that are recurrent in nature. While motifs that involve TFs were studied extensively, those including lncRNAs or epigenetic regulators (Sato et al. 2011) introduce overlooked dimensions to the role of miRNAs in cell physiology and pathology.
Regulation of gene expression is the key to maintaining homeostatic processes. Several databases aim to cope with miRNA-target interactions upon changing conditions (e.g., CSmiRTar (Wu et al. 2017). In this view, many events that involve cell pathological conditions are reflected by a shift in miRNA action in cells (e.g., miRwayDB (Das et al. 2018)). The dysregulation of miRNAs has been associated with many diseases such as type 2 diabetes (T2D) and cardiovascular diseases. The impact of miRNAs in other conditions such as arthritic diseases, Alzheimer, and several mental conditions becomes evident. The discovery of miRNA signaling by exosomes is a novel aspect of cell–cell regulation and is an attractive source of biomarkers.
5.10.1 Disease-Related miRNA Databases
Studying disease-related miRNAs is beneficial to understand disease mechanisms at the miRNA level. However, most current methodologies in miRNA research (Fig. 5.2) are limited to in vitro binding assays and cellular manipulation (Fig. 5.3). Several studies have developed networks of miRNAs and diseases (Gu et al. 2016). The validity of such networks was analyzed (Chen et al. 2018b), and proved to be useful for miRNA-disease relation predictions (You et al. 2017; Chen et al. 2018c).
HMDD v3.2 (1/2019) (Human microRNA Disease Database) is a database that curated experiment-supported evidence for miRNA-disease associations. The list of evidence (with 20 evidence codes) includes genetics, epigenetics, circRNAs, and miRNA-target interactions. HMDD bridges between observations from numerous experiments and disease-associated miRNAs. HMDD also covers GWAS results and copy number variations (CNV) leading to gain and loss of genomic miRNAs (Huang et al. 2019). The current version covers a manually collected list of 35.5 k miRNA-disease associations involving 1200 miRNAs and about 900 human diseases. The findings are supported by >19 k publications. The disease annotation is linked to ICD-10 that is the unified index used by the medical community. A connection to major disease ontology terms (e.g., DOID, MESH, OMIM, and HPO) is also provided (Huang et al. 2019).
miRNASNP-v3 (1/2020) is a rich resource that combines data on genetic variations in miRNAs and MBS with disease-related variations (DRVs). miRNASNP is used to determine the possible effect of SNVs on miRNA interactions. The resource analyzes >7 M germline and somatic SNVs and 0.5 M disease-related variations with respect to ~2600 mature miRNAs and > 18 k 3′ UTRs of human genes. miRNASNP compiled the set of SNVs from clinical samples (ClinVar and COSMIC) and population variation catalogs (dbSNP, GWAS Catalog). It provides a functional enrichment analysis of miRNA target gain/loss caused by SNPs/DRVs. Correlations between drug sensitivity and miRNA expression level are presented, with a special focus on potential targets in cancers (Liu et al. 2021).
miRandola (9/2017) is a database of extracellular ncRNAs that are attractive as noninvasive biomarkers from body fluids. miRandola collected data from 314 articles that reported on ~1000 miRNAs and other ncRNAs. The website provides a browsing capacity, name convertor, and details tabular information on the experiments and the nature of the carrier of miRNA (e.g., exosome and microparticle) (Russo et al. 2018).
5.10.2 Cancer-Related miRNA Databases
As miRNAs govern cell identity and physiology in many tissues, alterations in miRNAs signify all cancer types. Human cancer databases such as The Cancer Genome Atlas (TCGA) provide a rich resource for the expression levels of miRNAs and mRNAs for over 14 k cancer samples. Other collections include the OncomiR cancer database (e.g., (Sarver et al. 2018)). The expression levels of oncogenic miRNAs (oncomiRs) and those that act as tumor suppressors make them attractive sites for manipulation and a lead for cancer therapeutic methods.
dbDEMC 2.0 (1/2017) is a cancer-specific resource for storing and displaying differentially expressed miRNAs in human cancers. It uses a simple text search for human cancers from the GEO gene expression data collection. The latest version of dbDEMC contains ~2200 differentially expressed miRNAs identified for 36 cancer types (73 subtypes) from 436 experiments. From large-scale analyses of cancer samples (based on ~150 publications), a collection of 49 k miRNA–cancer associations is provided (Yang et al. 2017). For example, based on TCGA, a list of miRNAs in colon cancer (total 2100) is split into those induced or suppressed relative to the healthy tissue. A unique feature is a meta-profiling representation that allows the user to provide an input set of miRNAs and retrieves as an output their differential expression trend by a heatmap according to broad clinical characteristics (e.g., metastasis, high and low grades) (Yang et al. 2017).
miRCancer (1/2013) provides a comprehensive collection of miRNA expression profiles in various human cancers that are automatically extracted from published literature using text-mining approaches. It utilizes rule-based techniques for mining key sentences regarding the expression trend in cancer and control cells. Manual revision is applied after auto-extraction to improve precision. miRCancer reports on 236 miRNAs and 79 human cancer types from 26 k publications. A unique feature is the constant updating of the information by analyzing the literature in PubMed (Xie et al. 2013).
miRNACancerMAP (9/2018) is a user-friendly web server with integrated data sources and a computational workflow for exhaustive searching of miRNA-cancer information. Specifically, one can ask for the common miRNA-gene regulation networks in multiple cancers using context-dependent expression evidence. The resource allows identifying the sponge regulations by lncRNAs in a clinical setting. The interactive interface allows merging of public data (e.g., TCGA and GEO) with user results such as cancer-derived miRNA–mRNA expression data. Therefore, for the known pathways (e.g., KEGG and Reactome) the impact of miRNA dysregulation on cancer is determined. It allows highlighting miRNAs acting as cancer drivers and tumor suppressors by the cancer hallmark database. A unique feature is the possibility to analyze the user miRNA data by providing interactive visualization tools, and activating multiple miRNA algorithms (Tong et al. 2018).
OncomiR (2/2018) is a user-friendly resource for exploring miRNA dysregulation in cancer. OncomiR covers ~1200 mature miRNA and 30 k mRNA transcripts from ~10 k patients spanning 30 cancer types, along with statistical analysis. OncomiR consists of a database and a dynamic web server. Key functions of OncomiR are the identification of cancer-relevant miRNAs and de novo analysis based on miRNA expression. The unique functionality of OncomiR is in providing the most significant miRNAs for any specific cancer type. Moreover, it allows listing potential miRNAs for a survival signature with Kaplan–Meier (KM) survival curve representation available for a given cancer type (Wong et al. 2018).
5.11 Summary and Future Perspectives
The field of experimental and computational miRNA research has been gradually evolving over the last 18 years (Fig. 5.2). To this end, hundreds of stand-alone, online tools (e.g., multiMiR package) (Ru et al. 2014), algorithms, and databases have been developed for miRNA research. The main task was to provide simple rules for the miRNA regulation in living cells and at the organism level. Unfortunately, the degree of inconsistency remains high among the many miRNA-target prediction tools (Riffo-Campos et al. 2016). Therefore, selecting suitable databases and tools for researchers became increasingly challenging. In this chapter, we briefly discuss tools and databases for assisting miRNA-focused research according to major categories (Fig. 5.3). Notably, the improved HTP technologies such as deep sequencing led to an increase in the number of miRNA candidates, with many of them still awaiting experimental validation. It became clear that in living cells, examining miRNA profiles is not limited to simple miRNA-target pairing rules. Instead, an integration of different regulation layers (TFs, epigenetics, translation, and lncRNAs) is essential. Currently, tools for quantifying key players (i.e., miRNAs, TFs, mRNAs, and proteins) and their dynamics in living cells are missing (Mahlab-Aviv et al. 2019). Such measurements are essential for evaluating the degree of competition and cooperativity among miRNAs in cellular systems (Balaga et al. 2012). The use of medical informatics to determine genetic variations and their impact on diseases allowed to bridge between miRNA research and research in human health (Fig. 5.4). Refining experimental methods for miRNAs and collecting accurate data in databases and well-maintained and undated online tools will continue to advance the field. Specifically, designing solid benchmarks for comparing the tools’ performance is a pressing need. The current knowledge on miRNA regulation in health and disease will benefit from modern statistical methods (e.g., deep learning) and further development of integrative approaches.

A comparative table for selected set of disease-oriented online tools and databases. Each of the tools is indicated by its focus (any/specific disease or cancer), the use of large-scale experimental results, the capacity for downloading the pre-processed data and whether the resource is restricted to human or covers other species (marks as minus and plus sign, respectively). The year of the last major update is noted according to the primary publication
Abbreviations
- CAGE:
-
Cap-based expression analysis
- ceRNA:
-
Competing endogenous RNA
- ChIP:
-
Chromatin immunoprecipitation
- circRNA:
-
Circular RNA
- CLASH:
-
Cross linking, ligation and sequencing of hybrids
- CLIP:
-
Cross-linking immunoprecipitation
- CNV:
-
Copy number variation
- DRV:
-
Disease-related variation
- FFL:
-
Feed-forward loop
- GEO:
-
Gene expression omnibus
- GO:
-
Gene ontology
- GWAS:
-
Genome wide association study
- HTP:
-
High throughput
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- lncRNA:
-
Long non-coding RNAs
- LTP:
-
Low throughput
- MBS:
-
miRNA-binding sites
- miRNA:
-
microRNA
- ML:
-
Machine learning
- mRNA:
-
Messenger RNA
- MS:
-
Mass spectrometry
- MTI:
-
miRNA-target interaction
- RISC:
-
RNA-induced silencing complex
- RPM:
-
Reads per million
- Seq:
-
Sequencing
- smRNA:
-
Small RNA
- SNV:
-
Single nucleotide variation
- SVM:
-
Support vector machine
- TCGA:
-
The Cancer Genome Atlas
- TF:
-
Transcription factor
- TFBS:
-
TF binding sites
- TSS:
-
Transcription start sites
- UTR:
-
Untranslated region
References
Abugessaisa I, Ramilowski JA, Lizio M, Severin J, Hasegawa A, Harshbarger J, Kondo A, Noguchi S, Yip CW, Ooi JLC (2021) FANTOM enters 20th year: expansion of transcriptomic atlases and functional annotation of non-coding RNAs. Nucleic Acids Res 49:D892–D898
Agarwal V, Bell GW, Nam JW, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. elife 4
Aghaee-Bakhtiari SH, Arefian E, Lau P (2018) miRandb: a resource of online services for miRNA research. Brief Bioinform 19:254–262
Ahadi A, Sablok G, Hutvagner G (2017) miRTar2GO: a novel rule-based model learning method for cell line specific microRNA target prediction that integrates Ago2 CLIP-Seq and validated microRNA–target interaction data. Nucleic Acids Res 45:e42–e42
Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, García Girón C, Hourlier T (2016) The Ensembl gene annotation system. Database 2016
Akhtar MM, Micolucci L, Islam MS, Olivieri F, Procopio AD (2016) Bioinformatic tools for microRNA dissection. Nucleic Acids Res 44:24–44
Alexiou P, Vergoulis T, Gleditzsch M, Prekas G, Dalamagas T, Megraw M, Grosse I, Sellis T, Hatzigeorgiou AG (2010) miRGen 2.0: a database of microRNA genomic information and regulation. Nucleic Acids Res 38:D137–D141
Alvarez-Garcia I, Miska EA (2005) MicroRNA functions in animal development and human disease. Development 132:4653–4662
Andres-Leon E, Gonzalez Pena D, Gomez-Lopez G, Pisano DG (2015) miRGate: a curated database of human, mouse and rat miRNA-mRNA targets. Database (Oxford) bav035
Aparicio-Puerta E, Lebron R, Rueda A, Gomez-Martin C, Giannoukakos S, Jaspez D, Medina JM, Zubkovic A, Jurak I, Fromm B, Marchal JA, Oliver J, Hackenberg M (2019) smRNAbench and smRNAtoolbox 2019: intuitive fast small RNA profiling and differential expression. Nucleic Acids Res 47:W530–W535
Backes C, Kehl T, Stöckel D, Fehlmann T, Schneider L, Meese E, Lenhof H-P, Keller A (2016) miRPathDB: a new dictionary on microRNAs and target pathways. Nucleic Acids Res gkw926
Backes C, Fehlmann T, Kern F, Kehl T, Lenhof H-P, Meese E, Keller A (2018) miRCarta: a central repository for collecting miRNA candidates. Nucleic Acids Res 46:D160–D167
Balaga O, Friedman Y, Linial M (2012) Toward a combinatorial nature of microRNA regulation in human cells. Nucleic Acids Res 40:9404–9416
Bhartiya D, Laddha SV, Mukhopadhyay A, Scaria V (2011) miRvar: a comprehensive database for genomic variations in microRNAs. Hum Mutat 32:E2226–E2245
Bhattacharya A, Cui Y (2016) SomamiR 2.0: a database of cancer somatic mutations altering microRNA–ceRNA interactions. Nucleic Acids Res 44:D1005–D1010
Bhattacharya A, Ziebarth JD, Cui Y (2014) PolymiRTS database 3.0: linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res 42:D86–D91
Biggar KK, Storey KB (2015) Insight into post-transcriptional gene regulation: stress-responsive microRNAs and their role in the environmental stress survival of tolerant animals. J Exp Biol 218:1281–1289
Bruno AE, Li L, Kalabus JL, Pan Y, Yu A, Hu Z (2012) miRdSNP: a database of disease-associated SNPs and microRNA target sites on 3′UTRs of human genes. BMC Genomics 13:1–7
Cai Y, Yu X, Hu S, Yu J (2009) A brief review on the mechanisms of miRNA regulation. Genomics Proteomics Bioinformatics 7:147–154
Chen Y, Wang X (2020) miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res 48:D127–D131
Chen L, Heikkinen L, Wang C, Yang Y, Knott KE, Wong G (2018a) miRToolsGallery: a tag-based and rankable microRNA bioinformatics resources database portal. Database (Oxford)
Chen X, Wang C-C, Yin J, You Z-H (2018b) Novel human miRNA-disease association inference based on random forest. Molecular Therapy-Nucleic Acids 13:568–579
Chen X, Yin J, Qu J, Huang L (2018c) MDHGI: matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput Biol 14:e1006418
Chen L, Heikkinen L, Wang C, Yang Y, Sun H, Wong G (2019) Trends in the development of miRNA bioinformatics tools. Brief Bioinform 20:1836–1852
Cho S, Jang I, Jun Y, Yoon S, Ko M, Kwon Y, Choi I, Chang H, Ryu D, Lee B, Kim VN, Kim W, Lee S (2013) MiRGator v3.0: a microRNA portal for deep sequencing, expression profiling and mRNA targeting. Nucleic Acids Res 41:D252–D257
Chou CH, Lin FM, Chou MT, Hsu SD, Chang TH, Weng SL, Shrestha S, Hsiao CC, Hung JH, Huang HD (2013) A computational approach for identifying microRNA-target interactions using high-throughput CLIP and PAR-CLIP sequencing. BMC Genomics 14(Suppl 1):S2
Das SS, Saha P, Chakravorty N (2018) miRwayDB: a database for experimentally validated microRNA-pathway associations in pathophysiological conditions. Database (Oxford)
Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK (2018) The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res 46:D794–D801
Djuranovic S, Nahvi A, Green R (2011) A parsimonious model for gene regulation by miRNAs. Science 331:550–553
Dweep H, Gretz N (2015) miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat Methods 12:697
Friard O, Re A, Taverna D, De Bortoli M, Cora D (2010) CircuitsDB: a database of mixed microRNA/transcription factor feed-forward regulatory circuits in human and mouse. BMC Bioinformatics 11:435
Friedman Y, Karsenty S, Linial M (2014) miRror-suite: decoding coordinated regulation by microRNAs. Database (Oxford)
Fromm B, Billipp T, Peck LE, Johansen M, Tarver JE, King BL, Newcomb JM, Sempere LF, Flatmark K, Hovig E, Peterson KJ (2015) A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu Rev Genet 49:213–242
Fromm B, Domanska D, Hoye E, Ovchinnikov V, Kang W, Aparicio-Puerta E, Johansen M, Flatmark K, Mathelier A, Hovig E, Hackenberg M, Friedlander MR, Peterson KJ (2020) MirGeneDB 2.0: the metazoan microRNA complement. Nucleic Acids Res 48:D1172
Fu G, Brkić J, Hayder H, Peng C (2013) MicroRNAs in human placental development and pregnancy complications. Int J Mol Sci 14:5519–5544
Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A (2010) The UCSC genome browser database: update 2011. Nucleic Acids Res 39:D876–D882
Gebert LF, MacRae IJ (2019) Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 20:21–37
Gerlach D, Kriventseva EV, Rahman N, Vejnar CE, Zdobnov EM (2009) miROrtho: computational survey of microRNA genes. Nucleic Acids Res 37:D111–D117
Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H (2019) Next-generation characterization of the cancer cell line encyclopedia. Nature 569:503–508
Glogovitis I, Yahubyan G, Wurdinger T, Koppers-Lalic D, Baev V (2020) isomiRs-hidden soldiers in the miRNA regulatory Army, and how to find them? Biomol Ther 11
Gomes CPDC, Cho J-H, Hood LE, Franco OL, Pereira RWD, Wang K (2013) A review of computational tools in microRNA discovery. Front Genet 4:81
Griffiths-Jones S (2004) The microRNA registry. Nucleic Acids Res 32:D109–D111
Gu C, Liao B, Li X, Li K (2016) Network consistency projection for human miRNA-disease associations inference. Sci Rep 6:1–10
Guo Z, Maki M, Ding R, Yang Y, Xiong L (2014) Genome-wide survey of tissue-specific microRNA and transcription factor regulatory networks in 12 tissues. Sci Rep 4:1–9
Hamed M, Spaniol C, Nazarieh M, Helms V (2015) TFmiR: a web server for constructing and analyzing disease-specific transcription factor and miRNA co-regulatory networks. Nucleic Acids Res 43:W283–W288
Huang Z, Shi J, Gao Y, Cui C, Zhang S, Li J, Zhou Y, Cui Q (2019) HMDD v3. 0: a database for experimentally supported human microRNA–disease associations. Nucleic Acids Res 47:D1013–D1017
Huang HY, Lin YC, Li J, Huang KY, Shrestha S, Hong HC, Tang Y, Chen YG, Jin CN, Yu Y, Xu JT, Li YM, Cai XX, Zhou ZY, Chen XH, Pei YY, Hu L, Su JJ, Cui SD, Wang F, Xie YY, Ding SY, Luo MF, Chou CH, Chang NW, Chen KW, Cheng YH, Wan XH, Hsu WL, Lee TY, Wei FX, Huang HD (2020) miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res 48:D148–D154
Ison J, Ienasescu H, Chmura P, Rydza E, Ménager H, Kalaš M, Schwämmle V, Grüning B, Beard N, Lopez R (2019) The bio. tools registry of software tools and data resources for the life sciences. Genome Biol 20:1–4
Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y (2009) miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res 37:D98–D104
Kalvari I, Argasinska J, Quinones-Olvera N, Nawrocki EP, Rivas E, Eddy SR, Bateman A, Finn RD, Petrov AI (2018) Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res 46:D335–D342
Kamanu TK, Radovanovic A, Archer JA, Bajic VB (2013) Exploration of miRNA families for hypotheses generation. Sci Rep 3:1–8
Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, Vlachos IS, Tastsoglou S, Kanellos I, Papadimitriou D, Kavakiotis I, Maniou S, Skoufos G, Vergoulis T, Dalamagas T, Hatzigeorgiou AG (2018) DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res 46:D239–D245
Kehl T, Kern F, Backes C, Fehlmann T, Stockel D, Meese E, Lenhof HP, Keller A (2020) miRPathDB 2.0: a novel release of the miRNA pathway dictionary database. Nucleic Acids Res 48:D142–D147
Kozomara A, Griffiths-Jones S (2011) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39:D152–D157
Kozomara A, Birgaoanu M, Griffiths-Jones S (2019) miRBase: from microRNA sequences to function. Nucleic Acids Res 47:D155–D162
Kuenne C, Preussner J, Herzog M, Braun T, Looso M (2014) MIRPIPE: quantification of microRNAs in niche model organisms. Bioinformatics 30:3412–3413
Kyrollos DG, Reid B, Dick K, Green JR (2020) RPmirDIP: reciprocal perspective improves targeting prediction. Sci Rep 10:11770
Li J-H, Liu S, Zhou H, Qu L-H, Yang J-H (2014) starBase v2. 0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 42:D92–D97
Liu CJ, Fu X, Xia M, Zhang Q, Gu Z, Guo AY (2021) miRNASNP-v3: a comprehensive database for SNPs and disease-related variations in miRNAs and miRNA targets. Nucleic Acids Res 49:D1276–D1281
Loher P, Rigoutsos I (2012) Interactive exploration of RNA22 microRNA target predictions. Bioinformatics 28:3322–3323
Lukasik A, Zielenkiewicz P (2019) An overview of miRNA and miRNA target analysis tools. Methods Mol Biol 1932:65–87
Lukasik A, Wojcikowski M, Zielenkiewicz P (2016) Tools4miRs - one place to gather all the tools for miRNA analysis. Bioinformatics 32:2722–2724
Mahlab-Aviv S, Linial N, Linial M (2019) A cell-based probabilistic approach unveils the concerted action of miRNAs. PLoS Comput Biol 15:e1007204
Maselli V, Di Bernardo D, Banfi S (2008) CoGemiR: a comparative genomics microRNA database. BMC Genomics 9:457
Mendes ND, Freitas AT, Sagot MF (2009) Current tools for the identification of miRNA genes and their targets. Nucleic Acids Res 37:2419–2433
Militello G, Weirick T, John D, Döring C, Dimmeler S, Uchida S (2017) Screening and validation of lncRNAs and circRNAs as miRNA sponges. Brief Bioinform 18:780–788
Monga I, Kumar M (2019) Computational resources for prediction and analysis of functional miRNA and their targetome. Methods Mol Biol 1912:215–250
Morel L, Regan M, Higashimori H, Ng SK, Esau C, Vidensky S, Rothstein J, Yang Y (2013) Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J Biol Chem 288:7105–7116
Müller S, Rycak L, Afonso-Grunz F, Winter P, Zawada AM, Damrath E, Scheider J, Schmäh J, Koch I, Kahl G (2014) APADB: a database for alternative polyadenylation and microRNA regulation events. Database
Naamati G, Friedman Y, Balaga O, Linial M (2012) Susceptibility of the human pathways graphs to fragmentation by small sets of microRNAs. Bioinformatics 28:983–990
Panwar B, Omenn GS, Guan Y (2017) miRmine: a database of human miRNA expression profiles. Bioinformatics 33:1554–1560
Paraskevopoulou MD, Georgakilas G, Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C, Dalamagas T, Hatzigeorgiou AG (2013) DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res 41:W169–W173
Paraskevopoulou MD, Vlachos IS, Karagkouni D, Georgakilas G, Kanellos I, Vergoulis T, Zagganas K, Tsanakas P, Floros E, Dalamagas T (2016) DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res 44:D231–D238
Perdikopanis N, Georgakilas GK, Grigoriadis D, Pierros V, Kavakiotis I, Alexiou P, Hatzigeorgiou A (2021) DIANA-miRGen v4: indexing promoters and regulators for more than 1500 microRNAs. Nucleic Acids Res 49:D151–D159
Peterson SM, Thompson JA, Ufkin ML, Sathyanarayana P, Liaw L, Congdon CB (2014) Common features of microRNA target prediction tools. Front Genet 5
Preusse M, Theis FJ, Mueller NS (2016) miTALOS v2: analyzing tissue specific microRNA function. PLoS One 11:e0151771
Re A, Caselle M, Bussolino F (2017) MicroRNA-mediated regulatory circuits: outlook and perspectives. Phys Biol 14:045001
Riffo-Campos AL, Riquelme I, Brebi-Mieville P (2016) Tools for sequence-based miRNA target prediction: what to choose? Int J Mol Sci 17
Ru Y, Kechris KJ, Tabakoff B, Hoffman P, Radcliffe RA, Bowler R, Mahaffey S, Rossi S, Calin GA, Bemis L, Theodorescu D (2014) The multiMiR R package and database: integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res 42:e133
Rueda A, Barturen G, Lebron R, Gomez-Martin C, Alganza A, Oliver JL, Hackenberg M (2015) smRNAtoolbox: an integrated collection of small RNA research tools. Nucleic Acids Res 43:W467–W473
Russo F, Di Bella S, Vannini F, Berti G, Scoyni F, Cook HV, Santos A, Nigita G, Bonnici V, Lagana A, Geraci F, Pulvirenti A, Giugno R, De Masi F, Belling K, Jensen LJ, Brunak S, Pellegrini M, Ferro A (2018) miRandola 2017: a curated knowledge base of non-invasive biomarkers. Nucleic Acids Res 46:D354–D359
Sarver AL, Sarver AE, Yuan C, Subramanian S (2018) OMCD: Oncomir cancer database. BMC Cancer 18:1–6
Sato F, Tsuchiya S, Meltzer SJ, Shimizu K (2011) MicroRNAs and epigenetics. FEBS J 278:1598–1609
Schmitz U, Wolkenhauer O (2013) Web resources for microRNA research. Adv Exp Med Biol 774:225–250
Shaker F, Nikravesh A, Arezumand R, Aghaee-Bakhtiari SH (2020) Web-based tools for miRNA studies analysis. Comput Biol Med 127:104060
Shirdel EA, Xie W, Mak TW, Jurisica I (2011) NAViGaTing the micronome—using multiple microRNA prediction databases to identify signalling pathway-associated microRNAs. PLoS One 6:e17429
Shuang C, Maozu G, Chunyu W, Xiaoyan L, Yang L, Xuejian W (2016) MiRTDL: a deep learning approach for miRNA target prediction. IEEE/ACM Trans Comput Biol Bioinform 13:1161–1169
Shukla V, Varghese VK, Kabekkodu SP, Mallya S, Satyamoorthy K (2017) A compilation of web-based research tools for miRNA analysis. Brief Funct Genomics 16:249–273
Solomon J, Kern F, Fehlmann T, Meese E, Keller A (2020) HumiR: web services, tools and databases for exploring human microRNA data. Biomolecules 10
Sticht C, De La Torre C, Parveen A, Gretz N (2018) miRWalk: an online resource for prediction of microRNA binding sites. PLoS One 13:e0206239
The RNAcentral Consortium, Petrov AI, Kay SJE, Kalvari I, Howe KL, Gray KA, Bruford EA, Kersey PJ, Cochrane G, Finn RD, Bateman A, Kozomara A, Griffiths-Jones S, Frankish A, Zwieb CW, Lau BY, Williams KP, Chan PP, Lowe TM, Cannone JJ, Gutell R, Machnicka MA, Bujnicki JM, Yoshihama M, Kenmochi N, Chai B, Cole JR, Szymanski M, Karlowski WM, Wood V, Huala E, Berardini TZ, Zhao Y, Chen R, Zhu W, Paraskevopoulou MD, Vlachos IS, Hatzigeorgiou AG, Ma L, Zhang Z, Puetz J, Stadler PF, McDonald D, Basu S, Fey P, Engel SR, Cherry JM, Volders P-J, Mestdagh P, Wower J, Clark MB, Quek XC, Dinger ME (2017) RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic Acids Res 45:D128–D134
Thomson DW, Bracken CP, Goodall GJ (2011) Experimental strategies for microRNA target identification. Nucleic Acids Res 39:6845–6853
Tokar T, Pastrello C, Rossos AE, Abovsky M, Hauschild A-C, Tsay M, Lu R, Jurisica I (2018) mirDIP 4.1—integrative database of human microRNA target predictions. Nucleic Acids Res 46:D360–D370
Tong Y, Ru B, Zhang J (2018) miRNACancerMAP: an integrative web server inferring miRNA regulation network for cancer. Bioinformatics 34:3211–3213
Tong Z, Cui Q, Wang J, Zhou Y (2019) TransmiR v2.0: an updated transcription factor-microRNA regulation database. Nucleic Acids Res 47:D253–D258
Van Peer G, Lefever S, Anckaert J, Beckers A, Rihani A, Van Goethem A, Volders PJ, Zeka F, Ongenaert M, Mestdagh P, Vandesompele J (2014) miRBase tracker: keeping track of microRNA annotation changes. Database (Oxford)
Vejnar CE, Blum M, Zdobnov EM (2013) miRmap web: comprehensive microRNA target prediction online. Nucleic Acids Res 41:W165–W168
Wang P, Zhi H, Zhang Y, Liu Y, Zhang J, Gao Y, Guo M, Ning S, Li X (2015) miRSponge: a manually curated database for experimentally supported miRNA sponges and ceRNAs. Database
Wienholds E, Plasterk RH (2005) MicroRNA function in animal development. FEBS Lett 579:5911–5922
Wong NW, Chen Y, Chen S, Wang X (2018) OncomiR: an online resource for exploring pan-cancer microRNA dysregulation. Bioinformatics 34:713–715
Wu W-S, Tu B-W, Chen T-T, Hou S-W, Tseng JT (2017) CSmiRTar: condition-specific microRNA targets database. PLoS One 12:e0181231
Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T (2009) miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res 37:D105–D110
Xie B, Ding Q, Han H, Wu D (2013) miRCancer: a microRNA–cancer association database constructed by text mining on literature. Bioinformatics 29:638–644
Yang J-H, Li J-H, Jiang S, Zhou H, Qu L-H (2013) ChIPBase: a database for decoding the transcriptional regulation of long non-coding RNA and microRNA genes from ChIP-Seq data. Nucleic Acids Res 41:D177–D187
Yang Z, Wu L, Wang A, Tang W, Zhao Y, Zhao H, Teschendorff AE (2017) dbDEMC 2.0: updated database of differentially expressed miRNAs in human cancers. Nucleic Acids Res 45:D812–D818
You Z-H, Huang Z-A, Zhu Z, Yan G-Y, Li Z-W, Wen Z, Chen X (2017) PBMDA: a novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput Biol 13:e1005455
Yue D, Liu H, Huang Y (2009) Survey of computational algorithms for MicroRNA target prediction. Curr Genomics 10:478–492
Zou Q, Li J, Hong Q, Lin Z, Wu Y, Shi H, Ju Y (2015) Prediction of microRNA-disease associations based on social network analysis methods. BioMed Res Int
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Blass, I., Zohar, K., Linial, M. (2022). Turning Data to Knowledge: Online Tools, Databases, and Resources in microRNA Research. In: Schmitz, U., Wolkenhauer, O., Vera-González, J. (eds) Systems Biology of MicroRNAs in Cancer. Advances in Experimental Medicine and Biology, vol 1385. Springer, Cham. https://doi.org/10.1007/978-3-031-08356-3_5
Download citation
DOI: https://doi.org/10.1007/978-3-031-08356-3_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-08355-6
Online ISBN: 978-3-031-08356-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)