
Abstract
Improving the anti-tumour T cell response as a consequence of immunotherapy can result in eradication of tumour burden, however, the majority of patients fail with current treatment regimens and so novel immunotherapies with greater efficacy and improved tolerability are needed. The phosphoinositide-3-kinase (PI3K) family members that are directly involved in cell signalling comprise PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ, with the latter two isoforms expressed primarily by leukocytes. The survival and optimal function of regulatory T cells (Treg) and myeloid-derived suppressor cells (MDSCs) is dependent on PI3Kδ, whereas tumour-associated macrophages (TAMs), use PI3Kγ. Blocking these signalling isoforms can boost development of effective anti-cancer immune responses and result in control of tumour burden. The dependence on different PI3K isoforms in immune cells makes targeting this pathway an attractive approach for tumour immunotherapy. Herein, we discuss how inhibiting specific PI3K isoforms in pro-tumoural Tregs, MDSCS and TAMs can unleash a powerful anti-tumour immune response, driven by CD8+ T cells, capable of controlling tumour burden and consider how the immune response to therapy needs careful investigation, to identify both the correlates of successful treatment and those that impede the generation of robust anti-tumour responses. Furthermore, we review how combination immunotherapy approaches with both PI3K inhibitors and subsequent immune checkpoint blockade can potentiate the efficacy of monotherapy. Finally, we discuss the recent advances in the use of PI3K isoform-specific inhibitors as an immunotherapy for solid tumours in clinical trials.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Tumour
- Immunotherapy
- Regulatory T cell
- Tumour-associated macrophage
- Myeloid-derived suppressor cell
- CD8+ T cell
1 Introduction
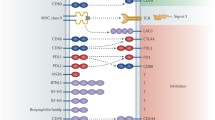
Aberrant PI3K signalling is known to drive cancer progression and common cancers often comprise mutations in PIK3CA and loss of PTEN function (reviewed in Thorpe et al. (2015)). A huge effort has been made to develop inhibitors of three broad types; dual PI3K-mTOR inhibitors, pan-class I inhibitors and isoform-specific inhibitors, developed to directly inhibit cancer cell proliferation and survival (reviewed in Janku et al. (2018)), with several currently undergoing testing in clinical studies either alone or in combination with other therapies (Fig. 1). Whilst there is extensive data indicating cancer cell-intrinsic effects of inhibiting PI3K signalling, it is becoming increasingly clear that indirect effects of inhibiting the pathway also contribute to control of tumour growth.

PI3K isoform-specific inhibitors. Schematic of the PI3K isoform-specific inhibitors currently in various stages of development and their tumour-specific targets
Immunotherapy as a cancer-targeting approach dates back to 1891 when William B. Coley first sought to harness the power of the immune system to control solid tumours (Kienle 2012). The administration of Coley’s toxins, a bacterial vaccine, designed to induce a large infection at the tumour site, resulted in clearance of both the infection and regression of the tumour in a proportion of patients. Despite these striking results, the lack of understanding at the time of how the immune system may control cancer, led to these first immunotherapy approaches falling from favour, as surgical, chemotherapy and radiotherapy treatments began to significantly improve cancer outcomes.
The field of immunotherapy has been revived in recent years, with the advent of new treatment strategies such as immune checkpoint blockade (ICB), adoptive transfer T cell therapy (ACT) and tumour vaccine therapy (Reviewed in Waldman et al. 2020). These therapeutic modalities are designed to reinvigorate the anti-tumour immune response, by either directly targeting the T cells themselves, to improve their anti-tumour response, or indirectly, by targeting immuno-suppressive populations, such as regulatory T cells (Tregs), tumour-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), within the tumour microenvironment that typically subdues the T cell response to cancer. Whilst immunotherapy can result in striking control of tumour burden, the number of patients who successfully respond to these therapies remains small. Typically, patients fail to mount a sufficient response to therapy as a consequence of T cells becoming exhausted and dysfunctional prior to or during treatment. As the scientific field continues to elucidate the impediments to T cell anti-tumour responses it is becoming clear that different approaches are still needed, and that a combination of therapies may offer the greatest potential for tumour control.
As discussed in previous chapters, the different PI3K isoforms have critical roles in the signalling of T cells, B cells and innate cells of myeloid origin, such as macrophages. Given the success and tolerability of isoform-specific inhibitors such as the PI3Kδ inhibitor, Idelalisib, for haematological malignancies (Yang et al. 2015), the focus of much research and drug development programmes in recent years has been to understand how other immune cell populations can be targeted via isoform-specific PI3K inhibitors.
Herein, we firstly discuss the data surrounding PI3K isoform-specific inhibition, through genetic inactivation and pharmacological blockade, to target immune cell populations within the tumour microenvironment in preclinical models (Fig. 1). Finally, we discuss the data reported from clinical trials to date, using different inhibitors in solid malignancies.
2 Immune Mechanisms of Action in Preclinical Studies
2.1 PI3Kδ Inhibitors as a Treg Targeted Therapy
FoxP3+ Tregs suppress CD8+ and conventional CD4+ T cells thereby helping to maintain tolerance and prevent autoimmunity. However, these mechanisms in the context of cancer can prevent the host from generating a successful anti-tumour T cell response. Shimizu et al. were the first to demonstrate that removal of regulatory T cells led to the development of potent anti-tumoural CD8+ T cell responses and control of tumour burden (Shimizu et al. 1999). However, complete depletion of regulatory T cells is not considered feasible as a clinical therapy due to the significant presentation of autoimmune side effects and so approaches to selectively inhibit Treg function have been sought. Due to the contrast in reliance on the PI3Kδ isoform for effector T cell and Treg signalling (discussed in Chap. 8 and by Ahmad et al. 2017), PI3Kδ inhibition has been suggested as an attractive approach for Treg-specific cancer immunotherapy.
Initial preclinical studies utilised a strain of mice with either a global or a Treg-specific inactivation of the PI3Kδ isoform (δD910A) to examine the effect that impaired T cell signalling had upon tumour growth. A seminal study by Ali and colleagues demonstrated that δD910A mice were better able to control tumour growth in a number of mouse tumour models (Ali et al. 2014). Adoptive transfer experiments of PI3Kδ-inactive Tregs into wild type tumour-bearing hosts established that the reduced immunosuppression mediated by δD910A Tregs was responsible for reduced tumour growth. The deletion of the CD8+ effector T cell population abrogated tumour control in δD910A mice, demonstrating that tumour control is dependent on both the loss of Treg-mediated immunosuppression and the generation of a robust anti-tumour CD8+ T cell response as a consequence (Ali et al. 2014; Lim et al. 2018). The authors confirmed the therapeutic potential of PI3Kδ, by treating mice with a small molecule inhibitor of PI3Kδ, PI-3065 and demonstrated partial tumour control in both breast and pancreatic cancers in vivo. This proof-of-concept study in solid tumours has been strengthened by a number of other groups (Carnevalli et al. 2018; Lauder et al. 2020) who have sought to delineate the mechanism by which PI3Kδ inhibition confers tumour control either alone or in combination with other therapeutic modalities.
Idelalisib, also known as CAL-101 or GS-1101, an approved PI3Kδ inhibitor for haematological cancers (discussed in Chap. 23) and similar PI3Kδ inhibitors are being redeployed by several groups as a therapy designed to specifically target Tregs in solid malignancies. Ahmad and colleagues reported that CAL-101 blockade of PI3Kδ signalling in vitro was critical for Treg suppression and survival, whereas effector CD4+ T cells could utilise PI3Kα and PI3Kβ to maintain their function (Ahmad et al. 2017). Using the Treg-dependent tumour model, TC-1 (lung carcinoma), the authors demonstrated similar findings with modest control of tumour burden and partial improvement in survival time observed following CAL-101 treatment. However, when they boosted the antigen-specific response using an E7 tumour-specific vaccine in combination with CAL-101, they observed a significant reduction in tumour burden and improved long-term survival compared to either treatment strategy alone. Phenotypic analysis of the anti-tumour immune response in combination-treated animals demonstrated similar findings to the original study by Ali and colleagues (Ali et al. 2014), collectively pointing to a reduction in Treg response and the generation of a robust CD8+ T cell response is necessary for potent tumour control.
Lauder and colleagues expanded on previous studies and demonstrated that whilst all treated mice exhibited a level of tumour control following PI-3065 treatment, there was a dichotomy in the response to therapy, with complete tumour regression occurring in a small proportion of treated animals (Lauder et al. 2020, 2021). Detailed analysis of the anti-tumour immune response generated following PI-3065 treatment supported the previous studies that eradication of tumour burden was reliant on the dampening of the Treg response and the generation of a robust antigen-specific CD8+ T cell response. Whilst all treated mice had a reduced number of tumoural Tregs, those that exhibited only partial control developed a pool of dysfunctional Tregs characterised by increased Ki67, CD69 and LAG3 expression and a reduced number of tumour antigen-specific CD8+ T cells (Fig. 2). Combination treatment with PI-3065 and subsequent anti-LAG3 antibody therapy resulted in significant tumour control in all treated mice (Lauder et al. 2020). However, the greatest significance of the aforementioned study is that it highlighted the tumour-specific impediments to PI3Kδ inhibitors as a therapy. An essential requisite for tumour control was the development of an increased CD8+ T cell: Treg ratio in PI3Kδ responsive tumours. In the absence of an increased ratio, PI3Kδ unresponsive tumours, such as the MC38 colon cancer model, remained unresponsive despite treatment with a secondary immune checkpoint therapy (Lauder et al. 2020, 2021).

Immune mechanisms of PI3K specific isoform action. a Pharmacological blockade of PI3Kδ in vitro leads to the development of CD8+ T cells with superior anti-tumour activity when adoptively transferred into tumour-bearing hosts. b In vivo therapeutic targeting of PI3Kδ results in a reduced Treg: CD8+ T cell ratio and an enrichment in antigen-specific CD8+ T cells with improved anti-tumour function. c Specific targeting of PI3Kγ controls tumour growth by inducing immune gene signature switching of macrophages in vivo, from pro-tumoural to anti-tumoural
In preclinical models that are resistant to Treg-specific inhibition such as MC38, genetic inactivation of PI3Kδ signalling has been reported to enhance tumour growth as a consequence of reduced CD8+ T cell function in vivo (Putz et al. 2012). These conflicting data indicate that simply targeting Tregs may not be sufficient in every tumour type. Indeed, combination immunotherapy approaches designed to target multiple tumour resident populations namely Tregs and TAMs were able to control tumour growth in the previously unresponsive MC38 model (Gyori et al. 2018). Dual PI3K isoform inhibitors offer the potential to target multiple cell types within the tumour microenvironment with a single treatment. Carnevalli et al. compared tumour control in PI3Kδ responsive (CT26 and 4T1) and unresponsive (MC38) tumours to either a PI3Kδ single inhibitor (PI-3065) or a dual PI3Kα/δ inhibitor (AZD8835) (Carnevalli et al. 2018). In all models, AZD8835 offered superior control of tumour growth and prolonged survival. Unlike the previously discussed studies where PI3Kδ isoform inhibitors were routinely administered daily for the duration of the study, the robust anti-tumour response reported with AZD8835 was a consequence of an intermittent dosing regimen, with 2 days on treatment/5 days off treatment. Although not as pronounced as the changes with continual PI-3065 treatment, this intermittent therapeutic approach resulted in a reduction in Tregs and increased CD8+ T cell: Treg ratio within the tumour. However, AZD8835 conferred enhanced transcriptional and phenotypic changes resulting in CD8+ T cells with increased expression of Ki67, CD25, Granzyme B, IFNγ and a reduced susceptibility to exhaustion as determined by reduced PD1 expression. Furthermore, the intermittent interruption of PI3K signalling in CD8+ T cells appears to promote an IL-2 autocrine signalling loop within the tumour that drives T cell effector function and survival, which ultimately supports tumour control (Carnevalli et al. 2018).
2.2 PI3Kδ Inhibitors as an Adjuvant to Improve T Cell Therapy in Cancer
The role of the PI3Kδ isoform in CD8+ T cell signalling during proliferation and effector functions are described in detail in Chap. 12. It is known that pharmacological inhibition of PI3Kδ skews activated CD8+ T cells to develop progeny with a self-renewing phenotype characterised by the expression of the transcription factor TCF1 (Lin et al. 2015; Nish et al. 2017). Several recent studies have demonstrated that the presence of CD8+TCF1+ stem-like T cells within the tumour microenvironment is associated with tumour control and successful responses to checkpoint therapy (Siddiqui et al. 2019; Kurtulus et al. 2019; Sade-Feldman et al. 2018; Baharom et al. 2021).
Using a preclinical model of melanoma, Bowers and colleagues demonstrated that expansion of CD8+ T cells in the presence of CAL-101 prior to adoptive cell transfer (ACT) into B16F10 tumour-bearing hosts, resulted in significantly reduced tumour burden and prolonged survival (Bowers et al. 2017). RNA-sequencing revealed that CAL-101-treated T cells had improved anti-tumour capacity driven by enhanced expression of TCF1 and a stem-like memory phenotype characterised by increased expression of CD62L, CD127 and CCR7 (Fig. 2). These findings have the capacity to significantly improve cellular immunotherapy approaches such as ACT or chimeric antigen receptor (CAR) T cell therapy, which despite their potential to reinvigorate the anti-tumour immune response, typically fail as a consequence of the transferred cells becoming exhausted and dysfunctional. The generation of human CAR-T cells in the presence of the PI3Kδ inhibitors TGR-1202 or CAL-101 resulted in cells with a less differentiated phenotype compared to untreated cells but with increased cytotoxic capacity in vitro (Dwyer et al. 2020). Such encouraging findings in the preclinical setting warrant the development of clinical trials in patients to determine if PI3Kδ blockade during the manufacture of CAR-T cells can result in a superior tumour control following transfer into patients.
PI3Kδ inhibitors have been also shown to reinvigorate the existing T cell population independently of ACT. Therapeutic administration of the PI3Kδ inhibitor, PI-3065, to mice bearing 4T1 breast tumours promoted the development of a population of stem-like memory T cells, identified by their expression of TCF1, that had superior anti-tumour capacity (Lauder et al. 2020).
2.3 Inhibition of Myeloid Populations Within the Tumour Microenvironment via Specific PI3K Isoform Inhibitors
As discussed in Chap. 6, cells of myeloid origins, such as macrophages, monocytes and neutrophils are reliant on the PI3Kγ isoform for signalling and downstream functions. Within solid tumours, TAMs and MDSCs are typically considered to elicit a pro-tumoural role as they mediate immunosuppression, promote angiogenesis and aid tumour invasion and metastasis (reviewed in Cassetta and Pollard 2018; Groth et al. 2019). High frequencies of TAMs and/or MDSCs within the tumour microenvironment are routinely associated with poor clinical prognosis in a number of cancers (Zhang et al. 2012). Using PI3Kγ specific blockade to target these populations and improve tumour outcomes has been widely studied in recent years with favourable outcomes at the preclinical stage.
Schmid and colleagues were the first to demonstrate that PI3Kγ promotes myeloid cell recruitment to a range of murine tumours driven by the expression of the integrin α4β1 (Schmid et al. 2011). This macrophage recruitment to tumours could be blocked through genetic inactivation (PI3Kγ−/−) or pharmacological inactivation with the PI3Kγ specific inhibitor, TG100-115. Consequently, reducing the infiltration of macrophages into the tumour significantly reduced tumour burden. Subsequent studies have sought to delineate further how PI3Kγ inhibition of TAMs contributes to tumour control. Using a mouse model of pancreatic ductal adenocarcinoma (PDAC), TG100-115 was employed to successfully control both primary tumour burden and metastasis (Kaneda et al. 2016a). Kaneda and colleagues demonstrated that inhibition of PI3Kγ altered the immune signature of TAMs, shifting them from an immune-suppressive phenotype by reducing arginase-1, TGFβ, IL-10 and PDGF-BB expression, to a pro-inflammatory phenotype by increasing IFNγ and IL-12. These phenotypic changes to the TAMs resulted in an elevated CD8+ T cell infiltrate into PDAC tumours that promoted tumour control. A parallel study by the same group demonstrated that inhibition of macrophage PI3Kγ promoted NFκB activation whilst impeding C/EBPβ activation. This alternation in transcriptional programme resulted in a switch away from the normal immune suppression driven by TAMS to an anti-tumour immune-stimulatory phenotype (Kaneda et al. 2016b). Whilst PI3Kγ inhibition does not directly target T cells, the switch to a pro-inflammatory phenotype in PI3Kγ-inhibited macrophages indirectly augments the development of a robust anti-tumour T cell response, characterised by increased CD8+ T cell cytotoxicity and Th1 responses driven by increased granzyme B and IFNγ expression and reduced IL-10 (Fig. 2). This enhanced anti-tumour T cell response increased sensitivity to immune checkpoint blockade, with combination therapy (TG100-115 and anti-PD1 antibodies) inducing significantly greater tumour regression (≥80% of treated mice) and long-term survival in comparison to either therapy alone. This sensitivity to immune checkpoint blockade was recapitulated in a study by De Henau and colleagues who demonstrated that high tumoural infiltration of MDSCs was associated with resistance to immunotherapies such as anti-PD1 and anti-CTLA4 antibodies in a number of preclinical models (Henau et al. 2016). Targeting the MDSCs using the specific PI3Kγ inhibitor, IPI-549, restored sensitivity to checkpoint immunotherapy and improved tumour control. Gene expression analysis revealed that IPI-549 treatment resulted in MDSCs with a reduced immune-suppressive phenotype that promoted CD8+ T cell infiltration and an increased CD8+ T cell: Treg ratio within the tumour.
As the studies by Kaneda and colleagues have indicated, not all macrophages within the TME are pro-tumoural. Macrophages with a pro-inflammatory phenotype are considered to be anti-tumoural and contentiously referred to as M1 macrophages whilst M2 macrophages elicit anti-inflammatory effects that promote tumour growth (reviewed in Mantovani et al. 2021). With the previously discussed studies demonstrating that PI3Kγ blockade could switch the transcriptional profile of TAMs from pro-tumoural to anti-tumoural, Lee and colleagues expanded these findings in colon cancer whereby TG100-115 treatment of mice bearing CT26 tumours resulted in a significant reduction in tumour growth (Lee et al. 2020). Analysis of the tumour microenvironment demonstrated an increased infiltration of M1 macrophages and a reduction in M2 macrophages. Retrospective analysis of a cohort of colorectal cancer patients found that patients with an increased ratio of M1 to M2 macrophages had significantly improved progression-free and overall survival (Lee et al. 2020).
Taken together these studies support the use of PI3Kγ-specific inhibitors as an immunotherapy to reduce both the total number of tumoural TAMS, but also to skew the resident TAM population in favour of M1 macrophages. As PI3Kγ inhibitors reach clinical trials, elucidating the immune contexture of different tumours will highlight the potential for combination therapy with other immunotherapies such as checkpoint inhibitors that could significantly potentiate the effect of either treatment alone.
Despite their preference for PI3Kγ signalling, TAMs and MDSCs can also be therapeutically targeted by blocking the PI3Kδ isoform. In vitro studies demonstrated that CSF-1-induced migration and degradation of the extracellular matrix by TAMs was reduced in the presence of the PI3Kδ inhibitor GS-1101 (Mouchemore et al. 2013). Furthermore, Ali et al. demonstrated that the MDSC expansion driven by the breast tumour cell line, 4T1, is significantly abrogated in D910A mice and the ex vivo capacity of D910A MDSCs to suppress CD8+ T cell proliferation is also potently reduced (Ali et al. 2014). We have found that the therapeutic administration of PI-3065 to 4T1 tumour mice, results in a significant reduction in peripheral expansion of MDSCs (unpublished findings, manuscript in preparation). Furthermore, genetic inactivation of PI3Kδ or oral administration of the PI3Kδ inhibitor IC87114 resulted in reduced recruitment of TAMs to the breast tumour microenvironment, conferring partial tumour control (Goulielmaki et al. 2018). A note of caution when considering these findings is that tumours can drive expansion of MDSCs, therefore, reduced MDSCs in treated animals may be an indirect effect of PI3Kδ inhibition resulting in better control of tumour growth rather than inhibition of PI3Kδ in MDSCs. In addition, Tregs can also drive MDSC expansion hence a reduction in MDSCs may reflect inhibition of Tregs rather than direct effects of the PI3Kδ inhibitor on MDSCs.
Oncolytic viral therapy is a novel cancer immunotherapy approach, whereby viruses are genetically manipulated to specifically target and kill the tumour (reviewed in Harrington et al. 2019). However, despite their high specificity when administered intratumorally, their clinical promise falls short due to low levels of virus reaching the tumour when delivered intravenously (reviewed in Cook and Chauhan 2020). A ground-breaking study by Ferguson and colleagues demonstrated that in vitro treatment of macrophages with the PI3Kδ inhibitor, IC87114 prevented oncolytic viral attachment to the macrophages (Ferguson et al. 2020). Pre-treatment of tumour-bearing mice with IC87114 3 hours prior to administration of the tumour-specific oncolytic virus, resulted in significantly reduced tumour burden, prolonged survival and the development of enhanced anti-tumour immunity as characterised by increased CD4+ and CD8+ T cell tumoural infiltration and elevated numbers of IFNγ+ CD8+ T cells.
3 Clinical Trials and Human Studies
Following the success of the first-in-class PI3Kδ isoform-specific inhibitor Idelalisib as a therapy for B cell-derived haematological malignancies, and the preclinical data showing efficacy in solid tumours, multiple clinical trials are now underway with a range of PI3K specific inhibitors in both haematological and solid cancers. Many of the trials for solid tumours use pan-PI3K or isoform-specific inhibitors to treat cancers with activating mutations in the PI3K pathway or loss of PTEN. Details of all trials can be found using the clinical databases: clinicaltrials.gov or eudract.ema.europa.eu; the key trials for isoform-specific inhibitors are also listed in Table 1. However, several trials are now utilising either PI3K inhibitors as an immunotherapy, where the target is the immune cells within the tumour rather than the cancer itself, or as an adjuvant to improve existing immunotherapy approaches, and these trials are discussed in detail below.
3.1 PI3Kδ Inhibitor: Idelalisib
Given the clear role of PI3Kδ in Treg-mediated suppression within the tumour, the potential for repurposing Idelalisib as a therapy for solid tumours has been an attractive prospect. A study sponsored by Gilead (NCT02468557) recently reported its findings from a phase 1 study in pancreatic ductal adenocarcinoma (PDAC). The study intended to primarily assess the safety and adverse event incidence of Idelalisib alone and in combination with other chemotherapy drugs. The study also sought to determine the efficacy of treatment as determined by overall response rate, progression-free and overall survival and immune phenotyping of the tumour environment, specifically the effect Idelalisib had on CD8+ T cells and FoxP3+ Tregs within the tumour. However, the study was terminated early due to two progression-associated deaths and three serious adverse events in the 12 participants enrolled and treated in the Idelalisib only arm (Borazanci et al. 2020). All 12 participants reported adverse events. The toxicity reported is unlikely to be specific to PDAC patients, as serious off-target effects have been widely reported in patients treated with Idelalisib in haematological cancers (reviewed in Cuneo et al. 2019; Hanlon 2020).
A second trial aims to reinvigorate the T cell response in patients who have failed on immunotherapy by using Idelalisib to target immunosuppression within the tumour. Patients with non-small cell lung cancer who have become refractory to anti-PD1 immunotherapy will be treated with a combination of Idelalisib and the anti-PD1 monoclonal antibody, pembrolizumab, to determine if response rates can be improved by dual therapy. As a phase 2 study (NCT03257722), the safe and tolerable dose of Idelalisib in combination with a standard dose of pembrolizumab that results in optimal Treg suppression will be established and the efficacy of dual therapy will be measured by the overall response rate to treatment. This study is still in the early stages of recruiting patients and is yet to report any data or safety concerns.
Given its unquestionable clinical success for CLL and NHL, many drug companies have invested heavily in developing next-generation PI3Kδ inhibitors that replicate Idelalisib’s efficacy but with reduced toxicity. Many of these inhibitors are now being trialled with success for haematological cancers; those that are now being tested in solid tumours are discussed in detail below.
3.2 PI3Kδ Inhibitor: Parsaclisib
Parsaclisib (INCB050465) is an Incyte-developed structurally unique, next-generation PI3Kδ inhibitor, that offers significantly less side effects than first-generation PI3Kδ inhibitors such as Idelalisib (Yue et al. 2019). As a therapy, parsaclisib has shown efficacy in the preclinical Pfeiffer DLBCL model of B cell lymphoma (Shin et al. 2015). A number of clinical trials have reported both efficacy and improved tolerability in haematological cancers (Forero-Torres et al. 2019; Coleman et al. 2021) so its therapeutic potential in solid cancers is now being trialled in patients.
The Incyte sponsored phase 1 trial (NCT02646748), is a two-stage combination therapy trial. In the first stage, escalating doses of parsaclisib were given alongside pembrolizumab to evaluate the safety and tolerability in patients with a range of solid cancers (colorectal, endometrial, breast, pancreatic, lung, head and neck cancer, melanoma). In stage two, efficacy will be assessed in patients with either small cell or non-small cell lung cancer and urothelial cancer. Secondary outcomes will examine how treatment alters the immune contexture of the tumour, specifically examining how the intra-tumoural CD8+ T cell: Treg ratio is altered. The study is due for completion in December 2021, however, preliminary findings indicate that combined therapy significantly reduces the number of intra-tumoural Tregs and increases the CD8+ T cell: Treg ratio ( Kirkwood et al. 2018). Furthermore, analysis of both tumour and PBMCs showed increased T cell activation in patients administered combination therapy.
A parallel trial (NCT03589651), also seeks to determine the efficacy of parsaclisib as combination therapy, with the Incyte-developed anti-PD1 monoclonal antibody therapy, retifanlimab, which has shown promise in other clinical trials for solid tumours (Berton-Rigaud et al. 2020). A third trial by the same sponsors (NCT02559492), was designed to determine both the tolerability and efficacy as measured by tumour response rate, progression-free survival and duration of response, to a combination therapy of parsaclisib and the JAK1 inhibitor, itacitinib, in patients with metastatic cancer. After the primary outcome of measuring safety and tolerability of combined treatment groups was completed the study was terminated early, although no data has been published to date.
3.3 Novel PI3Kδ Inhibitors
A Cancer Research UK sponsored phase 2 trial (NCT02540928), sought to examine changes to the CD8+ T cell infiltrate of head and neck squamous cell carcinoma before and after treatment with the PI3Kδ inhibitor, AMG319. An initial report published in 2018, demonstrated between 50 and 88% inhibition of pAKT following treatment with AMG319 (Ottensmeier et al. 2018). However, of the 22 participants recruited, 10 patients reported skin and gut-associated adverse events, with nine participants terminating treatment early. These adverse events are similar to those seen in an earlier clinical trial in CLL and NHL patients treated with AMG319 (Lanasa et al. 2013). As a consequence of the adverse events reported, this study has subsequently been terminated early due to safety concerns, with full results yet to be reported.
The Shanghai Yingli Pharmaceutical developed inhibitor, YY-20394 or Linperlisib, is currently being studied in a number of clinical trials for lymphoma and leukaemia (NCT04108325, NCT04379167, NCT04370405, NCT04279405, NCT04705090, NCT04500561). Interim results from these studies suggest YY-20394 may offer an improved safety profile with less adverse events reported to date (Qiu et al. 2019). YY-20394 is reported to be structurally unique to other PI3Kδ inhibitors, such as Idelalisib. Patients with advanced cancers are currently being recruited to a phase 1 trial (NCT04049929), designed to assess primarily the safety profile of YY-20394 and secondly the efficacy as determined by tumour progression rate.
Finally, the iOnctura developed inhibitor, IOA-244, has reportedly shown great therapeutic potential as a Treg and MDSC targeting therapy in a preclinical model of colon cancer with high Treg: CD8+ T cell ratio (Johnson et al. 2019). IOA-244 combination therapy with either anti-PD1 or anti-PD-L1 significantly inhibited tumour growth. IOA-244 is now being studied in a phase 1 trial (NCT04328844) as a monotherapy and in combination with the chemotherapeutics pemetrexed/cisplatin in a range of advanced solid tumour indications. This first-in-human study will involve a dose escalation to determine the safety profile of IOA-244. The second stage of the study will determine both tolerability and efficacy of IOA-244 as either a mono or combined therapy and will examine changes to the immune phenotype of lymphocytes in peripheral blood. Results from this study are not expected until the middle of 2023 at the earliest.
3.4 PI3Kγ Inhibitor: IPI-549
Given the success of targeting macrophages via PI3Kγ and improving tumour control in the preclinical models discussed earlier, a number of drug companies have advanced PI3Kγ inhibitors into human trials. Infinity pharmaceuticals were the first to test IPI-549, eganelisib, in a cohort of over 200 patients with a range of solid tumours. The first part of the MARIO-1 (Macrophage Reprogramming in Immuno-Oncology) phase 1 trial (NCT02637531) sought to test the safety and tolerability of eganelisib, in a dose-escalation study, with the efficacy of eganelisib as either a monotherapy or in combination with nivolumab measured in stage two. Full data sets from the study which was due to finish mid-2021 are yet to be released, however, preliminary findings were reported in 2017 and 2018. These preliminary data suggest that eganelisib and nivolumab combination therapy was generally well tolerated with patients typically only experiencing grade 1–2 adverse events (Sullivan et al. 2018; Tolcher et al. 2017). Blood samples taken during the treatment phase indicated T cell activation and reduced immune suppression in peripheral blood. The study sponsors have subsequently commenced two additional trials with eganelisib in 2019. The MARIO-3 phase 2 trial (NCT03961698) is a multi-arm trial in patients with triple-negative breast cancer (TNBC) or renal cell cancer, designed to test the efficacy of targeting macrophages with eganelisib. Patients will receive eganelisib, in combination with either an anti-PD1 (Atezolizumab) or anti-VEGF therapy (Bevacizumab) and the primary outcome of complete response to therapy will be measured over an 18-month period. The secondary outcomes will determine the safety profile, progression-free survival and duration of response. The third Infinity pharmaceuticals sponsored trial is the MARIO-275, phase 2 trial (NCT03980041). Similarly to the MARIO-1 and -3 studies, the efficacy of eganelisib as a monotherapy or in combination with nivolumab will be tested in immunotherapy-naïve advanced urothelial cancer patients.
The efficacy of IPI-549 in a checkpoint inhibitor-independent setting will be tested in patients with TNBC or ovarian cancer (NCT03719326). This two-part dose escalation or expansion study will measure the safety and tolerability of IPI-549, in combination with the dual adenosine receptor antagonist, etrumadenant and the chemotherapy, doxorubicin. Secondary outcomes intend to determine the efficacy of therapy with respect to progression-free and overall survival, duration of response and immune phenotyping of peripheral blood during the study to determine the effect of treatment on the immune response.
A further study, independent of Infinity pharmaceuticals, is designed as a ‘poof-of-concept’ study to test the hypothesis that macrophage phenotype switching occurs in humans in response to the PI3Kγ inhibitor, IPI-549, as previously reported in preclinical models (Kaneda et al. 2016a, 2016b; Henau et al. 2016). This phase 2 window trial (NCT03795610) in a small cohort of patients with head and neck cancer will take tumour biopsies before and after IPI-549 treatment to allow comparison of the immune signature of TAMs. The secondary objectives aim to determine the safety and tolerability of IPI-549 and to examine changes to the myeloid and T cell tumoural infiltrate following treatment.
3.5 Dual PI3Kδ/γ Inhibitor: Duvelisib
IPI-145, Duvelisib, is one of the next-generation PI3K isoform inhibitors, designed to target both the immune cell dominant PI3Kδ and γ isoforms. A number of clinical trials have reported encouraging results for haematological cancers (O'Brien et al. 2018; Flinn et al. 2019, 2018). Given the success shown with individual δ and γ isoform inhibitors in preclinical models described earlier, the ability to target immunosuppressive Tregs, MDSCs and TAMs with a single agent may offer advanced efficacy but without the toxicity of pan-PI3K inhibitors.
Two parallel studies are currently underway to determine the incidence of adverse events and overall efficacy of Duvelisib and anti-PD1 treatment in patients with head and neck cancer (NCT04193293) or unresectable melanoma (NCT04688658). The effect of Duvelisib and Nivolumab treatment on immune cell function and phenotype in both the tumour and the periphery will be established before, during and after treatment in melanoma patients. Furthermore, the development of PD1 resistance mechanisms will be established by examining the gene signature of tumour-infiltrating immune cells. To date neither study has reported any findings, however, a recent study demonstrated that treatment with IPI-145 provided no control of tumour growth in a mouse model of melanoma (Dwyer et al. 2020). In comparison to treatment with the single δ inhibitors CAL-101 or TGR-1202 or the γ inhibitor IPI-549, T cells treated with IPI-145 were functionally impaired, with less cytokine production and reduced persistence in vivo, suggesting that IPI-145 treatment prevented the generation of a sufficient anti-tumour T cell response capable of controlling tumour burden.
4 Future Perspectives
Targeting the PI3K pathway using isoform-specific inhibitors has demonstrated promise at the preclinical stage, either as a novel monotherapy or in combination with other treatments. Similar to the first-generation immune checkpoint inhibitors that target PD1 and CTLA4 (Nivolumab, Pembrolizumab and Ipilimumab), only tumours that have high numbers of immunosuppressive cells can be successfully treated with PI3K isoform inhibitors. As PI3K isoform-specific inhibitors move into the clinical phase of testing, elucidating the effect of PI3Kδ or PI3Kγ inhibition on the immune response within each tumour type will be critical in determining which patient cohorts are likely to be responsive to PI3K specific therapy. A detailed phenotypic analysis of the tumour-infiltrating lymphocytes has the potential to identify which co-inhibitory receptors could be targeted with additional immunotherapies that could potentiate the clinical response to PI3K inhibitors. However, given the immune-related toxicity reported with both checkpoint inhibitors and first-generation PI3K inhibitors such as Idelalisib, it remains to be seen if PI3Kδ- and PI3Kγ-specific inhibitors will be tolerated sufficiently to enable their future use as a first-line treatment regimen.
References
Abramson VG, Oliveira M, Cervantes A, Wildiers H, Patel MR, Bauer TM, Bedard PL, Becerra C, Richey S, Wei MC, Reyner E, Bond J, Cui N, Wilson TR, Moore HM, Saura C, Krop IE (2019) A phase Ib, open-label, dose-escalation study of the safety and pharmacology of taselisib (GDC-0032) in combination with either docetaxel or paclitaxel in patients with HER2-negative, locally advanced, or metastatic breast cancer. Breast Cancer Res Treat 178(1):121–133. https://doi.org/10.1007/s10549-019-05360-3
Ahmad S, Abu-Eid R, Shrimali R, Webb M, Verma V, Doroodchi A, Berrong Z, Samara R, Rodriguez PC, Mkrtichyan M, Khleif SN (2017) Differential PI3Kδ signaling in CD4. Cancer Res 77(8):1892–1904. https://doi.org/10.1158/0008-5472.CAN-16-1839
Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H, Friedman L, Turner M, Okkenhaug K, Vanhaesebroeck B (2014) Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 510(7505):407–411. https://doi.org/10.1038/nature13444
André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, Yamashita T, Lu YS, Inoue K, Takahashi M, Pápai Z, Longin AS, Mills D, Wilke C, Hirawat S, Juric D, Group S-S (2019) Alpelisib for. N Engl J Med 380(20):1929–1940. https://doi.org/10.1056/NEJMoa1813904
Arkenau H-T, Mateo J, Lemech CR, Infante JR, Burris HA, Bang Y-J, Eder JP, Herbst RS, Sharma S, Chung HC, Decordova S, Swales KE, Garrett MD, Loftiss JI, Durante M, Russo MW, Suttle BB, Motwani M, Kumar R, Bono JSD (2014) A phase I/II, first-in-human dose-escalation study of GSK2636771 in patients (pts) with PTEN-deficient advanced tumors. In: ASCO, vol 15. Journal of Clinical Oncology
Baharom F, Ramirez-Valdez RA, Tobin KKS, Yamane H, Dutertre CA, Khalilnezhad A, Reynoso GV, Coble VL, Lynn GM, Mulè MP, Martins AJ, Finnigan JP, Zhang XM, Hamerman JA, Bhardwaj N, Tsang JS, Hickman HD, Ginhoux F, Ishizuka AS, Seder RA (2021) Intravenous nanoparticle vaccination generates stem-like TCF1. Nat Immunol 22(1):41–52. https://doi.org/10.1038/s41590-020-00810-3
Berton-Rigaud D, Vergote I, Pautier P, Kryzhanivska A, Selle AADKMD-SCTNBF A phase 1 study of retifanlimab (INCMGA00012), a PD-1 inhibitor. In: patients with advanced solid tumors: preliminary results in recurrent MSI-high or dMMR endometrial cancer (POD1UM-101). In: SITC, Online, 2020. Journal for Imunotherapy of Cancer. https://doi.org/10.1136/jitc-2020-SITC2020.0268
Bono JSd, Hansen A, Choudhury AD, Cook N, Heath EI, Higano C, Linch M, Martin-Liberal J, Rathkopf DE, Wisinski KB, Barry S, Bruin Ed, Brugger W, Colebrook S, Klinowska T, Moschetta M, Mortimer PGS, Siu LL, Shapiro G (2018) AZD8186, a potent and selective inhibitor of PI3Kb/d, as monotherapy and in combination with abiraterone acetate plus prednisone (AAP), in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC). In: ESMO, vol 8. Annals of Oncology
Borazanci E, Pishvaian MJ, Nemunaitis J, Weekes C, Huang J, Rajakumaraswamy N (2020) A phase Ib study of single-agent idelalisib followed by idelalisib in combination with chemotherapy in patients with metastatic pancreatic ductal adenocarcinoma. Oncologist 25(11):e1604–e1613. https://doi.org/10.1634/theoncologist.2020-0321
Bowers JS, Majchrzak K, Nelson MH, Aksoy BA, Wyatt MM, Smith AS, Bailey SR, Neal LR, Hammerbacher JE, Paulos CM (2017) PI3Kδ inhibition enhances the antitumor fitness of adoptively transferred CD8. Front Immunol 8:1221. https://doi.org/10.3389/fimmu.2017.01221
Carnevalli LS, Sinclair C, Taylor MA, Gutierrez PM, Langdon S, Coenen-Stass AML, Mooney L, Hughes A, Jarvis L, Staniszewska A, Crafter C, Sidders B, Hardaker E, Hudson K, Barry ST (2018) PI3Kα/δ inhibition promotes anti-tumor immunity through direct enhancement of effector CD8. J Immunother Cancer 6(1):158. https://doi.org/10.1186/s40425-018-0457-0
Cassetta L, Pollard JW (2018) Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov 17(12):887–904. https://doi.org/10.1038/nrd.2018.169
Choueiri TK, Porta C, Rodriguez CS, Alter R, Czaykowski P, Duran I, Gross-Goupil M, Kalinka-Warzocha E, Melichar B, Patel C, Neuwirth R, Enke A, Zohren F, Powles T (2017) A phase 2 study of investigational TORC1/2 inhibitor TAK-228 and TAK-228 plus investigational PI3Ka-selective inhibitor TAK-117 vs everolimus in adults with advanced or metastatic clear-cell renal cell carcinoma (ccRCC) that has progressed on VEGF-targeted therapy. In: ESMO, vol 5. Annals of Oncology. https://doi.org/10.1093/annonc/mdx371.079
Coleman M, Belada D, Casasnovas RO, Gressin R, Lee HP, Mehta A, Munoz J, Verhoef G, Corrado C, DeMarini DJ, Zhao W, Li J, Fay K (2021) Phase 2 study of parsaclisib (INCB050465), a highly selective, next-generation PI3Kδ inhibitor, in relapsed or refractory diffuse large B-cell lymphoma (CITADEL-202). Leuk Lymphoma 62(2):368–376. https://doi.org/10.1080/10428194.2020.1832660
Cook M, Chauhan A (2020) Clinical application of oncolytic viruses: a systematic review. Int J Mol Sci 21(20). https://doi.org/10.3390/ijms21207505
Cuneo A, Barosi G, Danesi R, Fagiuoli S, Ghia P, Marzano A, Montillo M, Poletti V, Viale P, Zinzani PL (2019) Management of adverse events associated with idelalisib treatment in chronic lymphocytic leukemia and follicular lymphoma: a multidisciplinary position paper. Hematol Oncol 37(1):3–14. https://doi.org/10.1002/hon.2540
De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, Douglas M, Tibbitts T, Sharma S, Proctor J, Kosmider N, White K, Stern H, Soglia J, Adams J, Palombella VJ, McGovern K, Kutok JL, Wolchok JD, Merghoub T (2016) Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 539(7629):443–447. https://doi.org/10.1038/nature20554
Dent S, Cortés J, Im YH, Diéras V, Harbeck N, Krop IE, Wilson TR, Cui N, Schimmoller F, Hsu JY, He J, De Laurentiis M, Sousa S, Drullinsky P, Jacot W (2021) Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: the SANDPIPER trial. Ann Oncol 32(2):197–207. https://doi.org/10.1016/j.annonc.2020.10.596
Dwyer CJ, Arhontoulis DC, Rangel Rivera GO, Knochelmann HM, Smith AS, Wyatt MM, Rubinstein MP, Atkinson C, Thaxton JE, Neskey DM, Paulos CM (2020) Ex vivo blockade of PI3K gamma or delta signaling enhances the antitumor potency of adoptively transferred CD8. Eur J Immunol 50(9):1386–1399. https://doi.org/10.1002/eji.201948455
Ferguson MS, Chard Dunmall LS, Gangeswaran R, Marelli G, Tysome JR, Burns E, Whitehead MA, Aksoy E, Alusi G, Hiley C, Ahmed J, Vanhaesebroeck B, Lemoine NR, Wang Y (2020) Transient inhibition of PI3Kδ enhances the therapeutic effect of intravenous delivery of oncolytic vaccinia virus. Mol Ther 28(5):1263–1275. https://doi.org/10.1016/j.ymthe.2020.02.017
Filho OM, Goel S, Barry WT, Hamilton EP, Tolaney SM, Yardley DA, Rees R, Demeo M, Mills C, Hafner M, Winer EP, Zhao J, Krop IE (2017) A mouse-human phase I co-clinical trial of taselisib in combination with TDM1 in advanced HER2-positive breast cancer (MBC). In: ASCO, vol 15. Journal of Clinical Oncology
Flinn IW, Hillmen P, Montillo M, Nagy Z, Illés Á, Etienne G, Delgado J, Kuss BJ, Tam CS, Gasztonyi Z, Offner F, Lunin S, Bosch F, Davids MS, Lamanna N, Jaeger U, Ghia P, Cymbalista F, Portell CA, Skarbnik AP, Cashen AF, Weaver DT, Kelly VM, Turnbull B, Stilgenbauer S (2018) The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood 132(23):2446–2455. https://doi.org/10.1182/blood-2018-05-850461
Flinn IW, Cherry MA, Maris MB, Matous JV, Berdeja JG, Patel M (2019) Combination trial of duvelisib (IPI-145) with rituximab or bendamustine/rituximab in patients with non-Hodgkin lymphoma or chronic lymphocytic leukemia. Am J Hematol 94(12):1325–1334. https://doi.org/10.1002/ajh.25634
Forero-Torres A, Ramchandren R, Yacoub A, Wertheim MS, Edenfield WJ, Caimi P, Gutierrez M, Akard L, Escobar C, Call J, Persky D, Iyer S, DeMarini DJ, Zhou L, Chen X, Dawkins F, Phillips TJ (2019) Parsaclisib, a potent and highly selective PI3Kδ inhibitor, in patients with relapsed or refractory B-cell malignancies. Blood 133(16):1742–1752. https://doi.org/10.1182/blood-2018-08-867499
Goulielmaki E, Bermudez-Brito M, Andreou M, Tzenaki N, Tzardi M, de Bree E, Tsentelierou E, Makrigiannakis A, Papakonstanti EA (2018) Pharmacological inactivation of the PI3K p110δ prevents breast tumour progression by targeting cancer cells and macrophages. Cell Death Dis 9(6):678. https://doi.org/10.1038/s41419-018-0717-4
Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, Umansky V (2019) Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer 120(1):16–25. https://doi.org/10.1038/s41416-018-0333-1
Gyori D, Lim EL, Grant FM, Spensberger D, Roychoudhuri R, Shuttleworth SJ, Okkenhaug K, Stephens LR, Hawkins PT (2018) Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight 3(11). https://doi.org/10.1172/jci.insight.120631
Hanlon A (2020) Brander DM (2020) managing toxicities of phosphatidylinositol-3-kinase (PI3K) inhibitors. Hematology Am Soc Hematol Educ Program 1:346–356. https://doi.org/10.1182/hematology.2020000119
Hansen AR, Shapiro G, Do KT, Kumar R, Martin-Liberal J, Higano CS, Wisinski KB, Dean EJ, Heath EI, Rathkopf DE, Linch MD, Barry ST, Brugger W, Bruin ED, Colebrook S, Klinowska T, Mitchell PD, Moorthy G, Bono JSD, Siu LL (2017) A first in human phase I study of AZD8186, a potent and selective inhibitor of PI3K in patients with advanced solid tumours as monotherapy and in combination with the dual mTORC1/2 inhibitor vistusertib (AZD2014) or abiraterone acetate. In: ASCO, vol 15. Journal of Clinical Oncology
Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC (2019) Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov 18(9):689–706. https://doi.org/10.1038/s41573-019-0029-0
Henegouwen JMvB, Hoes LR, Wijngaart Hvd, Velden DLVD, Huitema A, Cuppen EPJG, Lugtenburg EJ, Vos FYFLD, Bloemendal H, Grunberg K, Verheul HMW, Hans Gelderblom, Voest EE (2019) Update on the drug rediscovery protocol: expanded use of existing anticancer drugs in patients with a known molecular profile. In: ASCO, vol 15. Journal of Clinical Oncology
Jakubowski C, Collins NB, Sugar EA, Berg M, Cao H, Giannakis M, Jaffee EM, Azad NS (2020) A phase I/II study of PI3Kinase inhibition with copanlisib combined with the anti-PD-1 antibody nivolumab in relapsed/refractory solid tumors with expansions in MSS colorectal cancer. In: ASCO, vol 15. Journal of Clinical Oncology
Janku F, Yap TA, Meric-Bernstam F (2018) Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol 15(5):273–291. https://doi.org/10.1038/nrclinonc.2018.28
Johnson Z, Papakonstanti E, Tsapara A, Macqueen A, Shah P, Veen LVd, Lahn M (2019) Preclinical development of a novel, highly selective PI3Kdinhibitor, IOA-244, for the treatment of solid malignancies. In: ESMO, vol 7. Annals of Oncology, p 93p
Juric D, de Bono JS, LoRusso PM, Nemunaitis J, Heath EI, Kwak EL, Macarulla Mercadé T, Geuna E, Jose de Miguel-Luken M, Patel C, Kuida K, Sankoh S, Westin EH, Zohren F, Shou Y, Tabernero J (2017) A first-in-human, Phase I, dose-escalation study of TAK-117, a selective PI3Kα isoform inhibitor, in patients with advanced solid malignancies. Clin Cancer Res 23(17):5015–5023. https://doi.org/10.1158/1078-0432.CCR-16-2888
Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, Middleton MR, Berlin J, Schuler M, Gil-Martin M, Rugo HS, Seggewiss-Bernhardt R, Huang A, Bootle D, Demanse D, Blumenstein L, Coughlin C, Quadt C, Baselga J (2018) Phosphatidylinositol 3-Kinase α-selective inhibition with Alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol 36(13):1291–1299. https://doi.org/10.1200/JCO.2017.72.7107
Juric D, Janku F, Rodón J, Burris HA, Mayer IA, Schuler M, Seggewiss-Bernhardt R, Gil-Martin M, Middleton MR, Baselga J, Bootle D, Demanse D, Blumenstein L, Schumacher K, Huang A, Quadt C, Rugo HS (2019) Alpelisib plus fulvestrant in PIK3CA-Altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: a phase 1b clinical trial. JAMA Oncol 5(2):e184475. https://doi.org/10.1001/jamaoncol.2018.4475
Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, Schmid MC, Sun P, Mose E, Bouvet M, Lowy AM, Valasek MA, Sasik R, Novelli F, Hirsch E, Varner JA (2016a) Macrophage PI3Kγ drives pancreatic ductal adenocarcinoma progression. Cancer Discov 6(8):870–885. https://doi.org/10.1158/2159-8290.CD-15-1346
Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, Schmid MC, Pink M, Winkler DG, Rausch M, Palombella VJ, Kutok J, McGovern K, Frazer KA, Wu X, Karin M, Sasik R, Cohen EE, Varner JA (2016b) PI3Kγ is a molecular switch that controls immune suppression. Nature 539(7629):437–442. https://doi.org/10.1038/nature19834
Kienle GS (2012) Fever in cancer treatment: Coley’s therapy and epidemiologic observations. Glob Adv Health Med 1(1):92–100. https://doi.org/10.7453/gahmj.2012.1.1.016
Kirkwood JM, Iannotti N, Cho D, O'Day S, Gibney G, Stephen Hodi F, Munster P, Hoyle P, Owens S, Smith M, Mettu N (2018) Effect of JAK/STAT or PI3Kδ plus PD-1 inhibition on the tumor microenvironment: biomarker results from a phase Ib study in patients with advanced solid tumors. In: AACR annual meeting, Chicago, vol 13. Cancer Research. https://doi.org/10.1158/1538-7445.AM2018-CT176
Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, Yardley DA, Melichar B, Forero-Torres A, Lee SC, de Boer R, Petrakova K, Vallentin S, Perez EA, Piccart M, Ellis M, Winer E, Gendreau S, Derynck M, Lackner M, Levy G, Qiu J, He J, Schmid P (2016) Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 17(6):811–821. https://doi.org/10.1016/S1470-2045(16)00106-6
Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, Kuchroo VK, Regev A, Anderson AC (2019) Checkpoint blockade immunotherapy induces dynamic changes in PD-1. Immunity 50(1):181-194.e186. https://doi.org/10.1016/j.immuni.2018.11.014
Lanasa MC, Glenn M, Mato AR, Allgood SD, Wong S, Amore B, Means G, Stevens E, Yan C, Friberg G, Goy A (2013) First-in-human study of AMG 319, a highly selective, small molecule inhibitor of PI3Kδ. In: Adult patients with relapsed or refractory lymphoid malignancies. In: 55th ASH, vol 21. Blood, p 678. https://doi.org/10.1182/blood.V122.21.678.678
Langer CJ, Redman MW, Wade JL, Aggarwal C, Bradley JD, Crawford J, Stella PJ, Knapp MH, Miao J, Minichiello K, Herbst RS, Kelly K, Gandara DR, Papadimitrakopoulou VA (2019) SWOG S1400B (NCT02785913), a Phase II study of GDC-0032 (Taselisib) for previously treated PI3K-positive patients with Stage IV squamous cell lung cancer (Lung-MAP sub-study). J Thorac Oncol 14(10):1839–1846. https://doi.org/10.1016/j.jtho.2019.05.029
Lauder SN, Vanhaesebroeck B, Gallimore A (2021) Sequential targeting of PI3Kδ and LAG3 as an effective anti-cancer approach. Br J Cancer. https://doi.org/10.1038/s41416-021-01285-1
Lauder SN, Smart K, Kersemans V, Allen D, Scott J, Pires A, Milutinovic S, Somerville M, Smart S, Kinchesh P, Lopez-Guadamillas E, Hughes E, Jones E, Scurr M, Godkin A, Friedman LS, Vanhaesebroeck B, Gallimore A (2020) Enhanced antitumor immunity through sequential targeting of PI3Kδ and LAG3. J Immunother Cancer 8(2). https://doi.org/10.1136/jitc-2020-000693
Lee YS, Song SJ, Hong HK, Oh BY, Lee WY, Cho YB (2020) The FBW7-MCL-1 axis is key in M1 and M2 macrophage-related colon cancer cell progression: validating the immunotherapeutic value of targeting PI3Kγ. Exp Mol Med 52(5):815–831. https://doi.org/10.1038/s12276-020-0436-7
Lehmann BD, Abramson VG, Sanders ME, Mayer EL, Haddad TC, Nanda R, Van Poznak C, Storniolo AM, Nangia JR, Gonzalez-Ericsson PI, Sanchez V, Johnson KN, Abramson RG, Chen SC, Shyr Y, Arteaga CL, Wolff AC, Pietenpol JA, Consortium TBCR (2020) TBCRC 032 IB/II multicenter study: molecular insights to AR antagonist and PI3K inhibitor efficacy in patients with AR. Clin Cancer Res 26(9):2111–2123. https://doi.org/10.1158/1078-0432.CCR-19-2170
Leong S, Moss RA, Bowles DW, Ware JA, Zhou J, Spoerke JM, Lackner MR, Shankar G, Schutzman JL, van der Noll R, Voest EE, Schellens JHM (2017) A Phase I dose-escalation study of the safety and pharmacokinetics of pictilisib in combination with erlotinib in patients with advanced solid tumors. Oncologist 22(12):1491–1499. https://doi.org/10.1634/theoncologist.2017-0090
Lim EL, Cugliandolo FM, Rosner DR, Gyori D, Roychoudhuri R, Okkenhaug K (2018) Phosphoinositide 3-kinase δ inhibition promotes antitumor responses but antagonizes checkpoint inhibitors. JCI Insight 3 (11). https://doi.org/10.1172/jci.insight.120626
Lin WH, Adams WC, Nish SA, Chen YH, Yen B, Rothman NJ, Kratchmarov R, Okada T, Klein U, Reiner SL (2015) Asymmetric PI3K signaling driving developmental and regenerative cell fate bifurcation. Cell Rep 13(10):2203–2218. https://doi.org/10.1016/j.celrep.2015.10.072
Lopez JS, SelviMiralles M, Ameratunga M, Minchom A, Pascual J, Banerji U, Bye H, Raynaud FI, Swales KE, Malia J, Hubank M, Garcia-Murillas I, Parmar M, Ward SE, Finneran L, Hall E, Turner AJ, Bono JSD, Yap TA, Turner NC (2019) PIPA: a phase Ib study of selective ß-isoform sparing phosphatidylinositol 3-kinase (PI3K) inhibitor taselisib (T) plus palbociclib (P) in patients (pts) with advanced solid cancers—safety, tolerability, pharmacokinetic (PK), and pharmacodynamic (PD) analysis of the doublet combination. In: ASCO, vol 15. Journal of Clinical Oncology
Mantovani A, Marchesi F, Jaillon S, Garlanda C, Allavena P (2021) Tumor-associated myeloid cells: diversity and therapeutic targeting. Cell Mol Immunol 18(3):566–578. https://doi.org/10.1038/s41423-020-00613-4
Mouchemore KA, Sampaio NG, Murrey MW, Stanley ER, Lannutti BJ, Pixley FJ (2013) Specific inhibition of PI3K p110δ inhibits CSF-1-induced macrophage spreading and invasive capacity. FEBS J 280(21):5228–5236. https://doi.org/10.1111/febs.12316
Narayan P, Prowell TM, Gao JJ, Fernandes LL, Li E, Jiang X, Qiu J, Fan J, Song P, Yu J, Zhang X, King-Kallimanis BL, Chen W, Ricks TK, Gong Y, Wang X, Windsor K, Rhieu SY, Geiser G, Banerjee A, Chen X, Reyes Turcu F, Chatterjee DK, Pathak A, Seidman J, Ghosh S, Philip R, Goldberg KB, Kluetz PG, Tang S, Amiri-Kordestani L, Theoret MR, Pazdur R, Beaver JA (2021) FDA approval summary: alpelisib plus fulvestrant for patients with HR-positive, HER2-negative, PIK3CA-mutated, advanced or metastatic breast cancer. Clin Cancer Res 27(7):1842–1849. https://doi.org/10.1158/1078-0432.CCR-20-3652
Nish SA, Zens KD, Kratchmarov R, Lin WW, Adams WC, Chen YH, Yen B, Rothman NJ, Bhandoola A, Xue HH, Farber DL, Reiner SL (2017) CD4+ T cell effector commitment coupled to self-renewal by asymmetric cell divisions. J Exp Med 214(1):39–47. https://doi.org/10.1084/jem.20161046
O’Brien S, Patel M, Kahl BS, Horwitz SM, Foss FM, Porcu P, Jones J, Burger J, Jain N, Allen K, Faia K, Douglas M, Stern HM, Sweeney J, Kelly P, Kelly V, Flinn I (2018) Duvelisib, an oral dual PI3K-δ, γ inhibitor, shows clinical and pharmacodynamic activity in chronic lymphocytic leukemia and small lymphocytic lymphoma in a phase 1 study. Am J Hematol 93(11):1318–1326. https://doi.org/10.1002/ajh.25243
Oliveira M, Baird R, Rossum Av, Beelen K, Garcia-Corbacho J, Mandjes I, Vallier A, Werkhoven Ev, Garrigós L, Kumar S, Tinteren Hv, Muñoz S, Linossi C, Rosing H, Miquel J, Schrier M, Schultink AdV, Saura C, Gallagher W, Bernards R, Tabernero J, Cortés J, Caldas C, Linn S (2016) Abstract OT2-01-11: Phase II of POSEIDON: a phase Ib/randomized phase II trial of tamoxifen plus taselisib or placebo in hormone receptor positive, HER2 negative, metastatic breast cancer patients with prior exposure to endocrine treatment. In: San Antonio breast cancer symposium, vol 4. Cancer Research
Ottensmeier CHH, Jones T, Sacco JJ, McCaul JA, Brennan P, Paterson C, Schache AG, Shaw R, Singh RP, Davies JH, Chudley L, Jeffrey D, Thomas G, Loadman P, Dobbs NA, Essame L, Acton G, Katugampola S, Vanhaesebroeck B, King EA (2018) randomised, double-blind, placebo-controlled phase IIa trial of AMG319 given orally as neoadjuvant therapy in patients with human papillomavirus (HPV) positive and negative head and neck squamous cell carcinoma (HNSCC). In: ASCO, vol 15. Journal of Clinical Oncology, pp 6068–6068
Putz EM, Prchal-Murphy M, Simma OA, Forster F, Koenig X, Stockinger H, Piekorz RP, Freissmuth M, Müller M, Sexl V, Zebedin-Brandl E (2012) PI3Kδ is essential for tumor clearance mediated by cytotoxic T lymphocytes. PLoS ONE 7(7):e40852. https://doi.org/10.1371/journal.pone.0040852
Qiu L, Qi J, Song Y, Jiang B, Tu M, Ping L, Li Z, Zhu J, Xu Y, Bao H, Xu Z (2019) A phase I study of a selective PI3Kδ inhibitor YY-20394 in patients with relapsed or refractory B-cell malignancies. In: ASCO, 2019. vol 15. Journal of Clinical Oncology, pp 7563–7563
Razak ARA, Ahn M-J, Yen C-J, Solomon BJ, Lee S-H, Wang H-M, Munster PN, Herpen CML-V, Gilbert J, Pal RR, Blumenstein L, Gomez-Carrillo B, Coughlin CM, Lim W-T (2014) Phase lb/ll study of the PI3Kα inhibitor BYL719 in combination with cetuximab in recurrent/metastatic squamous cell cancer of the head and neck (SCCHN). In: ASCO, vol 15. Journal of Clinical Oncology. https://doi.org/10.1200/jco.2014.32.15_suppl.6044
Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, Freeman SS, Reuben A, Hoover PJ, Villani AC, Ivanova E, Portell A, Lizotte PH, Aref AR, Eliane JP, Hammond MR, Vitzthum H, Blackmon SM, Li B, Gopalakrishnan V, Reddy SM, Cooper ZA, Paweletz CP, Barbie DA, Stemmer-Rachamimov A, Flaherty KT, Wargo JA, Boland GM, Sullivan RJ, Getz G, Hacohen N (2018) Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175(4):998-1013.e1020. https://doi.org/10.1016/j.cell.2018.10.038
Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, Clarke PA, Raynaud FI, Levy G, Ware JA, Mazina K, Lin R, Wu J, Fredrickson J, Spoerke JM, Lackner MR, Yan Y, Friedman LS, Kaye SB, Derynck MK, Workman P, de Bono JS (2015) First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res 21(1):77–86. https://doi.org/10.1158/1078-0432.CCR-14-0947
Saura C, Hlauschek D, Oliveira M, Zardavas D, Jallitsch-Halper A, de la Peña L, Nuciforo P, Ballestrero A, Dubsky P, Lombard JM, Vuylsteke P, Castaneda CA, Colleoni M, Santos Borges G, Ciruelos E, Fornier M, Boer K, Bardia A, Wilson TR, Stout TJ, Hsu JY, Shi Y, Piccart M, Gnant M, Baselga J, de Azambuja E (2019) Neoadjuvant letrozole plus taselisib versus letrozole plus placebo in postmenopausal women with oestrogen receptor-positive, HER2-negative, early-stage breast cancer (LORELEI): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 20(9):1226–1238. https://doi.org/10.1016/S1470-2045(19)30334-1
Scambia G, Han SN, Oza AM, Colombo N, Oaknin A, Raspagliesi F, Wenham RM, Braicu EI, Jewell A, Makker V, Guerra EM, Moxley KM, Baurain J-F, Su Z, Neuwirth R, Vincent S, Sedarati F, Faller DV, Krell J (2020) Randomized phase II study of sapanisertib (SAP) + paclitaxel (PAC) versus PAC alone in patients (pts) with advanced, recurrent, or persistent endometrial cancer. In: ASCO, vol 15. Journal of Clinical Oncology
Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JR, Song X, Wrasidlo W, Blair SL, Ginsberg MH, Cheresh DA, Hirsch E, Field SJ, Varner JA (2011) Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell 19(6):715–727. https://doi.org/10.1016/j.ccr.2011.04.016
Schöffski P, Cresta S, Mayer IA, Wildiers H, Damian S, Gendreau S, Rooney I, Morrissey KM, Spoerke JM, Ng VW, Singel SM, Winer E (2018) A phase Ib study of pictilisib (GDC-0941) in combination with paclitaxel, with and without bevacizumab or trastuzumab, and with letrozole in advanced breast cancer. Breast Cancer Res 20(1):109. https://doi.org/10.1186/s13058-018-1015-x
Shimizu J, Yamazaki S, Sakaguchi S (1999) Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol 163(10):5211–5218
Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE, Held W (2019) Intratumoral Tcf1. Immunity 50(1):195-211.e110. https://doi.org/10.1016/j.immuni.2018.12.021
Soares HP, Al-Toubah TE, Kim RD, Kim J, Lewis NK, Mahipal A (2018) Final report: a phase I trial of BYL719 in combination with gemcitabine and nab-paclitaxel in locally advanced and metastatic pancreatic cancer. In: Gastrointestinal cancers symposium, vol 4. Journal of Clinical Oncology. https://doi.org/10.1200/JCO.2018.36.4_suppl.398
Shin N, Koblish H, Covington M, Li Y, Wang K, Wang Q, Feldman P, Hall L, O'Connor S, He X, Katiyar K, Li Y, Yue EW, Maduskuie TP, Douty B, Mei S, Li Y-L, Xue C-B, Combs A, Yao W, Diamond-Fosbenner S, Yeleswaram S, Newton R, Vaddi K, Huber R (2015) INCB050465, a novel PI3Kδ inhibitor, synergizes with PIM protein kinase inhibition to cause tumor regression in a model of DLBCL. In: AACR 106th annual meeting 2015, Philadelphia, PA, USA, vol 15, Suppl, AACR. Cancer Res. https://doi.org/10.1158/1538-7445.AM2015-2671
Sullivan RJ, Hong DS, Tolcher AW, Patnaik A, Shapiro G, Chmielowski B, Ribas A, Brail LH, Roberts J, Lee L, O'Connell B, Kutok JL, Mahabhashyam S, Ullmann CD, Postow MA, Wolchok JD (2018) Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. In: ASCO, vol 15. Journal of Clinical Oncology, p 3013
Tabernero J, Geel RV, Guren TK, Yaeger RD, Spreafico A, Faris JE, Yoshino T, Yamada Y, Kim TW, Bendell JC, Schuler MH, Lenz H-J, Eskens F, Desai J, Hochster HS, Avsar E, Demuth T, Sandor V, Elez E, Schellens JHM (2016) Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). In: ASCO, vol 15. Journal of Clinical Oncology
Tawbi HA-H, Peng W, Phillips S, Milton DR, Amaria RN, Diab A, Glitza IC, Patel SP, Wong MKK, Yee C, McQuade JL, Tetzlaff MT, Lazar AJ, Shephard M, Cain S, Burton EM, Hwu P, Davies MA (2020) Safety results from phase I/II study of the PI3Kβ inhibitor GSK2636771 (G) in combination with pembrolizumab (P) in patients (pts) with PD-1 refractory metastatic melanoma (MM) and PTEN loss. In: ASCO, vol 15. Journal of Clinical Oncology
Thorpe LM, Yuzugullu H, Zhao JJ (2015) PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15(1):7–24. https://doi.org/10.1038/nrc3860
Tolcher A, Hong D, Sullivan R, Mier J, Shapiro G, Chmielowski B, Ribas A, Postow M, Pearlberg J, Brail L, Lee L, Ullmann CAD, Wolchok JD (2017) PI-549-01—A phase 1/1b, first-in-human study of IPI-549, a PI3K-γ inhibitor, as monotherapy and in combination with nivolumab in patients with advanced solid tumors. In: AACR annual meeting 2017, Washington, vol 13. Cancer Research. https://doi.org/10.1158/1538-7445.AM2017-CT089
Vuylsteke P, Huizing M, Petrakova K, Roylance R, Laing R, Chan S, Abell F, Gendreau S, Rooney I, Apt D, Zhou J, Singel S, Fehrenbacher L (2016) Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann Oncol 27(11):2059–2066. https://doi.org/10.1093/annonc/mdw320
Waldman AD, Fritz JM, Lenardo MJ (2020) A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 20(11):651–668. https://doi.org/10.1038/s41577-020-0306-5
Williams CB, Williams KA, Krie AK, De P, Dey N, Leyland-Jones B, Starks D, Rojas-Espaillat LA (2020) Results of a phase Ib trial evaluating the safety and clinical activity of sapanisertib (TAK 228) in combination with serabelisib (TAK 117) and paclitaxel in patients with advanced ovarian, endometrial, or breast cancer. In: ASCO, vol 15. Journal of Clinical Oncology
Yang Q, Modi P, Newcomb T, Quéva C, Gandhi V (2015) Idelalisib: first-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin Cancer Res 21(7):1537–1542. https://doi.org/10.1158/1078-0432.CCR-14-2034
Yue EW, Li YL, Douty B, He C, Mei S, Wayland B, Maduskuie T, Falahatpisheh N, Sparks RB, Polam P, Zhu W, Glenn J, Feng H, Zhang K, Li Y, He X, Katiyar K, Covington M, Feldman P, Shin N, Wang KH, Diamond S, Koblish HK, Hall L, Scherle P, Yeleswaram S, Xue CB, Metcalf B, Combs AP, Yao W (2019) INCB050465 (Parsaclisib), a novel next-generation inhibitor of Phosphoinositide 3-Kinase Delta (PI3Kδ). ACS Med Chem Lett 10(11):1554–1560. https://doi.org/10.1021/acsmedchemlett.9b00334
Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, Zhao YW, Wei YQ (2012) Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE 7(12):e50946. https://doi.org/10.1371/journal.pone.0050946
Zhou Q, Zhang X-C, Tu H-Y, Gan B, Wang B-C, Xu C-R, Chen H-J, Zheng M-Y, Wang Z, Bai X-Y, Sun Y-L, Myers A, Lv X, Chakrabcorti Y, Zhao S, Yang J-J, Wu Y-L (2018) Biomarker-integrated study of single agent targeting molecular alterations of PI3KCA, MET, ALK, ROS1, KRAS, NRAS or BRAF in advanced NSCLC: Phase 2 umbrella trial in China (CTONG1505). In: ESMO Asia Congress, vol 9. Annals of Oncology. https://doi.org/10.1093/annonc/mdy441.002
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Scott, J., Rees, L., Gallimore, A., Lauder, S.N. (2022). PI3K Isoform Immunotherapy for Solid Tumours. In: Dominguez-Villar, M. (eds) PI3K and AKT Isoforms in Immunity . Current Topics in Microbiology and Immunology, vol 436. Springer, Cham. https://doi.org/10.1007/978-3-031-06566-8_16
Download citation
DOI: https://doi.org/10.1007/978-3-031-06566-8_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-06565-1
Online ISBN: 978-3-031-06566-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)