
Abstract
Purpose
This open-label, phase Ib, dose-escalation, and dose-expansion study (NCT01862081) evaluated taselisib with a taxane in locally advanced or metastatic breast cancer (BC) and/or non-small cell lung cancer (NSCLC).
Methods
Patients received taselisib (2–6 mg tablet or 3–6 mg capsule) plus docetaxel or paclitaxel. Primary endpoints were safety, dose-limiting toxicities, maximum tolerated dose, and identification of a recommended phase II dose. Secondary endpoints included pharmacokinetics and antitumor activity assessment.
Results
Eighty patients (BC: 72; NSCLC: 7; BC/NSCLC: 1) were enrolled (docetaxel-receiving arms: 21; paclitaxel-receiving arms: 59). Grade ≥ 3 adverse events (AEs), serious AEs, and AEs leading to death were reported in 90.5%, 42.9%, and 14.3% of patients, respectively (docetaxel-receiving arms), and 78.9%, 40.4%, and 3.5% of patients, respectively (paclitaxel-receiving arms). Eight patients experienced dose-limiting toxicities. The maximum tolerated dose was exceeded with 3 mg taselisib (capsule) for 21 consecutive days plus 75 mg/m2 docetaxel and not exceeded with 6 mg taselisib (tablet) for 5 days on/2 days off plus 80 mg/m2 paclitaxel. Objective response rates and clinical benefit rates were 35.0% and 45.0%, respectively (docetaxel-receiving arms), and 20.4% and 27.8%, respectively (paclitaxel-receiving arms). Exposure for paclitaxel or docetaxel plus taselisib was consistent with the single agents.
Conclusions
Taselisib in combination with a taxane has a challenging safety profile. Despite evidence of antitumor activity, the benefit–risk profile was deemed not advantageous. Further development is not planned.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There is an unmet need to improve treatment options for patients with relapsed human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer (BC) and non-small cell lung cancer (NSCLC) [1, 2]. Current standard treatment options for HER2-negative locally recurrent/metastatic BC include the single-agent taxanes paclitaxel and docetaxel [1]. When our study was initiated, docetaxel was a standard option for NSCLC following progression with first-line chemotherapy [3, 4].
The phosphatidylinositol 3-kinase (PI3K) signaling pathway is commonly altered in cancer [5, 6] and may be activated by gain-of-function mutations and/or amplification of the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) gene [7,8,9]. PIK3CA encodes the α-isoform of the catalytic subunit of PI3K (PI3Kα) [8] and is mutated in ~40% of hormone receptor (HR)-positive BCs [10,11,12]. Increased PI3K signaling occurs frequently in lung cancer [13], including mutations in PIK3CA (2–3%) [14,15,16] and loss/low expression of the PI3K pathway suppressor, phosphatase, and tensin homolog (PTEN) (39–48%) [17].
Taselisib (GDC-0032), a potent and selective inhibitor of class I PI3Kα-, δ-, and γ-isoforms, has greater activity against tumor cells harboring PIK3CA mutations versus wild-type PIK3CA [18,19,20,21]. In PIK3CA-mutant BC models, taselisib plus docetaxel or paclitaxel enhanced activity versus taxanes alone [22]. A phase I dose-escalation study of single-agent taselisib suggested activity in PIK3CA-mutant BC and NSCLC [23] and a tolerable safety profile, with expected PI3K inhibitor class adverse events (AEs) [23,24,25,26]. Based on these results, the recommended single-agent dose of taselisib was 9 mg (capsule) [23]. The tablet formulation of taselisib has higher bioavailability than the capsule and 6 mg provides equivalent exposure to a 9 mg capsule [27].
Taselisib has clinical activity in patients with PIK3CA-mutant, estrogen receptor (ER)-positive, HER2-negative BC [28, 29]. In the neoadjuvant LORELEI study, addition of taselisib to letrozole significantly improved the overall response rate in ER-positive, HER2-negative early BC (vs. placebo plus letrozole; intention-to-treat and PIK3CA-mutant populations) [28]. In SANDPIPER, addition of taselisib to fulvestrant significantly increased progression-free survival (PFS) in patients with ER-positive, HER2-negative advanced BC, and PIK3CA mutations (vs. placebo plus fulvestrant) [29]. Tolerability was considered challenging, with frequent gastrointestinal toxicities and hyperglycemia, and a higher proportion of discontinuation due to AEs in the taselisib arm (vs. the placebo arm) [29].
This open-label, phase Ib study evaluated the safety and pharmacology of taselisib plus docetaxel or paclitaxel in locally advanced/metastatic BC or NSCLC, including PIK3CA-mutated cancers.
Methods
Patients
Eligible patients were ≥ 18 years and had histologically/cytologically documented breast adenocarcinoma with locally recurrent/metastatic disease (paclitaxel- or docetaxel-containing arms) or histologically documented advanced (stage IV)/recurrent NSCLC (docetaxel-containing arms). Patients with HR-positive BC had disease progression after ≥ 1 prior endocrine therapy in the adjuvant or metastatic setting. Patients with NSCLC had ≥ 1 prior anticancer regimen in an advanced setting and had docetaxel considered as appropriate per local guidelines. Patients had evaluable/measurable disease [Response Evaluation Criteria in Solid Tumours (RECIST) v.1.1], life expectancy ≥ 12 weeks, Eastern Cooperative Oncology Group performance status (ECOG PS) 0/1 at screening, and adequate hematologic and end-organ function (Supplementary methods).
This study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. Approval for the protocol and any material provided to the patient was obtained from independent ethics committees at participating institutions (Supplementary methods). All patients provided written informed consent.
Study design and treatment
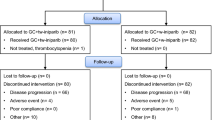
This was an open-label, phase Ib, dose-escalation study (NCT01862081). Each arm had two stages: dose-escalation (stage 1) and dose-expansion (stage 2). Patients received taselisib with docetaxel (Arms A, C, D, and E) or taselisib with paclitaxel (Arms B and F), with taselisib at different dosing schedules (Fig. 1 and Supplementary methods). Patients in Arms A, C, D, and E received intravenous docetaxel (75 mg/m2) on Day 1 of each 21-day cycle and premedication with oral corticosteroids (per institutional guidelines and docetaxel treatment guidelines). Patients in Arms B and F received intravenous paclitaxel (80 mg/m2) on Days 1, 8, 15, and 22 of each 28-day cycle and could receive premedication with dexamethasone, diphenhydramine, and either ranitidine or famotidine (per institutional practice).

Participant flow diagrams. Study schema for patients treated with a taselisib plus docetaxel and b taselisib plus paclitaxel. AE adverse event, DLT dose-limiting toxicities, G-CSF granulocyte-colony stimulating factor, HER2 human epidermal growth factor receptor 2, MBC metastatic breast cancer, MTD maximum tolerated dose, NSCLC non-small cell lung carcinoma, qd once-daily, qw once-weekly, q3w every 3 weeks. Arms B and F enrolled 20 and 21 patients, respectively, in the expansion cohorts; however, the taselisib dosing information was not verifiable for two patients, and therefore these patients were not included in the safety-evaluable population
Once the maximum tolerated dose (MTD), or maximum assessed dose in the absence of an MTD, of taselisib had been established in an arm from dose escalation, additional patients with each combination could be enrolled in stage 2 to confirm a potential recommended dose for future studies (Supplementary methods).
Outcomes
The primary endpoints were safety and tolerability, dose-limiting toxicities (DLTs), MTD, and identification of a recommended phase II dosing regimen. Secondary endpoints were pharmacokinetics (PK: taselisib and docetaxel; taselisib and paclitaxel; 6α-OH-paclitaxel), and preliminary assessment of the antitumor activity of taselisib plus docetaxel or paclitaxel. Exploratory endpoints are listed in the Supplementary methods.
Safety assessment
Safety was evaluated by monitoring all AEs, laboratory abnormalities, vital signs, and treatment exposure (Supplementary methods). AEs were defined and graded per National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.0. All patients with a verifiable record of taselisib dosing were included. Investigators assessed whether DLTs (Supplementary methods) that occurred within the DLT assessment window (Arms A, C, D, and E: Cycle 1, Days 1–21; Arms B and F: Cycle 1, Days 1–28) were possibly related to study treatment. The MTD was exceeded and a lower dose level evaluated if a DLT was observed in ≥ 33% of patients in a cohort (Supplementary methods). The Supplementary methods list AEs of special interest (AESIs).
Tumor assessments performed per RECIST v1.1 were recorded during the last week of the cycle and before the start of treatment in the next cycle (Supplementary methods). Objective responses were confirmed by repeat assessment ≥ 4 weeks after initial documentation. Clinical benefit rate (CBR) was defined as confirmed complete response, confirmed partial response, or stable disease lasting ≥ 6 months in all patients.
PK analysis
Blood samples were collected for PK characterization of taselisib, docetaxel, and paclitaxel. Individual plasma concentration versus time data and summary statistics were tabulated by treatment arm, stage, cycle, and dose level. The plasma concentration–time data were analyzed using non-compartmental methods employing Phoenix WinNonlin software (Certara USA, Inc., Princeton, NJ, USA) (Supplementary methods).
Biomarker assessments
PIK3CA mutation status was determined using local or central testing, with retrospective central confirmation using the Roche cobas® PIK3CA Mutation Test (Roche Diagnostics, Indianapolis, IN, USA) (Supplementary methods) [30]. Tumors were classified as PIK3CA-mutated if a positive result was obtained, PIK3CA-mutation not detected (MND) if no mutations were detected, or PIK3CA mutation status unknown if there was an assay failure/no tissue available for central confirmation.
Analysis of PIK3CA mutations in plasma circulating tumor DNA (ctDNA) was performed using the BEAMing Digital PCR platform (Sysmex Inostics GmbH, Hamburg, Germany) (Supplemental methods).
Tumors were classified as PIK3CA-mutated if a mutation was detected by central tissue or plasma ctDNA testing.
Statistical methods
Safety analysis was based on patients who received ≥ 1 taselisib dose (safety-evaluable population) and patient data were collected until study discontinuation. Efficacy analysis was based on the intention-to-treat population of all patients enrolled. Data were summarized (medians and standard deviations) and Kaplan–Meier methodology used for time-to-event analysis. Analyses were performed by PIK3CA status using tissue samples (docetaxel arms) or both ctDNA and tissue samples (paclitaxel arms); both sample types were analyzed in the paclitaxel arms to better understand the impact of PIK3CA mutation status. Data cutoff was the last patient last visit (June 9, 2017).
Results
Patient characteristics
Eighty patients were enrolled from 11 centers [USA (n = 7), Spain [2], Belgium [1], and Canada [1]] into either docetaxel-containing (n = 21) or paclitaxel-containing arms (n = 59) (Fig. 1). The taselisib safety-evaluable population comprised all patients who received docetaxel and 57/59 who received paclitaxel; taselisib dosing information was not verifiable for one patient each from the expansion cohorts of Arms B and F. At the time of data cutoff, all patients had discontinued taselisib and were no longer on -study (Fig. 1).
In the docetaxel-containing arms, 15 patients had BC and six had NSCLC (Table 1). In the paclitaxel-containing arms, 56 safety-evaluable patients had BC, and of these, 45 had HR-positive BC, while 11 had triple-negative BC. One patient with NSCLC was inadvertently enrolled into a paclitaxel-containing arm (expansion cohort F) and allowed to continue on study treatment.
In the docetaxel-containing arms, study discontinuation was most commonly due to progressive disease (90.5%) and death (9.5%); disease progression (95.2%) was the most common reason for taselisib discontinuation. In the paclitaxel-containing arms, study discontinuation was most commonly due to disease progression (75.4%), AEs (12.3%), and withdrawal of consent (7.0%); disease progression (68.4%) and AEs (22.8%) were the most common reasons for taselisib discontinuation.
Safety
In the docetaxel-containing arms, 2/4 patients who received the 3 mg taselisib capsule plus docetaxel (Arm A) had DLTs (one experienced Grade 4 neutropenia; one had both Grade 3 stomatitis and Grade 4 decreased neutrophil count). The MTD was exceeded in Arm A with 3 mg taselisib capsules (equivalent to 2 mg tablets) once-daily (qd) for 21 consecutive days and Arm A was closed to enrollment. In Arm C, there were no DLTs, and the taselisib tablet was escalated from 2 mg to 4 mg; however, Grade 4 neutropenia was reported in both the 2 mg (3 cases) and 4 mg (1 case) cohorts. In Arm D, while no DLTs were reported, there were two cases of Grade 4 neutropenia in the 2 mg tablet cohort. Arm D was therefore closed, and Arm E opened with mandatory granulocyte-colony stimulating factor (G-CSF) prophylaxis, and taselisib escalated from the 2 mg to the 4 mg tablet; there were no DLTs and no cases of Grade 4 neutropenia reported.
In the docetaxel-containing arms, all patients experienced ≥ 1 AE (Table 2). The most common treatment-emergent AEs (≥ 40%) were fatigue (52.4%), diarrhea (47.6%), and alopecia, nausea, and neutropenia (42.9%, each) (Table S1). All patients had ≥ 1 taselisib-related AE, most commonly diarrhea and fatigue (47.6% each). Grade ≥ 3 AEs were common (90.5%), with neutropenia and decreased neutrophil count the most frequent (33.3% each) (Table S2). Nine patients (42.9%) experienced ≥ 1 serious AE (SAE), with the majority (23.8%) being Grade 3. SAEs reported in ≥ 2 patients were BC progression and pneumonia (Table S3). Two patients (9.5%) experienced taselisib-related SAEs; Grade 2 gastritis in one, and Grade 4 respiratory failure and Grade 5 sepsis in another. Three patients (14.3%) died (Table 2), one due to sepsis (considered to be related to taselisib) and the other two due to disease progression. One patient (4.8%) had Grade 4 respiratory failure that led to withdrawal of taselisib, while 28.6% had AEs that led to withdrawal of docetaxel (Table 2). AESIs reported were diarrhea (47.6%), hyperglycemia (28.6%), and rash-AE group term (AEGT) (28.6%); all were Grade 1–2.
In the taselisib plus paclitaxel arms, four DLTs were reported: one patient in Arm B, Cohort 1 had Grade 3 maculopapular rash, one in Arm B, Cohort 2 had Grade 3 mucosal inflammation. In Arm B, the MTD was not reached and an expansion cohort was opened (4 mg tablet qd dose level). Two DLTs were reported in Arm F (Grade 3 rash, Grade 4 decreased neutrophil count). The MTD was exceeded at the 6 mg tablet 5 days on/2 days off schedule, and an Arm F expansion cohort was opened at the 4 mg tablet 5 days on/2 days off schedule.
In the taselisib plus paclitaxel arms, all patients experienced ≥ 1 AE (Table 2). The most common treatment-emergent AEs were diarrhea (61.4%), nausea (47.4%), fatigue (43.9%), and alopecia (42.1%) (Table S4). Fifty-five patients (96.5%) had ≥ 1 taselisib-related AE, with diarrhea (54.4%) and fatigue (38.6%) the most frequent. Grade ≥ 3 AEs were frequent (78.9%), with hyperglycemia and decreased neutrophil count (14.0% each) the most common (Table S5). Twenty-three patients (40.4%) had ≥ 1 SAE, which were mostly Grade 3 (29.8%). The most common SAEs were pneumonitis (7.0%) and BC progression (3.5%) (Table S6); two patients (3.5%) died in the paclitaxel-containing arms, both due to BC progression. Fourteen patients (24.6%) had AEs that led to withdrawal of taselisib, most commonly due to pneumonitis (7.0%), followed by diarrhea (3.5%). The rate of AEs leading to taselisib discontinuation was 36.8% in the Arm B and 15% in the Arm F expansion cohorts. Sixteen patients (28.1%) had AEs that led to withdrawal of paclitaxel, most commonly neurotoxicity, peripheral sensory neuropathy, and pneumonitis (5.3% each). The AESIs reported were diarrhea (61.4%), rash-AEGT (52.6%), hyperglycemia (28.1%), pneumonitis (14.0%), and colitis-AEGT (5.3%). In the Arm B expansion cohort, 6/19 (31.6%) patients had pneumonitis/interstitial lung disease, including 4/19 (21.1%) at Grade 3. In the Arm F expansion cohort, the pneumonitis rate was 5% (Grade 3 event).
Exposure
In the docetaxel-containing arms, median duration of taselisib exposure was 3.9 months (range 0.5–15.2) (Table S7a), median number of doses was 56 (range 8–460), and mean total cumulative dose for all patients was 208.3 mg (range 32–1004). Median duration of docetaxel exposure was 3.5 months (range 0.7–12.5). The mean total number of doses for all patients was 6.3 (range 2–19; median 6.0).
In the paclitaxel-containing arms, median duration of taselisib exposure was 3.2 months (range 0.2–39.1) (Table S7b). In all patients, the total mean number of doses was 128.7 (range 5–789, median 68) and the mean total cumulative dose was 415.1 mg (range 20–2552). The median duration of paclitaxel exposure was 3.0 months (range 0.0–27.4). The mean total number of doses for all patients was 13.9 (range 1–70; median 12).
PK analysis
Based upon the relatively sparse sampling schedule for all study drugs, taselisib displayed moderate absorption and demonstrated approximately proportional and linear increases in maximum observed plasma concentration (Cmax), area under the curve from time 0 to the last measurable concentration (AUC0–last), and area under the curve during 24 h (AUC0–24) after a single dose and at steady state (following 15 days of daily dosing), with increasing dose levels from 2 mg to 6 mg capsule or tablet equivalent. On average, steady-state qd exposures (AUC0–24) were consistent with predicted efficacious exposures at dose levels of 2–6 mg. Data from the expansion cohorts for Arms B and F (4 mg tablet dose) indicated that the Cycle 1, Day 15 pre-dose (Cmax) levels of taselisib were 33.1 ng/mL (68.8 ng/mL) and 15.4 ng/mL (31.5 ng/mL), respectively. Docetaxel [31] and paclitaxel [32] demonstrated PK characteristics similar to previously reported values at all taselisib dose levels.
Clinical activity
Clinical activity was assessed in the intention-to-treat population who received ≥ 1 dose of study drug (including the two patients excluded from the taselisib safety-evaluable population), with response rates reported in those with measurable disease at baseline.
Across all doses of taselisib plus docetaxel, 7/20 patients (35.0%) with baseline measurable disease had partial responses and none had a complete response (Table 3). Objective response rate was 35.0% (7/20) and CBR was 45.0% (9/20; Table 3). In the seven patients who responded to taselisib plus docetaxel, median duration of response (DoR) was 5.5 months [95% confidence interval (CI) 3.1–5.5] (Table 3). DoR was 2.6 months and 3.1 months in the two patients with PIK3CA MND, while in the four with detectable PIK3CA mutations, DoR ranged from 3.5 to 12.5 months. In the 21 patients who received any dose of taselisib plus docetaxel, median PFS was 4.1 months (95% CI 2.7–6.8) (Table 3). PFS ranged from 1.2 (censored) to 8.3 months in 11 patients with PIK3CA MND and from 6.2 to 15.1 months in five with PIK3CA mutations.
Across all doses of taselisib plus paclitaxel, 1/54 patients (1.9%) with baseline measurable disease had a complete response and 10/54 (18.5%) had partial responses (Table 3; Fig. S1a). Objective response rate was 20.4% (11/54 patients) and CBR was 27.8% (15/54) (Table 3). In the 11 patients who responded to taselisib and paclitaxel, median DoR was 7.3 months (95% CI 4.4–12.7) (Table 3). Among seven patients with PIK3CA MND, DoR ranged from 2.1 to 36.6 months, while in four with detectable PIK3CA mutations, DoR ranged from 1.9 (censored) to 13.3 months (Table 3). In the 59 patients who received any dose of taselisib plus paclitaxel, median PFS was 4.1 months (95% CI 3.0–7.1) (Table 3). PFS by PIK3CA mutation status is shown in Table 3 and Fig. S1b. Table S8 shows efficacy in the paclitaxel-containing arm by PIK3CA mutation status by tumor tissue central analysis.
Among all 18 responders treated with taselisib plus taxanes, eight had received prior taxane therapy.
Discussion
This was an open-label, multicenter, phase Ib dose-escalation study of oral taselisib in combination with either docetaxel (Arms A, C, D, and E) or paclitaxel (Arms B and F) in patients with HER2-negative, locally recurrent/metastatic BC or NSCLC. The observed safety profiles of taselisib, paclitaxel, and docetaxel were generally consistent with the known safety profiles of PI3K-AKT-mTOR inhibitors or taxanes; however, pneumonitis was higher than expected (14%) in the taselisib plus paclitaxel arms.
DLTs were reported in the 3 mg taselisib capsule (qd for 21 consecutive days) plus docetaxel arm. DLTs were not observed in the other investigated dosing schedules and the MTD was not reached for other schedules. However, Grade 4 neutropenia was commonly observed with this combination, requiring G-CSF support throughout dose escalation and preventing the opening of the expansion cohorts.
The MTD was not reached for 3 mg and 6 mg taselisib capsules (qd for 28 consecutive days) plus paclitaxel. The 4 mg taselisib tablet (qd for 28 consecutive days in a 28-day cycle) plus paclitaxel was not recommended for further investigation based on discontinuations due to AEs and the higher number of patients with pneumonitis in Arm B. The 31.6% rate of pneumonitis/interstitial lung disease (21% at Grade 3) observed in the Arm B expansion cohort with qd taselisib was numerically higher than observed in other studies with single-agent paclitaxel or taselisib [23, 33].
The 4 mg taselisib tablet (5 days on/2 days off) plus paclitaxel (Arm F) was better tolerated than the qd schedule (Arm B), with a lower rate of discontinuation due to AEs and pneumonitis. The observed decrease in trough concentration (and Cmax) on Cycle 1, Day 15 for the 5 days on/2 days off dosing suggested that lowered exposure over the 2-day dosing holiday provides a sufficient break to improve drug safety. Plasma concentrations of taselisib, combined with docetaxel or paclitaxel, were generally consistent with studies where single-agent taselisib was administered. Paclitaxel and docetaxel exposures when given with taselisib were consistent with single-agent therapies.
Addition of PI3K inhibitors to taxanes is challenging. In BELLE-4, there was no improvement in PFS with the pan-PI3K inhibitor buparlisib plus paclitaxel versus placebo plus paclitaxel in patients with HER2-negative advanced BC (overall or PI3K-activated population), with a higher rate of discontinuations due to AEs in the buparlisib arm [34]. In PEGGY, pictilisib plus paclitaxel (vs. placebo plus paclitaxel) did not improve PFS in the intention-to-treat or PIK3CA-mutated subgroup, in patients with HR-positive, HER2-negative locally recurrent/metastatic BC, and there were a significant number of dose modifications due to AEs in the pictilisib arm [35].
In our study, and despite its relative selectivity for mutant PI3Kα, combining taselisib with paclitaxel or docetaxel was challenging. Alpelisib (BYL719), another PI3Kα inhibitor, plus nab–paclitaxel, showed encouraging efficacy and manageable toxicity in HER2-negative metastatic BC, particularly in PIK3CA-mutant tumors [36]. Highly selective PI3Kα inhibitors, such as GDC-0077, which showed tumor regression in PIK3CA-mutant breast tumor xenografts, may circumvent the narrow therapeutic index of PI3K inhibitors seen in recent clinical trials [37].
Inhibition of downstream effectors of PI3K may improve safety and/or efficacy. Ipatasertib, an AKT inhibitor, plus docetaxel or paclitaxel, was well tolerated, with improved PFS and overall survival [38, 39]. PFS was longer in patients with triple-negative BC who received ipatasertib plus paclitaxel, versus placebo plus paclitaxel [40]. In addition, paclitaxel plus AZD5363, a highly selective small-molecule AKT inhibitor, significantly improved PFS and overall survival, versus paclitaxel plus placebo, in first-line triple-negative BC [41]; the most common Grade ≥ 3 AEs were diarrhea, infection, neutropenia, rash, and fatigue [41].
In this study, taselisib plus taxanes showed preliminary evidence of antitumor activity, including partial responses in advanced BC. Our study was not randomized, but the median PFS for taselisib plus paclitaxel was less than for the paclitaxel control arms from two prior randomized studies [34, 35]. Since this was a non-randomized study in a heterogenous patient population, efficacy results should be interpreted with caution. The small population studied and multiple potential confounding factors, including tumor type (triple-negative BC; HR-positive, HER2-negative BC; NSCLC), line of therapy, dose, and schedule, mean that no firm conclusions can be reached on whether taselisib adds to the antitumor activity of taxanes or whether tumor PIK3CA mutation status plays a role.
In this phase Ib study, taselisib plus taxanes had a challenging safety profile that may be partly mitigated by intermittent dosing. While there was some evidence of antitumor activity in locally recurrent or metastatic HER2-negative BC, the overall benefit–risk profile was not clearly advantageous, and no further development of taselisib plus taxanes is currently planned.
Data availability
Qualified researchers may request access to individual patient-level data through the clinical study data request platform: www.clinicalstudydatarequest.com. Further details on Roche’s criteria for eligible studies are available here: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here: https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
Abbreviations
- AE:
-
Adverse event
- AEGT:
-
Adverse event group term
- AESI:
-
Adverse event of special interest
- AUC0–last :
-
Area under the curve from time 0 to the last measurable concentration
- AUC0–24 :
-
Area under the curve during 24 h
- BC:
-
Breast cancer
- CBR:
-
Clinical benefit rate
- CI:
-
Confidence interval
- C max :
-
Maximum observed plasma concentration
- C min :
-
Minimum observed plasma concentration
- CR:
-
Complete response
- ctDNA:
-
Circulating tumor DNA
- DLT:
-
Dose-limiting toxicity
- DoR:
-
Duration of response
- ECOG PS:
-
Eastern Cooperative Oncology Group performance status
- ER:
-
Estrogen receptor
- G-CSF:
-
Granulocyte-colony stimulating factor
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hormone receptor
- MBC:
-
Metastatic breast cancer
- MedDRA:
-
Medical Dictionary for Regulatory Activities
- MND:
-
Mutation not detected
- MTD:
-
Maximum tolerated dose
- NE:
-
Not evaluable
- NCI-CTCAE:
-
National Cancer Institute-Common Terminology Criteria for Adverse Events
- NSCLC:
-
Non-small cell lung cancer
- PCR:
-
Polymerase chain reaction
- PD:
-
Progressive disease
- PFS:
-
Progression-free survival
- PgR:
-
Progesterone receptor
- PIK3CA :
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- PI3K:
-
Phosphatidylinositol 3-kinase
- PK:
-
Pharmacokinetics
- PR:
-
Partial response
- PTEN:
-
Phosphatase and tensin homolog
- qd:
-
Once-daily
- qw:
-
Once-weekly
- q3w:
-
Every 3 weeks
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
- SAE:
-
Serious adverse event
- SD:
-
Stable disease
- SLD:
-
Sum of the longest diameter
- t max :
-
Time to maximum observed plasma concentration
References
National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): Breast Cancer. Version 1. 2018. https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. Accessed 12 Dec 2018
National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): Non-Small Cell Lung Cancer. Version 5. 2018. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed 12 Dec 2018
Fossella FV, DeVore R, Kerr RN et al (2000) Randomized phase III trial of docetaxel versus vinorelbine or ifosfamide in patients with advanced non-small-cell lung cancer previously treated with platinum-containing chemotherapy regimens. The TAX 320 Non-Small Cell Lung Cancer Study Group. J Clin Oncol 18:2354–2362. https://doi.org/10.1200/JCO.2000.18.12.2354
Shepherd FA, Dancey J, Ramlau R et al (2000) Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy. J Clin Oncol 18:2095–2103. https://doi.org/10.1200/JCO.2000.18.10.2095
Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27:5497–5510. https://doi.org/10.1038/onc.2008.245
Engelman JA (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 9:550–562. https://doi.org/10.1038/nrc2664
Fruman DA, Rommel C (2014) PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13:140–156. https://doi.org/10.1038/nrd4204
Samuels Y, Wang Z, Bardelli A et al (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554. https://doi.org/10.1126/science.1096502
Zhang Y, Ng PK, Kucherlapati M et al (2017) A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell 31:820–832.e3. https://doi.org/10.1016/j.ccell.2017.04.013
Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490:61–70. https://doi.org/10.1038/nature11412
Ciriello G, Gatza ML, Beck AH et al (2015) Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163:506–519. https://doi.org/10.1016/j.cell.2015.09.033
Sabine VS, Crozier C, Brookes CL et al (2014) Mutational analysis of PI3K/AKT signaling pathway in tamoxifen exemestane adjuvant multinational pathology study. J Clin Oncol 32:2951–2958. https://doi.org/10.1200/JCO.2013.53.8272
Sarris EG, Saif MW, Syrigos KN (2012) The biological role of PI3K pathway in lung cancer. Pharmaceuticals (Basel) 5:1236–1264. https://doi.org/10.3390/ph5111236
Yamamoto H, Shigematsu H, Nomura M et al (2008) PIK3CA mutations and copy number gains in human lung cancers. Cancer Res 68:6913–6921. https://doi.org/10.1158/0008-5472.CAN-07-5084
Wang L, Hu H, Pan Y et al (2014) PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS ONE 9:e88291. https://doi.org/10.1371/journal.pone.0088291
Barlesi F, Mazieres J, Merlio JP et al (2016) Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 387:1415–1426. https://doi.org/10.1016/S0140-6736(16)00004-0
Panagiotou I, Tsiambas E, Lazaris AC et al (2012) PTEN expression in non small cell lung carcinoma based on digitized image analysis. J BUON 17:719–723
Olivero AG, Heffron TP, Baumgardner M et al (2013) Discovery of GDC-0032: a beta-sparing PI3K inhibitor active against PIK3CA mutant tumors. Cancer Res. https://doi.org/10.1158/1538-7445.AM2013-DDT02-01
Wallin JJ, Edgar KA, Guan J et al (2013) The PI3K inhibitor GDC-0032 is selectively potent against PIK3CA mutant breast cancer cell lines and tumors. Cancer Res. https://doi.org/10.1158/0008-5472.SABCS13-P2-17-01
Ndubaku CO, Heffron TP, Staben ST et al (2013) Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a β-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J Med Chem 56:4597–4610. https://doi.org/10.1021/jm4003632
Edgar KA, Song K, Schmidt S et al (2016) The PI3K inhibitor, taselisib (GDC-0032), has enhanced potency in PIK3CA mutant models through a unique mechanism of action. Cancer Res. https://doi.org/10.1158/1538-7445.AM2016-370
Sampath D, Nannini MA, Jane G et al (2013) The PI3K inhibitor GDC-0032 enhances the efficacy of standard of care therapeutics in PI3K alpha mutant breast cancer models. Cancer Res. https://doi.org/10.1158/0008-5472.SABCS13-P4-15-02
Juric D, Krop I, Ramanathan RK et al (2017) Phase I dose-escalation study of taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors. Cancer Discov 7:704–715. https://doi.org/10.1158/2159-8290.CD-16-1080
Bendell JC, Rodon J, Burris HA et al (2012) Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 30:282–290. https://doi.org/10.1200/JCO.2011.36.1360
Gopal AK, Kahl BS, de Vos S et al (2014) PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med 370:1008–1018. https://doi.org/10.1056/NEJMoa1314583
Sarker D, Ang JE, Baird R et al (2015) First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res 21:77–86. https://doi.org/10.1158/1078-0432.CCR-14-0947
Faber KP, Borin MT, Cheeti S et al (2016) Impact of formulation and food on taselisib (GDC-0032) bioavailability: powder-in-capsule formulation represents unique drug development challenge. Clin Pharmacol Ther. https://doi.org/10.1002/cpt.310
Saura C, de Azambuja E, Hlauschek D et al (2017) Primary results of LORELEI: a phase II randomized, double-blind study of neoadjuvant letrozole (LET) plus taselisib versus LET plus placebo (PLA) in postmenopausal patients (pts) with ER +/HER2-negative early breast cancer (EBC). Ann Oncol. https://doi.org/10.1093/annonc/mdx440.001
Baselga J, Dent SF, Cortés J et al (2018) Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): primary analysis from SANDPIPER. J Clin Oncol. https://doi.org/10.1200/JCO.2018.36.18_suppl.LBA1006
Dickler MN, Saura C, Richards D et al (2018) Phase II study of taselisib (GDC-0032) in combination with fulvestrant in patients with HER2-negative, hormone receptor-positive advanced breast cancer. Clin Cancer Res 24:4380–4387. https://doi.org/10.1158/1078-0432.CCR-18-0613
Kenmotsu H, Tanigawara Y (2015) Pharmacokinetics, dynamics and toxicity of docetaxel: why the Japanese dose differs from the Western dose. Cancer Sci 106:497–504. https://doi.org/10.1111/cas.12647
Huizing M, Vermorken J, Rosing H et al (1995) Pharmacokinetics of paclitaxel and three major metabolites in patients with advanced breast carcinoma refractory to anthracycline therapy treated with a 3-hour paclitaxel infusion: a European Cancer Centre (ECC) trial. Ann Oncol 6:699–704. https://doi.org/10.1093/oxfordjournals.annonc.a059287
Paridaens R, Biganzoli L, Bruning P et al (2000) Paclitaxel versus doxorubicin as first-line single-agent chemotherapy for metastatic breast cancer: a European Organization for Research and Treatment of Cancer Randomized Study with cross-over. J Clin Oncol 18:724–733. https://doi.org/10.1200/JCO.2000.18.4.724
Martín M, Chan A, Dirix L et al (2016) A randomized adaptive phase II/III study of buparlisib, a pan-class I PI3K inhibitor, combined with paclitaxel for the treatment of HER2–advanced breast cancer (BELLE-4). Ann Oncol 28:313–320. https://doi.org/10.1093/annonc/mdw562
Vuylsteke P, Huizing M, Petrakova K et al (2016) Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann Oncol 27:2059–2066. https://doi.org/10.1093/annonc/mdw320
Sharma P, Abramson VG, O’Dea A et al (2018) Clinical and biomarker results from phase I/II study of PI3K inhibitor BYL 719 (alpelisib) plus nab-paclitaxel in HER2-negative metastatic breast cancer. J Clin Oncol. https://doi.org/10.1200/JCO.2018.36.15_suppl.1018
Staben ST (2017) Discovery of GDC-0077, a highly isoform selective inhibitor of PI3Kα that promotes selective loss of mutant-p110α. Cancer Res. https://doi.org/10.1158/1538-7445.AM2017-DDT02-01
Isakoff SJ, Bendell JC, Cervantes A et al (2015) Phase Ib dose-escalation study of an Akt inhibitor ipatasertib (Ipat) in combination with docetaxel (Doc) or paclitaxel (Pac) in patients (pts) with metastatic breast cancer (MBC). Cancer Res. https://doi.org/10.1158/1538-7445.sabcs14-p6-12-02
Dent R, Im S, Espie M et al (2018) Overall survival (OS) update of the double-blind placebo (PBO)-controlled randomized phase 2 LOTUS trial of first-line ipatasertib (IPAT) + paclitaxel (PAC) for locally advanced/metastatic triple-negative breast cancer (mTNBC). J Clin Oncol. https://doi.org/10.1200/JCO.2018.36.15_suppl.1008
Kim S, Dent R, Im S et al (2017) Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 18:1360–1372. https://doi.org/10.1016/S1470-2045(17)30450-3
Schmid P, Abraham J, Chan S et al (2018) AZD5363 plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (PAKT): a randomised, double-blind, placebo-controlled, phase II trial. J Clin Oncol. https://doi.org/10.1200/JCO.2018.36.15_suppl.1007
Acknowledgements
We would like to thank the patients, their families, the nurses, and the investigators who participated in this study. The authors would like to thank Thomas J. Stout, PhD, Jerry Y. Hsu, MD, PhD, Jing He, MD, Alison Cardenas, RN, Deanna Wilson, MS, and Jiaheng Qiu, PhD (Genentech, Inc., South San Francisco, CA, USA), for input during the study. This study was funded by Genentech, Inc./F. Hoffmann-La Roche Ltd. F. Hoffmann-La Roche Ltd funded the study, provided study drugs, and was involved in the study design, protocol development, regulatory and ethics approvals, safety monitoring and reporting, data management, and data analysis and interpretation. Support for third-party writing assistance, furnished by Islay Steele, PhD, of Health Interactions, was provided by F. Hoffmann-La Roche Ltd.
Funding
This study was funded by Genentech, Inc./F. Hoffmann-La Roche Ltd. F. Hoffmann-La Roche Ltd funded the study, provided study drugs, and was involved in the study design, protocol development, regulatory and ethics approvals, safety monitoring and reporting, data management, and data analysis and interpretation.
Author information
Authors and Affiliations
Contributions
VGA and IEK were involved in the conception and design of the study. VGA, MCW, and ER were involved in development of the methodology used. VGA, MO, AC, HW, MRP, TMB, PLB, CB, SR, HMM, CS, and IEK were involved in the acquisition of data (acquired and managed patients, provided facilities, etc.). HW, MCW, ER, JB, NC, TRW, and HMM were involved in analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis). MCW, ER, JB, NC, TRW, and HMM were involved in administrative, technical, or material support (i.e., reporting or organizing data, constructing databases). MCW was involved in study supervision. All authors were involved in the writing, review, and/or revision of this manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
VGA has received research funding from Genentech, Astellas, and Lilly, and has consulted for Eisai and Novartis. MO has received remuneration from Roche, consulting or advisory fees from Roche, GSK, PUMA Biotechnology, and funding from AstraZeneca, Philips Healthcare, Genentech, Roche, Novartis, Immunomedics, Seattle Genetics, GSK, Boehringer-Ingelheim, PUMA Biotechnology (all to the Institution). AC has received consulting or advisory fees from Merck Serono, Roche, Beigene, Bayer, Servier, Lilly, Novartis, Takeda and Astelas, and funding from Genentech, Merck Serono, Roche, Beigene, Bayer, Servier, Lilly, Novartis, Takeda, Astelas, Fibrogen, Amcure, Sierra Oncology, Astra Zeneca, Medimmune, BMS, and MSD. HW has received travel support from Roche, TRM Oncology, Puma Biotechnology, and Pfizer (outside of the submitted work), and his institution has received consulting fees and honoraria from Roche, AstraZeneca, Amgen, Lilly, Novartis, AbbVie, Vifor Pharma, Pfizer, Celldex Therapeutics, Janssen-Cilag, TRM Oncology, Puma Biotechnology, Orion Corporation, and an unrestricted research grant from Roche (outside of the submitted work). MRP has received consulting or advisory fees from Exelixis, Pfizer, EMD Serono, Pharmacyclics, Genentech, and Celgene. TMB has received remuneration for employment Tennessee Oncology, remuneration for speakers’ bureau from Bayer (personal), remuneration for travel from Astellas Pharma, AstraZeneca, Celgene, Clovis Oncology, EMD Serono, Genentech, Lilly, Merck, Novartis, Ignyta, Pharmacyclics, Loxo, Bayer, Guardant Health, Moderna Therapeutics and Sysmex, consulting or advisory fees from Guardant Health (personal), Loxo (personal), Pfizer (personal), Leap Therapeutics (institutional), Ignyta (Institutional), Moderna Therapeutics (Institutional), Bayer (personal), Guardant Health and Pfizer (personal and Institutional), and Institutional funding from: Daiichi Sankyo, Medpacto, Inc., Incyte, Mirati Therapeutics, MedImmune, Abbvie, AstraZeneca, Leap Therapeutics, MabVax, Stemline Therapeutics, Merck, Lilly, GlaxoSmithKline, Novartis, Pfizer, Genentech/Roche, Deciphera, Merrimack, Immunogen, Millennium, Ignyta, Calithera Biosciences, Kolltan Pharmaceuticals, Principa Biopharma, Peleton, Immunocore, Aileron Therapeutics, Bristol-Myers Squibb, Amgen, Moderna Therapeutics, Sanofi, Boehringer Ingelheim, Astellas Pharma, Five Prime Therapeutics, Jacobio, Top Alliance BioScience, Loxo, Janssen, Clovis Oncology, Takeda, Karyopharm Therapeutics, Onyx, Phosplatin Therapeutics, Foundation Medicine and ARMO BioSciences and Bayer. PLB reports institutional grants from Bristol-Myers Squibb, Sanofi, AstraZeneca, Genentech/Roche, Servier, Merck, Nektar, Mersana, Novartis, GlaxoSmithKline, SignalChem, PTC Therapeutics (all during the conduct of the study), and is the current Chair of the Investigational New Drug Committee, Canadian Clinical Trials Group, an Executive Board Member for the Breast International Group, a Steering Committee Member for the American Association for Cancer Research Project GENIE, and a member of the NCI-BIO Breast Cancer Immunotherapy Task Force. CB has received honoraria from Taiho Pharmaceutical, consulting or advisory fees from SOBI, Ipsen, Takeda, Bayer, HERON, and Agenus, and fees from speakers’ bureau from Taiho Pharmaceutical, Bristol-Myers Squibb, and Celgene. SR has received remuneration from AstraZeneca, Genentech Inc., Genzyme Corporation, consulting or advisory fees from AstraZeneca, and funding from Acerta Pharma LLC, Eisai Inc., Eli Lilly and Company, EMD Serono, Inc., E.R. Squibb & Sons, L.L.C., Genentech, Inc., Ipsen Biopharmaceuticals, Inc., Janssen Research & Development, LLC., Janssen Research & Development, LLC., Novartis Pharmaceuticals Corporation, Tesaro, Inc. MCW, ER, JB, NC, TRW, and HMM are employees of Genentech and hold stock in Roche. CS has received travel grants from Puma Biotechnology, Pfizer, Roche, Astra Zeneca, Celgene, Daiichi Sankyo, Eisai, Genomyc Health, Novartis, Pierre Fabre, and Synthon Biopharmaceuticals, consulting or advisory fees from Puma Biotechnology, Pfizer, Roche, Astra Zeneca, Celgene, Daiichi Sankyo, Eisai, Genomyc Health, Novartis, Pierre Fabre, and Synthon Biopharmaceuticals, and funding from Roche-Genentech, Macrogenics, Pfizer, Piqur Therapeutics, Puma Biotechnology, Synthon Biopharmaceuticals, and Novartis. IEK has received grants and personal fees from Genentech/Roche (during the conduct of the study), grants from Pfizer, and personal fees from Macrogenics, Amgen, Taiho, Context Therapeutics, Seattle Genetics, and Daiichi Sankyo (outside of the submitted work). All authors received support for third-party writing assistance for this manuscript from Genentech, Inc./F. Hoffmann-La Roche Ltd.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (Supplementary methods) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Abramson, V.G., Oliveira, M., Cervantes, A. et al. A phase Ib, open-label, dose-escalation study of the safety and pharmacology of taselisib (GDC-0032) in combination with either docetaxel or paclitaxel in patients with HER2-negative, locally advanced, or metastatic breast cancer. Breast Cancer Res Treat 178, 121–133 (2019). https://doi.org/10.1007/s10549-019-05360-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-019-05360-3