
Abstract
Enteropathogenic Escherichia coli (EPEC) is considered one of the most important enteric pathogens infecting children and one of the main causes of diarrhea worldwide. EPEC uses a type 3 secretion system (T3SS) to inject effector proteins into host intestinal epithelial cells, causing diarrhea. Through a coordinated action of virulence factors, EPEC translocates effectors into host cells, resulting in the perturbation of cellular structures and functions by altering cell signaling pathways. Epithelial cells are held together by apical junctional complexes, including tight junctions (TJs), adherens junctions (AJs), and desmosomes. TJs contribute to the establishment of barrier function and maintenance of apico-basal cell polarity. TJ integrity relies on several cell structures and functions including the actin cytoskeleton, microtubule networks, membrane integrity, inflammation, and cell survival. EPEC perturbs TJ structure and function, leading to impairment of the intestinal barrier. This chapter summarizes the various mechanisms employed by EPEC that contribute to TJ disruption.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Enteropathogenic E. coli
- Tight junctions
- Microtubules
- Intestinal permeability
- Apico-basal polarity
- Transepithelial electrical resistance
Introduction
Historically, enteropathogenic Escherichia coli (EPEC) has been considered an important cause of infantile diarrhea in developing countries. EPEC, however, are divided into typical and atypical strains. Typical EPEC expresses bundle-forming pili (BFP) encoded on the E. coli adherence factor plasmid (EAF). These structures allow for microcolony formation and enhanced efficiency of T3SS effector delivery into host cells via BFP retraction [1]. Atypical EPEC now accounts for ~95% of isolated clinical EPEC strains [2]. Atypical EPEC causes diarrhea not only in children but also in adults. Interestingly, up to 22% of healthy asymptomatic individuals test positive for EPEC by multiplex PCR assays [2,3,4]. Most of the studies regarding the effect of EPEC on tight junctions have used typical EPEC strains, most notably E2348/69. Therefore, the information presented here concerns typical EPEC.
EPEC adheres to the apical surface of host intestinal epithelial cells and produce attaching and effacing (A/E) lesions, characterized by intimate attachment and the loss of microvilli [5, 6]. The factors responsible for A/E lesion formation are encoded by a 35-kilobase pathogenicity island called the locus of enterocyte effacement (LEE) [7, 8]. The LEE encodes both the T3SS and bacterial effector proteins it delivers into host cells. The molecular structure of the T3SS shows that EspB and EspD form a pore in the host membrane and EspA forms a hollow filamentous structure that assembles as a physical bridge between bacteria and the host cell surface, allowing the translocation of EPEC effectors into host cells [9, 10]. LEE EPEC-secreted effector proteins, including translocated intimin receptor (Tir), EspF, EspG, EspH, mitochondrial-associated protein (Map), among others, interfere with a variety of host cells functions. EPEC also uses effectors encoded outside of LEE, known as non-LEE-encoded effectors such as NleA, NleB, NleC, NleH, EspG2, EspM, and EspT, among others. The synergistic effects of effectors, either encoded inside or outside of LEE, contribute to EPEC pathogenesis [11,12,13,14,15].
Molecular Structure and Function of Epithelial Tight Junctions
Tight junctions (TJs) are part of the cell-cell adhesion complex localized to the most apical portion of the lateral membrane of epithelial cells. Ultrathin-section electron microscopy reveals that TJs are discrete membrane fusions, involving the outer leaflet of the plasma membrane of adjacent cells sealing the intercellular space in epithelial and endothelial cellular sheets [16]. Freeze-fracture electron microscopy shows that TJs are a set of continuous intramembranous particle strands composed of integral membrane proteins [17, 18]. TJ strands contribute to the establishment of a permeable-selective barrier for charged ions and uncharged solutes through the paracellular space [19, 20]. TJs are also involved in creating and establishing apical and basolateral membrane domains, inhibiting the free movement of proteins and lipids through the different membrane surfaces [21].
At the molecular level, TJs consist of transmembrane cell-cell adhesion molecules located between cells in epithelial sheets (occludin, claudins, tricelulin, JAMs, CAR, etc.), and scaffold submembrane proteins (ZO-1, −2, −3, cingulin, paracingulin, MAGI-1-3, MUPPI-1, etc.), associated with integral TJ membrane proteins through multiple interactions. In this chapter, we briefly describe the adhesion proteins involved in regulating barrier function that are targeted by EPEC.
Occludin was the first transmembrane TJ protein identified [22,23,24]. Although occludin appears to not be involved in TJ strand formation or in intestinal barrier function, occludin-deficient mice show histological abnormalities in several tissues [24]. The claudin family of proteins is composed of 27 four-transmembrane domain proteins and constitutes the backbone of TJ strands [25,26,27]. Intracellular interactions between claudins create the paracellular barrier and/or channels with specific characteristics that define TJ function [28,29,30,31,32,33]. Tricellulin is a transmembrane protein preferentially localized at the vertically oriented TJs of tricellular contacts. Tricellulin has been reported to be critical in the formation of the epithelial barrier and the organization of bi- and tricellular TJ contacts [34]. Junctional adhesion molecules (JAMs) are single-span TJ membrane proteins. JAM proteins contain immunoglobulin (Ig)-like ectodomains and are found in epithelial cells, endothelial cells, leukocytes, and myocytes. JAM-A has been reported to coordinate TJ development and epithelial polarity [35, 36].
The TJ scaffold proteins ZO-1, ZO-2, and ZO-3 are multidomain proteins that belong to the membrane-associated guanylate kinase homologs (MAGUKs) consisting of PDZ, SH3, and enzymatically inactive guanylate kinase (GUK) domains [37,38,39]. ZO-1 and ZO-2 are required for TJ strand assembly and epithelial polarity [36, 40]. Cingulin is a coil-coiled domain peripheral membrane protein that binds directly to ZO-1 [41, 42]. Cingulin is involved in the recruitment of GTPase regulatory proteins to TJs; its phosphorylation promotes the junctional association of microtubules [43,44,45]. Cingulin has also been reported to regulate gene expression and cell proliferation [46, 47]. Afadin is localized at both TJ and AJs and has a critical role in the early polarization of the apical junctional complex [48, 49]. MAGI (membrane-associated guanylate kinase inverted-1 and 3) and MPDZ (multiple PDZ domain protein, MUPPI-1) are PDZ domain scaffolding proteins that interact with a large number of TJ proteins [50,51,52]. MAGI-3 has been implicated in the regulation and phosphorylation of the JNK signaling pathway [53]. Although there is vast evidence regarding its numerous protein interactions, the role of these multiple PDZ molecules in TJ physiology is yet not clear.
Impact of EPEC Effectors on TJs
TJs are dynamic structures under normal physiological conditions and are altered in a variety of disease states. Infection with microorganisms, such as bacteria, viruses, parasites, and bacterial toxins, can disrupt TJ barrier. Increased intestinal TJ permeability may contribute to diarrhea and to inflammatory responses. EPEC perturbs intestinal epithelial cell (IEC) function as demonstrated by increased permeability to ions and solutes and loss of transepithelial electrical resistance (TEER), a conventional barrier assessment used to detect changes in paracellular barrier properties. These changes are associated with the redistribution of TJ proteins. EPEC effectors cause such changes by activating a variety signaling pathways that induce cytoskeletal rearrangements that impact the localization and functionality of membrane-associated proteins (e.g., adhesion components, cotransporters, channels, microtubules, polarity complexes, etc.). In this chapter, we focus only on the EPEC effectors that play an important role in the disruption of TJs.
In vivo and in vitro studies show that EPEC changes the localization of TJ proteins and perturbs intestinal barrier function. EPEC infection diminishes ileal and colonic mucosal barrier function in murine models and augments paracellular permeability demonstrated by the ability of the molecular tracer biotin to traverse the intestinal epithelium into the lamina propria. These changes correlate with the redistribution of occludin, claudin-1, and ZO-1 from cell-cell contacts into cytoplasm [54,55,56]. Citrobacter rodentium , an A/E-inducing pathogen of mice, has similar pathogenic mechanisms as EPEC and shows the redistribution of claudin-1, claudin-3, and claudin-5 requires EspF [57]. In vitro, EPEC effectors exert synergistic effects to increase intestinal permeability, redistribute TJ proteins, and alter transport functions [58,59,60,61].
Tir is delivered into the plasma membrane of host cells by the T3SS and serves as a receptor for the EPEC outer membrane adhesion protein, intimin, a bacterial surface adhesion encoded by the eae gene housed in the LEE pathogenicity island [11]. Initial Tir/intimin binding promotes intimate EPEC adhesion to host cells and A/E lesion formation. A/E lesions are characterized by effacement of microvilli in the area of attached EPEC and a dense concentration of actin microfilaments beneath intimately attached bacteria [6]. EPEC infection stimulates the production of inositol phosphates such as PI(4,5)P2 and PI (3,4,5)P3, which accumulate beneath EPEC microcolonies and are required for EPEC adherence to the host cell surface and actin pedestal development in a Tir-dependent manner [62,63,64,65,66,67,68,69].
Tir is phosphorylated at its COOH-terminus by host tyrosine kinases [70, 71]. Tir phosphorylation (Y474) facilitates binding to the SH2 domain of Nck, a host adaptor molecule, and the activation of Neural Wiskott-Aldrich syndrome protein (N-WASP) and Arp2/3 actin-nucleation complex, promoting reorganization of the host cytoskeleton leading to the formation EPEC pedestals [72,73,74,75,76,77]. The NH-2 terminus of Tir binds to cytoskeletal components found within pedestals, such as α-actinin, talin, vinculin, and ezrin [73, 78,79,80,81,82]. Additionally, Tir interacts with and recruits components of intermediate filaments (IF) , CK8 and CK18, to pedestals [83]. EPEC effectors also redistribute TJ proteins to these structures. N-WASP activation triggered by Tir/intimin binding contributes to the recruitment of ZO-1 to EPEC pedestals via its proline-rich region (PRR) [84]. These findings suggest that Tir promotes the local accumulation of inositol phosphates beneath EPEC pedestals and, through its protein-protein interactions, stabilizes pedestals by anchoring EPEC to the cytoskeleton of host cells. The recruitment of TJ proteins mediated by Tir to pedestals may be an important step to indirectly destabilize the structure and function of TJs.
EPEC effectors cooperate in a coordinated manner to cause TJ disruption. For example, Tir and intimin interactions play a crucial role in mediating EPEC intestinal epithelial TJ disruption [85, 86]. Infection of intestinal cell monolayers with a tir deletion strain does not alter TEER, suggesting that the Tir/intimin interaction is needed for attachment and delivery of EPEC effectors into host cells that ultimately impact permeability. In addition, EPEC alters the distribution of β1-integrin, which is typically restricted to the basolateral membrane. Interestingly, the apical positioning of β1-integrin from the basal domain upon EPEC infection allows it to interact with Tir, substituting for the natural EPEC ligand intimin. The interaction of β1-integrin with Tir results in impaired barrier function [85]. However, EPEC-mediated loss of TEER does not require Tir as expression of intimin alone can induce intestinal barrier dysfunction and remove occludin from TJs. The absence of Tir, however, does not prevent the delivery of Map or EspF effectors into host cells, two well-known effectors associated with TJ disruption [59, 60, 87, 88]. These data suggest that both Tir and intimin are required to initiate the downstream signals that perturb TJs function, and that intimin, probably by interacting with host cell proteins, contributes to the ability of EPEC effectors to disrupt intestinal barrier function in a Tir-independent manner.
EspF has multiple functions in host cells including nucleoli disruption, multinucleation, and cell hypertrophy [89, 90]. The NH-2 region of EspF functions as a mitochondrial targeting signal (MTS) that targets host cell mitochondria and induces cell death [91,92,93,94]. The COOH-terminus of EspF contains binding sites for the SH3 domain of the endocytic modulator sorting nexin 9 (SNX9) through its RxAPxxP motif [95, 96]. EspF also interacts with SNX18 and SNX33, and WIPF1 (WAS/WASL interacting protein family member 1) proteins [97]. EspF possesses N-WASP-binding sites and directly stimulates the actin-polymerizing activity of N-WASP [96, 98]. EspF from rabbit EPEC (REPEC) associates with N-WASP and Arp2/3 and recruits ZO-1, ZO-2, occludin, and claudins to actin-pedestals, promoting pedestal biogenesis [99]. In addition, EspF induces the sequential removal of ZO-1 and afadin from cell-cell contacts with relocalization to actin-pedestals, driving pedestal maturation [100]. Recent evidence demonstrates that EspF, through its binding partners SNX9 and N-WASP, promotes the redistribution of ZO-1 and ZO-2 from TJs to actin-pedestals [101]. Interestingly, although mutations in neither SNX9- or N-WASP-binding domains of EspF affect EPEC actin-pedestal formation, the binding of EspF to SNX9 alone is sufficient to contribute to the actin-pedestal organization and increased colocalization of aPKCζ and F-actin in those structures [101, 102].
The essential role of EspF in EPEC pathogenesis has been demonstrated. Infection of mice with an EPEC strain deficient of espF has no effect on barrier function or on ileal TJ morphology. Only minor alterations of colonic TJ structure are seen at 1-day postinfection, but these defects disappear at later times postinfection [54]. In polarized intestinal epithelial cell culture monolayers, delivery of EspF correlates with a decrease in TEER, increased intestinal permeability, and redistribution of occludin [103]. Although the mechanisms by which EspF perturbs TJs are still unknown, data suggest that the EspF chaperone CesF plays a crucial role in the disruption of intestinal epithelial barrier function [104, 105].
Although EPEC is characterized as a noninvasive pathogen, EPEC utilizes membrane microdomains (lipid rafts) of the host cell to invade IECs, leading to the disruption of TJ structure, altered composition of TJ proteins associated with lipid raft-membrane fractions, and a drastic drop in TEER [96, 106, 107]. EPEC invasion has been reported to be dependent on EspF, which accumulates in patches at the cell surface and colocalizes with clathrin. EspF interacts with SNX9, causing the formation of elongated plasma membrane tubules, as well as the internalization of EPEC into IECs [96, 107]. Recent evidence shows that EPEC prompts the recruitment of clathrin and AP2, early (Rab5a, Rab7, and EEA1) and recycling (Rab4a, Rab11a, Rab11b, FIP2, Myo5b) endocytic proteins, to the sites of bacterial attachment [97, 108]. Additionally, transferrin receptor (TfR), β1-integrin, Exo70, a major component of the exocyst complex, and the basolateral protein VAMP3, involved in docking and fusion vesicles containing basolateral cargo, and aquaporins (AQPs) , are also recruited to pedestals. The recruitment of these proteins to sites of infection correlates with increased endocytosis, recycling, and transcytosis to the infected plasma membrane [97, 108]. The movement of endosomes and associated endocytic proteins in polarized epithelial cells depend on EspF. SNX9 is recruited to clathrin-coated pits and, in conjunction with N-WASP and associated proteins (dynamin, Arp2/3 and AP2), promotes the endocytosis and recycling of plasma membrane proteins [96, 97, 109,110,111,112,113]. For example, Crumbs3 (Crb3), a polarity protein, is internalized via Rab5 vesicles and it is driven to the lysosome compartment in an EspF- and dynamin-dependent endocytic process. The association of EspF with SNX9 causes displacement of Crb3 from the membrane surface to the cytoplasm [114]. EspF/SNX9 interaction also alters the localization of occludin, ZO-1, and JAM-A/Ser285, a crucial phosphorylation step for TJ assembly. Ablation of the EspF/SNX9 interaction or mutations in the SNX9- and N-WASP-binding sites restore the junctional localization of these TJ proteins [35, 101, 102, 114].
Several studies have independently demonstrated EspF binding to SNX9 and N-WASP and their impact on TJs. An espF-SNX9-binding-deficient mutant strain disrupts barrier function in polarized T84 cells but does not alter the membrane localization of occludin, suggesting that this interaction may be dispensable for EPEC-induced junction disruption [96]. However, recent studies have shown that although mutations in either the SNX9- or N-WASP-binding domain of EspF, or an espF mutant, failed to bind SNX9, both attenuated EPEC-induced TJ disruption and do not alter the junctional localization of occludin in infected cells [101, 102]. The discrepancy in these data suggests that redundancy in sorting nexin proteins in colon cancer cell lines may influence the impact of EPEC on barrier function. The ability of EspF to bind both SNX9 and N-WASP is important, however, for the full effects of EPEC on intestinal permeability.
EspF also regulates the expression of TJ proteins through transcriptional and posttranslational mechanisms. Ectopic expression of EspF disrupts TJ integrity and prevents the recruitment of occludin, claudin-1, and − 4 and ZO-1 into TJs during junction assembly, leading to their cytoplasmic accumulation and eventual removal by lysosomes [115]. These data suggest that EspF promotes EPEC invasion of IECs, regulates the transcription of TJ proteins, and induces endocytosis, recycling, and trafficking of vesicles destined for the basolateral membrane to the apical sites of bacterial attachment. This may contribute to EPEC attachment and subsequent microcolony growth.
Besides its role in TJ perturbation, EspF dismantles IF architecture although other secreted proteins may be involved. EPEC infection increases the solubility of CK18, a component of IF cytoskeleton, and promotes its binding with the adaptor protein 14–3-3ζ in infected cells. EspF interacts with host cell protein CK18 and it has been suggested that EspF forms a complex with CK18 and 14–3-3ζ during EPEC infection [116].
Map is a 203 amino acid protein that contains a mitochondrial-targeting sequence that directs it to mitochondria, causing dysfunction [117, 118]. Map also possesses a WxxxE motif that induces filopodia formation through the activation of Cdc42, functioning as a GEF [14, 119, 120]. A functional PDZ ligand, the TRL motif, at the COOH-terminus, is also involved in filopodia formation and remodeling and elongation of the brush border in infected cells [119, 121]. This motif allows Map to interact with the host cell protein NHERF. During EPEC infection, NHERF1 is recruited to sites of bacterial attachment colocalizing with ezrin, which is activated by EPEC, resulting in altered barrier function [121, 122]. The binding of NHERF1 to ezrin functions as a molecular scaffold that links Map to the actin cytoskeleton. This interaction assembles a positive feedback loop that amplifies Cdc42 signaling within membrane microdomains [123].
Map has been shown to play a major role in disruption of intestinal barrier function as map deletion strains have less impact on both TEER and loss of occludin from cell junctions. Although the mechanisms by which Map disrupts TJs are not known, it likely occurs through modulation of the actin cytoskeleton. Map cooperates with EspF to mediate TJ disruption [88]. Epithelial cells constitutively expressing EspF and Map have been used to elucidate the role of these effectors on TJs. Ectopic expression of Map increases epithelial permeability and prevents junctional recruitment of TJ proteins, ZO-1, occludin, claudin-1, and claudin-4 into TJs during de novo TJ assembly. In addition, expression of EspF and Map downregulates the expression of claudin-1, claudin-4, and occludin and inhibits their junctional recruitment, leading to depletion at the plasma membrane. Interestingly, Map significantly downregulates the transcription of claudin-1. Map interacts with all isoforms of nonmuscle myosin II and actin, and EspF binds to ZO-1 and actin [115]. These data suggest that these EPEC effectors regulate TJs by interacting with cytoskeletal proteins and regulating contractility of the actomyosin cytoskeleton.
EspG is an EPEC effector with multiple functions. EspG binds to host ADP-ribosylation factor (ARF), p21-activated kinases (PAKs), and Rac/Cdc42-binding domain of PAK1, all important regulators of the actin cytoskeleton and cell motility [124,125,126]. Recent findings indicate that EspG, via binding to ARF and PAK, plays an essential role in EPEC attachment and pedestal formation in host cells [127, 128]. EPEC activates Rac and Cdc42 RhoA GTPases in an EspG-dependent manner. EPEC promotes the activation of Rho GTPases by recruiting Fabrin, a host Cdc42-specific GEF and important regulator of the actin cytoskeleton, to actin-pedestals [128, 129]. PAK activation, pedestal formation, and bacterial attachment are reduced in Fabrin-deficient cells. EspG localizes to actin-pedestals via Arf6 binding. The recruitment of Fabrin to sites of EPEC attachment depends on EspG and requires the enrichment of PIP2 and host ARF6 [128]. These data suggest that EspG contributes to pedestal biogenesis by binding and activating cellular GTPases required for actin pedestal formation.
It has been demonstrated that microtubule (MT) networks contribute to TJ maintenance. Disruption of MT prevents the movement of occludin-containing vesicles to the plasma membrane and leads to a disruption of cell barrier function [130, 131]. In addition, disruption of MT or loss of myosin IIA and B results in impaired TJ function [132]. EspG and its homolog Orf3 (EspG2) cause progressive fragmentation and loss of the MT network and induction of stress fibers formation, which impact intestinal barrier function [133,134,135,136,137,138]. EspG and Orf3 induce disruption of the MT network beneath adherent bacteria by direct association with tubulins, stimulating MT destabilization. The regulation of MT disassembly is mediated by activation of the RhoA/ROCK signaling pathway, which enhances myosin contractility and actin stress fiber formation [139,140,141]. EPEC activates RhoA in an EspG- or EspG2-dependent manner [133, 134]. GEF-H1, a RhoA-specific guanine nucleotide exchange factor, binds to MT in its inactive form and switches to an active form when released from MT. [142] Interestingly, both EspG and Orf3 release GEF-H1 from the cytoskeleton into the cytosol, possibly increasing its activity. EPEC-induced actin stress fiber formation is prevented by expression of dominant-negative form of GEF-H1 and RhoA and by ROCK inhibition, suggesting that EspG and Orf3 disrupt MT integrity, triggering activation of the RhoA-ROCK signaling pathway via GEF-H1 activity [133]. EspG1/G2 causes the progressive movement of occludin, claudin-1, and ZO-1 away from the membrane into the cytosol and reduces the expression of tricellulin, leading to epithelial barrier disruption [134, 135, 139, 143]. Interestingly, EspG1/G2 regulates size-selective paracellular permeability without altering the TJ architecture during EPEC infection in MDCK monolayers [134]. In contrast, in polarized intestinal epithelial cells, depletion of espG1/G2 induces a gradual loss of TEER and eradication of EPEC with gentamicin promotes barrier function recovery [138]. These data suggest that the effects of EspG1/G2 on MT delay the recovery of TJs damaged by EPEC infection, thus perpetuating the loss of barrier function. In addition to its effect on MT and TJ disruption, EspG also contributes to the arrest of vesicle trafficking and blocks the recycling of vesicle cargo to the cell surface [144, 145].
EspH is translocated into host cells by the T3SS and localizes beneath EPEC microcolonies. EspH decreases filopodia formation, disrupts stress fibers, and modulates pedestal formation and pedestal elongation during EPEC infection, suggesting its potential function in modulating actin dynamics [15, 146]. EspH associates with the plasma membrane of host cells and causes disruption of filamentous actin structures. EspH binds directly to the guanine nucleotide exchange factor for Rho, p115RhoGEF. This binding prevents Rho activation, thereby inhibiting downstream Rho signaling and actin cytoskeleton dynamics [147]. Recently, it was found that EspH cooperates with Tir to promote actin polymerization at the bacterial attachment sites. EspH then promotes the recruitment of N-WASP and Arp2/3 to bacterial attachment sites via a mechanism involving the COOH-terminus of Tir and the WH1 domain of N-WASP [146]. Additionally, WASP-interacting protein (WIP), which binds the N-WASP WH1 domain, is crucial in EspH-mediated actin polymerization. EspH induces the colocalization of Tir, WIP, and N-WASP at actin-rich structures. These data suggest that Tir- and EspH-mediated actin signaling pathways contribute to pedestal biogenesis.
Interestingly, EPEC controls Rho GTPase activity by translocating one effector to inactivate mammalian RhoGEFs and replacing them with bacterial RhoGEFs that promote cell survival. EspH induces focal adhesion disassembly, cell detachment, and induces cytotoxicity. EPEC translocates EspH, which inactivates mammalian RhoGEFs, and, at the same time, translocates the bacterial RhoGEFs, EspM2, and EspT, which inhibit the EspH-induced focal adhesion disassembly, cell adhesion, and survival [148]. Furthermore, EspH perturbs desmosomes structures, which are intercellular junctions that are connected to IF components of the cytoskeleton. It has been reported that desmosomal proteins, desmoglein, desmocollin, and desmoplakin, as well as desmosome morphology, are unaltered during EPEC infection [149]. However, recent evidence demonstrates that EPEC induces a dramatic separation of apposing lateral membranes of adjacent cells by perturbing desmosomes, compromising cell-cell adhesion and barrier function of IECs. The EPEC effector EspH is responsible for these changes [150]. EspH inhibits RhoA GTPase, resulting in actin depolymerization, in turn perturbing IF stability and ultimately leading to destabilization of desmosomes and desmoglein-2 (DSG2) downregulation. EspH induces DSG2 redistribution and subsequent degradation within lysosomes. These changes precede the loss of occludin from cell-cell contacts [150]. EspH-induced DSG2 loss and desmosomal perturbation compromise epithelial monolayer integrity, leading to loss of epithelial barrier function.
NleA effector (non-LEE-encoded effector A) binds and inhibits mammalian COPII through direct interaction with the COPII component Sec24 [151]. COPII is a protein complex that mediates the packaging and trafficking of proteins in the endoplasmic reticulum (ER). NleA inhibits protein secretion from the ER to the Golgi by direct interaction with Sec24. NleA increases intestinal permeability by disrupting occludin and ZO-1 from cell-cell contacts [152]. Knockdown of COPII components in epithelial cells is sufficient to disrupt TJ-associated occludin and ZO-1, suggesting that NleA inhibits the transport of newly synthesized TJ proteins, thus disrupting the barrier. Murine models infected with C. rodentium show that NleA mutants unable to bind Sec24 have normal TJs and their fecal water content is similar to that of uninfected mice [153]. Therefore, NleA and its interaction with components of the COPII complex promotes TJ disassembly and diarrhea in murine models.
EspM is an effector harboring WxxxE motifs, thereby possessing the ability to modulate actin cytoskeleton dynamics [154, 155]. EspM induces the formation of actin pedestals and stress fibers that are linked to the plasma membrane through focal adhesions. Actin stress fiber formation is regulated by the GTP-binding protein RhoA. Infection of cells expressing RhoAN19, a mutant that inhibits activation of the small GTPases, leads to the formation of typical actin-rich pedestals, but there is a mark reduction in formation of stress fibers, suggesting that formation of stress fibers by EspM is RhoA-dependent [154, 155]. Additionally, EspM increases the phosphorylation of cofilin, a protein that binds actin filaments, and is a downstream ROCK target, suggesting that EspM regulates stress fiber formation through RhoA-ROCK-cofilin signaling. EspM has also been demonstrated to be an important modulator of pedestal formation. Ablation of both espM1 and espM2 induces highly developed actin pedestals, whereas those induced by the wild-type strain were poorly developed [156]. Importantly, EspM causes dramatic changes in TJ architecture in infected polarized monolayers. In addition, cells infected with EPEC expressing EspM2 show disruption of ZO-1 from TJs to the apical and basolateral membrane surface but without a reduction in barrier function.
EPEC Perturbs Cytoskeletal Networks and Adhesion Complexes Affecting Barrier Function
EPEC Infection Stimulates Contraction of the Actomyosin Perijunctional Ring
TJ permeability is regulated in part by contraction of the perijunctional actomyosin ring, leading to decreased TEER [157]. Phosphorylation of myosin light chain (MLC) stimulates contraction of the actomyosin ring, thereby increasing TJ permeability. EPEC infection enhances the phosphorylation of MLC and its association with the cytoskeleton [158,159,160,161]. MLC is distributed between cytosolic and cytoskeletal cell fractions; its association with the cytoskeleton increases with the duration of EPEC infection. Analysis of phosphopeptide mapping indicates that MLC is phosphorylated at different sites, strongly suggesting that PKC and MLC kinase (MLCK) are involved in MLC phosphorylation in response to EPEC infection [160]. PKC activators or ectopic expression of the catalytic domain of MLCK increase MLC phosphorylation in a manner similar to levels observed during EPEC infection [162, 163]. Phosphorylation of MLC by MLCK is associated with increased intestinal TJ permeability during EPEC infection [161]. Inhibition of MLCK with ML-7/9 pretreatment, or a membrane-permeant inhibitor of MLCK (PIK), or the neuropeptide vasoactive intestinal peptide (VIP), which regulates epithelial paracellular permeability, decreases intracellular MLC phosphorylation, prevents the redistribution of occludin, claudin-3, and ZO-1, and ameliorates EPEC-induced disruption of the colonic epithelial barrier [161, 164,165,166]. These data suggest that MLC phosphorylation by MLCK is involved in the perturbation of TJs by EPEC.
EPEC Disrupts AJs by Activation of PKCα
AJs are linked to the actin cytoskeleton through β and α-catenins. EPEC dissociates β-catenin from the membrane and moves it to the cytosol, thus increasing intestinal epithelial paracellular permeability [167]. EPEC phosphorylates PKCα, which induces its association with E-cadherin, thus dissociating the E-cadherin/β-catenin complex [167, 168]. Expression of dominant-negative PKC or treatment with a PKCα-inhibitory peptide blocks these effects. Therefore, targeting of AJs by EPEC may undermine the integrity of intestinal epithelial barrier function [167].
EPEC Alters the Phosphorylation State of Adhesion and Cytoskeleton Molecules
Phosphorylation of occludin is required for its association with TJ complex. EPEC induces a progressive decrease in occludin phosphorylation, correlating with an increase in the nonphosphorylated form and its subsequent dissociation from TJs. These changes correlate with a leaky intestinal barrier [87]. Inhibition of serine/threonine phosphatases prevents EPEC-induced changes in both occludin and TEER, implicating their involvement in the regulation of TJs. EPEC promotes the accumulation of ezrin in A/E lesions, increasing its activity and association with the actin cytoskeleton by promoting ezrin phosphorylation [122]. Ezrin belongs to the Ezrin-Radixin-Moesin (ERM) protein family, which mediates the dynamic linkage between the plasma membrane and cortical actin. Ezrin is reorganized by EPEC infection, leading to impaired TJs [78]. EPEC-induced association of ezrin with the cytoskeleton as well as the drop in TEER depend on the presence of EspB and EspF. Expression of dominant-negative ezrin attenuates the effect of EPEC on ZO-1 and the drop in TEER [122]. These findings suggest that EPEC regulates the phosphorylation states of several proteins important in mediating EPEC-induced signals that result in perturbation of the TJ barrier.
EPEC Perturbs Apico-Basal Polarity
TJs are crucial for the establishment and maintenance of epithelial apico-basal polarity , which is controlled by Crb (Crumbs3/Pals1/Patj), Par (Par3/Par6/aPKC/Cdc42), and Scribble (Scrib/Lgl/Dlg) polarity complexes. Apico-basal polarity contributes to cell morphology, directional vesicle transportation, ion and solute transport, and specific localization of proteins and lipids to different membrane domains [169]. The impact of EPEC on intestinal epithelial polarity has been studied. EPEC alters the distribution of β1-integrin and Na+/K+ ATPase, which are typically restricted to the basolateral membrane. EPEC infection caused the movement of these proteins to the apical surface, resulting in impaired barrier function [85, 114]. Furthermore, cells infected with EPEC expressing EspM exhibit apical β1-integrin staining at bacteria attachment sites and at the cell-cell contacts, presumably just above TJs [156]. These data suggest that changes in apical domain morphology induced by EspM are associated with redistribution of some basolateral membrane proteins. EPEC also recruits basolateral endocytic and recycling membrane proteins (Rabs), AQPs, Tnf, Exo70, and VAMP3 to the apical surface beneath sites of EPEC microcolony attachment [97, 108]. In addition, EPEC alters plasma membrane lipids by inducing the formation of PI(3,4,5)P3 at actin pedestals [69]. In polarized MDCK monolayers, PI(3,4,5)P3 is restricted to the basolateral membrane domain where it functions as a regulator of the basolateral membrane formation [170]. Therefore, it appears that basolateral proteins are specifically trafficked to EPEC microcolonies at the apical membrane by intracellular vesicle transport, indicating failed fence function in host cells.
In addition to the redistribution of basolateral proteins, EPEC infection induces the relocalization of occludin, claudin-1, and ZO-1 from the TJ region to the lateral membrane and cytoplasmic compartment, impacting the architecture and function of TJs [171]. Freeze-fracture replicas of EPEC-infected monolayers reveal aberrant strands containing claudin-1 and occludin extending down the lateral membrane surface well below the TJ area. These structural changes correlate with both increased paracellular permeability and decreased TEER [171]. Interestingly, EspM causes host cells to take on an abnormal round shape, with the main mass of the cell body bulging out and the TJs located at the lower part of the cell, close to the basal membrane. In addition, EspM changes the localization of ZO-1 from the TJs to the apical and basolateral membranes but without a reduction in barrier function [156]. These findings suggest that EPEC-induced perturbation of apico-basal polarity and TJ structure allow the free diffusion of cytoplasmic and membrane proteins to inappropriate cellular domains, further contributing to EPEC pathogenesis.
The impact of EPEC on apico-basal polarity complexes was reported recently. Par6 and aPKCζ, both Par polarity members, are crucial for the establishment of cell polarity and TJ formation. Par6 is a scaffolding protein that interacts with all polarity complexes, thus allowing aPKCζ to phosphorylate its kinase substrates. EPEC displaces Par6 and aPKCζ, but not Par3, from the cell-cell contacts to the cytoplasm and basolateral membrane of IECs [102]. The interaction of Par6 with Cdc42-GTP activates aPKCζ to phosphorylate Par3, which binds 14–3-3 protein; this interaction regulates cell polarity and TJ formation [172,173,174,175,176,177]. Par6 interacts with Pals1 and Crb3, the latter being target of aPKCζ, and this complex is crucial for TJ formation [178,179,180]. Detailed in vitro and in vivo analyses show that EPEC redistributes Crb3 and Pals1, but not Patj, from cell-cell contacts to the cytoplasm of IECs [114]. Both espF and map are involved in this phenotype. Interestingly, in a cyst morphogenesis assay, ectopic expression of EspF leads to the formation of 3D cysts with multiple lumens, indicating disruption of the formation of cell polarity [114]. These findings indicate that EspF perturbs cell polarity in intestinal epithelial cells.
Several studies have demonstrated the direct effect of EPEC on the activation and translocation of aPKCζ in infected epithelia. EPEC significantly increases aPKCζ activity in T84, but not SKCO-15, polarized monolayers. Inhibition of aPKCζ with a pseudosubstrate protects against disruption of the TJ barrier by EPEC, suggesting that aPKCζ is involved in EPEC-induced barrier disruption. EPEC also induces the translocation of aPKCζ from the cytoplasm to membrane and from cell-cell contacts to actin-rich pedestals of infected monolayers; both situations correlate with loss of barrier function [102, 165, 181, 182]. Interestingly, aPKCζ recruitment to EPEC pedestals is not diminished by deletion of map or espF or by mutation of the SNX9-binding domain of EspF. However, the interaction of EspF with SNX9 plays an important role in actin-pedestal organization and contributes to the colocalization of active aPKCζ and F-actin to the plasma membrane and within pedestals in a cell-specific manner [102].
EPEC impairs both cell polarity and intestinal epithelial TJ barrier function. Recent evidence indicates that the temporal sequence of TJ disruption following EPEC infection correlates with the redistribution of polarity proteins. EPEC induces the recruitment of aPKCζ to pedestals colocalizing with actin almost immediately upon bacterial attachment (5–15 min). TJ proteins are then removed from cell-cell contacts to cytoplasm. JAM-A S285 is internalized by 30 min and TEER significantly decreases 45 min postinfection. Phosphorylation of JAM-A at tyrosine 280, which is related to loss of barrier function, is detectable after 1–2 h post EPEC infection, corresponding with leaky TJs [102, 183]. Occludin and ZO-1 disruption occurs at 1–2 h postinfection, respectively, corresponding with a more significant drop in TEER. aPKCζ has direct and indirect roles in the formation and maintenance of polarity and TJ structure and function. aPKCζ phosphorylates Crb3, occludin, claudins, JAM-A, ZO-1, and ZO-2 to establish and maintain TJ structure and barrier function [35, 184,185,186,187]. EPEC alters the phosphorylation state and localization of these proteins, leading to a loss of barrier function [87, 102, 114]. The displacement of aPKCζ away from TJs results in perturbation of barrier function, suggesting that the progressive dismantling of TJ by EPEC and the corresponding impact on barrier function occur after the early recruitment of aPKCζ to actin pedestals [102].
EPEC Activates Various Signaling Pathways
Inflammation in response to infection by enteric pathogens contributes to TJ disruption. For example, IL-13, a pro-inflammatory cytokine, which is increased in the mucosa of patients with ulcerative colitis and Crohn’s disease, is able to induce expression of claudin-2, which increases TJ permeability to both ions and small nonionic solutes [188, 189]. Interferon (IFN)-γ and tumor necrosis factor (TNF)-α, both pro-inflammatory cytokines, are critical regulators of barrier function via MLC phosphorylation and TJ remodeling [164, 190,191,192]. TNF-α induces occludin internalization, paracellular barrier loss, and diarrhea [190, 193]. Infection of T84 cells by enteric bacterial pathogens has been shown to increase the expression of both TNF-α and IL-1β pro-inflammatory molecules [194]. EPEC and REPEC infection increases TNF-α, IL-1β, IL-8, and IL-6 cytokines [195, 196]. EPEC infection induces only a modest inflammatory response due in part to the presence of anti-inflammatory EPEC effectors including non-LEE-encoded NleB, NleC, NleD, NleE and the homologs NleH1 and NleH2 [197,198,199,200]. EPEC infection also activates the mitogen-activated protein (MAP) kinases ERK1/2, p38 and JNK, and nuclear factor-κB (NF-κB) signaling pathways that upregulate expression of the pro-inflammatory cytokine IL-8 [201,202,203,204]. aPKCζ has also been reported to be involved in ERK1/2 and NF-κB activation; both pathways are activated by EPEC [202,203,204,205,206,207,208,209]. aPKCζ regulates NF-κB activation by binding and activating IκB kinase (IKK), which phosphorylates the NF-κB inhibitor IκB, triggering its degradation. Inhibition of aPKCζ or expression of a dominant-negative mutant significantly suppresses EPEC-induced IκBα phosphorylation but does not impact the EPEC-induced stimulation of ERK1/2 [181]. Interestingly, inhibition of ERK1/2 and p38 attenuates the phosphorylation and degradation of IκBα and expression of IL-8 but does not affect A/E lesion formation or protect against the decrease in barrier function associated with EPEC infection [202, 204]. These data suggest that EPEC-activated ERK1/2 and aPKCζ signaling pathways contribute to the inflammatory response during EPEC pathogenesis.
EPEC Alters Intestinal Transport
EPEC-induced loss of intestinal barrier function is believed to contribute to diarrhea by disrupting intestinal epithelial ion and fluid transport. The loss of TJ barrier function would prevent the creation of ion gradients needed for effective ion and solute transport. In addition, infectious diarrhea can be caused by increased chloride secretion, decreased NaCl absorption, or both. EPEC infection attenuates secretagogue-induced net ion transport and impacts chloride secretion [210,211,212]. Both electroneutral Na+/H+ and Cl−/OH− exchange activities are altered by EPEC [213]. EPEC modulates intestinal epithelial cell electrolyte transport, reducing the expression of the Na+/H+ exchanger-3 (NHE3), the major intestinal transporter of Na+ absorption in an EspF-dependent manner, and stimulating the apical NHE2 and basolateral NHE1 activity [213, 214]. Analysis of the signal transduction cascades responsible for the increased NHE2 activity during EPEC infection shows that PLC, PKCα, and PKCε signaling pathways are implicated [215]. Map interacts with NHERF1 (Na+/H+ exchanger regulatory factor I), Map and NleH1 bind to NHERF2 altering its function [121, 216]. EPEC infection decreases Cl− absorption in intestinal cells by reducing the activity of the Cl−/HCO3− exchanger, SLC26A3 (downregulated in Adenoma, DRA), resulting in a reduced Cl− uptake and its accumulation in the lumen, driving water loss. Mortality in C. rodentium-infected mice is associated with downregulation of DRA and other genes involved in intestinal transport, decreased uptake of chloride, and fatal diarrhea [217,218,219]. EPEC reduces DRA protein expression and redistributes its localization from the apical membrane surface to intracellular compartments. This process is mediated by EspG1/G2 [220, 221]. EPEC alters the activity of the sodium-D-glucose cotransporter (SGLT-1), a major water pump in the small intestine [222]. EPEC causes a rapid movement of SGLT-1 from the apical membrane into intracellular vesicles. EspF, Map, Tir, and intimin cooperate to decrease activity of SGLT-1. AQPs function as a water channels to maintain the dehydration of fecal contents. C. rodentium infection alters the localization of AQP2 and AQP3, from the lateral membrane to the cytoplasm. The change in localization correlates with the diarrhea in infected mice [223]. The altered distribution of AQPs is partially dependent on EspF and EspG effectors, suggesting that AQPs may also contribute to diarrhea during bacterial infection. These data suggest that during EPEC infection, the ability of the intestinal epithelium to regulate absorption and secretion of ions and water is compromised, contributing to diarrhea.
Conclusions
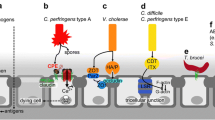
The findings presented in this chapter support the contention that EPEC-induced perturbation of TJs is a complex process driven by the downstream effect of multiple signaling pathways activated by EPEC effectors, disruption of cytoskeletal networks, increased endocytosis, changes in the localization of proteins to different cell compartments and membrane domains, and altered apico-basal polarity (Fig. 1a). EPEC-induced loss of intestinal epithelial barrier function is believed to contribute to diarrhea by disrupting intestinal epithelial ion and fluid transport (Fig. 1b). All of these changes converge disrupting intestinal epithelial host cell homeostasis, leading to TJ disruption that contributes to EPEC-associated diarrhea.

Model displaying the mechanisms by which EPEC perturbs TJs and induces diarrhea. EPEC injects a large repertoire of effector proteins into the host cell. EPEC effectors mediate actin rearrangement and contribute to pedestal formation. (a) EPEC induces the recruitment of several host proteins to actin pedestals immediately upon bacterial attachment. Effectors dismantle cytoskeletal cell structures (MTs and IF) and induce disruption of vesicle trafficking, impacting TJs function. Proteins from TJs, AJs, desmosomes, and polarity complexes are displaced from cell-cell contacts and internalized into the cytoplasm, and some are degraded by lysosomes. EPEC alters the apico-basal polarity, evidenced by redistribution of basolateral proteins (β-1 integrin and Na+/K+ ATPase) to the apical membrane, and TJ proteins to the lateral and apical domains (occludin, claudin-1, and ZO-1). All these events contribute to TJs disassembly. (b) EPEC alters intestinal transport. EPEC infection regulates apical Na+, Cl−, and glucose absorption, impacting the activity of the cotransporters NHE2 and NHE3, DRA, and SGLT1. EPEC also induces the endocytosis of DRA in an EspG-dependent manner. The basolateral water channel proteins AQP2 and AQP3 are redistributed to the cytoplasm after EPEC infection. EPEC alters the activity and localization of cotransporters, contributing to increased diarrhea during EPEC pathogenesis
References
Zahavi EE, Lieberman JA, Donnenberg MS, Nitzan M, Baruch K, Rosenshine I, et al. Bundle-forming pilus retraction enhances enteropathogenic Escherichia coli infectivity. Mol Biol Cell 2011 Jul 15;22(14):2436-2447.
Hu J, Torres AG. Enteropathogenic Escherichia coli: foe or innocent bystander? Clin Microbiol Infect 2015; 21(8):729-734.
Buss SN, Leber A, Chapin K, Fey PD, Bankowski MJ, Jones MK, et al. Multicenter evaluation of the BioFire FilmArray gastrointestinal panel for etiologic diagnosis of infectious gastroenteritis. J Clin Microbiol 2015; 53(3): 915-925.
Spina A, Kerr KG, Cormican M, Barbut F, Eigentler A, Zerva L, et al. Spectrum of enteropathogens detected by the FilmArray GI Panel in a multicentre study of community-acquired gastroenteritis. Clin Microbiol Infect 2015; 21(8); 719-728.
Moon HW, Whipp SC, Argenzio RA, Levine MM, Giannella RA. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect Immun 1983 Sep;41(3):1340-1351.
Knutton S, Lloyd DR, McNeish AS. Adhesion of enteropathogenic Escherichia coli to human intestinal enterocytes and cultured human intestinal mucosa. Infect Immun 1987 Jan;55(1):69-77.
McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci U S A 1995 Feb 28;92(5):1664-1668.
Elliott SJ, Wainwright LA, McDaniel AU, Jarvis KG, Deng YK, Lai LC, et al. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol Microbiol 1998 Apr;28(1):1-4.
Knutton S, Rosenshine I, Pallen MJ, Nisan I, Neves BC, Bain C, et al. A novel EspA-associated surface organelle of enteropathogenic Escherichia coli involved in protein translocation into epithelial cells. EMBO J 1998 Apr 15;17(8):2166-2176.
Sekiya K, Ohishi M, Ogino T, Tamano K, Sasakawa C, Abe A. Supermolecular structure of the enteropathogenic Escherichia coli type III secretion system and its direct interaction with the EspA-sheath-like structure. Proc Natl Acad Sci U S A 2001 Sep 25;98(20):11638-11643.
Kenny B, DeVinney R, Stein M, Reinscheid DJ, Frey EA, Finlay BB. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 1997 Nov 14;91(4):511-520.
McNamara BP, Donnenberg MS. A novel proline-rich protein, EspF, is secreted from enteropathogenic Escherichia coli via the type III export pathway. FEMS Microbiol Lett 1998 Sep 1;166(1):71-78.
Elliott SJ, Krejany EO, Mellies JL, Robins-Browne RM, Sasakawa C, Kaper JB. EspG, a novel type III system-secreted protein from enteropathogenic Escherichia coli with similarities to VirA of Shigella flexneri. Infect Immun 2001 Jun;69(6):4027-4033.
Kenny B. Mechanism of action of EPEC type III effector molecules. Int J Med Microbiol 2002 Feb;291(6-7):469-477.
Tu X, Nisan I, Yona C, Hanski E, Rosenshine I. EspH, a new cytoskeleton-modulating effector of enterohaemorrhagic and enteropathogenic Escherichia coli. Mol Microbiol 2003 Feb;47(3):595-606.
Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol 1963 May;17:375-412.
Staehelin LA. Further observations on the fine structure of freeze-cleaved tight junctions. J Cell Sci 1973 Nov;13(3):763-786.
Tsukita S, Furuse M. Pores in the wall: claudins constitute tight junction strands containing aqueous pores. J Cell Biol 2000 Apr 3;149(1):13-16.
Claude P, Goodenough DA. Fracture faces of zonulae occludentes from “tight” and “leaky” epithelia. J Cell Biol 1973 Aug;58(2):390-400.
Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, et al. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci 2008 Feb 1;121(Pt 3):298-305.
Rodriguez-Boulan E, Nelson WJ. Morphogenesis of the polarized epithelial cell phenotype. Science 1989 Aug 18;245(4919):718-725.
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 1993 Dec;123(6 Pt 2):1777-1788.
Ando-Akatsuka Y, Saitou M, Hirase T, Kishi M, Sakakibara A, Itoh M, et al. Interspecies diversity of the occludin sequence: cDNA cloning of human, mouse, dog, and rat-kangaroo homologues. J Cell Biol 1996 Apr;133(1):43-47.
Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 2000 Dec;11(12):4131-4142.
Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol 1998 Oct 19;143(2):391-401.
Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 1998 Jun 29;141(7):1539-1550.
Mineta K, Yamamoto Y, Yamazaki Y, Tanaka H, Tada Y, Saito K, et al. Predicted expansion of the claudin multigene family. FEBS Lett 2011 Feb 18;585(4):606-612.
Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, et al. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 2014 Apr 18;344(6181):304-307.
Suzuki H, Tani K, Tamura A, Tsukita S, Fujiyoshi Y. Model for the architecture of claudin-based paracellular ion channels through tight junctions. J Mol Biol 2015 Jan 30;427(2):291-297.
Suzuki H, Tani K, Fujiyoshi Y. Crystal structures of claudins: insights into their intermolecular interactions. Ann N Y Acad Sci 2017 Jun;1397(1):25-34.
Saitoh Y, Suzuki H, Tani K, Nishikawa K, Irie K, Ogura Y, et al. Tight junctions. Structural insight into tight junction disassembly by Clostridium perfringens enterotoxin. Science 2015 Feb 13;347(6223):775-778.
Shinoda T, Shinya N, Ito K, Ohsawa N, Terada T, Hirata K, et al. Structural basis for disruption of claudin assembly in tight junctions by an enterotoxin. Sci Rep 2016 Sep 20;6:33632.
Tsukita S, Tanaka H, Tamura A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem Sci 2019 Feb;44(2):141-152.
Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol 2005 Dec 19;171(6):939-945.
Iden S, Misselwitz S, Peddibhotla SS, Tuncay H, Rehder D, Gerke V, et al. aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J Cell Biol 2012 Mar 5;196(5):623-639.
Otani T, Nguyen TP, Tokuda S, Sugihara K, Sugawara T, Furuse K, et al. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity. J Cell Biol 2019 Oct 7;218(10):3372-3396.
Jesaitis LA, Goodenough DA. Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. J Cell Biol 1994 Mar;124(6):949-961.
Haskins J, Gu L, Wittchen ES, Hibbard J, Stevenson BR. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol 1998 Apr 6;141(1):199-208.
Willott E, Balda MS, Fanning AS, Jameson B, Van Itallie C, Anderson JM. The tight junction protein ZO-1 is homologous to the Drosophila discs-large tumor suppressor protein of septate junctions. Proc Natl Acad Sci U S A 1993 Aug 15;90(16):7834-7838.
Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, et al. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 2006 Aug 25;126(4):741-754.
Citi S, Sabanay H, Jakes R, Geiger B, Kendrick-Jones J. Cingulin, a new peripheral component of tight junctions. Nature 1988 May 19;333(6170):272-276.
Cordenonsi M, D’Atri F, Hammar E, Parry DA, Kendrick-Jones J, Shore D, et al. Cingulin contains globular and coiled-coil domains and interacts with ZO-1, ZO-2, ZO-3, and myosin. J Cell Biol 1999 Dec 27;147(7):1569-1582.
Aijaz S, D’Atri F, Citi S, Balda MS, Matter K. Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev Cell 2005 May;8(5):777-786.
Guillemot L, Guerrera D, Spadaro D, Tapia R, Jond L, Citi S. MgcRacGAP interacts with cingulin and paracingulin to regulate Rac1 activation and development of the tight junction barrier during epithelial junction assembly. Mol Biol Cell 2014 Jul 1;25(13):1995-2005.
Yano T, Matsui T, Tamura A, Uji M, Tsukita S. The association of microtubules with tight junctions is promoted by cingulin phosphorylation by AMPK. J Cell Biol 2013 Nov 25;203(4):605-614.
Spadaro D, Tapia R, Pulimeno P, Citi S. The control of gene expression and cell proliferation by the epithelial apical junctional complex. Essays Biochem 2012;53:83-93.
Guillemot L, Spadaro D, Citi S. The junctional proteins cingulin and paracingulin modulate the expression of tight junction protein genes through GATA-4. PLoS One 2013;8(2):e55873.
Yamamoto T, Harada N, Kano K, Taya S, Canaani E, Matsuura Y, et al. The Ras target AF-6 interacts with ZO-1 and serves as a peripheral component of tight junctions in epithelial cells. J Cell Biol 1997 Nov 3;139(3):785-795.
Mandai K, Nakanishi H, Satoh A, Obaishi H, Wada M, Nishioka H, et al. Afadin: A novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J Cell Biol 1997 Oct 20;139(2):517-528.
Ide N, Hata Y, Nishioka H, Hirao K, Yao I, Deguchi M, et al. Localization of membrane-associated guanylate kinase (MAGI)-1/BAI-associated protein (BAP) 1 at tight junctions of epithelial cells. Oncogene 1999 Dec 16;18(54):7810-7815.
Hamazaki Y, Itoh M, Sasaki H, Furuse M, Tsukita S. Multi-PDZ domain protein 1 (MUPP1) is concentrated at tight junctions through its possible interaction with claudin-1 and junctional adhesion molecule. J Biol Chem 2002 Jan 4;277(1):455-461.
Adamsky K, Arnold K, Sabanay H, Peles E. Junctional protein MAGI-3 interacts with receptor tyrosine phosphatase beta (RPTP beta) and tyrosine-phosphorylated proteins. J Cell Sci 2003 Apr 1;116(Pt 7):1279-1289.
Yao R, Natsume Y, Noda T. MAGI-3 is involved in the regulation of the JNK signaling pathway as a scaffold protein for frizzled and Ltap. Oncogene 2004 Aug 12;23(36):6023-6030.
Shifflett DE, Clayburgh DR, Koutsouris A, Turner JR, Hecht GA. Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Lab Invest 2005 Oct;85(10):1308-1324.
Zhang Q, Li Q, Wang C, Liu X, Li N, Li J. Enteropathogenic Escherichia coli changes distribution of occludin and ZO-1 in tight junction membrane microdomains in vivo. Microb Pathog 2010 Jan;48(1):28-34.
Zhang Q, Li Q, Wang C, Li N, Li J. Redistribution of tight junction proteins during EPEC infection in vivo. Inflammation 2012 Feb;35(1):23-32.
Guttman JA, Li Y, Wickham ME, Deng W, Vogl AW, Finlay BB. Attaching and effacing pathogen-induced tight junction disruption in vivo. Cell Microbiol 2006 Apr;8(4):634-645.
Spitz J, Yuhan R, Koutsouris A, Blatt C, Alverdy J, Hecht G. Enteropathogenic Escherichia coli adherence to intestinal epithelial monolayers diminishes barrier function. Am J Physiol 1995 Feb;268(2 Pt 1):G374-9.
Philpott DJ, McKay DM, Sherman PM, Perdue MH. Infection of T84 cells with enteropathogenic Escherichia coli alters barrier and transport functions. Am J Physiol 1996 Apr;270(4 Pt 1):G634-45.
Canil C, Rosenshine I, Ruschkowski S, Donnenberg MS, Kaper JB, Finlay BB. Enteropathogenic Escherichia coli decreases the transepithelial electrical resistance of polarized epithelial monolayers. Infect Immun 1993 Jul;61(7):2755-2762.
Viswanathan VK, Hodges K, Hecht G. Enteric infection meets intestinal function: how bacterial pathogens cause diarrhoea. Nat Rev Microbiol 2009 Feb;7(2):110-119.
Foubister V, Rosenshine I, Finlay BB. A diarrheal pathogen, enteropathogenic Escherichia coli (EPEC), triggers a flux of inositol phosphates in infected epithelial cells. J Exp Med 1994 Mar 1;179(3):993-998.
Dytoc M, Fedorko L, Sherman PM. Signal transduction in human epithelial cells infected with attaching and effacing Escherichia coli in vitro. Gastroenterology 1994 May;106(5):1150-1161.
Ismaili A, Philpott DJ, Dytoc MT, Sherman PM. Signal transduction responses following adhesion of verocytotoxin-producing Escherichia coli. Infect Immun 1995 Sep;63(9):3316-3326.
Guan Y, Xue L, Ye C, Zhang D. The transmembrane signal transduction in HEp-2 cells induced by bacterial adherence. Chin Med Sci J 2000 Mar;15(1):20-23.
Celli J, Olivier M, Finlay BB. Enteropathogenic Escherichia coli mediates antiphagocytosis through the inhibition of PI 3-kinase-dependent pathways. EMBO J 2001 Mar 15;20(6):1245-1258.
Zobiack N, Rescher U, Laarmann S, Michgehl S, Schmidt MA, Gerke V. Cell-surface attachment of pedestal-forming enteropathogenic E. coli induces a clustering of raft components and a recruitment of annexin 2. J Cell Sci 2002 Jan 1;115(Pt 1):91-98.
Rescher U, Ruhe D, Ludwig C, Zobiack N, Gerke V. Annexin 2 is a phosphatidylinositol (4,5)-bisphosphate binding protein recruited to actin assembly sites at cellular membranes. J Cell Sci 2004 Jul 15;117(Pt 16):3473-3480.
Sason H, Milgrom M, Weiss AM, Melamed-Book N, Balla T, Grinstein S, et al. Enteropathogenic Escherichia coli subverts phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate upon epithelial cell infection. Mol Biol Cell 2009 Jan;20(1):544-555.
Bommarius B, Maxwell D, Swimm A, Leung S, Corbett A, Bornmann W, et al. Enteropathogenic Escherichia coli Tir is an SH2/3 ligand that recruits and activates tyrosine kinases required for pedestal formation. Mol Microbiol 2007 Mar;63(6):1748-1768.
Swimm A, Bommarius B, Li Y, Cheng D, Reeves P, Sherman M, et al. Enteropathogenic Escherichia coli use redundant tyrosine kinases to form actin pedestals. Mol Biol Cell 2004 Aug;15(8):3520-3529.
Kenny B. Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modifications. Mol Microbiol 1999 Feb;31(4):1229-1241.
Gruenheid S, DeVinney R, Bladt F, Goosney D, Gelkop S, Gish GD, et al. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat Cell Biol 2001 Sep;3(9):856-859.
Lommel S, Benesch S, Rottner K, Franz T, Wehland J, Kuhn R. Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep 2001 Sep;2(9):850-857.
Campellone KG, Giese A, Tipper DJ, Leong JM. A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol Microbiol 2002 Mar;43(5):1227-1241.
Campellone KG, Rankin S, Pawson T, Kirschner MW, Tipper DJ, Leong JM. Clustering of Nck by a 12-residue Tir phosphopeptide is sufficient to trigger localized actin assembly. J Cell Biol 2004 Feb 2;164(3):407-416.
Frankel G, Phillips AD. Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: getting off the pedestal. Cell Microbiol 2008 Mar;10(3):549-556.
Finlay BB, Rosenshine I, Donnenberg MS, Kaper JB. Cytoskeletal composition of attaching and effacing lesions associated with enteropathogenic Escherichia coli adherence to HeLa cells. Infect Immun 1992 Jun;60(6):2541-2543.
Freeman NL, Zurawski DV, Chowrashi P, Ayoob JC, Huang L, Mittal B, et al. Interaction of the enteropathogenic Escherichia coli protein, translocated intimin receptor (Tir), with focal adhesion proteins. Cell Motil Cytoskeleton 2000 Dec;47(4):307-318.
Goosney DL, DeVinney R, Pfuetzner RA, Frey EA, Strynadka NC, Finlay BB. Enteropathogenic E. coli translocated intimin receptor, Tir, interacts directly with alpha-actinin. Curr Biol 2000 Jun 15;10(12):735-738.
Cantarelli VV, Takahashi A, Yanagihara I, Akeda Y, Imura K, Kodama T, et al. Talin, a host cell protein, interacts directly with the translocated intimin receptor, Tir, of enteropathogenic Escherichia coli, and is essential for pedestal formation. Cell Microbiol 2001 Nov;3(11):745-751.
Huang L, Mittal B, Sanger JW, Sanger JM. Host focal adhesion protein domains that bind to the translocated intimin receptor (Tir) of enteropathogenic Escherichia coli (EPEC). Cell Motil Cytoskeleton 2002 Aug;52(4):255-265.
Batchelor M, Guignot J, Patel A, Cummings N, Cleary J, Knutton S, et al. Involvement of the intermediate filament protein cytokeratin-18 in actin pedestal formation during EPEC infection. EMBO Rep 2004 Jan;5(1):104-110.
Hanajima-Ozawa M, Matsuzawa T, Fukui A, Kamitani S, Ohnishi H, Abe A, et al. Enteropathogenic Escherichia coli, Shigella flexneri, and Listeria monocytogenes recruit a junctional protein, zonula occludens-1, to actin tails and pedestals. Infect Immun 2007 Feb;75(2):565-573.
Muza-Moons MM, Koutsouris A, Hecht G. Disruption of cell polarity by enteropathogenic Escherichia coli enables basolateral membrane proteins to migrate apically and to potentiate physiological consequences. Infect Immun 2003 Dec;71(12):7069-7078.
Miyake M, Hanajima M, Matsuzawa T, Kobayashi C, Minami M, Abe A, et al. Binding of intimin with Tir on the bacterial surface is prerequisite for the barrier disruption induced by enteropathogenic Escherichia coli. Biochem Biophys Res Commun 2005 Nov 25;337(3):922-927.
Simonovic I, Rosenberg J, Koutsouris A, Hecht G. Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol 2000 Aug;2(4):305-315.
Dean P, Kenny B. Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol Microbiol 2004 Nov;54(3):665-675.
Dean P, Scott JA, Knox AA, Quitard S, Watkins NJ, Kenny B. The enteropathogenic E. coli effector EspF targets and disrupts the nucleolus by a process regulated by mitochondrial dysfunction. Plops Pathog 2010 Jun 24;6(6):e1000961.
Dean P, Kenny B. A bacterial encoded protein induces extreme multinucleation and cell-cell internalization in intestinal cells. Tissue Barriers 2013 Jan 1;1(1):e22639.
Crane JK, McNamara BP, Donnenberg MS. Role of EspF in host cell death induced by enteropathogenic Escherichia coli. Cell Microbiol 2001 Apr;3(4):197-211.
Nougayrede JP, Donnenberg MS. Enteropathogenic Escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway. Cell Microbiol 2004 Nov;6(11):1097-1111.
Nougayrede JP, Foster GH, Donnenberg MS. Enteropathogenic Escherichia coli effector EspF interacts with host protein Abcf2. Cell Microbiol 2007 Mar;9(3):680-693.
Nagai T, Abe A, Sasakawa C. Targeting of enteropathogenic Escherichia coli EspF to host mitochondria is essential for bacterial pathogenesis: critical role of the 16th leucine residue in EspF. J Biol Chem 2005 Jan 28;280(4):2998-3011.
Marches O, Batchelor M, Shaw RK, Patel A, Cummings N, Nagai T, et al. EspF of enteropathogenic Escherichia coli binds sorting nexin 9. J Bacteriol 2006 Apr;188(8):3110-3115.
Alto NM, Weflen AW, Rardin MJ, Yarar D, Lazar CS, Tonikian R, et al. The type III effector EspF coordinates membrane trafficking by the spatiotemporal activation of two eukaryotic signaling pathways. J Cell Biol 2007 Sep 24;178(7):1265-1278.
Kassa EG, Zlotkin-Rivkin E, Friedman G, Ramachandran RP, Melamed-Book N, Weiss AM, et al. Enteropathogenic Escherichia coli remodels host endosomes to promote endocytic turnover and breakdown of surface polarity. PLoS Pathog 2019 Jun 26;15(6):e1007851.
Campellone KG, Cheng HC, Robbins D, Siripala AD, McGhie EJ, Hayward RD, et al. Repetitive N-WASP-binding elements of the enterohemorrhagic Escherichia coli effector EspF(U) synergistically activate actin assembly. PLoS Pathog 2008 Oct;4(10):e1000191.
Peralta-Ramirez J, Hernandez JM, Manning-Cela R, Luna-Munoz J, Garcia-Tovar C, Nougayrede JP, et al. EspF Interacts with nucleation-promoting factors to recruit junctional proteins into pedestals for pedestal maturation and disruption of paracellular permeability. Infect Immun 2008 Sep;76(9):3854-3868.
Ugalde-Silva P, Navarro-Garcia F. Coordinated transient interaction of ZO-1 and afadin is required for pedestal maturation induced by EspF from enteropathogenic Escherichia coli. Microbiologyopen 2019 Sep 30:e931.
Garber JJ, Mallick EM, Scanlon KM, Turner JR, Donnenberg MS, Leong JM, et al. Attaching-and-Effacing Pathogens Exploit Junction Regulatory Activities of N-WASP and SNX9 to Disrupt the Intestinal Barrier. Cell Mol Gastroenterol Hepatol 2017 Dec 15;5(3):273-288.
Tapia R, Kralicek SE, Hecht GA. Enteropathogenic Escherichia coli (EPEC) Recruitment of PAR Polarity Protein Atypical PKCzeta to Pedestals and Cell-Cell Contacts Precedes Disruption of Tight Junctions in Intestinal Epithelial Cells. Int J Mol Sci 2020 Jan 14;21(2):https://doi.org/10.3390/ijms21020527.
McNamara BP, Koutsouris A, O’Connell CB, Nougayrede JP, Donnenberg MS, Hecht G. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J Clin Invest 2001 Mar;107(5):621-629.
Elliott SJ, O’Connell CB, Koutsouris A, Brinkley C, Donnenberg MS, Hecht G, et al. A gene from the locus of enterocyte effacement that is required for enteropathogenic Escherichia coli to increase tight-junction permeability encodes a chaperone for EspF. Infect Immun 2002 May;70(5):2271-2277.
Viswanathan VK, Koutsouris A, Lukic S, Pilkinton M, Simonovic I, Simonovic M, et al. Comparative analysis of EspF from enteropathogenic and enterohemorrhagic Escherichia coli in alteration of epithelial barrier function. Infect Immun 2004 Jun;72(6):3218-3227.
Li Q, Zhang Q, Wang C, Li N, Li J. Invasion of enteropathogenic Escherichia coli into host cells through epithelial tight junctions. FEBS J 2008 Dec;275(23):6022-6032.
Weflen AW, Alto NM, Viswanathan VK, Hecht G. E. coli secreted protein F promotes EPEC invasion of intestinal epithelial cells via an SNX9-dependent mechanism. Cell Microbiol 2010 Jul;12(7):919-929.
Pedersen GA, Jensen HH, Schelde AB, Toft C, Pedersen HN, Ulrichsen M, et al. The basolateral vesicle sorting machinery and basolateral proteins are recruited to the site of enteropathogenic E. coli microcolony growth at the apical membrane. PLoS One 2017 Jun 21;12(6):e0179122.
Badour K, McGavin MK, Zhang J, Freeman S, Vieira C, Filipp D, et al. Interaction of the Wiskott-Aldrich syndrome protein with sorting nexin 9 is required for CD28 endocytosis and cosignaling in T cells. Proc Natl Acad Sci U S A 2007 Jan 30;104(5):1593-1598.
Merrifield CJ, Qualmann B, Kessels MM, Almers W. Neural Wiskott Aldrich Syndrome Protein (N-WASP) and the Arp2/3 complex are recruited to sites of clathrin-mediated endocytosis in cultured fibroblasts. Eur J Cell Biol 2004 Feb;83(1):13-18.
Benesch S, Polo S, Lai FP, Anderson KI, Stradal TE, Wehland J, et al. N-WASP deficiency impairs EGF internalization and actin assembly at clathrin-coated pits. J Cell Sci 2005 Jul 15;118(Pt 14):3103-3115.
Yarar D, Waterman-Storer CM, Schmid SL. SNX9 couples actin assembly to phosphoinositide signals and is required for membrane remodeling during endocytosis. Dev Cell 2007 Jul;13(1):43-56.
Lundmark R, Carlsson SR. SNX9 - a prelude to vesicle release. J Cell Sci 2009 Jan 1;122(Pt 1):5-11.
Tapia R, Kralicek SE, Hecht GA. EPEC effector EspF promotes Crumbs3 endocytosis and disrupts epithelial cell polarity. Cell Microbiol 2017 Nov;19(11):https://doi.org/10.1111/cmi.12757. Epub 2017 Jul 27.
Singh AP, Sharma S, Pagarware K, Siraji RA, Ansari I, Mandal A, et al. Enteropathogenic E. coli effectors EspF and Map independently disrupt tight junctions through distinct mechanisms involving transcriptional and post-transcriptional regulation. Sci Rep 2018 Feb 27;8(1):3719-018-22017-1.
Viswanathan VK, Lukic S, Koutsouris A, Miao R, Muza MM, Hecht G. Cytokeratin 18 interacts with the enteropathogenic Escherichia coli secreted protein F (EspF) and is redistributed after infection. Cell Microbiol 2004 Oct;6(10):987-997.
Papatheodorou P, Domanska G, Oxle M, Mathieu J, Selchow O, Kenny B, et al. The enteropathogenic Escherichia coli (EPEC) Map effector is imported into the mitochondrial matrix by the TOM/Hsp70 system and alters organelle morphology. Cell Microbiol 2006 Apr;8(4):677-689.
Kenny B, Jepson M. Targeting of an enteropathogenic Escherichia coli (EPEC) effector protein to host mitochondria. Cell Microbiol 2000 Dec;2(6):579-590.
Alto NM, Shao F, Lazar CS, Brost RL, Chua G, Mattoo S, et al. Identification of a bacterial type III effector family with G protein mimicry functions. Cell 2006 Jan 13;124(1):133-145.
Huang Z, Sutton SE, Wallenfang AJ, Orchard RC, Wu X, Feng Y, et al. Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics. Nat Struct Mol Biol 2009 Aug;16(8):853-860.
Simpson N, Shaw R, Crepin VF, Mundy R, FitzGerald AJ, Cummings N, et al. The enteropathogenic Escherichia coli type III secretion system effector Map binds EBP50/NHERF1: implication for cell signalling and diarrhoea. Mol Microbiol 2006 Apr;60(2):349-363.
Simonovic I, Arpin M, Koutsouris A, Falk-Krzesinski HJ, Hecht G. Enteropathogenic Escherichia coli activates ezrin, which participates in disruption of tight junction barrier function. Infect Immun 2001 Sep;69(9):5679-5688.
Orchard RC, Kittisopikul M, Altschuler SJ, Wu LF, Suel GM, Alto NM. Identification of F-actin as the dynamic hub in a microbial-induced GTPase polarity circuit. Cell 2012 Feb 17;148(4):803-815.
Selyunin AS, Alto NM. Activation of PAK by a bacterial type III effector EspG reveals alternative mechanisms of GTPase pathway regulation. Small GTPases 2011 Jul;2(4):217-221.
Germane KL, Spiller BW. Structural and functional studies indicate that the EPEC effector, EspG, directly binds p21-activated kinase. Biochemistry 2011 Feb 15;50(6):917-919.
Dong N, Zhu Y, Lu Q, Hu L, Zheng Y, Shao F. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell 2012 Aug 31;150(5):1029-1041.
Singh V, Davidson A, Hume PJ, Koronakis V. Pathogenic Escherichia coli Hijacks GTPase-Activated p21-Activated Kinase for Actin Pedestal Formation. mBio 2019 Aug 20;10(4):https://doi.org/10.1128/mBio.01876-19.
Singh V, Hume PJ, Davidson A, Koronakis V. EPEC Recruits a Cdc42-Specific GEF, Frabin, To Facilitate PAK Activation and Host Cell Colonization. mBio 2020 Nov 3;11(6):https://doi.org/10.1128/mBio.01423-20.
Obaishi H, Nakanishi H, Mandai K, Satoh K, Satoh A, Takahashi K, et al. Frabin, a novel FGD1-related actin filament-binding protein capable of changing cell shape and activating c-Jun N-terminal kinase. J Biol Chem 1998 Jul 24;273(30):18697-18700.
Subramanian VS, Marchant JS, Ye D, Ma TY, Said HM. Tight junction targeting and intracellular trafficking of occludin in polarized epithelial cells. Am J Physiol Cell Physiol 2007 Nov;293(5):C1717-26.
Banan A, Choudhary S, Zhang Y, Fields JZ, Keshavarzian A. Oxidant-induced intestinal barrier disruption and its prevention by growth factors in a human colonic cell line: role of the microtubule cytoskeleton. Free Radic Biol Med 2000 Mar 1;28(5):727-738.
Sumigray KD, Foote HP, Lechler T. Noncentrosomal microtubules and type II myosins potentiate epidermal cell adhesion and barrier formation. J Cell Biol 2012 Oct 29;199(3):513-525.
Matsuzawa T, Kuwae A, Yoshida S, Sasakawa C, Abe A. Enteropathogenic Escherichia coli activates the RhoA signaling pathway via the stimulation of GEF-H1. EMBO J 2004 Sep 1;23(17):3570-3582.
Matsuzawa T, Kuwae A, Abe A. Enteropathogenic Escherichia coli type III effectors EspG and EspG2 alter epithelial paracellular permeability. Infect Immun 2005 Oct;73(10):6283-6289.
Tomson FL, Viswanathan VK, Kanack KJ, Kanteti RP, Straub KV, Menet M, et al. Enteropathogenic Escherichia coli EspG disrupts microtubules and in conjunction with Orf3 enhances perturbation of the tight junction barrier. Mol Microbiol 2005 Apr;56(2):447-464.
Shaw RK, Smollett K, Cleary J, Garmendia J, Straatman-Iwanowska A, Frankel G, et al. Enteropathogenic Escherichia coli type III effectors EspG and EspG2 disrupt the microtubule network of intestinal epithelial cells. Infect Immun 2005 Jul;73(7):4385-4390.
Hardwidge PR, Deng W, Vallance BA, Rodriguez-Escudero I, Cid VJ, Molina M, et al. Modulation of host cytoskeleton function by the enteropathogenic Escherichia coli and Citrobacter rodentium effector protein EspG. Infect Immun 2005 May;73(5):2586-2594.
Glotfelty LG, Zahs A, Hodges K, Shan K, Alto NM, Hecht GA. Enteropathogenic E. coli effectors EspG1/G2 disrupt microtubules, contribute to tight junction perturbation and inhibit restoration. Cell Microbiol 2014 Dec;16(12):1767-1783.
Enomoto T. Microtubule disruption induces the formation of actin stress fibers and focal adhesions in cultured cells: possible involvement of the rho signal cascade. Cell Struct Funct 1996 Oct;21(5):317-326.
Liu BP, Chrzanowska-Wodnicka M, Burridge K. Microtubule depolymerization induces stress fibers, focal adhesions, and DNA synthesis via the GTP-binding protein Rho. Cell Adhes Commun 1998 Jun;5(4):249-255.
Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J 1999 Feb 1;18(3):578-585.
Krendel M, Zenke FT, Bokoch GM. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol 2002 Apr;4(4):294-301.
Morampudi V, Graef FA, Stahl M, Dalwadi U, Conlin VS, Huang T, et al. Tricellular Tight Junction Protein Tricellulin Is Targeted by the Enteropathogenic Escherichia coli Effector EspG1, Leading to Epithelial Barrier Disruption. Infect Immun 2016 Dec 29;85(1). https://doi.org/10.1128/IAI.00700-16. Print 2017 Jan.
Clements A, Smollett K, Lee SF, Hartland EL, Lowe M, Frankel G. EspG of enteropathogenic and enterohemorrhagic E. coli binds the Golgi matrix protein GM130 and disrupts the Golgi structure and function. Cell Microbiol 2011 Sep;13(9):1429-1439.
Furniss RC, Slater S, Frankel G, Clements A. Enterohaemorrhagic E. coli modulates an ARF6:Rab35 signaling axis to prevent recycling endosome maturation during infection. J Mol Biol 2016 May 31, 428(17):3399-3407
Wong AR, Raymond B, Collins JW, Crepin VF, Frankel G. The enteropathogenic E. coli effector EspH promotes actin pedestal formation and elongation via WASP-interacting protein (WIP). Cell Microbiol 2012 Jul;14(7):1051-1070.
Dong N, Liu L, Shao F. A bacterial effector targets host DH-PH domain RhoGEFs and antagonizes macrophage phagocytosis. EMBO J 2010 Apr 21;29(8):1363-1376.
Wong AR, Clements A, Raymond B, Crepin VF, Frankel G. The interplay between the Escherichia coli Rho guanine nucleotide exchange factor effectors and the mammalian RhoGEF inhibitor EspH. mBio 2012 Jan 17;3(1). https://doi.org/10.1128/mBio.00250-11. Print 2012.
Guttman JA, Kazemi P, Lin AE, Vogl AW, Finlay BB. Desmosomes are unaltered during infections by attaching and effacing pathogens. Anat Rec (Hoboken) 2007 Feb;290(2):199-205.
Roxas JL, Monasky RC, Roxas BAP, Agellon AB, Mansoor A, Kaper JB, et al. Enteropathogenic Escherichia coli EspH-Mediated Rho GTPase Inhibition Results in Desmosomal Perturbations. Cell Mol Gastroenterol Hepatol 2018 Apr 27;6(2):163-180.
Kim J, Thanabalasuriar A, Chaworth-Musters T, Fromme JC, Frey EA, Lario PI, et al. The bacterial virulence factor NleA inhibits cellular protein secretion by disrupting mammalian COPII function. Cell Host Microbe 2007 Sep 13;2(3):160-171.
Thanabalasuriar A, Koutsouris A, Weflen A, Mimee M, Hecht G, Gruenheid S. The bacterial virulence factor NleA is required for the disruption of intestinal tight junctions by enteropathogenic Escherichia coli. Cell Microbiol 2010 Jan;12(1):31-41.
Thanabalasuriar A, Kim J, Gruenheid S. The inhibition of COPII trafficking is important for intestinal epithelial tight junction disruption during enteropathogenic Escherichia coli and Citrobacter rodentium infection. Microbes Infect 2013 Sep-Oct;15(10-11):738-744.
Arbeloa A, Bulgin RR, MacKenzie G, Shaw RK, Pallen MJ, Crepin VF, et al. Subversion of actin dynamics by EspM effectors of attaching and effacing bacterial pathogens. Cell Microbiol 2008 Jul;10(7):1429-1441.
Arbeloa A, Garnett J, Lillington J, Bulgin RR, Berger CN, Lea SM, et al. EspM2 is a RhoA guanine nucleotide exchange factor. Cell Microbiol 2010 May 1;12(5):654-664.
Simovitch M, Sason H, Cohen S, Zahavi EE, Melamed-Book N, Weiss A, et al. EspM inhibits pedestal formation by enterohaemorrhagic Escherichia coli and enteropathogenic E. coli and disrupts the architecture of a polarized epithelial monolayer. Cell Microbiol 2010 Apr 1;12(4):489-505.
Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol 1997 Oct;273(4):C1378-85.
Manjarrez-Hernandez HA, Amess B, Sellers L, Baldwin TJ, Knutton S, Williams PH, et al. Purification of a 20 kDa phosphoprotein from epithelial cells and identification as a myosin light chain. Phosphorylation induced by enteropathogenic Escherichia coli and phorbol ester. FEBS Lett 1991 Nov 4;292(1-2):121-127.
Manjarrez-Hernandez HA, Baldwin TJ, Aitken A, Knutton S, Williams PH. Intestinal epithelial cell protein phosphorylation in enteropathogenic Escherichia coli diarrhoea. Lancet 1992 Feb 29;339(8792):521-523.
Manjarrez-Hernandez HA, Baldwin TJ, Williams PH, Haigh R, Knutton S, Aitken A. Phosphorylation of myosin light chain at distinct sites and its association with the cytoskeleton during enteropathogenic Escherichia coli infection. Infect Immun 1996 Jun;64(6):2368-2370.
Yuhan R, Koutsouris A, Savkovic SD, Hecht G. Enteropathogenic Escherichia coli-induced myosin light chain phosphorylation alters intestinal epithelial permeability. Gastroenterology 1997 Dec;113(6):1873-1882.
Baldwin TJ, Brooks SF, Knutton S, Manjarrez Hernandez HA, Aitken A, Williams PH. Protein phosphorylation by protein kinase C in HEp-2 cells infected with enteropathogenic Escherichia coli. Infect Immun 1990 Mar;58(3):761-765.
Hecht G, Pestic L, Nikcevic G, Koutsouris A, Tripuraneni J, Lorimer DD, et al. Expression of the catalytic domain of myosin light chain kinase increases paracellular permeability. Am J Physiol 1996 Nov;271(5 Pt 1):C1678-84.
Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 2002 Jul;123(1):163-172.
Tomson FL, Koutsouris A, Viswanathan VK, Turner JR, Savkovic SD, Hecht G. Differing roles of protein kinase C-zeta in disruption of tight junction barrier by enteropathogenic and enterohemorrhagic Escherichia coli. Gastroenterology 2004 Sep;127(3):859-869.
Conlin VS, Wu X, Nguyen C, Dai C, Vallance BA, Buchan AM, et al. Vasoactive intestinal peptide ameliorates intestinal barrier disruption associated with Citrobacter rodentium-induced colitis. Am J Physiol Gastrointest Liver Physiol 2009 Oct;297(4):G735-50.
Malladi V, Shankar B, Williams PH, Balakrishnan A. Enteropathogenic Escherichia coli outer membrane proteins induce changes in cadherin junctions of Caco-2 cells through activation of PKCalpha. Microbes Infect 2004 Jan;6(1):38-50.
Crane JK, Oh JS. Activation of host cell protein kinase C by enteropathogenic Escherichia coli. Infect Immun 1997 Aug;65(8):3277-3285.
Rodriguez-Boulan E, Macara IG. Organization and execution of the epithelial polarity programme. Nat Rev Mol Cell Biol 2014 Apr;15(4):225-242.
Gassama-Diagne A, Yu W, ter Beest M, Martin-Belmonte F, Kierbel A, Engel J, et al. Phosphatidylinositol-3,4,5-trisphosphate regulates the formation of the basolateral plasma membrane in epithelial cells. Nat Cell Biol 2006 Sep;8(9):963-970.
Muza-Moons MM, Schneeberger EE, Hecht GA. Enteropathogenic Escherichia coli infection leads to appearance of aberrant tight junctions strands in the lateral membrane of intestinal epithelial cells. Cell Microbiol 2004 Aug;6(8):783-793.
Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol 2000 Aug;2(8):531-539.
Suzuki A, Yamanaka T, Hirose T, Manabe N, Mizuno K, Shimizu M, et al. Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J Cell Biol 2001 Mar 19;152(6):1183-1196.
Johansson A, Driessens M, Aspenstrom P. The mammalian homologue of the Caenorhabditis elegans polarity protein PAR-6 is a binding partner for the Rho GTPases Cdc42 and Rac1. J Cell Sci 2000 Sep;113 (Pt 18):3267-3275.
Hurd TW, Gao L, Roh MH, Macara IG, Margolis B. Direct interaction of two polarity complexes implicated in epithelial tight junction assembly. Nat Cell Biol 2003 Feb;5(2):137-142.
Hurd TW, Fan S, Liu CJ, Kweon HK, Hakansson K, Margolis B. Phosphorylation-dependent binding of 14-3-3 to the polarity protein Par3 regulates cell polarity in mammalian epithelia. Curr Biol 2003 Dec 2;13(23):2082-2090.
Izaki T, Kamakura S, Kohjima M, Sumimoto H. Phosphorylation-dependent binding of 14-3-3 to Par3beta, a human Par3-related cell polarity protein. Biochem Biophys Res Commun 2005 Apr 1;329(1):211-218.
Lemmers C, Michel D, Lane-Guermonprez L, Delgrossi MH, Medina E, Arsanto JP, et al. CRB3 binds directly to Par6 and regulates the morphogenesis of the tight junctions in mammalian epithelial cells. Mol Biol Cell 2004 Mar;15(3):1324-1333.
Sotillos S, Diaz-Meco MT, Caminero E, Moscat J, Campuzano S. DaPKC-dependent phosphorylation of Crumbs is required for epithelial cell polarity in Drosophila. J Cell Biol 2004 Aug 16;166(4):549-557.
Wei Z, Li Y, Ye F, Zhang M. Structural basis for the phosphorylation-regulated interaction between the cytoplasmic tail of cell polarity protein crumbs and the actin-binding protein moesin. J Biol Chem 2015 May 1;290(18):11384-11392.
Savkovic SD, Koutsouris A, Hecht G. PKC zeta participates in activation of inflammatory response induced by enteropathogenic E. coli. Am J Physiol Cell Physiol 2003 Sep;285(3):C512-21.
Zyrek AA, Cichon C, Helms S, Enders C, Sonnenborn U, Schmidt MA. Molecular mechanisms underlying the probiotic effects of Escherichia coli Nissle 1917 involve ZO-2 and PKCzeta redistribution resulting in tight junction and epithelial barrier repair. Cell Microbiol 2007 Mar;9(3):804-816.
Fan S, Weight CM, Luissint AC, Hilgarth RS, Brazil JC, Ettel M, et al. Role of JAM-A tyrosine phosphorylation in epithelial barrier dysfunction during intestinal inflammation. Mol Biol Cell 2019 Mar 1;30(5):566-578.
Jain S, Suzuki T, Seth A, Samak G, Rao R. Protein kinase Czeta phosphorylates occludin and promotes assembly of epithelial tight junctions. Biochem J 2011 Jul 15;437(2):289-299.
Andreeva AY, Krause E, Muller EC, Blasig IE, Utepbergenov DI. Protein kinase C regulates the phosphorylation and cellular localization of occludin. J Biol Chem 2001 Oct 19;276(42):38480-38486.
Aono S, Hirai Y. Phosphorylation of claudin-4 is required for tight junction formation in a human keratinocyte cell line. Exp Cell Res 2008 Nov 1;314(18):3326-3339.
Avila-Flores A, Rendon-Huerta E, Moreno J, Islas S, Betanzos A, Robles-Flores M, et al. Tight-junction protein zonula occludens 2 is a target of phosphorylation by protein kinase C. Biochem J 2001 Dec 1;360(Pt 2):295-304.
Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, et al. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005 Sep;85(9):1139-62.
Zeissig S, Bürgel N, Günzel D, Richter J, Mankertz J, Wahnschaffe U, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007 Jan;56(1):61-72.
Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest 2005 Oct;115(10):2702-15.
Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest 2006 Feb;86(2):191-201.
Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, et al. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci 2006 May 15;119(Pt 10):2095-106.
Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR 2nd, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol 2010 Apr 5;189(1):111-26.
Jung HC, Eckmann L, Yang SK, Panja A, Fierer J, Morzycka-Wroblewska E, et al. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest 1995 Jan;95(1):55-65.
Ramirez K, Huerta R, Oswald E, Garcia-Tovar C, Hernandez JM, Navarro-Garcia F. Role of EspA and intimin in expression of proinflammatory cytokines from enterocytes and lymphocytes by rabbit enteropathogenic Escherichia coli-infected rabbits. Infect Immun 2005 Jan;73(1):103-13.
Salazar-Gonzalez H, Navarro-Garcia F. Intimate adherence by enteropathogenic Escherichia coli modulates TLR5 localization and proinflammatory host response in intestinal epithelial cells. Scand J Immunol 2011 Apr;73(4):268-83.
Wong AR, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, et al. Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol Microbiol 2011 Jun;80(6):1420-1438.
Hemrajani C, Berger CN, Robinson KS, Marches O, Mousnier A, Frankel G. NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection. Proc Natl Acad Sci U S A 2010 Feb 16;107(7):3129-3134.
Royan SV, Jones RM, Koutsouris A, Roxas JL, Falzari K, Weflen AW, et al. Enteropathogenic E. coli non-LEE encoded effectors NleH1 and NleH2 attenuate NF-kappaB activation. Mol Microbiol 2010 Dec;78(5):1232-1245.
Kralicek SE, Nguyen M, Rhee KJ, Tapia R, Hecht G. EPEC NleH1 is significantly more effective in reversing colitis and reducing mortality than NleH2 via differential effects on host signaling pathways. Lab Invest 2018 Apr;98(4):477-488.
Savkovic SD, Koutsouris A, Hecht G. Activation of NF-kappaB in intestinal epithelial cells by enteropathogenic Escherichia coli. Am J Physiol 1997 Oct;273(4):C1160-7.
Savkovic SD, Ramaswamy A, Koutsouris A, Hecht G. EPEC-activated ERK1/2 participate in inflammatory response but not tight junction barrier disruption. Am J Physiol Gastrointest Liver Physiol 2001 Oct;281(4):G890-8.
Czerucka D, Dahan S, Mograbi B, Rossi B, Rampal P. Saccharomyces boulardii preserves the barrier function and modulates the signal transduction pathway induced in enteropathogenic Escherichia coli-infected T84 cells. Infect Immun 2000 Oct;68(10):5998-6004.
Czerucka D, Dahan S, Mograbi B, Rossi B, Rampal P. Implication of mitogen-activated protein kinases in T84 cell responses to enteropathogenic Escherichia coli infection. Infect Immun 2001 Mar;69(3):1298-1305.
Diaz-Meco MT, Berra E, Municio MM, Sanz L, Lozano J, Dominguez I, et al. A dominant negative protein kinase C zeta subspecies blocks NF-kappa B activation. Mol Cell Biol 1993 Aug;13(8):4770-4775.
Folgueira L, McElhinny JA, Bren GD, MacMorran WS, Diaz-Meco MT, Moscat J, et al. Protein kinase C-zeta mediates NF-kappa B activation in human immunodeficiency virus-infected monocytes. J Virol 1996 Jan;70(1):223-231.
Lozano J, Berra E, Municio MM, Diaz-Meco MT, Dominguez I, Sanz L, et al. Protein kinase C zeta isoform is critical for kappa B-dependent promoter activation by sphingomyelinase. J Biol Chem 1994 Jul 29;269(30):19200-19202.
Berra E, Diaz-Meco MT, Lozano J, Frutos S, Municio MM, Sanchez P, et al. Evidence for a role of MEK and MAPK during signal transduction by protein kinase C zeta. EMBO J 1995 Dec 15;14(24):6157-6163.
Berra E, Diaz-Meco MT, Dominguez I, Municio MM, Sanz L, Lozano J, et al. Protein kinase C zeta isoform is critical for mitogenic signal transduction. Cell 1993 Aug 13;74(3):555-563.
Collington GK, Booth IW, Knutton S. Rapid modulation of electrolyte transport in Caco-2 cell monolayers by enteropathogenic Escherichia coli (EPEC) infection. Gut 1998 Feb;42(2):200-7.
Collington GK, Booth IW, Donnenberg MS, Kaper JB, Knutton S. Enteropathogenic Escherichia coli virulence genes encoding secreted signalling proteins are essential for modulation of Caco-2 cell electrolyte transport. Infect Immun 1998 Dec;66(12):6049-53.
Hecht G, Koutsouris A. Enteropathogenic E. coli attenuates secretagogue-induced net intestinal ion transport but not Cl- secretion. Am J Physiol 1999 Mar;276(3):G781-8.
Hecht G, Hodges K, Gill RK, Kear F, Tyagi S, Malakooti J, et al. Differential regulation of Na+/H+ exchange isoform activities by enteropathogenic E. coli in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 2004 Aug;287(2):G370-8.
Hodges K, Alto NM, Ramaswamy K, Dudeja PK, Hecht G. The enteropathogenic Escherichia coli effector protein EspF decreases sodium hydrogen exchanger 3 activity. Cell Microbiol 2008 Aug;10(8):1735-1745.
Hodges K, Gill R, Ramaswamy K, Dudeja PK, Hecht G. Rapid activation of Na+/H+ exchange by EPEC is PKC mediated. Am J Physiol Gastrointest Liver Physiol 2006 Nov;291(5):G959-68.
Martinez E, Schroeder GN, Berger CN, Lee SF, Robinson KS, Badea L, et al. Binding to Na(+) /H(+) exchanger regulatory factor 2 (NHERF2) affects trafficking and function of the enteropathogenic Escherichia coli type III secretion system effectors Map, EspI and NleH. Cell Microbiol 2010 Dec;12(12):1718-1731.
Borenshtein D, Nambiar PR, Groff EB, Fox JG, Schauer DB. Development of fatal colitis in FVB mice infected with Citrobacter rodentium. Infect Immun 2007 Jul;75(7):3271-81.
Borenshtein D, McBee ME, Schauer DB. Utility of the Citrobacter rodentium infection model in laboratory mice. Curr Opin Gastroenterol. 2008 Jan;24(1):32-7.
Borenshtein D, Schlieper KA, Rickman BH, Chapman JM, Schweinfest CW, Fox JG, Schauer DB. Decreased expression of colonic Slc26a3 and carbonic anhydrase iv as a cause of fatal infectious diarrhea in mice. Infect Immun 2009 Sep;77(9):3639-50.
Gill RK, Borthakur A, Hodges K, Turner JR, Clayburgh DR, Saksena S, et al. Mechanism underlying inhibition of intestinal apical Cl/OH exchange following infection with enteropathogenic E. coli. J Clin Invest 2007 Feb;117(2):428-437.
Gujral T, Kumar A, Priyamvada S, Saksena S, Gill RK, Hodges K, et al. Mechanisms of DRA recycling in intestinal epithelial cells: effect of enteropathogenic E. coli. Am J Physiol Cell Physiol 2015 Dec 15;309(12):C835-46.
Dean P, Maresca M, Schuller S, Phillips AD, Kenny B. Potent diarrheagenic mechanism mediated by the cooperative action of three enteropathogenic Escherichia coli-injected effector proteins. Proc Natl Acad Sci U S A 2006 Feb 7;103(6):1876-1881.
Guttman JA, Samji FN, Li Y, Deng W, Lin A, Finlay BB. Aquaporins contribute to diarrhoea caused by attaching and effacing bacterial pathogens. Cell Microbiol 2007 Jan;9(1):131-41.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Tapia, R., Hecht, G. (2022). The Myriad Ways Enteropathogenic Escherichia coli (EPEC) Alters Tight Junctions. In: González-Mariscal, L. (eds) Tight Junctions. Springer, Cham. https://doi.org/10.1007/978-3-030-97204-2_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-97204-2_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-97203-5
Online ISBN: 978-3-030-97204-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)