
Abstract
It is well supported by the literature that the proliferation and migration of pulmonary arterial smooth muscle cells (PASMCs) are critical for the development of pulmonary arterial hypertension (PAH). Long intergenic noncoding RNA COX2 (lincRNA-COX2) is a regulator of inflammation and might be conducive to the progression of atherosclerosis, while its role in PAH is still unclear. This study was performed to explore the role and mechanism of lincRNA-COX2 in PASMCs proliferation and migration in an anaerobic environment. PASMCs were treated by hypoxia to construct PAH cell models. RT-PCR and western blot were recruited to evaluate the expression levels of lincRNA-COX2, miR-let-7a and STAT3. Their roles in proliferation and cell and migration of PASMCs were determined by the CCK-8 assay, wound-healing assay, and flow cytometry. In peripheral blood samples from PAH patients and hypoxic PASMCs, lincRNA-COX2 expression was enhanced. Silencing lincRNA-COX2 inhibited hypoxia-induced PASMCs proliferation by influencing the G2/M phase of the cell cycle. Meanwhile, lincRNA-COX2 regulated STAT3 through miR-let-7a and its effects on hypoxic PASMCs worked through miR-let-7a/STAT3 axis. To conclude, silencing lincRNA-COX2 attenuated the development of hypoxic PASMCs. LincRNA-COX2/miR-let-7a/STAT3 axis might be considered as a novel target to treat PAH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pulmonary arterial hypertension (PAH) refers to the mPAP (mean pulmonary artery pressure) greater than 25 mmHg in resting conditions [1, 2]. Patients often have high pulmonary artery pressure without obvious symptoms, resulting in right heart failure and even death. Pulmonary vascular remodeling and increased pulmonary artery resistance are the critical pathogenesis of PAH [3]. Pulmonary artery remodeling is mainly caused by abnormal growth, excessive proliferation and decreased anti-apoptotic ability of smooth muscle cells [4]. Therefore, the inhibition of pulmonary arteriole smooth muscle cells (PASMCs) proliferation or the induction of smooth muscle cell apoptosis is an effective therapeutic strategy for PAH.
Long noncoding RNAs (lncRNAs), which are more than 200 nt in length, form a newly discovered class of RNA molecules with strong biological regulatory capacity. Quinodoz et al. identified thousands of lncRNAs with conserved sequences in mammals, which play critical roles in the development of immune system diseases, cardiovascular system diseases, tumor, endocrine metabolic diseases, etc. [5]. For instance, lncRNA MIAT is involved in myocardial infarction by regulating miR-150-5p and vascular endothelial growth factor [6]. Through binding to intracellular MDM2 protein, lncRNA-p21 enhances the transcriptional activity of p53 protein, regulates apoptosis and proliferation of vascular endothelial cells, and participates in the occurrence and development of atherosclerotic diseases [7]. In addition, long intergenic noncoding COX2 (lincRNA-COX2) might contribute to the progression of atherosclerosis. In 2009, Guttman et al. found that lincRNA-COX2 expression increased by more than 1000 times in LPS-stimulated dendritic cells, and the locus analysis showed that the gene was located in the 51 kb upstream of oxidase reductase in prostaglandin [8]. In 2013, Carpenter et al. confirmed that lincRNA-COX2 was a pro-inflammatory molecule that can mediate the LPS-induced secretion of macrophages inflammatory factor CCL5 and activation of NF-κB inflammatory signaling pathway [9], whereas the research of lincRNA-COX2 in pulmonary hypertension is still limited.
MicroRNAs (miRNAs) regulate multiple pathophysiological processes, such as vascular remodeling, tissue repair, and lipid metabolism [10,11,12]. The abnormal expression of miRNAs has been shown in various lung diseases, including PAH [13]. miRNAs are correlated with the severity of PAH and could curb PAH pathological progression, offering a novel therapy target for PAH [14]. The significant downregulation of let-7a was observed in animals’ lungs with PAH [13]. A similar reduction in let-7a was also confirmed in cells exposed to chronic hypoxia, indicating a feasible role of let-7a in PAH development [15]. Furthermore, the expression of let-7 correlated negatively with the severity of PAH in patients who had systemic scleroderma [16]. Also, it was found that let-7a may be helpful for moderating PAH progression via curbing PASMC growth by targeting STAT3 signaling [17]. Numerous researches demonstrated that the dysregulation of STAT3 signaling plays a key role in PAH pathogenesis, and blocking STAT3 signaling with specific inhibitors such as plumbagin and dehydroepiandrosterone can reduced or reversed PAH symptoms [18,19,20].
The present study was designed to explore the function and mechanism of linc-COX2 in PAH. At the beginning of the study, we found that the expression of lincRNA-COX2 in the PAH cell model was significantly different from that of normal PASMCs and functioned as a sponge of let-7a. Thus, this study further tried to explore the function of lincRNA-COX2 in the development of PAH, with the involvement of the miR-let-7a/STAT3 axis.
Materials and methods
Human samples
Human peripheral blood samples were obtained from 34 patients admitted in structural heart disease department of the first affiliated hospital of Xi’an Jiaotong University. All patients in the case group underwent right cardiac catheterization, and the inspection results met the diagnostic criteria for PAH which referred that mPAP was above 25 mmHg, pulmonary arteriole wedge pressure (PAWP) was lower than 15 mmHg, and pulmonary vascular resistance was higher than 3 Wood units. And all of them were diagnosed as pulmonary hypertension. The control group included 19 healthy volunteers. All candidates with serious liver disease and kidney failure were ruled out. The peripheral venous blood (5–10 ml) of all selected subjects was collected in a labelled EDTA-K2 anticoagulation tube, and stored at − 80 °C. This experiment was approved by the ethics committee of the first affiliated hospital of Xi'an Jiaotong University. Informed consent forms were obtained from 19 healthy volunteers and 34 patients with a definite diagnosis of pulmonary hypertension before peripheral blood samples were taken.
Cell culture and treatment
Human PASMCs were obtained from Gibco (Life Technologies, Zug, Switzerland) and cultured in SmGM-2™ (smooth muscle cell growth medium-2) containing 5% FBS (S00725, Gibco BRL/Invitrogen Inc., Carlsbad, CA, USA), 0.5 ng/ml human recombinant epidermal growth factor, 2 ng/ml human recombinant fibroblast growth factor, 5 μg/ml insulin, 50 μg/ml gentamicin, 10 U/ml penicillin, and 10 μg/ml streptomycin in a humidified incubator (5% CO2, 37 °C).
sh-NC, sh-COX2, inhibitor NC, and miR-let-7a inhibitor were obtained from RiboBio (Guangdong, China). PASMCs were placed in six-well dishes for 24 h, and then all of the plasmids were treated with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). For constructing a hypoxic condition, human PASMCs were incubated for 0, 6, 12, 24, and 48 h with a gas mixture constituted with 92% N2, − 5% CO2, − 3% O2.
RNA extraction and quantification
Total RNA was extracted via the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and quantified by NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific, Rockford, IL). Reverse transcription reactions were completed using the PrimeScript RT reagent kit (Qiagen). Real-time PCR was performed using TransStart™ SYBR Green qPCR Supermix (TransGenBiotech, Beijing, China). The primers recruited were all synthesized by Huda Gene (Shenzhen Huada Gene Co., Ltd.,) (Supplementary Table 1). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or U6 was used as the internal reference for lncRNA gene, mRNA, or microRNA gene. The fold changes of genes were evaluated using the 2−∆∆Ct method.
Western blotting
Proteins were extracted with RIPA lysis buffer (Thermo scientific, Rockford, IL, USA), and the concentrations were measured using the BCA assay kit (Pierce, Rockford, IL, USA). Then, 12% SDS–PAGE was used to separate proteins before being transferred to PVDF membranes (Sigma) which were then blocked with 5% skim milk in TBST for 2 h. The membranes were incubated at 4 °C overnight with the primary antibodies against PCNA (proliferating cell nuclear antigen), Ki67, p-Rb, cyclin B1, MMP-2, and MMP-9 and then with HRP-conjugated secondary antibodies for 1 h. Chemiluminescence (Santa Cruz, CA, USA) was used to visualize the binding proteins and the Quantity One v4.6.2 software was employed for further analyzed.
Cell proliferation and migration assays
CCK-8 assay was employed to measure cell proliferation capacity. PASMCs (1 × 104 cells/well) were cultured in 96-well plates for 24 h. Then, 10 µl CCK-8 solution was added into each well and cells were maintained for 4 h at 37 °C. Finally, the absorbance (A) at 450 nm was evaluated by a microplate reader (Bio-Rad, Inc., Hercules, CA, USA).
Wound-healing assay was employed to assess cell migration. Briefly, the fused human PASMCs cultured on a six-well plate were scratched with pipette tips, and each well formed a 1-mm-wide acellular line. The separated cells were rinsed with PBS. Photographs were taken from the same areas as those recorded at zero time after 24 h incubation.
Cell cycle analysis
Flow cytometry was recruited to evaluate cell cycle analysis as described previously [21]. Briefly, cells were fixed in precooled 70% ethanol at 4 °C to begin with. Then, they were resuspended with PBS containing 0.25% Triton X-100 for 15 min on ice and stained in a propidium iodide solution (P4170, Sigma, St. Louis, MO, USA) with 100 µg/ml RNase for 30 min in the dark. Cell cycle analysis was completed via flow cytometry (Beckman Coulter, USA).
Luciferase assay
To confirm whether miR-let-7 targeted linc-COX2 directly, PCR was used to amplify the 3′-UTR (wild type and mutated type) of linc-COX2 which was then cloned it into the pmiRRB-Report vector. After 48 h of transfection, the luciferase activity was evaluated via the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA).
Statistical analysis
Results are shown as mean ± SD. SPSS 18.0 software was recruited to analyze data and perform comparisons between two groups via the paired t test and between multiple groups using one-way analysis of variance (ANOVA). The Pearson correlation was employed to evaluate the correlation between linc-Cox2 and miR-let-7a. P < 0.05 was considered statistically significant.
Results
LincRNA-COX2 is upregulated in hypoxic PASMCs
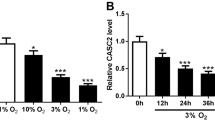
qRT-PCR was used to detect lincRNA-COX2 expression and the results indicated that lincRNA-COX2 expression was enhanced in peripheral blood samples from PAH patients (Fig. 1a) and hypoxic PASMCs in a time-dependent manner (Fig. 1b).

The expression of lincRNA-COX2 is enhanced in PAH. RT-PCR was used to detect the mRNA expression of linc-COX2 in a peripheral blood samples from PAH patients and b hypoxic PASMCs. *P < 0.05 vs. healthy controls or hypoxic treatment for 0 h
LincRNA-COX2 silencing inhibits hypoxia-induced PASMCs proliferation by influencing the G2/M phase of the cell cycle
Exorbitant PASMCs proliferation is one of the major pathological features of PAH and make contribution to the vascular remodeling [22, 23]. Thus, we explored the involvement of lincRNA-COX2 in PASMC proliferation. sh-COX2 and pcDNA-COX2 was transfected into human PASMCs to suppress or enhance lincRNA-COX2 expression with hypoxia treatment (Fig. 2a) (Supplementary Fig. 1a). CCK-8 assay results showed that cell proliferation was reduced by sh-COX2, while increased by pcDNA-COX2 (Fig. 2b) (Supplementary Fig. 1b). And cell cycle analysis showed that sh-COX2 moderated G2/M cell cycle arrest (Fig. 2c). Furthermore, protein expression of proliferation markers Ki67 and PCNA and G2/M cell cycle-related genes p-Rb and cyclin B1 were all significantly downregulated by sh-COX2 (Fig. 2d, e). All these data suggested lincRNA-COX2 was a crucial factor in regulating hypoxia-induced PASMCs proliferation and cell cycle.

Silencing lincRNA-COX2 curbs the proliferation of hypoxia-induced PASMCs. PASMCs were transfected with sh-COX2 and treated with hypoxia for 24 h. a RT-PCR was used to evaluate the transfection efficiency of sh-COX2. b The proliferation of hypoxic PASMCs was assessed by the CCK-8 assay. c The effect of sh-COX2 on the cell cycle was evaluated assessed via the flow cytometry assay. d, e Proliferation-related protein Ki-67 and PCNA and G2/M stage arrest-related proteins p-Rb and cyclin B1 expression were assessed by western blot. *P < 0.05 vs. sh-NC
LincRNA-COX2 silencing inhibits the migration of hypoxia-induced PASMCs
Subsequently, we analyzed the effects of lincRNA-COX2 on the migration capacity of human PASMC induced by hypoxia treatment, which was also a vital factor contributing to the development of cardiovascular diseases. Through the wound-healing assay, it was shown that there was an evident decrease in migration in sh-COX2 transfected cells (Fig. 3a). Similarly, the migration-related genes MMP-2 and MMP-9 were also downregulated by sh-COX2 transfection (Fig. 3b, c). Therefore, it was initially considered that sh-COX2 curbed migration capacity of hypoxia-induced PASMCs.

LincRNA-COX2 silencing reduces the migration of hypoxia-induced PASMCs. PASMCs were transfected with sh-COX2 and treated with hypoxia for 24 h. a Cell migration was analyzed via wound healing assays. b-c The protein expression of MMP-2 and MMP-9 was measured through western blotting. *P < 0.05 vs. sh-NC
LincRNA-COX2 functions as a sponge of miR-let-7a
Since miRNAs play critical roles in the effects of lincRNA, we tried to determine whether there was a miRNA bound with linc-COX2 to determine the underlying mechanism of linc-COX2 in PAH. RNAhybrid (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid/) was recruited to predict the binding sequences of linc-COX2 and miR-let-7a (Fig. 4a). Luciferase reporter plasmids containing the wild-type or mutated miR-let-7a binding sites of COX2 were constructed. The results showed that the aberrant expression of miR-let-7a evidently suppressed the luciferase activity of COX2-WT, while no significant differences were detected in the COX2-MUT group (Fig. 4b). RT-PCR results demonstrated that miR-let-7a expression was downregulated in both peripheral blood samples of PAH patients and hypoxic PASMCs (Fig. 4c, d). In addition, miR-let-7a expression was upregulated in hypoxic PASMCs transfected with sh-COX2 (Fig. 4e). Furthermore, the results of the Pearson correlation showed that lincRNA-COX2 and miR-let-7a expression was negatively correlated (Fig. 4f). In summary, all of these results showed that linc-COX2 directly targeted miR-let-7a.

LincRNA-COX2 functions as a sponge of miR-let-7a. PASMCs were transfected with sh-COX2 and treated with hypoxia for 24 h. a The predicted wild-type or mutated miR-let-7a binding sites in lincRNA-COX2. b The Luciferase reporter assay was used to detect the fluorescence intensity with mimic NC or miR-let-7a co-transfected with wt-COX2 or mut-COX2. *P < 0.05 vs. mimic NC. RT-PCR was recruited to detect miR-let-7a expression in c the peripheral blood samples of PAH patients and d hypoxic PASMCs. *P < 0.05 vs. Control. e The expression of miR-let-7a was assessed by qRT-PCR after sh-COX2 transfection. *P < 0.05 vs. sh-NC. f Pearson correlation was performed to evaluate the correlation between linc-COX2 and miR-let-7a
LincRNA-COX2 regulates STAT3 through miR-let-7a
Although we had already confirmed that miR-let-7a was a target of lincRNA-COX2, the role of miR-let-7a in lincRNA-COX2-mediated effects in hypoxic PASMCs remained unclear. Thus, we further explored whether lincRNA-COX2 regulates STAT3 through miR-let-7a. Firstly, the transfection efficiency of inhibitor NC and that of the miR-let-7a inhibitor were detected by qRT-PCR (Fig. 5a). In addition, the p-STAT3 protein level was decreased in the sh-COX2 group and rebounded by miR-let-7a inhibitor and sh-COX2 co-transfection (Fig. 5b, c). Above all, these data suggested that lincRNA-COX2 regulates STAT3 through miR-let-7a.

LincRNA-COX2 regulates STAT3 through miR-let-7a. sh-COX2 was transfected into PASMCs and then hypoxia treatment for 24 h was performed. a The transfection efficiency of miR-let-7a inhibition was evaluated by qRT-PCR. b, c The protein expression of p-STAT3 was assessed via western blot. *P < 0.05 vs. sh-NC; #P < 0.05 vs. sh-COX2+ inhibitor NC
LincRNA-COX2 regulates PAH development through the miR-let-7a/STAT3 axis
Activation of the STAT3 pathway has been reported in PAH animal models and it could regulate the growth of smooth muscle cells (SMCs) [14]. To confirm the underlying mechanism of linc-COX2 in PAH development, colivelin (STAT3 activator) was used to restore p-STAT3 expression. Notably, colivelin alleviated the inhibitory roles of sh-COX2 in proliferation (Fig. 6a), G2/M phase arrest of cell cycle (Fig. 6b) and cell migration (Fig. 6c) in hypoxic PASMCs. Overall, it was concluded that sh-COX2 might regulate PASMC proliferation and migration by miR-let-7a/STAT3 axis.

LincRNA-COX2 regulates PAH development through the miR-let-7a/STAT3 axis. PASMCs transfected with sh-COX2 were treated by hypoxia for 24 h and/or colivelin. a CCK-8 was recruited to analyze cell proliferation. b The cell cycle was assessed via flow cytometry. c Cell migration was evaluated by the wound-healing assay. *P < 0.05 vs. sh-NC, #P < 0.05 vs. sh-COX2
Discussion
The functional roles and molecular mechanisms of lncRNAs in pulmonary hypertension have been largely reported [24,25,26]. In this research, it was discovered that lincRNA-COX2 affected PAH development for the first time. The staining of several lincRNA-Cox2 KO mouse organs with a lacZ reporter cassette suggested that lincRNA-Cox2 expression was increased in lungs; RNA-seq analysis further validated this observation [27]. In addition, lincRNA-COX2 was highly expressed by multiple stimulations in various cells, including EV-induced microglia, TNFα-induced intestinal epithelial cells [28, 29], and LPS-stimulated macrophages [9]. Herein, we firstly found that lincRNA-COX2 expression was boosted in peripheral blood samples from PAH patients and hypoxia-treated PASMCs.
Pulmonary arterial hypertension has the characteristics of increased progression in pulmonary vascular resistance and obliterative pulmonary vascular remodeling with sophisticated and unclear mechanism. It was known that deviant proliferation and migration of PASMCs were crucial pathological features of pulmonary vascular remodeling in PAH pathogenesis [23]. The further characterization of lincRNA-Cox2 might be helpful for the exploration of new drug targets for atherosclerosis [9, 30]. The knockdown of lincRNA-Cox2 was reported to reverse the expression of LPS-induced cell cycle genes in mouse primary microglia leading to the inhibition of microglial proliferation in vitro [31]. In this study, it was observed that silencing lincRNA-COX2 not only inhibited hypoxia-induced PASMC proliferation by influencing the G2/M phase of the cell cycle, but also curbed PASMC migration.
Multiple researches have demonstrated that let-7, a tumor suppressor, was poorly expressed or lost in various human cancers. The restoration of let-7 expression was considered as a therapeutic option for cancer [32, 33]. Also, it was reported that let-7a was downregulated in lung injury and PAH in vivo models [13, 16, 34], which was in accordance with our results. Recently, the roles of let-7 in cardiovascular biology and disease were explored, and let-7 was viewed as a switch and regulator in the development of cardiovascular diseases [35]. Yang et al. demonstrated that the increased expression of let-7 stimulated cardiac hypertrophy by targeting cyclin D2 [36]. Furthermore, miR-let-7a has been reported to attenuate monocrotaline-stimulated pulmonary hypertension via curbing PASMCs growth by STAT3 signaling [17]. The role of STAT3 in PAH has been demonstrated these years [37]. STAT3 activation was supposed to be an early event in PAH etiology, at the origin of several signaling cascades and that it was vital in sustaining the pathologic phenotype [38]. Herein, we verified that lincRNA-COX2 regulated STAT3 by miR-let-7a and influenced PAH development through the miR-let-7a/STAT3 axis. All of these results suggest the important role of lincRNA-Cox2 in PAH.
In conclusion, this study indicated the regulatory role of lincRNA-COX2 in PAH development. Firstly, it was found that lincRNA-COX2 was enhanced in peripheral blood samples from PAH patients and hypoxic PASMCs in a time-dependent manner for the first time. In addition, we also found the miR-let-7a/STAT3 axis, which had been proven to be vital in PAH development, was regulated by lincRNA-COX2. Based on the above experiment results and previous researches, we hypothesized that lincRNA-COX2 might regulate PAH development through the miR-let-7a/STAT3 axis, which is of great significance for the exploration of targeted therapy for PAH.
Conclusion
We have shown that the silencing lincRNA-COX2 inhibits hypoxia-induced PASMC proliferation, the G2/M cell cycle, and migration through the miR-let-7a/STAT3 axis. Our research may be conducive to exploring more effective PAH therapeutic strategies.
References
Schulte C, Karakas M, Zeller T (2017) microRNAs in cardiovascular disease–clinical application. Clin Chem Lab Med 55:687–704. https://doi.org/10.1515/cclm-2016-0576
Bhan B, Koul A, Sharma D, Manzoor MM, Kaul S, Gupta S, Dhar MK (2019) Identification and expression profiling of miRNAs in two color variants of carrot (Daucus carota L.) using deep sequencing. PLoS ONE 14:e0212746. https://doi.org/10.1371/journal.pone.0212746
Chin KM, Rubin LJ (2008) Pulmonary arterial hypertension. J Am Coll Cardiol 51:1527–1538. https://doi.org/10.1016/j.jacc.2008.01.024
Tuder RM, Abman SH, Braun T, Capron F, Stevens T, Thistlethwaite PA, Haworth SG (2009) Development and pathology of pulmonary hypertension. J Am Coll Cardiol 54:S3–S9. https://doi.org/10.1016/j.jacc.2009.04.009
Quinodoz S, Guttman M (2014) Long noncoding RNAs: an emerging link between gene regulation and nuclear organization. Trends Cell Biol 24:651–663. https://doi.org/10.1016/j.tcb.2014.08.009
Li X, Chen Y, Fu C, Li H, Yang K, Bi J, Huo R (2020) Characterization of epigenetic and transcriptional landscape in infantile hemangiomas with ATAC-seq and RNA-seq. Epigenomics. https://doi.org/10.2217/epi-2020-0060
Tang SS, Zheng BY, Xiong XD (2015) LincRNA-p21: implications in human diseases. Int J Mol Sci 16:18732–18740. https://doi.org/10.3390/ijms160818732
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458:223. https://doi.org/10.1038/nature07672
Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB (2013) A long noncoding RNA mediates both activation and repression of immune response genes. Science 341:789–792. https://doi.org/10.1126/science.1240925
Gladka MM, van Rooij E (2015) AntimiR-34a to enhance cardiac repair after ischemic injury. Circ Res 117(5):395–397. https://doi.org/10.1161/circresaha.115.307066
Fang Y-C, Yeh C-H (2015) Role of microRNAs in vascular remodeling. Curr Mol Med 15:684–696. https://doi.org/10.2174/1566524015666150921105031
Maimaiti A, Maimaiti A, Yang Y, Ma Y (2016) MiR-106b exhibits an anti-angiogenic function by inhibiting STAT3 expression in endothelial cells. Lipids Health Dis 15:51. https://doi.org/10.1186/s12944-016-0216-5
Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, MacDonald RA, Greig JA, Robertson KE, Masson R (2010) Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol 30:716–723. https://doi.org/10.1161/atvbaha.109.202028
Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Côté J (2011) Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 208:535–548. https://doi.org/10.1083/jcb1924oia4
Kulshreshtha R, Davuluri R, Calin GA, Ivan M (2008) A microRNA component of the hypoxic response. Cell Death Differ 15:667–671. https://doi.org/10.1038/sj.cdd.4402310
Izumiya Y, Jinnn M, Kimura Y, Wang Z, Onoue Y, Hanatani S, Araki S, Ihn H, Ogawa H (2015) Expression of Let-7 family microRNAs in skin correlates negatively with severity of pulmonary hypertension in patients with systemic scleroderma. IJC Heart Vasc 8:98–102. https://doi.org/10.1016/j.cardfail.2015.08.311
Cheng G, Wang X, Li Y, He L (2017) Let-7a-transfected mesenchymal stem cells ameliorate monocrotaline-induced pulmonary hypertension by suppressing pulmonary artery smooth muscle cell growth through STAT3-BMPR2 signaling. Stem Cell Res Ther 8:34. https://doi.org/10.1186/s13287-017-0480-y
Courboulin A, Barrier M, Perreault T, Bonnet P, Tremblay V, Paulin R, Tremblay E, Lambert C, Jacob M, Bonnet SN (2012) Plumbagin reverses proliferation and resistance to apoptosis in experimental PAH. Eur Respir J 40:618–629. https://doi.org/10.1183/09031936.00084211
Paulin R, Courboulin A, Meloche J, Mainguy V, La Roque EDD, Saksouk N, Cote J, Provencher S, Sussman MA, Bonnet S (2011) Signal transducers and activators of transcription-3/Pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 123:1205–1215. https://doi.org/10.1161/circulationaha.110.963314
Paulin R, Meloche J, Jacob M, Bisserier M, Courboulin A, Bonnet S (2011) Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am J Physiol-Heart Circ Physiol 301(5):H1798–H1809. https://doi.org/10.1152/ajpheart.00654.2011
Li XW, Hu CP, Wu WH, Zhang WF, Zou XZ, Li YJ (2012) Inhibitory effect of calcitonin gene-related peptide on hypoxia-induced rat pulmonary artery smooth muscle cells proliferation: role of ERK1/2 and p27. Eur J Pharmacol 679:117–126. https://doi.org/10.1016/j.ejphar.2012.01.015
Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ, Nicolls MR (2012) Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur Respir J 40:1555–1565. https://doi.org/10.1183/09031936.00046612
Chai S, Wang W, Liu J, Guo H, Zhang Z, Wang C, Wang J (2015) Leptin knockout attenuates hypoxia-induced pulmonary arterial hypertension by inhibiting proliferation of pulmonary arterial smooth muscle cells. Transl Res 166:772–782. https://doi.org/10.1016/j.trsl.2015.09.007
Gu S, Li G, Zhang X, Yan J, Gao J, An X, Liu Y, Su P (2015) Aberrant expression of long noncoding RNAs in chronic thromboembolic pulmonary hypertension. Mol Med Rep 11:2631–2643. https://doi.org/10.3892/mmr.2014.3102
Zhuo Y, Zeng Q, Zhang P, Li G, Xie Q, Cheng Y (2017) Functional polymorphism of lncRNA MALAT1 contributes to pulmonary arterial hypertension susceptibility in Chinese people. Clin Chem Lab Med 55:38–46. https://doi.org/10.1515/cclm-2016-0056
Zhu B, Gong Y, Yan G, Wang D, Qiao Y, Wang Q, Liu B, Hou J, Li R, Tang C (2018) Down-regulation of lncRNA MEG3 promotes hypoxia-induced human pulmonary artery smooth muscle cell proliferation and migration via repressing PTEN by sponging miR-21. Biochem Biophys Res Commun 495:2125–2132. https://doi.org/10.1016/j.bbrc.2017.11.185
Elling R, Robinson EK, Shapleigh B, Liapis SC, Covarrubias S, Katzman S, Groff AF, Jiang Z, Agarwal S, Motwani M (2018) Genetic models reveal cis and trans immune-regulatory activities for lincRNA-Cox2. Cell Rep 25(1511–1524):e6. https://doi.org/10.1016/j.celrep.2018.10.027
Tong Q, Gong AY, Zhang XT, Lin C, Ma S, Chen J, Hu G, Chen XM (2015) LincRNA-Cox2 modulates TNF-α–induced transcription of Il12b gene in intestinal epithelial cells through regulation of Mi-2/NuRD-mediated epigenetic histone modifications. FASEB J 30:1187–1197. https://doi.org/10.1096/fj.15-279166
Hu G, Liao K, Niu F, Yang L, Dallon BW, Callen S, Tian C, Shu J, Cui J, Sun Z (2018) Astrocyte EV-induced lincRNA-Cox2 regulates microglial phagocytosis: implications for morphine-mediated neurodegeneration. Mol Ther-Nucleic Acids 13:450–463. https://doi.org/10.1016/j.omtn.2018.09.019
He J, Tu C, Liu Y (2018) Role of lncRNAs in aging and age-related diseases. Aging Med 1:158–175. https://doi.org/10.1002/agm2.12030
Liao K, Niu F, Dagur RS, He M, Tian C, Hu G (2019) Intranasal delivery of lincRNA-Cox2 siRNA loaded extracellular vesicles decreases lipopolysaccharide-induced microglial proliferation in mice. J Neuroimmune Pharmacol. https://doi.org/10.1007/s11481-019-09864-z
Boyerinas B, Park S-M, Hau A, Murmann AE, Peter ME (2010) The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer 17:F19–F36. https://doi.org/10.1677/erc-09-0184
Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, Wilson M, Wang X, Shelton J, Shingara J (2007) The let-7 microRNA represses cell proliferation pathways in human cells. Can Res 67:7713–7722. https://doi.org/10.1158/0008-5472.can-07-1083
Chen Z, Wang D, Gu C, Liu X, Pei W, Li J, Cao Y, Jiao Y, Tong J, Nie J (2015) Down-regulation of let-7 microRNA increased K-ras expression in lung damage induced by radon. Environ Toxicol Pharmacol 40:541–548. https://doi.org/10.1016/j.etap.2015.08.009
Bao M-H, Feng X, Zhang Y-W, Lou X-Y, Cheng Y, Zhou H-H (2013) Let-7 in cardiovascular diseases, heart development and cardiovascular differentiation from stem cells. Int J Mol Sci 14:23086–23102. https://doi.org/10.3390/ijms141123086
Yang Y, Ago T, Zhai P, Abdellatif M, Sadoshima J (2011) Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circ Res 108:305–313. https://doi.org/10.1161/circresaha.110.228437
Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand-Apte B, Erzurum SC (2007) Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol-Lung Cell Mol Physiol 293:L548–L554. https://doi.org/10.1152/ajplung.00428.2006
Paulin R, Meloche J, Bonnet S (2012) STAT3 signaling in pulmonary arterial hypertension. Jak-Stat 1:223–233. https://doi.org/10.4161/jkst.22366
Funding
The study was supported by the Natural Science Foundation of Shaanxi Province (2018JM7096).
Author information
Authors and Affiliations
Contributions
GC and YZ conceived the study, GC, LH and YZ performed the experiment and collected the data, LH analyzed the data, GC draft the manuscript, and YZ revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cheng, G., He, L. & Zhang, Y. LincRNA-Cox2 promotes pulmonary arterial hypertension by regulating the let-7a-mediated STAT3 signaling pathway. Mol Cell Biochem 475, 239–247 (2020). https://doi.org/10.1007/s11010-020-03877-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-020-03877-6