
Abstract
Genetic variation can only explain a small portion of risk to psychiatric disorders, including major depressive disorder, schizophrenia, and post-traumatic stress disorder. Epidemiological studies are increasingly showing a link between environmental factors and the development of various psychiatric disorders, mainly mediated by underlying epigenetic mechanisms. DNA methylation is one of the most studied epigenetic mechanisms in psychiatric disorders. Epigenome-wide association studies (EWAS) typically used to study changes in DNA methylation still face methodological challenges and limitations at both the fundamental, technical, and data analysis levels. In this chapter, we offer a brief overview of some EWAS studies in different psychiatric disorders and discuss the current challenges, pitfalls, and future considerations for this field.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Over the past two decades, a growing number of genome-wide association studies (GWAS) has shed light onto the etiology and progression of a range of psychiatric disorders. However, the estimates on the contribution of genetic variation obtained from these studies only explain a portion of the heritability (ranging from 40% to 60%) observed in psychiatric disorders such as major depressive disorder (MDD), anxiety disorders, and schizophrenia (SCZ) [1]. Whereas the symptomatology of these types of disorders is too heterogeneous to be linked to one gene as an underlying cause [2], a large number of epidemiological studies has revealed the importance of environmental factors, both pre- and postnatally, on the development of mental disorders [3, 4].
Epigenetic mechanisms are key players in mediating the complex interplay between genetic and environmental factors and, as such, might explain, at least in part, the missing heritability is psychiatric genetics. Epigenetic processes mediate dynamic alterations in the expression of genes without affecting the DNA sequence itself and are the result of developmental, environmental, or stochastic influences [2]. Epigenetic research offers a promising avenue in filling the knowledge gap in understanding the complex etiology and symptomatology of psychiatric disorders and could guide future research into the development of advanced treatment options.
This chapter provides a short overview of the recent literature on the involvement of epigenetic mechanisms in the development and course of psychiatric disorders, with a focus on the challenges associated with this line of research and perspectives for future research in this respect. The first section provides a brief summary of the most commonly studied epigenetic mechanisms, their relevance for brain development, and potential pitfalls associated with psychiatric epigenetics research. Subsequently, this chapter summarizes findings from recent epigenetic epidemiological research on psychiatric disorders, considering the potential challenges of this line of research and the extent to which these challenges have been addressed in existing studies. Finally, this chapter discusses future perspectives of psychiatric epigenetics research.
1.1 Epigenetic Mechanisms in Psychiatry
Epigenetic modifications consist of dynamic processes that can regulate gene expression, but without involving changes in the underlying DNA sequence [5]. They are crucial both for the development of the brain, as well as for experience-driven transcriptional changes [6, 7]. Such modifications affect every level of transcription, starting with regulating access of, e.g., the transcriptional machinery to the DNA by means of histone modifications or DNA methylation changes, as well as post-transcriptional modifications, mediated by, e.g., microRNAs (miRNAs), thereby affecting subsequent translation. Often, epigenetic regulation involves stable changes in the chromatin structure, which is comprised of DNA wrapped around a histone octamer. The nucleosome consists of two copies of the core histones H2A, H2B, H3, and H4 [5]. Chromatin structure is modified via remodeling enzymes such as histone acetyltransferases (HATs) and histone deacetylases (HDACs), causing either an open, i.e., active or closed, i.e., inactive chromatin state, respectively. HATs open up the chromatin by adding acetyl groups to lysine residues of the histone tail, which ultimately allows the transcriptional machinery access to the associated DNA sequence. HDACs, on the other hand, promote transcriptional repression by removing those acetyl groups [5]. In addition to acetylation, many other types of post-translational modifications have been found at histone residues, such as phosphorylation, sumoylation, methylation, and ubiquitination. The focus of this chapter will be on DNA methylation, mainly due to the fact that it is the most and best-studied epigenetic mechanism in relation to psychiatric disorders to date [8].
DNA methylation of the fifth position of cytosine is one of the best understood epigenetic modifications and targets the DNA bases directly [5, 9]. DNA is methylated via DNA methyltransferases (DNMTs), typically occurring at cytosine-phosphate-guanine (CpG) sites, resulting in the formation of 5-methylcytosine (5mC). DNA methylation is the most extensively studied mechanism of epigenetic regulation and has been implicated in the regulation of gene transcription, maintenance of genomic imprinting, X chromosome inactivation, chromatin structure, and the silencing of transposable elements [10]. Recent findings have shown the importance of another, closely related, type of DNA modification of the fifth position of cytosine, i.e., 5-hydroxymethylcytosine (5hmC). It occurs as a result of the oxidation of 5mC, catalyzed by the ten-eleven translocation (TET) family of enzymes. While 5hmC can be stable on its own, it can also contribute to the process of DNA demethylation, with two mechanisms that can convert 5hmC back into unmodified cytosine [11]. While 5hmC is detected in all tissue types, it is most abundant in the brain, suggesting its important role in brain function [10, 11]. Although an increasing number of studies suggest a role of 5hmC in the brain distinct from that of 5mC, traditionally used DNA methylation detection methods that make use of sodium bisulfite treatment cannot distinguish between 5mC and 5hmC. Whereas the great majority of those papers report the results as if reflecting true DNA methylation, i.e., 5mC, in reality, these data reflect the combined levels of 5mC and 5hmC.
In addition to histone modifications and DNA methylation, noncoding RNAs, specifically miRNAs, have been shown to exert an important role in epigenetic regulation by modifying protein levels [12]. MiRNAs are 18–25 nucleotide-long noncoding RNAs that regulate gene expression at the post-transcriptional stage, where they bind to the untranslated regions of mRNA molecules in order to suppress protein translation or to stimulate the breakdown of the associated mRNA [12]. Not only do miRNAs target key enzymes such as DNMTs, HDACs, and histone methyltransferases, but the expression of miRNAs is also regulated via epigenetic mechanisms, such as DNA methylation, RNA modifications and histone modification, creating a miRNA-epigenetic feedback loop [12, 13]. A single miRNA can regulate hundreds of mRNAs, while a single mRNA can also be targeted by numerous miRNAs, making the interpretation of findings from research in this area particularly challenging. Similarly to the area of DNA methylation, there are methodological challenges associated with the comparability of miRNA studies, such as the lack of consensus on the target tissue (such as peripheral blood, cerebrospinal fluid, or brain tissue), as well as the numerous techniques used for miRNA detection, such as quantitative reverse transcription polymerase chain reaction (RT-qPCR), microarray, Northern Blotting, and next-generation sequencing [14].
1.2 Methodological Considerations in Epigenome-Wide Studies
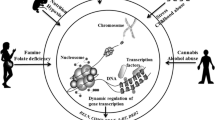
While the field of psychiatry epigenetics offers valuable insight into the underlying mechanisms of disorders extending beyond the genome, there are important challenges in epigenetics research that should be considered when conducting and interpreting epigenome-wide association studies (EWAS) to better understand how these changes impact key genes underlying psychiatric disorders [5, 15]. Such potential pitfalls can be found in every step of planning and conducting an EWAS study, starting with our basic understanding of the epigenome and study design, through specific methodological considerations regarding the laboratory analysis of samples and subsequent data pre-processing and statistical analyses. Understanding the limitations of epigenome-wide research is, therefore, crucial for both the careful interpretation of existing findings, as well as for the improvement of future studies in the field (Fig. 18.1).

Challenges of epigenetic-wide association studies (EWAS) in psychiatric disorders. Several challenges at the biological, technical, and data analysis levels aimed at unraveling epigenetic mechanisms, in particular DNA methylation in psychiatric disorders, remain. There are several avenues worth exploring in overcoming these challenges in order to allow optimal interpretation of the epigenetic data in psychiatry. See text for more details
1.2.1 Fundamental Understanding of the Epigenome and Study Design
Current understanding of the epigenome is still limited, which comes along with important considerations in terms of designing an EWAS and interpreting associated findings.
For example, mechanisms involving histone modifications are typically neglected likely due to challenges in either detecting them or in terms of interpreting the functional implications of the findings [16]. Accordingly, most of the current epigenome-wide research has focused on DNA methylation for both biological and practical reasons. While most challenges in terms of detecting DNA methylation have been overcome by now (see below), the functional relevance of differential methylation is still often unclear though. As such, differential methylation could indicate increased or reduced transcription depending on the location of the methylated position, such as the promoter region, which is commonly investigated. As in certain cases, differential methylation at intergenic CpG shores and intra-genic CpG islands may be even more relevant for phenotypic variation when compared to methylation changes at CpG islands located in promoter regions, selecting the appropriate method to detect DNA methylation, e.g., through arrays or by means of sequencing, is crucial in view of the desired coverage and genomic location.
Furthermore, as the epigenome is not dynamic, a prospective study design often offers the most robust findings, allowing for the interrogation of epigenetic changes, e.g., in response to environmental variation, over time [15]. The timing of sample collection is crucial in this respect, as epigenetic modifications can be transient in certain cases. In the case of research into the underlying effects of treatments, as well as exposure to certain environments, such as military deployment [17], longitudinal studies can offer valuable insight into the epigenetic modifications in human populations. Analysis of disease-discordant monozygotic (MZ) twins offers an excellent tool in the study of epigenetic modifications associated with the disease phenotype, while its combination with a longitudinal design can provide valuable insight into the environmental variation impacting upon the etiology of the disease [15]. Moreover, in view of the important role of the early environment in programming adult mental health and disease, applying a longitudinal design including the assessment of developmental epigenetic changes, could be of great added value [15]. A major challenge to this type of design, however, lies in the lack of detailed phenotyping, a thorough collection of detailed information regarding exposure to environmental variation and repetitive follow-up and sampling throughout life [15]. Of note, generally, DNA methylation changes originating during early development may have higher inter-tissue concordance than those induced later in life [16].
In addition, researchers must be wary that the disease process itself as well as its (e.g., pharmacological) treatment can also cause epigenetic changes and causal inference into the etiology of the disease, and is therefore problematic when based solely on findings from human populations [15]. For this purpose, additional in vitro or in vivo animal studies are often needed [16].
1.2.2 Technological Challenges
A major challenge of epigenetic research in humans lies in the access to the tissue of interest, i.e., the brain, which is typically not available. Unlike genetic research where the sample tissue is not as relevant, in epigenetics, differences in, e.g., DNA methylation can be confounded by the sample’s source and its cellular composition, especially in the case of whole blood samples [16]. While cells share the same DNA sequence, it is the epigenome that differentiates cell types and, therefore, cellular composition of the tissue of interest. This may in fact even differ between samples of the same type of tissue, which is of great importance in epigenetic analysis. Hence, taking into account potential differences in cell-type composition, either a priori by using cell sorting or post hoc using bioinformatics tools, is a must. Moreover, using peripheral tissue as a replacement of the brain can be problematic, especially that research shows that association between blood or saliva and the brain is quiet limited [18], and may respond differently to environmental challenges, such as exposure to stress [19]. One possible solution to optimize the translational ability of EWAS lies in the use of DNA methylation as a biomarker for mental illnesses [20].
The use of different techniques to detect DNA methylation raises an additional problem in comparability across studies [16]. Due to the demand for large sample sizes, researchers have to weigh the benefits of coverage versus precision. While there are various methods available to capture methylated DNA, the ‘golden standard’ in current epigenetic research is the use of bisulfite treatment, followed by either an array- or next-generation sequencing-based detection [16, 21, 22]. So far, based on the delicate balance between precision and coverage, most labs involved in EWAS have made use of commercially available Illumina 450k and EPIC (850k) Methylation Beadchip microarrays. The 450k array, which is not available anymore, assessed DNA methylation at over 480 000 CG dinucleotides, mainly covering promoter regions [22], and providing an affordable option for large-scale epidemiological findings. Its successor, the Infinium MethylationEPIC array, targets over 850 000 CpGs. While being of clear added value, next to the limited coverage, these platforms lack the possibility to thoroughly assess methylation at non-CpG sites [20]. Recently, more frequently, next-generation sequencing (NGS) is being used, which allows for flexibility in this respect, which evidently is more costly and time consuming. Whole-genome bisulfite sequencing (WGBS) offers almost full coverage [16, 21, 22]. Reduced representation bisulfite sequencing (RRBS), while granting single-base resolution as in WGBS, has lower coverage, including the great majority of promoters and CpG islands, implying a lower number of reads necessary to yield accurate sequencing, in addition to lower costs and processing time compared with WGBS. Other approaches, such as methylated DNA immunoprecipitation sequencing (MeDIP-Seq) or methyl-binding domain sequencing (MBD-Seq), make use of means to capture methylated pieces of DNA prior to sequencing. It is worth noting that while there is high correlation between different platforms, which is related to the fact that most of the genome is either simply methylated or unmethylated, there may be substantial differences within loci that display intermediate methylation levels [16].
As indicated above, accumulating evidence suggests an important role for other cytosine modifications such as 5hmC in the human brain. However, the great majority of EWAS published to date made use of sodium bisulfite-treated DNA, which does not allow to discriminate between 5mC and 5hmC, which may lead to incorrect interpretation of associated findings. Making use of oxidative-bisulfite DNA treatment (parallel to classical bisulfite treatment) now allows for simultaneously assessing DNA methylation and hydroxymethylation [23].
1.2.3 Data Analysis
There are various data analysis strategies available for analysing and interpreting epigenetic data and, similarly to the methodological problems raised earlier, to date, no clear consensus on the most optimal approach exists [15]. Most of the current studies in the literature have investigated individual CpGs to identify differentially methylated positions (DMPs), as that is also the method most suitable for the Illumina array-based platforms, owing to their modest coverage. Another approach is to explore differentially methylated regions (DMRs), which is based on the assumption that several differentially methylated cytosines in close proximity with each other are likely to affect chromatin formation and thus the transcription of the accompanying gene(s) [15]. Methods with greater coverage may be more suitable for the investigation of DMRs, as there is not clear consensus yet whether the number of CpGs covered by the Illumina arrays is sufficient for this type of analysis. The advantage of DMR analysis is the reduced risk of false-positives due to artifacts affecting single CpGs, as well as higher power because of the reduced multiple testing error [15]. Additionally, DMRs may be easier to be interpreted biologically. It should be noted though, that differential methylation at single CpG methylation sites can also have important functional consequences. In addition to these two commonly used methods, Mendelian randomization can be useful for causal inference and is being applied more regularly nowadays. As such, a methylation quantitative trait loci (mQTL) analysis aims at identifying genetic variants that affect DNA methylation patterns [24], knowledge of which is of extreme importance, as a certain degree of epigenetic variation may be caused by genetic variation. Another critical point to consider is accounting for cellular heterogeneity, especially in blood, which is crucial in EWAS. For instance, one study showed that cellular composition accounts for the majority of the detected variability in DNA methylation. It appears that DNA methylation profiles were both cell type- and age-dependent. This can be corrected for using a statistical approach as described in Ref. [25].
2 Epigenetic Profiles in Psychiatric Disorders
While the following sections include a discussion on numerous EWAS performed to date, it should be noted that this does not concern a systematic review of the available literature in this respect, as its aim is to highlight strengths and limitations of certain approaches in this respect.
2.1 Major Depressive Disorder
MDD is one of the most heterogeneous and prevalent psychiatric disorders and findings from EWAS studies suggest a wide variety of changes in epigenetic regulation which may underlie MDD [26]. A recent longitudinal study utilizing the 450k Illumina platform and examining tissue from the dorsolateral prefrontal cortex (DLPFC) of 608 participants showed epigenome-wide significant association between DNA methylation changes in certain genes and late-life MDD [27]. The most significant association found in this study was observed for the YOD1/PFKB2 locus. YOD1 has been found to be involved in the maintenance of the correct conformation of proteins, more specifically related to inflammatory responses. Although this study made use of a tissue-specific analysis and a longitudinal design, it is worth noting that the researchers employed tissue bulk analysis, which was later corrected for cell-type composition [27]. This notion is relevant, as cell-type composition can be different between, e.g., patients and controls in EWAS studies, e.g., due to cell loss, when assessing brain tissue, or inflammation, when assessing blood. This challenges the correct interpretation of results, as cell-type-specific modifications in one cell-type could, for example, be masked by changes in another.
In another study, on a cohort of 724 MZ Danish twins, making use of whole blood samples and the Illumina 450k platform, Starnawska and colleagues showed associations between DNA methylation and MDD for the gene encoding neuropsin, which is involved in synaptogenesis and has previously been implicated in schizophrenia and bipolar disorder (BP) [28]. The authors also found differential methylation for the DAZAP2 gene, which is known for inducing stress granule formation. Although the study utilized peripheral blood samples, it has the strong advantage of a discordant MZ design, as well as a relatively large sample size [28]. Additionally, the researchers investigated depression severity amongst the general population, as opposed to clinical cases. An additional strength of the study is that blood cell composition proportions were estimated using flow cytometry for a part of the individuals (n = 471). In another study using a relatively large sample size (N = 844), followed by a replication study (N = 1339), the relationship between umbilical cord DNA methylation and maternal depression throughout pregnancy was investigated, using the Illumina 450k platform [29]. Results from the first cohort showed a relationship between maternal depression at any point during pregnancy and 7 DMRs, which was, however, not replicated in the second cohort. The DMRs were located within genes related to brain development and the formation of the nervous system, as well as the LYNX1 gene, which was previously shown to be hypermethylated in the hippocampus of patients with MDD [29].
2.2 Suicide
Suicide is the fourth leading cause of death amongst 15–29 year old, with nearly one million deaths per year globally [30]. The heterogeneity of disorders accompanying suicide completion poses additional challenges in this line of research, where researchers investigating the epigenetic changes associated with suicide may either focus on a specific disorder, exclude individuals with a particular underlying disorder, or in some cases compare disorders. Additionally, depending on whether suicide attempts or suicide completion is being investigated, the sample methods differ, with tissue-specific samples being the typical choice in the latter case. It should be noted that while a history of suicide attempts is the strongest predictor of suicide completion, some studies focus on comparing individuals with a history of suicidal ideation versus healthy controls, whereas other studies examine post-mortem tissue in suicide completers. As there may be differences in epigenetic modifications between the two groups, findings should be interpreted with caution.
Due to high suicide attempt rates amongst individuals with BP, it is the most studied comorbid disorder in individuals with a history of suicide attempts [31]. In one such study, the authors investigated the prefrontal cortex of 23 individuals with BP who died of suicide, 27 who died of other causes, and 31 non-psychiatric controls [31]. Overall, BP subjects had more hypomethylated DMRs compared to controls, pooled for cause of death, whereas within the BP group, individuals who died of suicide showed increased methylation than those who did not. Of specific interest, ARHGEF38 was hypomethylated in BP associated with suicide, which, while relatively unknown, is believed to be involved in the GTPase cycle [31]. It should be noted that those findings were primarily driven by males with BP who died of suicide, which suggests that sex differences may be a factor that should be included in future studies. This is especially relevant since there are known sex differences in the rates and method of suicide, with males being more likely to die of suicide and opting for violent methods, whereas females tend to gravitate towards non-violent tools [32, 33]. Additionally, aggression and impulsivity are known risk factors for suicide [34], which may also be relevant as there have been sex differences demonstrated in aggressive behavior, impulsivity, and violent acts [35]. The difference in suicidal ideation and suicidal completion, therefore, with the former being more common in women, whereas the latter is more common in men, may suggest distinct molecular changes associated with each. Work on suicide is further complicated by the heterogeneity of disorders accompanying suicidal ideation, such as MDD, BP, SCZ, anxiety disorders, substance abuse, and personality disorders [31, 36, 37]. Finally, the authors showed differences in methylation in axonal guidance signaling pathways, cardiac beta-adrenergic signaling, and opioid signaling [31].
Another study by Jokinen and colleagues [37] investigated DNA methylation in whole blood of individuals with a history of suicide attempt. The researchers used both the Illumina 450k and the EPIC 850k platforms to investigate DMPs associated with suicide attempts. They excluded factors such as schizophrenia spectrum disorders, intravenous drug abuse, dementia, and mental retardation (N = 88 for cohort 1; N = 129 in the second cohort and N = 93 for the third cohort). Their findings suggest that individuals with a higher risk of suicide attempt had reduced levels of methylation in the promoter region of the CRH gene, which is crucial in HPA-axis regulation [37]. A strength of this study is that the initial findings were also replicated in the following cohort of adolescents. Yet again, most of the participants in the study were previously treated with antidepressants which also influences DNA methylation profiles [38], and there was no clear distinction in diagnosis in view of the underlying psychiatric illness(es).
In a comparison of depressed individuals who committed suicide versus non-psychiatric sudden-death controls (N = 75 in total), using brain tissue from Brodmann areas 11 and 25, Murphy and colleagues [39] showed methylation changes in several genes. The researchers observed hypomethylation in both cortical regions in MDD suicide cases for the PSORS1C3 gene, which is thought to be involved in the regulation of nearby immune system-related genes. In addition, two other DMRs related to antigen processing (TAPBP) and mitochondrial ATP synthase function (ATP5G2) were identified. It is worth to note that a limitation of this kind of design is the lack of medication data, which can be especially crucial when comparing groups with an underlying psychiatric illness with controls, as some of the methylation changes that are identified may be related to treatment or even result from the disease progress itself, as opposed to be causally associated with suicidal ideation specifically. A design including a group of individuals with MDD who died of other causes could have tackled those limitations. In addition to individuals with MDD and BP, individuals with SCZ are at increased risk for committing suicide. Bani-Fatemi and colleagues conducted two studies in peripheral blood investigating suicide attempts in individuals with SCZ, in the first of which they did not find changes in methylation between the group with a history of suicide attempts and those without [40]. Findings from the second study did show hypermethylation in both SLC20A1, a sodium-dependent phosphate transporter, and SMPD2, which encodes for the enzyme sphingomyelin phosphodiesterase 2, in those individuals displaying suicidal ideation. In both these studies, no records on medication were available, and a relatively small sample size was used (N = 123 and N = 107, respectively).
In addition, in another study investigating suicidal behavior in individuals with BP, the authors included not only a typical DMP and DMR analysis, but also correlated DNA methylation age with suicidal behavior [41]. In this study, individuals with a history of suicidal behavior displayed hypomethylation in MPP4, which is known to regulate the activity of membrane calcium ATPases, and TBC1D16, which represents a known activator of Rab4a involved in cell growth and survival, as well as hypermethylation in NUP133, which is known to be involved in spindle assembly and nuclear mRNA export [41]. Furthermore, age-related signatures of DNA methylation showed a weaker correlation in DNA methylation age and chronological age when compared to controls, where the DNA of individuals with a history of suicidal behavior was hypothesized to exhibit accelerated (epigenetic) aging. This study was the first to investigate the relationship between suicidal ideation and DNA methylation age, although it is worth to note that the authors did not specifically control for factors that have previously been associated with changes in DNA methylation age [42].
2.3 Schizophrenia
SCZ is a neurodevelopmental disorder characterized by psychosis and altered cognitive function, which was one of the first targets in the investigation of both genetic risk factors as well as epigenetic changes associated with disease risk and progression [43]. Initial twin and family studies have shown the heritability of the disease, although it does not fully explain its etiology. Together with MDD, it is also one of the disorders for which there are studies with relatively large sample sizes. In one such project, Hannon and colleagues [44] investigated methylation changes in three cohorts, the first of which was a discovery cohort (N = 675), followed by a replication one (N = 847), and an MZ pair cohort (N = 96 pairs). Findings show the top-ranked group of pathways associated with SCZ was related to immune functioning, with the second-ranked group involved in neuronal proliferation and brain development [45]. The large sample size, multiple phases and the inclusion of an MZ cohort make this project an excellent example of a study that aimed to address the methodological challenges of this line of research.
A more recent study by Wakeys and colleagues [46] examined 171 individuals, of which 57 diagnosed with SCZ, 59 with BP, and 55 healthy controls. In addition to the DMP analyses, the researchers computed poly-methylomic profile scores (PMPS), based on EWAS data and clinical status. All five PMPS computed were associated with SCZ, where individuals with SCZ showed heightened PMPS relative to controls. While similarly to the Hannon study [45], the researchers used peripheral blood for the methylation analyses and the sample size for this cohort was not very large, a strength of this study was the inclusion of polygenic risk score analysis, as well as the comparison between individuals with SCZ not only to controls, but also to BP patients.
A big challenge in the identification of epigenetic changes in SCZ is defining whether DNA methylation changes are a cause or a consequence of the disease [47]. That could also be due to the fact that antipsychotics also carry an effect on DNA methylation profiles [48, 49], and hence any observed EWAS association in SCZ could also be due to the effects of antipsychotics. That being said, future EWAS studies must work to include both medicated and drug-naïve patients to detect true disease phenotypes.
2.4 Post-Traumatic Stress Disorder
Post-traumatic stress disorder (PTSD) is a debilitating psychiatric disorder that typically develops as a result of direct or indirect exposure to a traumatic event [17]. While most individuals are resilient to the long-term effects of trauma exposure, some develop PTSD with no effective pharmacotherapy being developed to date. Epigenetic findings from this area can, therefore, not only shed light on risk and resilience factors, but also on the underlying mechanisms of the disorder directing further research into advanced treatment options. Due to the nature of the disorder and its cause, researchers often select military cohorts as their sample, often comprised exclusively of male individuals. The advantage of such a design is the possibility to analyze the methylome prior to, as well as in the short and long run after exposure to trauma, as well as the possibility to identify potential predictors of resilience. Additionally, the type of stressor that the individuals are exposed to is typically similar of nature, often related to combat. Evidently, there are also disadvantages related to the use of military samples, such as the higher number of male individuals, the potential underreporting of symptoms, as well as the relatively homogenous type of trauma. Samples drawn from the general population could shed more light on epigenetic mechanisms underlying PTSD following various types of traumas, as well as over different periods of time. Thus, while military cohorts may be more convenient from a methodological perspective, a well-devised longitudinal study of a community sample could also provide valuable insight into epigenetic modifications associated with the disorder.
In a large study including three military cohorts, Snijders and colleagues [17] investigated the peripheral blood of soldiers exposed to trauma, comparing those who did develop PTSD symptoms with resilient individuals and those not exposed to trauma. Participants with PTSD symptoms showed an association between DNA methylation profiles and post-deployment symptoms [17]. The top replicating DMP represented an intergenic site near a gene (SPRY4) that codes for a member of the Sprouty proteins involved in the inhibition of tyrosine kinase signaling. Additionally, the strongest association for the DMR analysis was in a region related to immune functioning [17]. It should be noted that the individuals in this study were males only, with those who developed PTSD having been exposed to more traumatic events. Another study that focused on adolescents (N = 39), the majority of which female (84.6%), showed two DMPs associated with PTSD symptom severity, one of which was in the MAML gene, which has been implicated in neuronal plasticity [50]. In addition, a DMR in the MOBP (myelin-associated oligodendrocyte basic protein) gene was identified, which has previously been associated with PTSD in the literature.
While the previous two studies both investigated peripheral blood, Logue and colleagues [51] used samples from two blood-based cohorts and a brain bank cohort (N = 513; replication cohort N = 1253; and brain tissue (prefrontal cortex) cohort N = 72). Analysing DMPs and DMRs, the authors found a negative correlation between the effect size estimations across the blood-based cohort and the brain bank samples, suggesting that effects investigated using peripheral blood should be interpreted with caution. This difference was mainly driven by one site in OR2AG1, which interestingly displayed the largest effect size in both blood and brain, but in the opposite direction. OR2AG1 is a gene coding for an olfactory receptor, whose activation carries consequences for serotonin release [52]. Additionally, in the brain bank cohort, a probe in the CHST11 gene, which was also among the top 10 results in the discovery cohort, was associated with PTSD. This gene is involved in, e.g., neuronal plasticity, fear learning, and neuroinflammation. Finally, an epigenome-wide association in the G0/G1 Switch 2 (GOS2) gene was identified, where higher methylation was associated with a PTSD diagnosis. Previous findings have shown cortisol to suppress G0S2 [53] and its expression has been linked to PTSD in the literature [54, 55]. It should be noted that while this study employs a much larger sample size and has the strength of multi-tissue type comparison, the researchers used different platforms for the discovery and replication cohort, namely Illumina 450k and EPIC 850k arrays, respectively.
In another recent study, Katrinli and colleagues [56] investigated methylation changes associated with PTSD in two large cohorts (N = 554 and 780), including correlating the levels of DNA methylation and gene expression. The authors found two DMRs associated with PTSD, for one of which (HLA-DPB1) the direction of the effect was in the opposite direction for the two cohorts, with this gene having been previously associated with PTSD in the literature [17] and is thought to be related to immune system dysregulation [57, 58]. The function of the other gene (SPATC1L) is thought to be related to the protection of cells from cell death induced by DNA-damaging alkylating agents [59], which are by-products of normal cellular function, as well as oxidative stress and chronic inflammation [60]. Similar to the previously discussed multi-cohort project, however, this study also used different platforms for the two cohorts investigated.
2.5 Anxiety Disorders
Due to the heterogeneity of anxiety disorders, the scope of studies that investigate epigenetic mechanisms associated with anxiety is relatively wide. Research in this area, therefore, varies both in terms of the type of anxiety investigated, with some studies including individuals with panic attacks, as well as in view of the stage of life at which they are being studied, where for example, some studies focus on transgenerational effects of maternal anxiety on the offspring.
Using a population-based (N = 1522) and a clinical cohort (N = 300), Emeny and colleagues [61] identified an association between severe anxiety and increased methylation at a CpG site located in the promoter of ASB1, a gene associated with ubiquitin degradation pathways, as well as scaffolding, neurogenesis, and neuroprotection [62]. While the population-based sample as well as the large sample size for both cohorts is a particular strength of this study, it should be noted that the authors used two different questionnaires to assess anxiety and there was no clear distinction between panic disorder and anxiety symptoms across the cohorts. In addition to studies that investigate epigenetic changes associated with anxiety in adults, some focus on the effects of maternal anxiety on DNA methylation in newborns. In one such study, Vangeel and colleagues [63] evaluated changes in DNA methylation derived from the umbilical cord blood of newborns whose mothers exhibited more symptoms of anxiety and correlated that with the cortisol awakening response measured at 2, 4, and 12 months of age of the infant. Of the 10 DMRs, the top DMR was found in the GABA-B receptor subunit 1 gene (GABBR1) and associated with prenatal anxiety especially in male newborns. GABBR1 methylation profile was also associated with newborn cortisol levels at 4 months. These findings highlight the role of GABBR1 in prenatal anxiety, particularly in influencing neuronal plasticity in the face of environmental stressors [64]. Another important observation points towards taking into account gender effects when analyzing differential methylation profiles [65].
3 Concluding Remarks
Epigenome-wide approaches have garnered much attention in the recent years, with the primary focus being on DNA methylation. While there is increasing interest in the role of epigenetic dysregulation in mental health and disease, results from EWAS studies should be interpreted with caution (Fig. 18.1). Most importantly, for various reasons, several studies still make use of low sample sizes. In addition, studies often make use of bulk brain tissue, which does not always allow correcting for cellular heterogeneity, an issue that can be overcome by, for example, cell sorting. Moreover, particularly when assessing DNA derived from brain tissue, specific approaches that are able to discriminate between 5mC and 5hmC should be employed. Future EWAS studies should also include sex as a determining factor owing to known sex differences in epigenetic mechanisms. Another consideration would be to include homogenous populations of a certain disorder, or addressing specific (e.g., behavioral) phenotypes (e.g., panic attacks) so as not to risk missing potential associations. In the near future, the field could benefit from validating candidate signatures, by for instance employing (e.g., CRISPR-dCas9-based) epigenetic editing tools, to determine whether signatures identified in EWAS in fact represent a causal association. Moreover, multi-omics approaches centered around an integrative analysis of various layers including genetic, epigenetic, proteomic, and metabolomic data show great promise in the development of novel treatment strategies and diagnostic and prognostic biomarkers for psychiatric disorders.
Abbreviations
- 5hmC:
-
5-hydroxymethylcytosine
- 5mC:
-
5-methylcytosine
- BP:
-
Bipolar disorder
- CpG:
-
Cytosine-phosphate-guanine
- CRISPR-dCas9:
-
Clustered regularly interspaced short palindromic repeat-deficient Cas9
- DLPFC:
-
Dorsolateral prefrontal cortex
- DMP:
-
Differentially methylated position
- DMR:
-
Differentially methylated region
- DNMT:
-
DNA methyltransferase
- EWAS:
-
Epigenome-wide association studies
- GABBR1:
-
GABA-B receptor subunit 1 gene
- GWAS:
-
Genome-wide association studies
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- MBD-Seq:
-
Methyl-binding domain sequencing
- MDD:
-
Major Depressive Disorder
- MeDIP-Seq:
-
Methylated DNA immunoprecipitation sequencing
- miRNA:
-
microRNA
- MOBP:
-
Myelin-associated oligodendrocyte basic protein
- mQTL:
-
Methylation quantitative trait loci
- MZ:
-
Monozygotic
- NGS:
-
Next-generation sequencing
- PMPS:
-
Poly-methylomic profile scores
- PTSD:
-
Post-traumatic stress disorder
- RRBS:
-
Reduced representation bisulfite sequencing
- RT-qPCR:
-
Reverse transcription quantitative polymerase chain reaction
- SCZ:
-
Schizophrenia
- TET:
-
Ten-eleven translocation
- WGBS:
-
Whole-genome bisulphite sequencing
References
Thibaut F (2019) Epigenetics: the missing link between genes and psychiatric disorders? Dialogues Clin Neurosci 21(4):337–338
Kular L, Kular S (2018) Epigenetics applied to psychiatry: clinical opportunities and future challenges. Psychiatry Clin Neurosci 72(4):195–211
Susser E et al (2006) Psychiatric epidemiology: searching for the causes of mental disorders. Oxford University Press
Cooper B (2001) Nature, nurture and mental disorder: old concepts in the new millennium. Br J Psychiatry 178(S40):s91–s101
Mahgoub M, Monteggia LM (2013) Epigenetics and psychiatry. Neurotherapeutics 10(4):734–741
Kravitz SN, Gregg C (2019) New subtypes of allele-specific epigenetic effects: implications for brain development, function and disease. Curr Opin Neurobiol 59:69–78
Nagy C, Vaillancourt K, Turecki G (2018) A role for activity-dependent epigenetics in the development and treatment of major depressive disorder. Genes Brain Behav 17(3):e12446
Starnawska A, Demontis D (2021) Role of DNA methylation in mediating genetic risk of psychiatric disorders. Front Psychiatry 12:596821
Smith ZD, Meissner A (2013) DNA methylation: roles in mammalian development. Nat Rev Genet 14(3):204–220
Wen L, Tang F (2014) Genomic distribution and possible functions of DNA hydroxymethylation in the brain. Genomics 104(5):341–346
Rustad SR, Papale LA, Alisch RS (2019) DNA methylation and hydroxymethylation and behavior. Curr Top Behav Neurosci 42:51–82
Yao Q, Chen Y, Zhou X (2019) The roles of microRNAs in epigenetic regulation. Curr Opin Chem Biol 51:11–17
Van den Hove DL et al (2014) Epigenetically regulated microRNAs in Alzheimer’s disease. Neurobiol Aging 35(4):731–745
Hunt EA et al (2015) MicroRNA detection: current technology and research strategies. Annu Rev Anal Chem 8(1):217–237
Mill J, Heijmans BT (2013) From promises to practical strategies in epigenetic epidemiology. Nat Rev Genet 14(8):585–594
Heijmans BT, Mill J (2012) Commentary: the seven plagues of epigenetic epidemiology. Int J Epidemiol 41(1):74–78
Snijders C et al (2020) Longitudinal epigenome-wide association studies of three male military cohorts reveal multiple CpG sites associated with post-traumatic stress disorder. Clin Epigenetics 12(1):11
Lin D et al (2018) Characterization of cross-tissue genetic-epigenetic effects and their patterns in schizophrenia. Genome Med 10(1):1–12
Jones MJ, Moore SR, Kobor MS (2018) Principles and challenges of applying epigenetic epidemiology to psychology. Annu Rev Psychol 69:459–485
Barker ED, Roberts S, Walton E (2019) Hidden hypotheses in ‘hypothesis-free’genome-wide epigenetic associations. Curr Opin Psychol 27:13–17
Khodadadi E et al (2021) Current Advances in DNA methylation analysis methods. Biomed Res Int 2021:8827516
Kurdyukov S, Bullock M (2016) DNA methylation analysis: choosing the right method. Biology 5(1):3
Zeng T et al (2015) Detection of 5-methylcytosine and 5-hydroxymethylcytosine in DNA via host–guest interactions inside α-hemolysin nanopores. Chem Sci 6(10):5628–5634
Gao X et al (2017) The impact of methylation quantitative trait loci (mQTLs) on active smoking-related DNA methylation changes. Clin Epigenetics 9(1):87
Jaffe AE, Irizarry RA (2014) Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 15(2):1–9
Lin E, Tsai S-J (2019) Epigenetics and depression: an update. Psychiatry Investig 16(9):654
Hüls A et al (2020) Association between DNA methylation levels in brain tissue and late-life depression in community-based participants. Transl Psychiatry 10(1):262
Starnawska A et al (2019) Epigenome-wide association study of depression symptomatology in elderly monozygotic twins. Transl Psychiatry 9(1):214
Viuff AC et al (2018) Maternal depression during pregnancy and cord blood DNA methylation: findings from the Avon longitudinal study of parents and children. Transl Psychiatry 8(1):244
Organization, W.H (2014) Preventing suicide: a global imperative. World Health Organization
Gaine ME et al (2019) Differentially methylated regions in bipolar disorder and suicide. Am J Med Genet B Neuropsychiatr Genet 180(7):496–507
Fox KR et al (2018) Examining the role of sex in self-injurious thoughts and behaviors. Clin Psychol Rev 66:3–11
Skogman K, Alsén M, Öjehagen A (2004) Sex differences in risk factors for suicide after attempted suicide. Soc Psychiatry Psychiatr Epidemiol 39(2):113–120
Coryell W et al (2018) Aggression, impulsivity and inflammatory markers as risk factors for suicidal behavior. J Psychiatr Res 106:38–42
Cross CP, Copping LT, Campbell A (2011) Sex differences in impulsivity: a meta-analysis. Psychol Bull 137(1):97
Hayashi N et al (2012) Post-hospitalization course and predictive signs of suicidal behavior of suicidal patients admitted to a psychiatric hospital: a 2-year prospective follow-up study. BMC Psychiatry 12(1):186
Jokinen J et al (2018) Epigenetic changes in the CRH gene are related to severity of suicide attempt and a general psychiatric risk score in adolescents. EBioMedicine 27:123–133
Li M et al (2019) What do DNA methylation studies tell us about depression? A systematic review. Transl Psychiatry 9(1):1–14
Murphy T et al (2017) Methylomic profiling of cortex samples from completed suicide cases implicates a role for PSORS1C3 in major depression and suicide. Transl Psychiatry 7(1):e989–e989
Bani-Fatemi A et al (2018) Epigenome-wide association study of suicide attempt in schizophrenia. J Psychiatr Res 104:192–197
Jeremian R et al (2017) Investigation of correlations between DNA methylation, suicidal behavior and aging. Bipolar Disord 19(1):32–40
Dhingra R et al (2018) DNA methylation age—Environmental influences, health impacts, and its role in environmental epidemiology. Curr Environ Health Rep 5(3):317–327
Viana J et al (2017) Schizophrenia-associated methylomic variation: molecular signatures of disease and polygenic risk burden across multiple brain regions. Hum Mol Genet 26(1):210–225
Hannon E, Dempster EL, Mansell G, Burrage J, Bass N, Bohlken MM, Corvin A, Curtis CJ, Dempster D, Di Forti M, Dinan TG, Donohoe G, Gaughran F, Gill M, Gillespie A, Gunasinghe C, Hulshoff HE, Hultman CM, Johansson V, Kahn RS, Kaprio J, Kenis G, Kowalec K, MacCabe J, McDonald C, McQuillin A, Morris DW, Murphy KC, Mustard CJ, Nenadic I, O'Donovan MC, Quattrone D, Richards AL, Rutten BP, St Clair D, Therman S, Toulopoulou T, Van Os J, Waddington JL, Wellcome Trust Case Control Consortium (WTCCC); CRESTAR consortium, Sullivan P, Vassos E, Breen G, Collier DA, Murray RM, Schalkwyk LS, Mill J (2021) DNA methylation meta-analysis reveals cellular alterations in psychosis and markers of treatment-resistant schizophrenia. Elife Feb 26;10:e58430. https://doi.org/10.7554/eLife.58430. PMCID: PMC8009672
Hannon E et al (2016) An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol 17(1):176
Watkeys OJ et al (2020) Derivation of poly-methylomic profile scores for schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry 101:109925
Cariaga-Martinez A, Saiz-Ruiz J, Alelú-Paz R (2016) From linkage studies to epigenetics: what we know and what we need to know in the neurobiology of schizophrenia. Front Neurosci 10:202
Ovenden ES et al (2018) DNA methylation and antipsychotic treatment mechanisms in schizophrenia: progress and future directions. Prog Neuro-Psychopharmacol Biol Psychiatry 81:38–49
Smigielski L et al (2020) Epigenetic mechanisms in schizophrenia and other psychotic disorders: a systematic review of empirical human findings. Mol Psychiatry 25(8):1718–1748
Sheerin CM et al (2021) Epigenome-wide study of posttraumatic stress disorder symptom severity in a treatment-seeking adolescent sample. J Trauma Stress 34:607–615
Logue MW et al (2020) An epigenome-wide association study of posttraumatic stress disorder in US veterans implicates several new DNA methylation loci. Clin Epigenetics 12(1):1–14
Maßberg D, Hatt H (2018) Human olfactory receptors: novel cellular functions outside of the nose. Physiol Rev 98:1739–1763
Stimson RH et al (2017) Acute physiological effects of glucocorticoids on fuel metabolism in humans are permissive but not direct. Diabetes Obes Metab 19(6):883–891
Daskalakis NP et al (2014) Expression profiling associates blood and brain glucocorticoid receptor signaling with trauma-related individual differences in both sexes. Proc Natl Acad Sci 111(37):13529–13534
Bam M et al (2016) Dysregulated immune system networks in war veterans with PTSD is an outcome of altered miRNA expression and DNA methylation. Sci Rep 6(1):1–13
Katrinli S et al (2021) PTSD is associated with increased DNA methylation across regions of HLA-DPB1 and SPATC1L. Brain Behav Immun 91:429–436
Michopoulos V et al (2017) Inflammation in fear-and anxiety-based disorders: PTSD, GAD, and beyond. Neuropsychopharmacology 42(1):254–270
Eraly SA et al (2014) Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry 71(4):423–431
Fry RC et al (2008) Genomic predictors of interindividual differences in response to DNA damaging agents. Genes Dev 22(19):2621–2626
La D, Upton P, Swenberg J (2010) Carcinogenic alkylating agents. Elsevier
Emeny RT et al (2018) Anxiety associated increased CpG methylation in the promoter of Asb1: a translational approach evidenced by epidemiological and clinical studies and a murine model. Neuropsychopharmacology 43(2):342–353
Leussis MP, Madison JM, Petryshen TL (2012) Ankyrin 3: genetic association with bipolar disorder and relevance to disease pathophysiology. Biol Mood Anxiety Disord 2(1):1–13
Vangeel EB et al (2017) Newborn genome-wide DNA methylation in association with pregnancy anxiety reveals a potential role for GABBR1. Clin Epigenetics 9(1):1–12
Bains JS, Cusulin JIW, Inoue W (2015) Stress-related synaptic plasticity in the hypothalamus. Nat Rev Neurosci 16(7):377–388
Bale TL (2011) Sex differences in prenatal epigenetic programing of stress pathways. Stress 14(4):348–356
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bassil, K., Ali, N., Pishva, E., van den Hove, D.L.A. (2022). Epigenome-Wide Association Studies in Psychiatry: Achievements and Problems. In: Michels, K.B. (eds) Epigenetic Epidemiology. Springer, Cham. https://doi.org/10.1007/978-3-030-94475-9_18
Download citation
DOI: https://doi.org/10.1007/978-3-030-94475-9_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-94474-2
Online ISBN: 978-3-030-94475-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)