
Abstract
This chapter summarizes how fatty acid (FA) oxidation is regulated in skeletal muscle during exercise and the role of obesity in regulation of FA oxidation in skeletal muscle. The substrates fueling increased FA oxidation in skeletal muscle during exercise are mainly circulating FAs, although hydrolysis of circulating triacylglycerol (TG) in very-low-density lipoproteins (VLDL-TG) and especially lipolysis of intramuscular TG (IMTG) also appear to contribute to some extent. Several steps are involved in FA uptake and oxidation in skeletal muscle and could all be of importance in the regulation of FA oxidation during exercise. Besides trans-sarcolemmal FA uptake via fatty acid transporters, it appears that intramyocellular carnitine content plays an important regulatory step in regulation of substrate selection during exercise. Interestingly, individuals with obesity exhibit a compromised ability to oxidize FAs and to increase FA oxidation in response to lipid exposure (reduced metabolic flexibility). Skeletal muscle mitochondrial function appears to be related to this defect. It remains controversial whether this impaired FA oxidative capacity in obesity diminishes the ability to increase and properly regulate FA oxidation during an acute, single exercise bout. However, despite these initial impairments in FA oxidation capacity in the obese situation, endurance exercise training can rescue the capacity for FA oxidation and the metabolic flexibility in the skeletal muscle of individuals with obesity at least to equivalent levels of their lean counterparts.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
8.1 Introduction
The work by Krogh and Lindhard and by Christensen and Hansen in the 1920s and 1930s demonstrated from measurements of the non-protein respiratory exchange ratio (RER) that fatty acid (FA) oxidation increased five- to tenfold above resting levels during mild-to-moderate exercise and decreased with increasing exercise intensities (Krogh and Lindhard 1920; Christensen and Hansen 1939). Today, it is well recognized that FA oxidation reaches its maximum at moderate intensities between 55 and 65% of maximal oxygen uptake (VO2peak) (Lundsgaard et al. 2018; Romijn et al. 1993). Beyond this level, a shift in fuel selection appears toward an increase in carbohydrate and a decrease in FA utilization (Lundsgaard et al. 2018; Romijn et al. 1993). Furthermore, the FA oxidation rate during mild-to-moderate exercise remains generally unchanged for about 60–90 min of exercise, but when exercise continues beyond this time point, a gradual increase in FA oxidation is induced at the expense of carbohydrate oxidation as fuel for energy (Romijn et al. 1993).
8.2 Fatty Acids as Energy Fuel in Skeletal Muscle
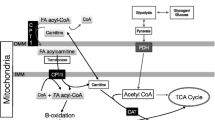
FAs as fuel for energy during exercise originate from three different sources: 1) Albumin-bound long-chain FAs liberated from lipolysis of triacylglycerol (TG) located in adipose tissue, 2) plasma FAs liberated from lipoprotein lipase (LPL)-dependent hydrolysis of TG situated in very-low density lipoprotein (VLDL-TG), and 3) FAs liberated by lipolysis of TG situated in lipid droplets in skeletal muscle (IMTG) (Fig. 8.1). The extent to which the different energy sources contribute during exercise is dependent on exercise intensity, duration, mode, and sex.

Proposed regulation of fatty acid oxidation in skeletal muscle during exercise. This figure illustrates how we propose that fatty acid (FA) oxidation is regulated in skeletal muscle during exercise. FA oxidation during exercise is mainly covered by uptake of circulating FAs, although hydrolysis of circulating triacylglycerol (TG) in very-low-density lipoproteins (VLDL-TG) and especially lipolysis of intramuscular TG (IMTG) also appears to contribute to some extent. Several steps are involved in FA oxidation and could all be of importance in the regulation of FA oxidation during exercise. Besides trans-sarcolemmal FA uptake via fatty acid transporters, it appears that intramyocellular carnitine content plays an important regulatory step in the mitochondrial import and hence FA oxidation during exercise
Findings indicate that approximately 55–65% of total whole-body FA utilization during moderate-intensity exercise, where FA oxidation is at its highest level, is derived from plasma FAs (Romijn et al. 1993; Friedlander et al. 1999; Helge et al. 2001; Roepstorff et al. 2002; van Loon et al. 2001) and that the contribution from plasma FAs to energy provision increases with time (van Loon et al. 2003).
8.2.1 Albumin-Bound Plasma FA
Plasma FA uptake into skeletal muscle is the product of blood flow and the arteriovenous FA concentration difference. The increase in blood flow during exercise, which increases up to 20-fold from rest to intense, dynamic exercise (Calbet and Lundby 2012; Radegran and Saltin 1998), is a main driver of the exercise-induced increased FA uptake. The other important determinant of FA uptake during exercise is the arterial FA concentration. At onset of exercise, a transient decrease in plasma FA concentration may appear followed by a slow increase (Roepstorff et al. 2002). If exercise is prolonged, the arterial concentration of FA may increase to levels approximately sixfold higher than resting levels (Romijn et al. 1993; Kiens and Richter 1998; Wolfe et al. 1990; van Hall et al. 2002; Bahr et al. 1991; Hagenfeldt and Wahren 1975). The initial drop in arterial FA concentration is caused by an imbalance between slow mobilization of FAs from the adipose tissue and a rapidly increased extraction of FAs in skeletal muscle. The following increase in plasma FA concentration during exercise is mainly caused by an increased release of FAs liberated from adipose tissue by lipolysis of TG. Accordingly, the whole-body lipolytic rate can be increased by up to fourfold compared with resting values during submaximal moderate-intensity exercise (Wolfe et al. 1990; Romijn et al. 2000). This exercise-induced increase in adipose tissue lipolysis is mediated by altered adrenergic stimulation of adipose tissue evidenced by a shift from predominant α-adrenergic suppression during rest toward predominant β-adrenergic stimulation during exercise (Arner et al. 1990). The primary adrenergic stimulus of adipose tissue during exercise originates from circulating adrenaline with only a minor contribution from noradrenaline released from sympathetic neurons (Stallknecht et al. 2001; de Glisezinski et al. 2009). Moreover, the natriuretic peptides appear to play an additional role in exercise-induced lipolysis in humans (Moro et al. 2004; de Glisezinski et al. 2009) and have been suggested to account for most of the non-adrenergic lipolytic signaling in adipose tissue during exercise (Moro et al. 2006; Lafontan et al. 2008). The regulation of adipose tissue lipolysis during exercise is described in more detail in another chapter of this eBook.
It appears that 60 to 76% in females and males, respectively, of the exercise-induced increase in whole-body FA removal from the circulation can be accounted for by uptake into working skeletal muscles (Kiens 2006). The question is how much of the FAs taken up by the working muscle is then directly oxidized? When FA uptake into skeletal muscle during exercise was directly measured, it was found that up to 100% and 84% of tracer-derived FA uptake were directly oxidized in trained females and males, respectively (Roepstorff et al. 2002; Hagenfeldt and Wahren 1968; Turcotte et al. 1992).
Another fat source generating energy during exercise may be hydrolysis of circulating TG. The liver is secreting endogenous TG enfolded into VLDL particles directly into the circulation. VLDLs are the main carriers of TG in the post-absorptive state. Hydrolysis of core TG in VLDL is mediated by the enzyme lipoprotein lipase (LPL), which in its active form is located at the luminal site of the endothelial cells in the capillary bed of various tissues such as skeletal muscle, heart, and adipose tissue (Fig. 8.1). By hydrolysis, FAs are liberated and taken up by the surrounding tissues. Oxidation of FAs from VLDL-TG may contribute to the total FA oxidation both at rest and during moderate-intensity exercise, though its relative contribution during exercise is less than albumin-bound FA oxidation. Hence, when VLDL-TG content was measured in the femoral artery and vein during knee-extensor exercise, a total net degradation of VLDL-TG was found during the 2 hours of exercise (Kiens et al. 1993), suggesting that circulating VLDL-TG contributes to the total FA oxidation during moderate exercise. Supporting these findings are recent elegant studies using infusion of labeled VLDL-TG in healthy, young humans. Data showed that FAs from labeled VLDL-TG comprised 3–6% of total energy utilization (Sondergaard et al. 2011) or 3–13% of total FA oxidation during moderate-intensity exercise in untrained and moderately trained men (Nellemann et al. 2014; Morio et al. 2004). Increased VLDL-TG hydrolysis during exercise is likely explained by the findings of increased activity of LPL in skeletal muscle in most (Kiens and Richter 1998; Perreault et al. 2004; Taskinen and Nikkila 1980), but not all, human studies (Lithell et al. 1979; Kiens et al. 1989; Søndergaard et al. 2017). The mechanisms driving translocation of newly synthesized LPL in the myocyte to the luminal side of the endothelial cells in the capillary bed during exercise remain to be elucidated. However, exercise-induced muscle LPL activity has been correlated to the IMTG and muscle glycogen concentrations (Kiens 2006), suggesting that the energy status of the myocyte might signal to LPL synthesis and translocation to the capillary to facilitate VLDL-TG hydrolysis during exercise. Importantly, LPL-derived VLDL-TG hydrolysis might be a greater contributor as substrate to cover the high FA oxidation during recovery from exercise (Kiens and Richter 1998; Morio et al. 2004; Lundsgaard et al. 2020).
8.2.2 Regulation of Fatty Acid Uptake into Skeletal Muscle
The increase in uptake of plasma FAs into skeletal muscle during exercise is regulated at several steps including transmembrane transport, cytosolic handling, mitochondrial membrane transport, and intra-mitochondrial FA oxidation (Fig. 8.1). Transport of plasma FAs across sarcolemma serves as the first step in myocellular FA uptake. Despite the fact that FAs can diffuse within biological membranes, membrane-bound lipid-binding proteins have been identified in human skeletal muscle, and evidence show that these proteins either individually or in complexes act as regulators of FA transmembrane transport (Fig. 8.1). The first suggestion for FA transporter limited FA uptake came from both human (Kiens et al. 1993) and rat studies (Turcotte et al. 1992) indicating that FA uptake is a saturable process. The FA translocase cluster of differentiation 36 (CD36) are most extensively studied for its importance in FA uptake during exercise. In the basal resting state, a large part of total muscle CD36 is stored in intramyocellular compartments (endosomes), whereas a small part is present at the sarcolemma, the outer membrane of the muscle cell, to mediate basal FA uptake (Chabowski et al. 2007). During exercise, CD36 reversibly translocates to sarcolemma (Jeppesen et al. 2011) shown in human skeletal muscle by a 75% higher sarcolemmal content of CD36 after prolonged submaximal exercise compared with rest (Bradley et al. 2012). In rat skeletal muscle, FA transport was 75% higher in contracted compared with rested rat skeletal muscle, which correlated with CD36 translocation to sarcolemma (Bonen et al. 2000). In addition, mice lacking CD36 exhibited lower FA oxidation compared with control mice during treadmill exercise at the same relative workload (McFarlan et al. 2012), whereas mice overexpressing CD36 protein exhibited greater contraction-stimulated FA oxidation than control mice (Ibrahimi et al. 1999). Together findings in both rodent and human skeletal muscle clearly show that translocation of CD36 seems vital in FA uptake in skeletal muscle during exercise.
Skeletal muscle contractions also induce translocation of other FA transporters, such as FA-binding protein at the plasma membrane (FABPpm) and FA transport 1 and 4 (FATP1/4), to the plasma membrane in human skeletal muscle (Bradley et al. 2012; Jain et al. 2009) (Fig. 8.1). Overexpression or inhibition of these proteins in rodent skeletal muscle has been shown to increase or decrease FA uptake, respectively, during resting conditions (Holloway et al. 2007b; Clarke et al. 2004; Turcotte et al. 2000). However, the role of these proteins in FA oxidation during acute exercise remains to be established.
The signaling mechanism(s) governing sarcolemmal FA transporter translocation during exercise are not clarified. Activation of various intracellular signaling pathways related to the energy status (e.g., AMP-activated protein kinase (AMPK)) (Jeppesen et al. 2011; Bonen et al. 2007), mechanical stress (e.g., extracellular signal-regulated kinase (ERK) 1/2 and p38 mitogen-activated protein (MAP) kinase signaling) (Raney and Turcotte 2006), ionic homeostasis (e.g., Ca2+/calmodulin-dependent protein kinases (CaMK)) (Lally et al. 2012; McFarlan et al. 2012), and other signaling molecules such as the Rab-GTPase activating proteins TBC1 domain family member 1 and 4 (TBC1D1 and -4) (Chadt et al. 2008; Benninghoff et al. 2020; Samovski et al. 2012) have all been proposed as regulators of FA uptake via CD36 translocation to sarcolemma during resting conditions. However, exercise-stimulated CD36 translocation, FA uptake, and/or FA oxidation in skeletal muscle were not impaired, when AMPK (Jeppesen et al. 2011; Dzamko et al. 2008; Hingst et al. 2020) and TBC1D1 (Whitfield et al. 2017) were genetically deleted in rodent muscles. Besides, contraction-induced CD36 translocation occurred prior to changes in phosphorylation of ERK1/2 and other major MAP kinases in mouse muscle (Jeppesen et al. 2011). Thus, it is presently unknown what signal that stimulates CD36 and other lipid-binding proteins to translocate to sarcolemma and hereby increase FA uptake in skeletal muscle during exercise. Systemic signaling molecules from outside the muscle cells could likely also contribute, e.g., circulating levels of the lipid 12,13-dihydroxy-9Zoctadecenoic acid (12,13-diHOME) originating from brown adipose tissue (BAT) increases during exercise and are shown to be able to induce skeletal muscle FA uptake and oxidation (Stanford et al. 2018). This emphasizes that not only intracellular mechanisms but likely also systemic signals could possibly together orchestrate the increased uptake of circulating FAs into skeletal muscle during exercise.
8.3 The Intracellular FA Source
Circulating FAs taken up into skeletal muscle can either be oxidized or stored in intramyocellular lipid droplets (IMTG) and add to the plasma-delivered FAs as a potential energy source during exercise (Fig. 8.1). To what degree IMTG is used during exercise is discussed heavily in the literature for several decades, and different methodologies have contributed to evaluate the contribution of IMTG as fuel for FA oxidation during exercise. Early studies with the muscle biopsy technique found a 25–30% IMTG reduction after 99–147 min submaximal exercise in men (Costill et al. 1979; Carlson et al. 1971), and such exercise-induced decrease in IMTG in men was later supported by more sophisticated freeze-dried dissection of muscle tissue after exercise protocols of ∼2 h (Essén 1978; Hurley et al. 1986; Phillips et al. 1996). However, other studies did not find detectable changes in IMTG during exercise in mainly shorter 60–120 min exercise protocols (Helge et al. 2001; Roepstorff et al. 2002; Kiens and Richter 1998; Kiens et al. 1993; Bergman et al. 1999; Starling et al. 1997; Steffensen et al. 2002). Lack of breakdown of IMTG was also shown indirectly by microdialysis during knee-extensor exercise at submaximal exercise in men (Stallknecht et al. 2004). This could emphasize that exercise needs to be prolonged—likely above 90 min to acquire IMTG as a major substrate to fuel FA oxidation during exercise.
From studies, where the 1H-MRS technique was applied, treadmill running at 50–70% of VO2peak for 2 h or until exhaustion decreased IMTG signal (IMCL in MRS terminology) by 22–33% in men and women (Krssak et al. 2000; Larson-Meyer et al. 2002; Decombaz et al. 2001). However, it is hard to exclude from those studies, where it takes considerable time to be positioned in the scanner and perform the measurements, whether the lower IMCL signal is due to use of IMTG in the early recovery period rather than during exercise.
Importantly, it seems that women of widely differing training backgrounds in contrast to matched men utilize a significant greater amount of IMTG in the vastus lateralis during prolonged bicycle exercise (Steffensen et al. 2002). The role of sex in regulation of lipid metabolism during exercise is described in more detail in a separate chapter in this eBook.
Estimations from tracer-infusion studies suggest that oxidation of FAs from plasma versus IMTG and other lipid sources such as VLDL-TG and TG between fibers comprise ∼60% and 30% of FA oxidation, respectively, during 2 h of moderate-intensity exercise (Romijn et al. 1993; van Loon et al. 2003). These findings support the notion that IMTG lipolysis is not likely to limit FA availability to oxidation in skeletal muscle during exercise.
Summarizing data which seems as quantitative importance of IMTG as an energy source during exercise depends on several factors as duration and intensity of exercise, exercise mode, and sex (Kiens 2006). Overall, it appears that FAs derived from hydrolysis of IMTG may contribute as energy fuel especially when exercise is prolonged—beyond 90 min, mainly when exercise is performed in the fasted state and to a larger extent in women than in men (Kiens 2006).
Interestingly, another key aspect in the necessity of IMTG as substrate for FA oxidation during exercise is a potential interdependency with the circulating FA availability. If circulating levels of FAs become limited, it could be speculated that there is an inverse interaction between plasma-derived FAs and those generated from IMTG lipolysis during exercise. Thus, when adipose tissue lipolysis was pharmacologically inhibited by nicotinic acid or acipimox prior to prolonged submaximal exercise in healthy individuals, the exercise-induced increase in plasma FA concentration was completely suppressed, while IMTG utilization was increased during exercise compared with when plasma FA availability was not limited (Watt et al. 2004b; van Loon et al. 2005). Therefore, the uptake of circulating FAs may interact with the regulation of IMTG lipolysis during exercise.
Another consideration about the importance of IMTG for FA oxidation during exercise is the fact that most investigations have focused on measuring net IMTG breakdown during exercise. From studies during resting conditions using pulse-chase methods by intravenous infusions of two distinct isotopically labelled FAs combined with mass spectrometry measurements of intramuscular lipids, it was shown that plasma FAs taken up by the muscle were not directly oxidized, but traversed the IMTG pool prior to oxidation in the resting state (Kanaley et al. 2009). At very-low-intensity exercise loads, a similar fate of circulating FA uptake undergoing esterification and then subsequent hydrolysis prior to mitochondrial entry may appear. Also, when the exercise load is low and only few muscle fibers are recruited, it could be speculated that FAs taken up into skeletal muscle are esterified and stored as IMTG in the non-recruited muscle fibers. Under such circumstances, it is difficult to evaluate IMTG breakdown during exercise with the available methods.
In terms of the molecular regulation of IMTG lipolysis during exercise and muscle contractions, this is orchestrated primarily by activation of the two lipases adipose triglyceride lipase (ATGL) (Alsted et al. 2013) and hormone-sensitive lipase (HSL) in skeletal muscle (Langfort et al. 2000; Watt et al. 2004a), which are catalyzing the conversion of TG to diacylglycerol and further to monoacylglycerol, respectively (Fig. 8.1). The signals regulating ATGL and HSL activity during exercise are complex and only scarcely understood in skeletal muscle. In this regard, ex vivo contractions in isolated skeletal muscle resulted in HSL translocation to lipid droplets (Prats et al. 2006). The translocation of HSL appears accredited to an intrinsic activation of HSL at different serine residues within the protein achieved by catecholamine-induced protein kinase A (PKA) activation (Talanian et al. 2006). The resultant translocation of HSL to the lipid droplet initiates lipolysis. In contrast, activation of ATGL does not seem to be PKA-dependent, but rather is requiring co-activation by comparative gene identification-58 (CGI-58) to achieve maximal hydrolase activity (Zechner et al. 2012) (Fig. 8.1).
The lipid droplet associated perilipins (PLINs) are also part of the lipolytic machinery in skeletal muscle, and PLIN3 and PLIN5 physically interact with HSL and ATGL (MacPherson et al. 2013; Smirnova et al. 2006) (Fig. 8.1). It remains to be further elucidated how the lipolytic regulation and/or intracellular trafficking of IMTG lipid droplets determine its quantitative importance for the FA oxidative rate in skeletal muscle during exercise.
8.4 Mitochondrial Regulation of FA Oxidation During Exercise
Plasma FAs taken into the cell or liberated from intracellular lipolysis must be activated in the cytosol to fatty acyl-CoAs by a family of acyl-CoA synthetases (ACSs) (Fig. 8.1). The active site of the ACSs has been located to the plasma membrane and mitochondria and in close proximity to lipid droplets. The isoform ACSL1 seems particularly important for partitioning FAs toward oxidation in skeletal muscle, which is emphasized by the findings in mice with muscle-specific ACSL1 deficiency exhibiting lower FA utilization during submaximal exercise compared with control mice (Li et al. 2015). To enter the mitochondria for β-oxidation, long-chain fatty acyl-CoAs (the primary FAs in humans) are converted to their fatty acyl carnitine derivatives, a reaction that requires carnitine. This reaction is catalyzed by the enzyme carnitine palmitoyl transferase 1 (CPT1), located at the outer mitochondrial membrane (Bonnefont et al. 2004) (Fig. 8.1). The importance of CPT1 for FA uptake into mitochondria during exercise is evidenced by reduced FA oxidation and increased lipid accumulation in mice with muscle-specific deletion of CPT1 (Wicks et al. 2015) and 50–90% decreased FA oxidation during ex vivo muscle contractions with concomitant pharmacological CPT1 inhibition (Dzamko et al. 2008). A role of CPT1 for regulation of long-chain FA oxidation is also displayed in humans, since oxidation of the medium-chain FA octanoate (C8), which is able to bypass CPT1, did not change when exercise intensity was shifted from 40 to 80% of VO2peak, as was the case for oleate, a CPT1-dependent long-chain FA (Sidossis et al. 1997).
CPT1 might not be the only step in mitochondrial import of FAs. CD36 was found to be located at the outer mitochondrial membrane in some (Campbell et al. 2004), but not all studies (Jeppesen et al. 2010). Exercise has been further demonstrated to induce CD36 translocation from intracellular depots to the mitochondrial membrane in rodent and human muscle (Monaco et al. 2015; Holloway et al. 2006), and it was suggested that mitochondrial CD36 interacts with ACSs and hereby regulates fatty acyl-CoA availability to CPT1 (Smith et al. 2011). The regulatory role of CD36 in mitochondrial FA import and oxidation during exercise needs to be further investigated.
8.4.1 CPT, Carnitine, and Mitochondrial Fatty Acid Import During Exercise
Since carnitine is substrate in the CPT1 reaction, changes in the free carnitine content in skeletal muscle during exercise could contribute to the regulation of mitochondrial transmembrane FA transport and hereby FA oxidation. Acetyl-CoAs are produced both from β-oxidation of FAs and from glycolysis-derived pyruvate by pyruvate dehydrogenase complex (PDC) (Harris et al. 2002) (Fig. 8.1). Free carnitine can buffer acetyl-CoA by forming acetyl-carnitine and free CoA (Friedman and Fraenkel 1955), a reaction catalyzed by the enzyme carnitine acetyltransferase (CAT) (Fig. 8.1). This entrapment of carnitine increases, when acetyl-CoA is generated in excess of its metabolism in the Krebs cycle. Consequently, lowered amount of free carnitine to the CPT1 reaction would be expected to diminish the supply of fatty acyl-CoA for β-oxidation and hence FA oxidation.
Such mechanism could play a role in regulation of substrate selection during exercise especially at increasing exercise intensities where an increasing carbohydrate oxidation in replacement for FA oxidation takes place.
It is well established that skeletal muscle PDC activity increases rapidly during exercise in an intensity-dependent manner (Constantin-Teodosiu et al. 1991), but decreases gradually when exercise is prolonged (Watt et al. 2002). To alleviate allosteric product inhibition of PDC activity by acetyl-CoA formed in the glycolysis during increasing exercise intensities, the CAT enzyme buffers excess acetyl-CoA into acetyl-L-carnitine. This CAT-mediated acetyl-CoA buffering will reduce cellular free carnitine content and thereby limits mitochondrial FA import and hence FA oxidation allowing for a high rate of pyruvate oxidation. In agreement, in mice with a muscle-specific deletion of CAT, a higher whole-body FA oxidation rate was demonstrated during graded submaximal treadmill running (Seiler et al. 2015). As the PDC reaction rate and the concomitant CAT activity regulate mitochondrial FA import and hence substrate availability to β-oxidation, this could point to lowering of FA oxidation during increasing exercise intensities as a secondary result of increased glucose flux and accelerated glycolysis rather than due to an initial lowering of FA oxidation within lipid metabolic machinery.
This notion is supported by the findings of a one- to threefold increase in muscle acetyl-CoA and acetyl-L-carnitine content at higher exercise intensities compared with rest or low-intensity exercise in untrained individuals (Sahlin 1990; Constantin-Teodosiu et al. 1991; Harris et al. 1987). This resulted in a decreased free carnitine content from comprising ~75% of muscle total carnitine at rest to ~20% at an exercise intensity of 90–100% of VO2peak (Sahlin 1990; Constantin-Teodosiu et al. 1991; Harris et al. 1987). Moreover, an association between lowering of free carnitine levels and increased acetyl-L-carnitine entrapment during exercise with increasing intensities is evident from several studies (van Loon et al. 2001; Odland et al. 1998; Ren et al. 2013). Importantly, when muscle carnitine content was enhanced by oral carnitine supplementation in healthy, young men, this enabled an increased use of acetyl-CoA from β-oxidation evidenced by a lower glycogen utilization and PDC activation during moderate-intensity exercise in the carnitine-supplemented state compared with the control (Wall et al. 2011). Recently, these findings were reproduced in older men, in which 25 weeks of carnitine supplementation resulted in a 20% increase in both muscle total carnitine content in total FA oxidation during a 1 h submaximal exercise bout at 50% of VO2peak (Chee et al. 2021).
A final line of evidence highlighting carnitine availability as a regulatory mechanism for FA oxidation is derived from studies where muscle glycogen content was manipulated to be either high or low prior to a moderate-intensity exercise bout in humans. During exercise with initially high glycogen stores, RER during exercise was high indicating a high carbohydrate oxidation. In contrast, when glycogen stores were low prior to exercise, FA oxidation was high during exercise. Accordingly, when carbohydrate oxidation was high during exercise, a 49% higher muscle acetyl-CoA content, 37% higher acetyl-carnitine levels, and 55% lower free carnitine content were observed, while muscle content of acetyl-CoA, free carnitine, and acetyl-carnitine remained unchanged, when FA oxidation rate was enhanced during exercise with a low initial muscle glycogen content (Roepstorff et al. 2005). A 20% increase in muscle free carnitine content was also observed during 10 min of high-intensity exercise in a glycogen-depleted state, whereas this free-carnitine decreased 60% during exercise in the high-glycogen conditions (Constantin-Teodosiu et al. 2004).
Collectively, there is a great body of evidence suggesting carnitine as an important regulatory step in regulation of substrate selection during exercise—especially during exercise with increasing intensities. Importantly, since exercise-induced PDC activation and hereby glycolysis-derived acetyl-CoA production are lowered during prolonged exercise (Watt et al. 2002), and carnitine entrapment into acetyl-carnitine is lowered during exercise with low muscle glycogen (Roepstorff et al. 2005; Constantin-Teodosiu et al. 2004), increased amounts of free carnitine to the CPT1 reaction, in turn increasing the supply of fatty acyl-CoA for FA oxidation, could likely be a regulatory mechanism responsible for the established gradual increase in FA oxidation during prolonged exercise above ⁓60–90 minutes, when muscle glycogen levels are being emptied.
In addition to carnitine availability, other contributing steps regulating FA oxidation in skeletal muscle during exercise must also be considered. Carnitine-independent regulation of CPT1 could likely be an additional regulatory step for muscle FA import into the mitochondria. Activation of the cellular energy sensor, AMPK, during exercise (Wojtaszewski et al. 2000)—previously proposed to increase FA oxidation via regulation of malonyl-CoA-mediated inhibition of CPT1 (McGarry et al. 1983; Smith et al. 2012; Rasmussen and Winder 1997)—has in recent years from observations in humans (Odland et al. 1996; Odland et al. 1998; Dean et al. 2000; McConell et al. 2020) and transgenic mouse models (Dzamko et al. 2008; Hingst et al. 2020; Miura et al. 2009; Lee-Young et al. 2009; Fritzen et al. 2015; O'Neill et al. 2015) been shown not to be essential for regulation of FA oxidation in skeletal muscle during exercise (Lundsgaard et al. 2018; McConell 2020). However, fatty acyl-CoA/malonyl-CoA ratio appears to be important for CPT1 catalytic activity and FA oxidation, rather than the total malonyl-CoA content per se (Smith et al. 2012), potentially by decreasing the affinity of CPT1 for malonyl-CoA binding (Kolodziej and Zammit 1990). Moreover, malonyl-CoA inhibition kinetics of CPT1 seems modulated by interaction between the cytoskeleton and mitochondria during exercise (Miotto et al. 2017). CPT1 activity could be regulated in also malonyl-CoA-independent mechanism during exercise by a reduction in muscle pH during intense exercise (Starritt et al. 2000). Lastly, ex vivo findings suggest that the reaction rate of β-oxidative enzymes, such as the β-ketoacyl-CoA thiolase, the enzyme catalyzing the final step in the β-oxidation, is feedback regulated by mitochondrial acetyl-CoA content (Eaton 2002) and enzymes in the β-oxidation could hence also be a contributing factor in fine-tuning regulation of FA oxidation during exercise.
8.5 Summarizing Remarks on the Regulation of FA Oxidation in Skeletal Muscle During Exercise
Whole-body FA oxidation is increased several fold during prolonged moderate exercise, but a shift in substrate selection toward increased carbohydrate and decreased relative FA oxidation takes place when exercise intensity is increased beyond 55–65%. Uptake of FAs from the circulation into the skeletal muscle is a major contributing substrate to FA oxidation during submaximal exercise and mostly derived from adipose tissue lipolysis liberated FAs. However, LPL-hydrolysis of circulating VLDL-TG and lipolysis of IMTG also seem to contribute as substrate to fueling the increased FA oxidation in skeletal muscle during exercise.
The increased oxidation of plasma FAs into skeletal muscle during exercise is regulated at several steps, and regulation of FA oxidation in skeletal muscle is not allocated to one single mechanism or signaling pathway, but is apparently orchestrated by a symphony of tightly coordinated molecular events reliant on the metabolic fluxes.
The increase in FA oxidation from rest to submaximal exercise is reliant on an increased transmembrane transport of FA, in which CD36 translocation to the sarcolemma has emerged to serve a pivotal role (Fig. 8.1). The fine-tuning of FA oxidation during exercise appears allocated to the regulation within the mitochondria, where the FA import into the mitochondrial matrix, via formation of fatty acyl carnitine by CPT1, appears to be a central regulatory stage in exercise FA oxidation. This process seems regulated by the intramitochondrial acetyl-CoA homeostasis in response to exercise duration and intensity, as the acetyl-CoA content determines the free carnitine availability for CPT1 during exercise (Fig. 8.1). In this process, the rate of glycolysis appears to be a central tenet for mitochondrial acetyl-CoA availability and hence regulation of FA oxidation. Accordingly, regulation of FA oxidation in skeletal muscle includes a chain of interdependent processes, and dysfunction in any of these can lead to metabolic impairment.
8.6 Are There Impairments in Fatty Acid Oxidation in Skeletal Muscle with Obesity?
In individuals with obesity, an impairment FA oxidation is observed at least during resting conditions. In a seminal study in Pima Indians examining 24-hour whole-body RER while consuming a eucaloric diet, the incidence of weight gain (>5 kg) over a subsequent 3-year period was higher in individuals with elevated 24-hour RERs, indicative of a preference for carbohydrate over FA oxidation (Zurlo et al. 1990). Similarly, other studies have reported that a higher RER is linked with subsequent weight gain (Marra et al. 2004; Seidell et al. 1992; Rogge 2009), while tracer methodology has revealed a decreased utilization of lipid at rest in individuals with severe obesity (Thyfault et al. 2004).
A possible mechanism explaining this inability to oxidize lipid may involve skeletal muscle. We have reported an ≈50% reduction in FA oxidation in two distinct muscle groups (rectus abdominus and vastus lateralis) in individuals with severe obesity (Hulver et al. 2003; Kim et al. 2000); these findings suggest that reductions in the capacity for FA oxidation with obesity could be evident in numerous muscle groups, which could in turn affect whole-body metabolism. Others have also reported a reduction in FA oxidation in human skeletal muscle in individuals with obesity but not severe obesity (Holloway et al. 2007a). A ≈ 50% reduction in FA oxidation in primary skeletal muscle cell cultures (myotubes) derived from individuals with obesity has also been observed (Consitt et al. 2010; Hulver et al. 2005; Maples et al. 2015b; Gaster 2009; Bell et al. 2010) as well as an increased proportion of incomplete FA oxidation (Løvsletten et al. 2020). These findings in cell cultures are indicative of a persistent and perhaps inherent defect, as the muscle cells proliferate (myoblasts) and differentiate into myotubes in the absence of factors which may affect FA oxidation in vivo such as the hormonal milieu and/or differences in contractile activity. An elevated RER with obesity was also reported in vivo across a muscle bed (Kelley et al. 1999).
Deficits in the activity and/or expression of enzymes involved in FA oxidation likely contribute to this impairment in FA oxidation with obesity during resting conditions; the roles of specific proteins involved with lipid transport and oxidation are discussed in review papers (Rogge 2009; Houmard 2008; Houmard et al. 2012; Fritzen et al. 2020; Houmard et al. 2011; Holloway et al. 2009). On the organelle scale, mitochondrial mass appears to be compromised with obesity as proteins indicative of mitochondrial mass are decreased at both moderate (Simoneau et al. 1999; Kriketos et al. 1996; Kelley et al. 2002) and higher ranges (Kim et al. 2000; Holloway et al. 2007a) of obesity. Holloway et al. (Holloway et al. 2007a; Holloway et al. 2009) reported a decline in skeletal muscle FA oxidation in muscle homogenates of subjects with obesity. However, when mitochondria were isolated, FA oxidation was equivalent between lean and obese individuals, indicating that the decline at the tissue level was due to compromised mitochondrial mass. Similarly, in primary myotubes, indices of mitochondrial content and FA oxidation were reduced with obesity, but when FA oxidation was normalized to mitochondrial content (mitochondrial DNA (mtDNA), cytochrome c oxidase subunit 4 (COXIV)), there were no differences between groups suggesting that mitochondrial volume may be the driving factor in the metabolic impairments seen with obesity (Consitt et al. 2010). There is also an increased percentage of glycolytic, low mitochondrial content Type IIb fibers in the skeletal muscle of severely obese subjects (Tanner et al. 2002). The mechanism(s) responsible for the reductions in mitochondrial mass with obesity are not readily evident; it was recently reported that mitochondrial network quality and morphology were altered in primary myotubes derived from individuals with severe obesity (Gundersen et al. 2020).
There is also evidence that the mitochondrial processes involved with FA oxidation are compromised with obesity. Indeed, both FA and glucose oxidation are reduced in the skeletal muscle of obese individuals (Jones et al. 2019). The confluence of FA and glucose oxidation is the TCA cycle, and flux through the TCA cycle is impaired in skeletal muscle both with obesity (Zou et al. 2019) and type 2 diabetes (Gaster 2012). A mechanism for this reduction in TCA cycle activity could involve posttranslational modifications (i.e., phosphorylation, acetylation) which would in turn impair flux (Gaster 2012; Gaster et al. 2012; Boyle et al. 2012). In agreement, there is an increase in blood lactate concentration with obesity which would result from a reduction in the partitioning of substrate to oxidative pathways via the TCA cycle in skeletal muscle (Jones et al. 2019; Broskey et al. 2020). Together, these and other findings (Dahlmans et al. 2016; Diaz-Vegas et al. 2020) indicate that mitochondrial dysfunction and a reduction in mitochondrial mass both contribute to the dampened ability to oxidize lipid in the skeletal muscle of individuals with obesity.
Impaired FA oxidation in individuals with obesity has not only been observed during resting, fasted conditions. An inability to increase FA oxidation in response to increased lipid availability has also been reported with obesity under a variety of conditions (e.g., high-fat feedings, fasting, lipid incubations of primary myotubes, lipid infusion), and findings are summarized in more detail elsewhere (Galgani et al. 2008; Storlien et al. 2004; Goodpaster and Sparks 2017). In reference to obesity, an inability to increase FA oxidation (24 hour RER) in response to a high-fat diet was predictive of greater weight gain in the subsequent year even in lean individuals (Begaye et al. 2020). A dampened ability to increase FA oxidation in individuals with obesity is evident after as little as 2 days of a high-fat diet and subsequently contributes to positive lipid balance (Galgani et al. 2008). Thus, an inability to appropriately increase FA oxidation, or lack of metabolic flexibility, is evident with obesity and individuals prone to obesity under a variety of free-living situations.
One of the mechanisms involved with this lack of metabolic flexibility may be an impaired ability to increase the oxidative machinery of skeletal muscle upon lipid exposure. In response to lipid incubation, primary myotubes from lean individuals increased mitochondrial DNA copy number and mRNA content of genes that upregulate FA oxidation (nuclear respiratory factor (NRF)-1, NRF-2, peroxisome proliferator-activated receptor (PPAR)α, PPARδ, and pyruvate dehydrogenase kinase 4 (PDK4, CPT1); in contrast, cells derived from individuals with obesity did not alter or actually decreased the expression of these genes (Maples et al. 2015b; Boyle et al. 2012; Maples et al. 2015a). Similarly, a 5-day high-fat diet elicited increases in the mRNA content of key genes involved in oxidative pathways such as peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α), PDK4, mitochondrial uncoupling protein (UCP)3, and PPARα. Moreover, a single high-fat feeding increased PPARα mRNA in skeletal muscle biopsies from lean individuals, while there were no changes in subjects with obesity (Boyle et al. 2011). These findings indicate a multifaceted lack of metabolic flexibility in terms of appropriately increasing FA oxidation in the skeletal muscle of individuals with obesity.
8.7 Exercise: Do the Alterations in Fatty Acid Oxidation in Skeletal Muscle with Obesity Alter Substrate Utilization During Exercise?
A bout of mild-to-moderate endurance exercise increases the rates of both carbohydrate and FA oxidation in order to meet the energy demands of increased contractile activity in skeletal muscle. This ability to increase oxidation in response to exercise is another index of metabolic flexibility (Galgani et al. 2008; Goodpaster and Sparks 2017). As indicated in previous sections, mitochondrial content and function are compromised in the skeletal muscle of individuals with obesity, which contributes to a dampened ability to oxidize lipid as well as an impairment in metabolic flexibility. However, in spite of these mitochondrial defects, acute exercise increases the rate of FA oxidation over resting conditions both during and after the exercise bout regardless of obesity status (Thyfault et al. 2004; Fritzen et al. 2020; Arad et al. 2020; Guesbeck et al. 2001; Hansen et al. 2005). This adjustment is critical, as exercise can subsequently contribute to a state of negative lipid balance which in turn can aid in minimizing ectopic lipid accumulation. However, it is not apparent if the lack of metabolic flexibility with obesity compromises the magnitude of the increase in FA oxidation compared to lean individuals, thus resulting in a lower absolute rate of FA oxidation during exercise.
Several studies have reported an impaired ability to appropriately increase FA oxidation in response to endurance exercise with obesity. Thyfault et al. (Thyfault et al. 2004) compared FA oxidation (infused [14C] palmitate and [14C] acetate) during 60 minutes of cycling exercise at 50% VO2peak in lean patients, patients with severe obesity, and patients who were previously severely obese and had lost >45 kg (bariatric surgery). FA oxidation was significantly lower in the subjects with obesity. Surprisingly, FA oxidation was also depressed during exercise in previously severely obese individuals even after profound weight loss (Thyfault et al. 2004). Similar findings were obtained in response to 10 minutes of exercise at either the same absolute (15 Watts) or relative (65% VO2peak) exercise intensities, as FA oxidation (via indirect calorimetry) was depressed in previously severely obese women at >1 year after bariatric surgery (gastric bypass) compared to age and BMI-matched controls. Eaves et al. (Eaves et al. 2012) examined pre-pubescent (≈11 years old) children of parents that were either lean or severely obese. FA oxidation/metabolic flexibility was determined during 10 minutes of mild-intensity cycle exercise. The children with a parent with severe obesity displayed a reduced rate of FA oxidation at the same absolute exercise workload of 15 watts (≈40% VO2peak). There were, however, no differences at the higher exercise intensity (65% VO2peak) between the two groups.
Other findings provide a more equivocal view of whether substrate utilization is altered during exercise in individuals with obesity. A comprehensive systematic review (Arad et al. 2020) concluded that majority of evidence indicated that individuals with obesity rely on the oxidation of lipid to a similar extent as lean subjects during exercise. This conclusion (Arad et al. 2020) was based upon analyses of 24 papers (out of 729 identified papers on the topic) which met rigorous inclusion criteria that included eliminating studies of older adults, individuals with chronic diseases, and/or subjects with exercise limitations. Upon further examination, ten of these 24 papers utilized incremental exercise bouts with relatively short (3–6 minutes) stages compared to those with longer stages where steady-state levels of substrate oxidation are more likely to be achieved. One study (Balcı 2012) also placed overweight and obese individuals into a single group rather than studying the effects of obesity alone. In the remaining studies, there were reduced (Mittendorfer et al. 2004), equivalent (Colberg et al. 1996; Devries et al. 2013; Ezell et al. 1999; Kanaley et al. 1993; Kanaley et al. 2001; Santiworakul et al. 2014; Slusher et al. 2015; Steffan et al. 1999), and even increased (Kanaley et al. 2001; Goodpaster et al. 2002; Horowitz and Klein 2000) rates of FA oxidation in obese compared to lean individuals, which prompted the conclusion that FA oxidation is not compromised in response to exercise with obesity.
It is difficult to determine why such disparate findings have been obtained. Factors such as duration of the exercise bout, exercise mode, exercise intensity, body fat distribution, time of day of testing, pre-exercise diet, method by which FA oxidation is determined (e.g., indirect calorimetry or tracer), comparison to appropriate control group, and numerous other factors could explain the conflicting results. There may also be racial differences as Caucasian women with obesity exhibited a reduction in FA oxidation during exercise at 65% VO2peak compared to their lean counterparts, while there were no differences in FA oxidation between African American women with normal BMI or obesity (Hickner et al. 2001). Another confounding variable may be the degree of obesity. A decline in FA oxidation during exercise has been reported in subjects with severe (BMI ≥ 40 kg/m2) obesity, subjects with severe obesity after weight loss, and the offspring of individuals with severe obesity (Thyfault et al. 2004; Guesbeck et al. 2001; Eaves et al. 2012). Studies indicating no differences (Colberg et al. 1996; Devries et al. 2013; Ezell et al. 1999; Kanaley et al. 1993, 2001; Santiworakul et al. 2014; Slusher et al. 2015; Steffan et al. 1999) or increases (Kanaley et al. 2001; Goodpaster et al. 2002; Horowitz and Klein 2000) in FA oxidation studied obese (BMI ranges of 30 to 39.9 kg/m2) but not individuals with severe obesity. Regardless, although FA oxidation consistently increases in response to exercise, it remains to be defined if the response is compromised in individuals with obesity.
8.8 Exercise Training: An Effective Intervention for the Reduction in Fatty Acid Oxidation in the Skeletal Muscle of Individuals with Obesity?
Endurance exercise training (e.g., repeated days of aerobic exercise) classically increases the ability of skeletal muscle to oxidize lipid, primarily by increasing mitochondrial function, but also by induction of several lipid metabolic proteins related to uptake, handling, and breakdown of FAs within the myocyte (Fritzen et al. 2020). However, it is important to realize there is heterogeneity in the responses to exercise training, with some individuals displaying little to no changes in health-related variables such as VO2peak, fasting insulin, or insulin sensitivity (Bouchard and Rankinen 2001; Bouchard et al. 2012; Stephens and Sparks 2015; Sparks 2017). In addition, individuals with obesity, particularly severe (BMI ≥40 kg/m2) obesity, exhibit compromised cardiorespiratory fitness which results in low absolute exercise workloads and may not optimally stimulate adaptations to training. Supporting the concept of “exercise resistance” (Sparks 2017) with obesity, the overexpression of PGC1α, a global regulator of exercise-training mediated adaptations, did not increase FA oxidation in primary myotubes from individuals with severe obesity to the same extent as in lean subjects (Consitt et al. 2010). There are also findings suggesting that the lesions in FA oxidation and metabolic flexibility with obesity are inherent (Hulver et al. 2005; Gaster 2009; Tanner et al. 2002; Zou et al. 2019; Boyle et al. 2012) which may minimize the impact of any interventions. In support, substantive (i.e., > 50 kg) weight loss did not alter FA oxidation in skeletal muscle (Berggren et al. 2004) at rest or during exercise (Thyfault et al. 2004; Guesbeck et al. 2001) in subjects with severe obesity. A smaller magnitude of weight loss (≈14 kg) in individuals with Class I obesity (BMI of 34 kg/m2) also did not alter resting FA oxidation determined in vivo across a muscle bed (Kelley et al. 1999). These findings suggest that the positive effects of exercise training on FA oxidation in the skeletal muscle of individuals with obesity cannot be assumed and warrant investigation.
In an attempt to minimize the confounding effects of concurrent weight loss, studies have implemented a short period (7–10 days) of exercise training (60 min/d, 50–75% VO2peak) where the energy deficit induced by exercise is minimal and no weight loss is evident. One such study examined subjects with severe obesity and subjects that were previously severely obese who had undergone bariatric surgery (1–2 y post-surgery). The 10-day training stimulus was sufficient to increase FA oxidation in the obese groups to the same rates as in lean subjects after 10 days of exercise (Berggren et al. 2004). A similar training regimen increased FA oxidation in skeletal muscle in Caucasian and African American women with severe obesity to the same levels as lean subjects (Cortright et al. 2006). Other longer-duration studies have reported equivalent increases in the expression or activity of proteins involved with FA oxidation in the skeletal muscle of lean subjects and individuals with obesity (Devries et al. 2013; Gillen et al. 2013; de Matos et al. 2018).
In contrast, 24 hours of contractile stimuli (electrical pulse stimulation, EPS) increased FA oxidation in primary human myotubes derived from lean subjects but not in myotubes from individuals with obesity or type 2 diabetes (Løvsletten et al. 2020). The cells from the subjects with obesity initially had higher rates of incomplete oxidation and reduced expression of complexes II, III, and IV of the respiratory chain (Løvsletten et al. 2020). A comparison of these in vivo (see preceding paragraph) and in vitro (Løvsletten et al. 2020) findings suggests that factors present during whole-body exercise (i.e., hormones, myokines, neural signaling, etc.) could be important in inducing the increases in FA oxidation seen with exercise training in individuals with obesity.
In relation to metabolic flexibility, the skeletal muscle of lean individuals increased FA oxidation, but there was no change in the muscle of obese subjects in response to a 3-day eucaloric high-fat diet (Battaglia et al. 2012). Both groups then exercised for 10 consecutive days (1 hour/day, 70% of VO2peak) and on the final 3 days of training consumed the same high-fat diet. The initial 7 days of exercise training was sufficient to increase FA oxidation in the skeletal muscle of both groups to the same extent, and both groups responded to the 3 day high-fat diet in a similar manner (Battaglia et al. 2012). Endurance exercise training also increased FA oxidation during exercise (an index of metabolic flexibility) in individuals with obesity (Fritzen et al. 2020; Devries et al. 2013; Goodpaster et al. 2003). Eaves et al. (Eaves et al. 2012) examined the effects of exercise training (4 weeks at ≈65% VO2peak, 3 days/week for 30 minutes for week 1 progressing to 60 minutes/day for weeks 2–4), and even with this relatively mild stimulus, there were trends (P = 0.06) for FA oxidation to increase during exercise to an equivalent degree in children of either lean parents or parents with obesity. These findings indicate that endurance exercise training increases metabolic flexibility to a similar extent in individuals with obesity compared to their lean counterparts.
8.9 Summarizing Remarks on the Role of Obesity in Regulation of FA Oxidation During Rest and Exercise and the Counter-regulatory Effect of Exercise Training
In summary, individuals with obesity display an impaired ability to oxidize lipid and to increase FA oxidation in the face of increased lipid exposure (metabolic flexibility). At the cellular level, these defects are evident in skeletal muscle mitochondria and likely contribute, at least in part, to positive lipid balance and ectopic lipid deposition. It is equivocal if these deficits impair the ability to increase FA oxidation in response to a single exercise bout. However, despite these initial impairments, endurance exercise training can rescue FA oxidation and metabolic flexibility in the skeletal muscle of individuals with obesity at least to equivalent levels of their lean counterparts. These findings indicate the efficacy of exercise training in treating the metabolic inflexibility in human skeletal muscle metabolism evident with obesity.
References
Alsted TJ, Ploug T, Prats C, Serup AK, Hoeg L, Schjerling P, Holm C, Zimmermann R, Fledelius C, Galbo H, Kiens B (2013) Contraction-induced lipolysis is not impaired by inhibition of hormone-sensitive lipase in skeletal muscle. J Physiol 591:5141–5155
Arad AD, Basile AJ, Albu J, DiMenna FJ (2020) No influence of overweight/obesity on exercise lipid oxidation: a systematic review. Int J Mol Sci 21:1614
Arner P, Kriegholm E, Engfeldt P, Bolinder J (1990) Adrenergic regulation of lipolysis in situ at rest and during exercise. J Clin Invest 85:893–898
Bahr R, Hostmark AT, Newsholme EA, Gronnerod O, Sejersted OM (1991) Effect of exercise on recovery changes in plasma levels of FFA, glycerol, glucose and catecholamines. Acta Physiol Scand 143:105–115
Balcı SS (2012) Comparison of substrate oxidation during walking and running in normal-weight and overweight/obese men. Obes Facts 5:327–338
Battaglia GM, Zheng D, Hickner RC, Houmard JA (2012) Effect of exercise training on metabolic flexibility in response to a high-fat diet in obese individuals. Am J Physiol Endocrinol Metab 303:E1440–E1445
Begaye B, Vinales KL, Hollstein T, Ando T, Walter M, Bogardus C, Krakoff J, Piaggi P (2020) Impaired metabolic flexibility to high-fat overfeeding predicts future weight gain in healthy adults. Diabetes 69:181–192
Bell JA, Reed MA, Consitt LA, Martin OJ, Haynie KR, Hulver MW, Muoio DM, Dohm GL (2010) Lipid partitioning, incomplete fatty acid oxidation, and insulin signal transduction in primary human muscle cells: effects of severe obesity, fatty acid incubation, and fatty acid translocase/CD36 overexpression. J Clin Endocrinol Metab 95:3400–3410
Benninghoff T, Espelage L, Eickelschulte S, Zeinert I, Sinowenka I, Müller F, Christina Schöndeling C, Batchelor H, Cames S, Zhou Z, Kotzka J, Chadt A, Al-Hasani H (2020) The Rab GAPs TBC1D1 and TBC1D4 control uptake of Long-chain fatty acids into skeletal muscle via fatty acid transporter SLC27A4/FATP4. Diabetes 69:2281–2293
Berggren JR, Hulver MW, Dohm GL, Houmard JA (2004) Weight loss and exercise: implications for muscle lipid metabolism and insulin action. Med Sci Sports Exerc 36:1191–1195
Bergman BC, Butterfield GE, Wolfel EE, Casazza GA, Lopaschuk GD, Brooks GA (1999) Evaluation of exercise and training on muscle lipid metabolism. Am J Phys 276:E106–E117
Bonen A, Han XX, Habets DD, Febbraio M, Glatz JF, Luiken JJ (2007) A null mutation in skeletal muscle FAT/CD36 reveals its essential role in insulin- and AICAR-stimulated fatty acid metabolism. Am J Physiol Endocrinol Metab 292(6):E1740–E1749
Bonen A, Luiken JJ, Arumugam Y, Glatz JF, Tandon NN (2000) Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem 275:14501–14508
Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J (2004) Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Asp Med 25(5–6):495–520
Bouchard C, Blair SN, Church TS, Earnest CP, Hagberg JM, Häkkinen K, Jenkins NT, Karavirta L, Kraus WE, Leon AS, Rao DC, Sarzynski MA, Skinner JS, Slentz CA, Rankinen T (2012) Adverse metabolic response to regular exercise: is it a rare or common occurrence? PLoS One 7:e37887
Bouchard C, Rankinen T (2001) Individual differences in response to regular physical activity. Med Sci Sports Exerc 33:S446–S451
Boyle KE, Canham JP, Consitt LA, Zheng D, Koves TR, Gavin TP, Holbert D, Neufer PD, Ilkayeva O, Muoio DM, Houmard JA (2011) A high-fat diet elicits differential responses in genes coordinating oxidative metabolism in skeletal muscle of lean and obese individuals. J Clin Endocrinol Metab 96:775–781
Boyle KE, Zheng D, Anderson EJ, Neufer PD, Houmard JA (2012) Mitochondrial lipid oxidation is impaired in cultured myotubes from obese humans. Int J Obes 36:1025–1031
Bradley NS, Snook LA, Jain SS, Heigenhauser GJ, Bonen A, Spriet LL (2012) Acute endurance exercise increases plasma membrane fatty acid transport proteins in rat and human skeletal muscle. Am J Physiol Endocrinol Metab 302:E183–E189
Broskey NT, Zou K, Dohm GL, Houmard JA (2020) Plasma lactate as a marker for metabolic health. Exerc Sport Sci Rev 48:119–124
Calbet JA, Lundby C (2012) Skeletal muscle vasodilatation during maximal exercise in health and disease. J Physiol 590:6285–6296
Campbell SE, Tandon NN, Woldegiorgis G, Luiken JJ, Glatz JF, Bonen A (2004) A novel function for fatty acid translocase (FAT)/CD36: involvement in long chain fatty acid transfer into the mitochondria. J Biol Chem 279:36235–36241
Carlson LA, Ekelund LG, Fröberg SO (1971) Concentration of triglycerides, phospholipids and glycogen in skeletal muscle and of free fatty acids and beta-hydroxybutyric acid in blood in man in response to exercise. Eur J Clin Investig 1:248–254
Chabowski A, Górski J, Luiken JJ, Glatz JF, Bonen A (2007) Evidence for concerted action of FAT/CD36 and FABPpm to increase fatty acid transport across the plasma membrane. Prostaglandins Leukot Essent Fatty Acids 77:345–353
Chadt A, Leicht K, Deshmukh A, Jiang LQ, Scherneck S, Bernhardt U, Dreja T, Vogel H, Schmolz K, Kluge R, Zierath JR, Hultschig C, Hoeben RC, Schurmann A, Joost HG, Al-Hasani H (2008) Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet 40:1354–1359
Chee C, Shannon CE, Burns A, Selby AL, Wilkinson D, Smith K, Greenhaff PL, Stephens FB (2021) Increasing skeletal muscle carnitine content in older individuals increases whole-body fat oxidation during moderate-intensity exercise. Aging Cell 20:e13303
Christensen EH, Hansen O (1939) Respiratorisher quotientund O2–aufname. Skand Arch Physiol 81:160–171
Clarke DC, Miskovic D, Han XX, Calles-Escandon J, Glatz JF, Luiken JJ, Heikkila JJ, Bonen A (2004) Overexpression of membrane-associated fatty acid binding protein (FABPpm) in vivo increases fatty acid sarcolemmal transport and metabolism. Physiol Genomics 17:31–37
Colberg SR, Hagberg JM, McCole SD, Zmuda JM, Thompson PD, Kelley DE (1996) Utilization of glycogen but not plasma glucose is reduced in individuals with NIDDM during mild-intensity exercise. J Appl Physiol (1985) 81:2027–2033
Consitt LA, Bell JA, Koves TR, Muoio DM, Hulver MW, Haynie KR, Dohm GL, Houmard JA (2010) Peroxisome proliferator-activated receptor-gamma coactivator-1alpha overexpression increases lipid oxidation in myocytes from extremely obese individuals. Diabetes 59:1407–1415
Constantin-Teodosiu D, Carlin JI, Cederblad G, Harris RC, Hultman E (1991) Acetyl group accumulation and pyruvate dehydrogenase activity in human muscle during incremental exercise. Acta Physiol Scand 143:367–372
Constantin-Teodosiu D, Peirce NS, Fox J, Greenhaff PL (2004) Muscle pyruvate availability can limit the flux, but not activation, of the pyruvate dehydrogenase complex during submaximal exercise in humans. J Physiol 561:647–655
Cortright RN, Sandhoff KM, Basilio JL, Berggren JR, Hickner RC, Hulver MW, Dohm GL, Houmard JA (2006) Skeletal muscle fat oxidation is increased in African-American and white women after 10 days of endurance exercise training. Obesity (Silver Spring) 14:1201–1210
Costill DL, Fink WJ, Getchell LH, Ivy JL, Witzmann FA (1979) Lipid metabolism in skeletal muscle of endurance-trained males and females. J Appl Physiol Respir Environ Exerc Physiol 47:787–791
Dahlmans D, Houzelle A, Schrauwen P, Hoeks J (2016) Mitochondrial dynamics, quality control and mi RNA regulation in skeletal muscle: implications for obesity and related metabolic disease. Clin Sci (Lond) 130:843–852
de Glisezinski I, Larrouy D, Bajzova M, Koppo K, Polak J, Berlan M, Bulow J, Langin D, Marques MA, Crampes F, Lafontan M, Stich V (2009) Adrenaline but not noradrenaline is a determinant of exercise-induced lipid mobilization in human subcutaneous adipose tissue. J Physiol 587:3393–3404
de Matos MA, Vieira DV, Pinhal KC, Lopes JF, Dias-Peixoto MF, Pauli JR, de Castro MF, Little JP, Rocha-Vieira E, Amorim FT (2018) High-intensity interval training improves markers of oxidative metabolism in skeletal muscle of individuals with obesity and insulin resistance. Front Physiol 9:1451
Dean D, Daugaard JR, Young ME, Saha A, Vavvas D, Asp S, Kiens B, Kim KH, Witters L, Richter EA, Ruderman N (2000) Exercise diminishes the activity of acetyl-CoA carboxylase in human muscle. Diabetes 49(8):1295–1300
Decombaz J, Schmitt B, Ith M, Decarli B, Diem P, Kreis R, Hoppeler H, Boesch C (2001) Postexercise fat intake repletes intramyocellular lipids but no faster in trained than in sedentary subjects. Am J Physiol Regul Integr Comp Physiol 281:R760–R769
Devries MC, Samjoo IA, Hamadeh MJ, McCready C, Raha S, Watt MJ, Steinberg GR, Tarnopolsky MA (2013) Endurance training modulates intramyocellular lipid compartmentalization and morphology in skeletal muscle of lean and obese women. J Clin Endocrinol Metab 98:4852–4862
Diaz-Vegas A, Sanchez-Aguilera P, Krycer JR, Morales PE, Monsalves-Alvarez M, Cifuentes M, Rothermel BA, Lavandero S (2020) Is mitochondrial dysfunction a common root of noncommunicable chronic diseases? Endocr Rev 41:491–517
Dzamko N, Schertzer JD, Ryall JG, Steel R, Macaulay SL, Wee S, Chen ZP, Michell BJ, Oakhill JS, Watt MJ, Jorgensen SB, Lynch GS, Kemp BE, Steinberg GR (2008) AMPK-independent pathways regulate skeletal muscle fatty acid oxidation. J Physiol 586(Pt 23):5819–5831
Eaton S (2002) Control of mitochondrial beta-oxidation flux. Prog Lipid Res 41:197–239
Eaves AD, Colon A, Dubose KD, Collier D, Houmard JA (2012) Substrate utilization during submaximal exercise in children with a severely obese parent. Nutr Metab (Lond) 9:38–39
Essén B (1978) Studies on the regulation of metabolism in human skeletal muscle using intermittent exercise as an experimental model. Acta Physiol Scand Suppl 454:1–32
Ezell DM, Geiselman PJ, Anderson AM, Dowdy ML, Womble LG, Greenway FL, Zachwieja JJ (1999) Substrate oxidation and availability during acute exercise in non-obese, obese, and post-obese sedentary females. Int J Obes Relat Metab Disord 23:1047–1056
Friedlander AL, Casazza GA, Horning MA, Usaj A, Brooks GA (1999) Endurance training increases fatty acid turnover, but not fat oxidation, in young men. J Appl Physiol (1985) 86:2097–2105
Friedman S, Fraenkel G (1955) Reversible enzymatic acetylation of carnitine. Arch Biochem Biophys 59:491–501
Fritzen AM, Lundsgaard AM, Jeppesen J, Christiansen ML, Bienso R, Dyck JR, Pilegaard H, Kiens B (2015) 5'-AMP activated protein kinase alpha2 controls substrate metabolism during post-exercise recovery via regulation of pyruvate dehydrogenase kinase 4. J Physiol 593:4765–4780
Fritzen AM, Lundsgaard AM, Kiens B (2020) Tuning fatty acid oxidation in skeletal muscle with dietary fat and exercise. Nat Rev Endocrinol 16:683–696
Galgani JE, Moro C, Ravussin E (2008) Metabolic flexibility and insulin resistance. Am J Physiol Endocrinol Metab 295:E1009–E1017
Gaster M (2009) Reduced lipid oxidation in myotubes established from obese and type 2 diabetic subjects. Biochem Biophys Res Commun 382:766–770
Gaster M (2012) Reduced TCA flux in diabetic Myotubes: determined by single defects? Biochem Res Int 2012:716056
Gaster M, Nehlin JO, Minet AD (2012) Impaired TCA cycle flux in mitochondria in skeletal muscle from type 2 diabetic subjects: marker or maker of the diabetic phenotype? Arch Physiol Biochem 118:156–189
Gillen JB, Percival ME, Ludzki A, Tarnopolsky MA, Gibala MJ (2013) Interval training in the fed or fasted state improves body composition and muscle oxidative capacity in overweight women. Obesity (Silver Spring) 21:2249–2255
Goodpaster BH, Katsiaras A, Kelley DE (2003) Enhanced fat oxidation through physical activity is associated with improvements in insulin sensitivity in obesity. Diabetes 52:2191–2197
Goodpaster BH, Sparks LM (2017) Metabolic flexibility in health and disease. Cell Metab 25:1027–1036
Goodpaster BH, Wolfe RR, Kelley DE (2002) Effects of obesity on substrate utilization during exercise. Obes Res 10:575–584
Guesbeck NR, Hickey MS, Mac Donald KG, Pories WJ, Harper I, Ravussin E, Dohm GL, Houmard JA (2001) Substrate utilization during exercise in formerly morbidly obese women. J Appl Physiol (1985) 90:1007–1012
Gundersen AE, Kugler BA, McDonald PM, Veraksa A, Houmard JA, Zou K (2020) Altered mitochondrial network morphology and regulatory proteins in mitochondrial quality control in myotubes from severely obese humans with or without type 2 diabetes. Appl Physiol Nutr Metab 45:283–293
Hagenfeldt L, Wahren J (1968) Human forearm muscle metabolism during exercise. II. Uptake, release and oxidation of individual FFA and glycerol. Scand J Clin Lab Invest 21:263–276
Hagenfeldt L, Wahren J (1975) Turnover of free fatty acids during recovery from exercise. J Appl Physiol 39:247–250
Hansen K, Shriver T, Schoeller D (2005) The effects of exercise on the storage and oxidation of dietary fat. Sports Med 35:363–373
Harris RA, Bowker-Kinley MM, Huang B, Wu P (2002) Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzym Regul 42:249–259
Harris RC, Foster CV, Hultman E (1987) Acetylcarnitine formation during intense muscular contraction in humans. J Appl Physiol (1985) 63:440–442
Helge JW, Watt PW, Richter EA, Rennie MJ, Kiens B (2001) Fat utilization during exercise: adaptation to a fat-rich diet increases utilization of plasma fatty acids and very low density lipoprotein-triacylglycerol in humans. J Physiol 537:1009–1020
Hickner RC, Privette J, McIver K, Barakat H (2001) Fatty acid oxidation in African-American and Caucasian women during physical activity. J Appl Physiol (1985) 90:2319–2324
Hingst JR, Kjøbsted R, Birk JB, Jørgensen NO, Larsen MR, Kido K, Larsen JP, Kjeldsen SAS, Fentz J, Frøsig C, Holm S, Fritzen AM, Dohlman TL, Larsen S, Foretz M, Viollet B, Schjerling P, Overby P, Halling JF, Pilegaard H, Hellsten Y, Wojtaszewski JF (2020) Inducible deletion of skeletal muscle AMPKα reveals that AMPK is required fornucleotide balance but dispensable for muscle glucose uptake and fat oxidation during exercise. Mol Metab 40:101028
Holloway GP, Bezaire V, Heigenhauser GJ, Tandon NN, Glatz JF, Luiken JJ, Bonen A, Spriet LL (2006) Mitochondrial long chain fatty acid oxidation, fatty acid translocase/CD36 content and carnitine palmitoyltransferase I activity in human skeletal muscle during aerobic exercise. J Physiol 571:201–210
Holloway GP, Bonen A, Spriet LL (2009) Regulation of skeletal muscle mitochondrial fatty acid metabolism in lean and obese individuals. Am J Clin Nutr 89:455S–462S
Holloway GP, Lally J, Nickerson JG, Alkhateeb H, Snook LA, Heigenhauser GJ, Calles-Escandon J, Glatz JF, Luiken JJ, Spriet LL, Bonen A (2007b) Fatty acid binding protein facilitates sarcolemmal fatty acid transport but not mitochondrial oxidation in rat and human skeletal muscle. J Physiol 582(Pt 1):393–405
Holloway GP, Thrush AB, Heigenhauser GJ, Tandon NN, Dyck DJ, Bonen A, Spriet LL (2007a) Skeletal muscle mitochondrial FAT/CD36 content and palmitate oxidation are not decreased in obese women. Am J Physiol Endocrinol Metab 292:E1782–E1789
Horowitz JF, Klein S (2000) Oxidation of nonplasma fatty acids during exercise is increased in women with abdominal obesity. J Appl Physiol (1985) 89:2276–2282
Houmard JA (2008) Intramuscular lipid oxidation and obesity. Am J Physiol Regul Integr Comp Physiol 294:R1111–R1116
Houmard JA, Pories WJ, Dohm GL (2011) Is there a metabolic program in the skeletal muscle of obese individuals? J Obes 2011:250496
Houmard JA, Pories WJ, Dohm GL (2012) Severe obesity: evidence for a deranged metabolic program in skeletal muscle? Exerc Sport Sci Rev 40:204–210
Hulver MW, Berggren JR, Carper MJ, Miyazaki M, Ntambi JM, Hoffman EP, Thyfault JP, Stevens R, Dohm GL, Houmard JA, Muoio DM (2005) Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab 2:251–261
Hulver MW, Berggren JR, Cortright RN, Dudek RW, Thompson RP, Pories WJ, Mac Donald KG, Cline GW, Shulman GI, Dohm GL, Houmard JA (2003) Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab 284:E741–E747
Hurley BF, Nemeth PM, Martin WH III, Hagberg JM, Dalsky GP, Holloszy JO (1986) Muscle triglyceride utilization during exercise: effect of training. J Appl Physiol (1985) 60:562–567
Ibrahimi A, Bonen A, Blinn WD, Hajri T, Li X, Zhong K, Cameron R, Abumrad NA (1999) Muscle-specific overexpression of FAT/CD36 enhances fatty acid oxidation by contracting muscle, reduces plasma triglycerides and fatty acids, and increases plasma glucose and insulin. J Biol Chem 274(38):26761–26766
Jain SS, Chabowski A, Snook LA, Schwenk RW, Glatz JF, Luiken JJ, Bonen A (2009) Additive effects of insulin and muscle contraction on fatty acid transport and fatty acid transporters, FAT/CD36, FABPpm, FATP1, 4 and 6. FEBS Lett 583:2294–2300
Jeppesen J, Albers PH, Rose AJ, Birk JB, Schjerling P, Dzamko N, Steinberg GR, Kiens B (2011) Contraction-induced skeletal muscle FAT/CD36 trafficking and FA uptake is AMPK independent. J Lipid Res 52(4):699–711
Jeppesen J, Mogensen M, Prats C, Sahlin K, Madsen K, Kiens B (2010) FAT/CD36 is localized in sarcolemma and in vesicle-like structures in subsarcolemma regions but not in mitochondria. J Lipid Res 51:1504–1512
Jones TE, Pories WJ, Houmard JA, Tanner CJ, Zheng D, Zou K, Coen PM, Goodpaster BH, Kraus WE, Dohm GL (2019) Plasma lactate as a marker of metabolic health: implications of elevated lactate for impairment of aerobic metabolism in the metabolic syndrome. Surgery 166:861–866
Kanaley JA, Cryer PE, Jensen MD (1993) Fatty acid kinetic responses to exercise. Effects of obesity, body fat distribution, and energy-restricted diet. J Clin Invest 92:255–261
Kanaley JA, Shadid S, Sheehan MT, Guo Z, Jensen MD (2009) Relationship between plasma free fatty acid, intramyocellular triglycerides and long-chain acylcarnitines in resting humans. J Physiol 587:5939–5950
Kanaley JA, Weatherup-Dentes MM, Alvarado CR, Whitehead G (2001) Substrate oxidation during acute exercise and with exercise training in lean and obese women. Eur J Appl Physiol 85:68–73
Kelley DE, Goodpaster B, Wing RR, Simoneau JA (1999) Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Phys 277:E1130–E1141
Kelley DE, He J, Menshikova EV, Ritov VB (2002) Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51:2944–2950
Kiens B (2006) Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol Rev 86(1):205–243
Kiens B, Essen-Gustavsson B, Christensen NJ, Saltin B (1993) Skeletal muscle substrate utilization during submaximal exercise in man: effect of endurance training. J Physiol 469:459–478
Kiens B, Lithell H, Mikines KJ, Richter EA (1989) Effects of insulin and exercise on muscle lipoprotein lipase activity in man and its relation to insulin action. J Clin Invest 84(4):1124–1129
Kiens B, Richter EA (1998) Utilization of skeletal muscle triacylglycerol during postexercise recovery in humans. Am J Phys 275:E332–E337
Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA (2000) Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab 279:E1039–E1044
Kolodziej MP, Zammit VA (1990) Re-evaluation of the interaction of malonyl-CoA with the rat liver mitochondrial carnitine palmitoyltransferase system by using purified outer membranes. Biochem J 267:85–90
Kriketos AD, Pan DA, Lillioja S, Cooney GJ, Baur LA, Milner MR, Sutton JR, Jenkins AB, Bogardus C, Storlien LH (1996) Interrelationships between muscle morphology, insulin action, and adiposity. Am J Phys 270:R1332–R1339
Krogh A, Lindhard J (1920) The relative value of fat and carbohydrate as sources of muscular energy: with appendices on the correlation between standard metabolism and the respiratory quotient during rest and work. Biochem J 14(3–4):290–363
Krssak M, Petersen KF, Bergeron R, Price T, Laurent D, Rothman DL, Roden M, Shulman GI (2000) Intramuscular glycogen and intramyocellular lipid utilization during prolonged exercise and recovery in man: a 13C and 1H nuclear magnetic resonance spectroscopy study. J Clin Endocrinol Metab 85:748–754
Lafontan M, Moro C, Berlan M, Crampes F, Sengenes C, Galitzky J (2008) Control of lipolysis by natriuretic peptides and cyclic GMP. Trends Endocrinol Metab 19:130–137
Lally JS, Jain SS, Han XX, Snook LA, Glatz JF, Luiken JJ, McFarlan J, Holloway GP, Bonen A (2012) Caffeine-stimulated fatty acid oxidation is blunted in CD36 null mice. Acta Physiol (Oxf) 205:71–81
Langfort J, Ploug T, Ihlemann J, Holm C, Galbo H (2000) Stimulation of hormone-sensitive lipase activity by contractions in rat skeletal muscle. Biochem J 351:207–214
Larson-Meyer DE, Newcomer BR, Hunter GR (2002) Influence of endurance running and recovery diet on intramyocellular lipid content in women: a 1H NMR study. Am J Physiol Endocrinol Metab 282:E95–E106
Lee-Young RS, Griffee SR, Lynes SE, Bracy DP, Ayala JE, McGuinness OP, Wasserman DH (2009) Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J Biol Chem 284(36):23925–23934
Li LO, Grevengoed TJ, Paul DS, Ilkayeva O, Koves TR, Pascual F, Newgard CB, Muoio DM, Coleman RA (2015) Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis. Diabetes 64:23–35
Lithell H, Hellsing K, Lundqvist G, Malmberg P (1979) Lipoprotein-lipase activity of human skeletal-muscle and adipose tissue after intensive physical exercise. Acta Physiol Scand 105(3):312–315
Løvsletten NG, Rustan AC, Laurens C, Thoresen GH, Moro C, Nikolić N (2020) Primary defects in lipid handling and resistance to exercise in myotubes from obese donors with and without type 2 diabetes. Appl Physiol Nutr Metab 45:169–179
Lundsgaard AM, Fritzen AM, Kiens B (2018) Molecular regulation of fatty acid oxidation in skeletal muscle during aerobic exercise. Trends Endocrinol Metab 29:18–30
Lundsgaard AM, Fritzen AM, Kiens B (2020) The importance of fatty acids as nutrients during post-exercise recovery. Nutrients 12:nu12020280
MacPherson RE, Vandenboom R, Roy BD, Peters SJ (2013) Skeletal muscle PLIN3 and PLIN5 are serine phosphorylated at rest and following lipolysis during adrenergic or contractile stimulation. Physiol Rep 1:e00084
Maples JM, Brault JJ, Shewchuk BM, Witczak CA, Zou K, Rowland N, Hubal MJ, Weber TM, Houmard JA (2015a) Lipid exposure elicits differential responses in gene expression and DNA methylation in primary human skeletal muscle cells from severely obese women. Physiol Genomics 47:139–146
Maples JM, Brault JJ, Witczak CA, Park S, Hubal MJ, Weber TM, Houmard JA, Shewchuk BM (2015b) Differential epigenetic and transcriptional response of the skeletal muscle carnitine palmitoyltransferase 1B (CPT1B) gene to lipid exposure with obesity. Am J Physiol Endocrinol Metab 309:E345–E356
Marra M, Scalfi L, Contaldo F, Pasanisi F (2004) Fasting respiratory quotient as a predictor of long-term weight changes in non-obese women. Ann Nutr Metab 48:189–192
McConell GK (2020) It's well and truly time to stop stating that AMPK regulates glucose uptake and fat oxidation during exercise. Am J Physiol Endocrinol Metab 318:E564–E567
McConell GK, Wadley GD, Le PK, Linden KC (2020) Skeletal muscle AMPK is not activated during 2h of moderate intensity exercise at 65% VO2 peak in endurance trained men. J Physiol 598:3859–3870
McFarlan JT, Yoshida Y, Jain SS, Han XX, Snook LA, Lally J, Smith BK, Glatz JF, Luiken JJ, Sayer RA, Tupling AR, Chabowski A, Holloway GP, Bonen A (2012) In vivo, fatty acid translocase (CD36) critically regulates skeletal muscle fuel selection, exercise performance, and training-induced adaptation of fatty acid oxidation. J Biol Chem 287(28):23502–23516
McGarry JD, Mills SE, Long CS, Foster DW (1983) Observations on the affinity for carnitine, and malonyl-CoA sensitivity, of carnitine palmitoyltransferase I in animal and human tissues. Demonstration of the presence of malonyl-CoA in non-hepatic tissues of the rat. Biochem J 214:21–28
Miotto PM, Steinberg GR, Holloway GP (2017) Controlling skeletal muscle CPT-I malonyl-CoA sensitivity: the importance of AMPK-independent regulation of intermediate filaments during exercise. Biochem J 474:557–569
Mittendorfer B, Fields DA, Klein S (2004) Excess body fat in men decreases plasma fatty acid availability and oxidation during endurance exercise. Am J Physiol Endocrinol Metab 286:E354–E362
Miura S, Kai Y, Kamei Y, Bruce CR, Kubota N, Febbraio MA, Kadowaki T, Ezaki O (2009) Alpha 2-AMPK activity is not essential for an increase in fatty acid oxidation during low-intensity exercise. Am J Physiol Endocrinol Metab 296(1):E47–E55
Monaco C, Whitfield J, Jain SS, Spriet LL, Bonen A, Holloway GP (2015) Activation of AMPKalpha2 is not required for mitochondrial FAT/CD36 accumulation during exercise. PLoS One 10:e0126122
Morio B, Holmback U, Gore D, Wolfe RR (2004) Increased VLDL-TAG turnover during and after acute moderate-intensity exercise. Med Sci Sports Exerc 36:801–806
Moro C, Crampes F, Sengenes C, de Glisezinski I, Galitzky J, Thalamas C, Lafontan M, Berlan M (2004) Atrial natriuretic peptide contributes to physiological control of lipid mobilization in humans. FASEB J 18:908–910
Moro C, Polak J, Hejnova J, Klimcakova E, Crampes F, Stich V, Lafontan M, Berlan M (2006) Atrial natriuretic peptide stimulates lipid mobilization during repeated bouts of endurance exercise. Am J Physiol Endocrinol Metab 290:E864–E869
Nellemann B, Sondergaard E, Jensen J, Pedersen SB, Jessen N, Jorgensen JO, Nielsen S (2014) Kinetics and utilization of lipid sources during acute exercise and acipimox. Am J Physiol Endocrinol Metab 307:E199–E208
Odland LM, Heigenhauser GJ, Lopaschuk GD, Spriet LL (1996) Human skeletal muscle malonyl-CoA at rest and during prolonged submaximal exercise. Am J Phys 270:E541–E544
Odland LM, Howlett RA, Heigenhauser GJ, Hultman E, Spriet LL (1998) Skeletal muscle malonyl-CoA content at the onset of exercise at varying power outputs in humans. Am J Phys 274:E1080–E1085
O'Neill HM, Lally JS, Galic S, Pulinilkunnil T, Ford RJ, Dyck JR, van Denderen BJ, Kemp BE, Steinberg GR (2015) Skeletal muscle ACC2 S212 phosphorylation is not required for the control of fatty acid oxidation during exercise. Physiol Rep 3(7):e12444
Perreault L, Lavely JM, Kittelson JM, Horton TJ (2004) Gender differences in lipoprotein lipase activity after acute exercise. Obes Res 12:241–249
Phillips SM, Green HJ, Tarnopolsky MA, Heigenhauser GJ, Grant SM (1996) Progressive effect of endurance training on metabolic adaptations in working skeletal muscle. Am J Phys 270:E265–E272
Prats C, Donsmark M, Qvortrup K, Londos C, Sztalryd C, Holm C, Galbo H, Ploug T (2006) Decrease in intramuscular lipid droplets and translocation of HSL in response to muscle contraction and epinephrine. J Lipid Res 47:2392–2399
Radegran G, Saltin B (1998) Muscle blood flow at onset of dynamic exercise in humans. Am J Phys 274:H314–H322
Raney MA, Turcotte LP (2006) Regulation of contraction-induced FA uptake and oxidation by AMPK and ERK1/2 is intensity dependent in rodent muscle. Am J Physiol Endocrinol Metab 291:E1220–E1227
Rasmussen BB, Winder WW (1997) Effect of exercise intensity on skeletal muscle malonyl-CoA and acetyl-CoA carboxylase. J Appl Physiol (1985) 83:1104–1109
Ren J, Lakoski S, Haller RG, Sherry AD, Malloy CR (2013) Dynamic monitoring of carnitine and acetylcarnitine in the trimethylamine signal after exercise in human skeletal muscle by 7T 1H-MRS. Magn Reson Med 69:7–17
Roepstorff C, Halberg N, Hillig T, Saha AK, Ruderman NB, Wojtaszewski JF, Richter EA, Kiens B (2005) Malonyl-CoA and carnitine in regulation of fat oxidation in human skeletal muscle during exercise. Am J Physiol Endocrinol Metab 288:E133–E142
Roepstorff C, Steffensen CH, Madsen M, Stallknecht B, Kanstrup IL, Richter EA, Kiens B (2002) Gender differences in substrate utilization during submaximal exercise in endurance-trained subjects. Am J Physiol Endocrinol Metab 282:E435–E447
Rogge MM (2009) The role of impaired mitochondrial lipid oxidation in obesity. Biol Res Nurs 10:356–373
Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E, Wolfe RR (1993) Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Phys 265(3 Pt 1):E380–E391
Romijn JA, Coyle EF, Sidossis LS, Rosenblatt J, Wolfe RR (2000) Substrate metabolism during different exercise intensities in endurance-trained women. J Appl Physiol (1985) 88:1707–1714
Sahlin K (1990) Muscle carnitine metabolism during incremental dynamic exercise in humans. Acta Physiol Scand 138:259–262
Samovski D, Su X, Xu Y, Abumrad NA, Stahl PD (2012) Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a Rab GTPase. J Lipid Res 53:709–717
Santiworakul A, Chuaychoo B, Kriengsinyos W, Saengsirisuwan V, Jalayondeja W (2014) Substrate utilization during and after high intensity exercise in healthy lean and obese men. J Med Assoc Thail 97(Suppl 7):S50–S54
Seidell JC, Muller DC, Sorkin JD, Andres R (1992) Fasting respiratory exchange ratio and resting metabolic rate as predictors of weight gain: the Baltimore longitudinal study on aging. Int J Obes Relat Metab Disord 16:667–674
Seiler SE, Koves TR, Gooding JR, Wong KE, Stevens RD, Ilkayeva OR, Wittmann AH, DeBalsi KL, Davies MN, Lindeboom L, Schrauwen P, Schrauwen-Hinderling VB, Muoio DM (2015) Carnitine acetyltransferase mitigates metabolic inertia and muscle fatigue during exercise. Cell Metab 22:65–76
Sidossis LS, Gastaldelli A, Klein S, Wolfe RR (1997) Regulation of plasma fatty acid oxidation during low- and high-intensity exercise. Am J Phys 272:E1065–E1070
Simoneau JA, Veerkamp JH, Turcotte LP, Kelley DE (1999) Markers of capacity to utilize fatty acids in human skeletal muscle: relation to insulin resistance and obesity and effects of weight loss. FASEB J 13:2051–2060
Slusher AL, Whitehurst M, Zoeller RF, Mock JT, Maharaj A, Huang CJ (2015) Brain-derived neurotrophic factor and substrate utilization following acute aerobic exercise in obese individuals. J Neuroendocrinol 27:370–376
Smirnova E, Goldberg EB, Makarova KS, Lin L, Brown WJ, Jackson CL (2006) ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep 7:106–113
Smith BK, Jain SS, Rimbaud S, Dam A, Quadrilatero J, Ventura-Clapier R, Bonen A, Holloway GP (2011) FAT/CD36 is located on the outer mitochondrial membrane, upstream of long-chain acyl-CoA synthetase, and regulates palmitate oxidation. Biochem J 437:125–134
Smith BK, Perry CG, Koves TR, Wright DC, Smith JC, Neufer PD, Muoio DM, Holloway GP (2012) Identification of a novel malonyl-CoA IC (50) for CPT-I: implications for predicting in vivo fatty acid oxidation rates. Biochem J 448:13–20
Søndergaard E, Andersen IR, Sørensen LP, Gormsen LC, Nielsen S (2017) Lipoprotein lipase activity does not predict very low-density lipoprotein-triglyceride fatty acid oxidation during exercise. Scand J Med Sci Sports 27:474–481
Sondergaard E, Rahbek I, Sorensen LP, Christiansen JS, Gormsen LC, Jensen MD, Nielsen S (2011) Effects of exercise on VLDL-triglyceride oxidation and turnover. Am J Physiol Endocrinol Metab 300:E939–E944
Sparks LM (2017) Exercise training response heterogeneity: physiological and molecular insights. Diabetologia 60:2329–2336
Stallknecht B, Kiens B, Helge JW, Richter EA, Galbo H (2004) Interstitial glycerol concentrations in human skeletal muscle and adipose tissue during graded exercise. Acta Physiol Scand 180:367–377
Stallknecht B, Lorentsen J, Enevoldsen LH, Bulow J, Biering-Sorensen F, Galbo H, Kjaer M (2001) Role of the sympathoadrenergic system in adipose tissue metabolism during exercise in humans. J Physiol 536:283–294
Stanford KI, Lynes MD, Takahashi H, Baer LA, Arts PJ, May FJ, Lehnig AC, Middelbeek RJW, Richard JJ, So K, Chen EY, Gao F, Narain NR, Distefano G, Shettigar VK, Hirshman MF, Ziolo MT, Kiebish MA, Tseng YH, Coen PM, Goodyear LJ (2018) 12,13-diHOME: an exercise-induced Lipokine that increases skeletal muscle fatty acid uptake. Cell Metab 27:1111–1120
Starling RD, Trappe TA, Parcell AC, Kerr CG, Fink WJ, Costill DL (1997) Effects of diet on muscle triglyceride and endurance performance. J Appl Physiol (1985) 82:1185–1189
Starritt EC, Howlett RA, Heigenhauser GJ, Spriet LL (2000) Sensitivity of CPT I to malonyl-CoA in trained and untrained human skeletal muscle. Am J Physiol Endocrinol Metab 278:E462–E468
Steffan HG, Elliott W, Miller WC, Fernhall B (1999) Substrate utilization during submaximal exercise in obese and normal-weight women. Eur J Appl Physiol Occup Physiol 80:233–239
Steffensen CH, Roepstorff C, Madsen M, Kiens B (2002) Myocellular triacylglycerol breakdown in females but not in males during exercise. Am J Physiol Endocrinol Metab 282:E634–E642
Stephens NA, Sparks LM (2015) Resistance to the beneficial effects of exercise in type 2 diabetes: are some individuals programmed to fail? J Clin Endocrinol Metab 100:43–52
Storlien L, Oakes ND, Kelley DE (2004) Metabolic flexibility. Proc Nutr Soc 63:363–368
Talanian JL, Tunstall RJ, Watt MJ, Duong M, Perry CG, Steinberg GR, Kemp BE, Heigenhauser GJ, Spriet LL (2006) Adrenergic regulation of HSL serine phosphorylation and activity in human skeletal muscle during the onset of exercise. Am J Physiol Regul Integr Comp Physiol 291:R1094–R1099
Tanner CJ, Barakat HA, Dohm GL, Pories WJ, Mac Donald KG, Cunningham PR, Swanson MS, Houmard JA (2002) Muscle fiber type is associated with obesity and weight loss. Am J Physiol Endocrinol Metab 282:E1191–E1196
Taskinen MR, Nikkila EA (1980) Effect of acute vigorous exercise on lipoprotein lipase activity of adipose tissue and skeletal muscle in physically active men. Artery 6:471–483
Thyfault JP, Kraus RM, Hickner RC, Howell AW, Wolfe RR, Dohm GL (2004) Impaired plasma fatty acid oxidation in extremely obese women. Am J Physiol Endocrinol Metab 287:E1076–E1081
Turcotte LP, Richter EA, Kiens B (1992) Increased plasma FFA uptake and oxidation during prolonged exercise in trained vs. untrained humans. Am J Phys 262:E791–E799
Turcotte LP, Swenberger JR, Tucker MZ, Yee AJ, Trump G, Luiken JJ, Bonen A (2000) Muscle palmitate uptake and binding are saturable and inhibited by antibodies to FABP (PM). Mol Cell Biochem 210:53–63
van Hall G, Sacchetti M, Radegran G, Saltin B (2002) Human skeletal muscle fatty acid and glycerol metabolism during rest, exercise and recovery. J Physiol 543:1047–1058
van Loon LJ, Greenhaff PL, Constantin-Teodosiu D, Saris WH, Wagenmakers AJ (2001) The effects of increasing exercise intensity on muscle fuel utilisation in humans. J Physiol 536:295–304
van Loon LJ, Koopman R, Stegen JH, Wagenmakers AJ, Keizer HA, Saris WH (2003) Intramyocellular lipids form an important substrate source during moderate intensity exercise in endurance-trained males in a fasted state. J Physiol 553:611–625
van Loon LJ, Thomason-Hughes M, Constantin-Teodosiu D, Koopman R, Greenhaff PL, Hardie DG, Keizer HA, Saris WH, Wagenmakers AJ (2005) Inhibition of adipose tissue lipolysis increases intramuscular lipid and glycogen use in vivo in humans. Am J Physiol Endocrinol Metab 289:E482–E493
Wall BT, Stephens FB, Constantin-Teodosiu D, Marimuthu K, Macdonald IA, Greenhaff PL (2011) Chronic oral ingestion of L-carnitine and carbohydrate increases muscle carnitine content and alters muscle fuel metabolism during exercise in humans. J Physiol 589:963–973
Watt MJ, Heigenhauser GJ, Dyck DJ, Spriet LL (2002) Intramuscular triacylglycerol, glycogen and acetyl group metabolism during 4 h of moderate exercise in man. J Physiol 541(Pt 3):969–978
Watt MJ, Holmes AG, Steinberg GR, Mesa JL, Kemp BE, Febbraio MA (2004b) Reduced plasma FFA availability increases net triacylglycerol degradation, but not GPAT or HSL activity, in human skeletal muscle. Am J Physiol Endocrinol Metab 287(1):E120–E127
Watt MJ, Steinberg GR, Chan S, Garnham A, Kemp BE, Febbraio MA (2004a) Beta-adrenergic stimulation of skeletal muscle HSL can be overridden by AMPK signaling. FASEB J 18(12):1445–1446
Whitfield J, Paglialunga S, Smith BK, Miotto PM, Simnett G, Robson HL, Jain SS, Herbst EAF, Desjardins EM, Dyck DJ, Spriet LL, Steinberg GR, Holloway GP (2017) Ablating TBC1D1 impairs contraction-induced sarcolemmal glucose transporter type 4 redistribution, but not insulin-mediated responses in rats. J Biol Chem 292(40):16653–16664
Wicks SE, Vandanmagsar B, Haynie KR, Fuller SE, Warfel JD, Stephens JM, Wang M, Han X, Zhang J, Noland RC, Mynatt RL (2015) Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc Natl Acad Sci U S A 112:E3300–E3309
Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, Kiens B (2000) Isoform-specific and exercise intensity-dependent activation of 5'-AMP-activated protein kinase in human skeletal muscle. J Physiol 528(Pt 1):221–226
Wolfe RR, Klein S, Carraro F, Weber JM (1990) Role of triglyceride-fatty acid cycle in controlling fat metabolism in humans during and after exercise. Am J Phys 258(2 Pt 1):E382–E389
Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F (2012) FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell Metab 15:279–291
Zou K, Hinkley JM, Park S, Zheng D, Jones TE, Pories WJ, Hornby PJ, Lenhard J, Dohm GL, Houmard JA (2019) Altered tricarboxylic acid cycle flux in primary myotubes from severely obese humans. Int J Obes 43:895–905
Zurlo F, Lillioja S, Esposito-Del PA, Nyomba BL, Raz I, Saad MF, Swinburn BA, Knowler WC, Bogardus C, Ravussin E (1990) Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. Am J Phys 259:E650–E657
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Fritzen, A.M., Broskey, N.T., Lundsgaard, A.M., Dohm, G.L., Houmard, J.A., Kiens, B. (2022). Regulation of Fatty Acid Oxidation in Skeletal Muscle During Exercise: Effect of Obesity. In: McConell, G. (eds) Exercise Metabolism. Physiology in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-94305-9_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-94305-9_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-94304-2
Online ISBN: 978-3-030-94305-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)