
Abstract
Low-cost and widely available Next-Generation Sequencing (NGS) is revolutionizing clinical practice, paving the way for the realization of precision medicine. Applying NGS to clinical practice requires establishing a complex loop involving sample collection and sequencing, computational processing of the NGS outputs to identify variants, and the interpretation of the variants to establish their significance for the condition being treated. The computational tools that perform variant calling have been extensively used in bioinformatics, but there are few attempts to integrate them in a comprehensive, production-grade, Cloud-native infrastructure able to scale to national levels. Furthermore, there are no established interfaces for closing the loop between NGS machines, computational infrastructure, and variant interpretation experts.
We present here the platform developed for the Greek National Precision Medicine Network for Oncology. The platform integrates bioinformatics tools and their orchestration, makes provisions for both experimental and clinical usage of variant calling pipelines, provides programmatic interfaces for integration with NGS machines and for analytics, and provides user interfaces for supporting variant interpretation. We also present benchmarking results and discuss how these results confirm the soundness of our architectural and implementation choices.
The work described here has received funding from the Greek General Secretariat for Research and Innovation in the context of the Hellenic Network of Precision Medicine on Cancer. See also https://oncopmnet.gr for more details.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Low-cost and widely available Next-Generation Sequencing (NGS) is revolutionizing clinical practice and paves the way for the realization of precision medicine. Applying NGS to clinical practice requires establishing a complex loop involving sample collection and sequencing, variant calling, the computational processing of the NGS outputs to identify variants, and variant annotation, the interpretation of the variants to establish their significance for the condition being treated.
Computational tools for variant calling are computationally challenging processes, developed with parallelisation being a major design consideration. Both multi-threading of individual processes and the Map/Reduce paradigm have been extensively explored [1]. The advent of Cloud computing has also seen the appearance of specialized omics Cloud services that offer flexible scalability by porting these tools to remote infrastructures. Such service include those offered by the European ELIXIR project,Footnote 1 commercial services such as DNAnexus,Footnote 2 and services provided by the manufacturers of NGS engines such as Illumina.Footnote 3 Each of these services emphasises different aspects and features, ranging from open interfaces and software, ability to extend the bioinformatics toolset with custom tools, to tracking the lineage and provenance of data and experiments, and integrating with variant annotation databases and environments.
But there are few attempts to integrate a comprehensive, production-grade infrastructure able to scale to national levels. Furthermore, there are no established interfaces for closing the loop between NGS machines, computational infrastructure, and variant interpretation experts. Even more so, when one want to also include in the loop secondary medical research usage of the data produced by clinical usage.
In this paper we present an architecture that integrates bioinformatics tools and their orchestration, makes provisions for both experimental and clinical usage of variant calling pipelines, provides programmatic interfaces for integration with NGS machines and for medical analytics, and provides user interfaces for supporting variant interpretation.
We also present the implementation of this architecture as the end-to-end variant calling and annotation platform developed for the Greek National Precision Medicine Network for Oncology, and is in the final stages before entering productive clinical use.
2 Use Case-Driven Platform Design
The design of our platform was based on common processes followed by labs that support NGS processing for Precision Medicine applications. Our analysis for the distinct user roles in this context has identified the following user profiles and service needs:
-
Clinicians have access to samples and NGS machines and need to get the NGS data processed by executing a variant calling workflow in order to get a VCF data structure that represents all variants found in the sample’s DNA along with useful metadata (e.g., coverage, statistical significance). Subsequently, they annotate each variant according to its clinical significance.
-
Bioinformaticians provide the definition of the variant calling processing. In particular, they determine and configure the steps of the workflows and then test them by applying them on benchmark datasets and/or on the inputs from past clinical usage. They evaluate alternative tools and workflows on both their efficacy in correctly calling variants and their computational efficiency.
-
Medical researchers seek to correlate variants, diseases, treatments, and outcomes and, in general, to perform data analytics queries on the repository of results of the platform. They provide access to the clinical data the need to join with the VCF data computed using this platform.
Based on these user profiles, in the next paragraphs, we will present use cases and the relevant requirements for our precision medicine platform.

Services and interfaces required in clinical use.
In clinical usage (Fig. 1), the user needs to load the outputs of the NGS machine into the platform and select among parameter presets, such as the clinical purpose of the processing. The platform is pre-configured with the appropriate processing and the clinician cannot affect the workflows and tools used. This is facilitated by an Operator API through which the desktop computer directly attached to the NGS machine can upload inputs to the platform and the user can select and initiate the processing. A minimal Operator UI also provides a graphical environment for this usage.
When processing terminates, the infrastructure adds the resulting VCF data to the Results Repository that constitutes the clinical usage record of the infrastructure and also makes them available for annotation in a Variant Annotation Environment. These environments initialize the annotation by accessing databases of variants known to be significant for the specific condition and allow the user to edit these initial annotations. These manual annotations are also added to the Results Repository, available both for reporting and for medical research.

Services and interfaces required to execute bioinformatics benchmarks and maintain the clinical pipelines.
Bioinformatics usage (Fig. 2) ensures the efficacy and efficiency of the pre-configured processing made available to clinicians. Following standard practice in bioinformatics, this processing is organized as the application to the input data of a workflow that is the composition of several tools. A Workflow Orchestrator reads the definition of the workflow and then ensures that each tool referenced in the definition is given as input the output of the previous step in the pipeline and is appropriately parameterized and invoked. The software that implements each step is retrieved from a Software Repository and inputs and outputs are retrieved from and stored to a Data Repository.
Bioinformaticians update and evaluate reference data (effectively, inputs that are the same for all runs), bioinformatics tools, and workflows. Testing is performed by applying alternative workflows on benchmark data and on previous clinical data, so the infrastructure needs to clearly distinguish between clinical and experimental workflow execution. The former are restricted to the pre-configured workflow and write their output to the Results Repository; There is only one such output associated with each input. The latter are unrestricted computational experiments, are not meant to be used for any medical purposes, and there will be multiple runs and outputs for each input.
Bioinformaticians have access to several components that are internal from the perspective of clinicians: They can directly access the data repository to provide benchmark and reference data, they can add and update the bioinformatics tools and workflows in the Software Repository, and they can read detailed Operational Logs from the execution of workflows so that they can debug and benchmark tools and workflows.

Services and interfaces required for medical research.
It should be noted at this point that the data in the platform does not constitute personally identifiable information (PII) either by itself or when combined with other information: The number of variants needed for precision medicine purposes is too small to identify individuals. Furthermore, NGS data, VCF data, and annotations are internally linked by a unique key, but this key is created by infrastructure and unrelated to any external PII or key.
Medical research users need to combine access to external data with access to the platform’s Results Repository to extract statistics about how variants correlate with clinical and, possibly, other data (Fig. 3). This is facilitated by foreseeing linking the internal index used by the platform with an external index, such as a medical exam referral number. Medical research users who are authorized to access clinical and other sensitive PII can use this link to join variants with the clinical or personal data, without having to store PII in this platform.
3 Platform Description
The abstract architecture shaped by our functional requirements draws a landscape where a large number of different services, addressing the needs of different users, cluster around the core functionality of applying variant calling workflows. Accordingly, we will start by establishing how this core functionality will be implemented and then use this to drive the elicitation of technical requirements and the decisions on concrete software systems that implement the abstract components in Fig. 3.
As discussed in Sect. 1, the bioinformatics community has converged to organizing processing in pipelines of tools. Containerization technologies allow both the pipeline orchestrator and the individual tools to be deployed in Cloud computing infrastructures and to scale up or down the allocated computational resources. At the same time, CromwellFootnote 4 is a well-supported and ubiquitously-used orchestrator of containerized tools. Cromwell also supports the CWL workflow description language, the de facto standard in the bioinformatics community.
On the other hand, the Kubernetes container deployment, scaling, and management systemFootnote 5 and the various tools developed around it are the dominant open-source ecosystem for Cloud-native applications. Although Cromwell is not developed as a Cloud-native application, it offers the extremely useful abstraction of task execution schemas (TES)Footnote 6 that specify how Cromwell can deploy batch execution tasks. Although not officially supported by Cromwell, TESKFootnote 7 implements the TES API on Kubernetes.

Systems and components integrated to realize the architecture.
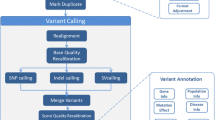
3.1 Variant Calling
The entry point of the Variant Calling sub-system is the REST API. Through this API a user can submit pipelines, either provided by the system and stored in platform’s software repository or custom pipelines defined by them. Either way the pipelines should be defined in CWL. The REST API is also the interface of the metadata subsystem so users can be informed about the status of pipelines submitted by them or their group and retrieve other pipeline metadata such as the exact pipeline inputs, the location of the output files in the object store, and pipelines logs.
Upon pipeline submission, the REST API submits the pipeline to Cromwell which handles the workflow orchestration. The orchestrator reads the CWL definition and starts the execution of every job defined there. Cromwell monitors the health of every job and handles job failures by retrying failed jobs whenever appropriate. It also ensures that each job is given the appropriate inputs from previous job outputs if needed. The metadata sub-system polls Cromwell to retrieve information about the status of the workflow, workflow logs and output files location in the object store (Fig. 4).
Task execution is handled by TESK. A task defines a set of input files, a set of (Docker) containers and commands to run, a set of output files, and some other logging and metadata. TESK implements TES on Kubernetes backends so each task is executed in a Kubernetes pod. It handles task execution, failure recovery, task resource assignments (CPU, RAM, block disk size), and housekeeping. Every task is scheduled by Kubernetes on nodes with available resources, is assigned block storage from NFS for temporary file storage and retrieves input files and stores output files in the system’s object store.
Finally, we use an instance of Gitlab installed on our Kubernetes infrastructureFootnote 8 as our software repository and identity server. By storing the platform’s predefined pipelines on a Git server we support version control, branch access controls, merge reviews, and merge approvals. Pipeline definitions are fully specified and reproducible as they refer to specific container images, served by the Gitlab Docker registry.
The sophisticated access control and software review mechanisms included in Gitlab are used to ensure the appropriate access rights and reviews before committing to the branches that the system trusts as the correct pipeline definitions and tool implementations for the clinical pipelines. The system is configured to recognize certain repositories and branches as pertaining to clinical, non-experimental usage, and to only write the VCF outputs from these pipelines to the results database. Images for these branches are built and served automatically using Gitlab CI/CD tools, so that updating the clinical pipelines amounts to merging into the appropriate (protected) branches, without any further platform administration actions. This approach facilitates using the infrastructure to also carry out bioinformatics experimentation and benchmarking with the data and tools that accumulate in the platform: Users can be given access to repositories and groups that are not recognized by the system as clinical to submit arbitrary workloads; these workloads will yield results and benchmarking measurements but will not update the clinical results database.
Besides enforcing access rights and review policies for tools and pipelines, Gitlab is also the identity and role-based access service across the system. Access from the internet goes through Keycloak/NGINX and the Kubernetes load balancer, which use Gitlab as an identity server. Roles that do not directly map to repository access (such as accessing the results repository) are mapped to a hierarchy of Gitlab groups that do not contain any repositories. Keycloak/NGINX knows how to map membership in these special groups to internal services that are made available or masked for each user. This allows extremely flexible administration as, for instance, a user can be made the administrator of the group that maps to accessing the results database; that user can invite further members without having to interact with the administration of the platform as a whole.
3.2 Variant Annotation
The Variant Annotation component of our platform automates and facilitates gathering genomic annotation data from external databases and combining them with VCF data coming from the Variant Calling step of the platform (Sect. 3.1). The integrated results are then stored in the platform’s data space and they are ready to be used both for the production of patient reports and for answering research questions posed by medical researchers (see also Sect. 3.3).
In the core of this component, lies a deployment of the open-source Ensembl Variant Effect Predictor (VEP) software [3]. This tool can be configured to make use of data gathered from a set of desired external databases to analyse, annotate, and prioritize genomic variants in coding and non-coding regions. In our case, VEP is run with the default configuration that includes a variety of variant (e.g., COSMIC, dbSNP), protein (UniProt), algorithmic variant effect prediction (e.g., SIFT, Polyphen), variant allele frequency (e.g. gnomAD), clinical significance (ClinVar), and scientific literature (PubMed) databases.Footnote 9 Further, we include data from the LOVD variation database (using VEP’s LOVD plugin),Footnote 10 as well as variant nomenclature from HGVS. For efficiency reasons, the data of each source are stored locally in our platform’s data space in the form of a collection of annotation database cache files, which can optionally be installed alongside VEP itself.
It is worth mentioning that for clinicians it is very crucial to have access to the most recent information; the results of new research may identify previously unknown variants or reveal and correct data for already studied ones. To make sure that the most recent information is used during the annotation step, we have developed a custom module which, at runtime, examines if the information gathered from important external databases (like ClinVar) is up-to-date. This module compares the cached version of the respective database to the most recently published one and loads the more recent version in the platform’s data space, if needed.
By default, the output of VEP is given in the form of an annotated VCF file which contains integrated information from the input VCF and the selected external databases. This file is stored by itself in the Results Repository, however, to better support basic functionalities of the Variant Annotation component the same information is also loaded into a relational database schema. This database schema is a custom extension of LOVD [2], particularly tailored to accommodate storing and indexing VCF annotation data. This is achieved by the extension, modification, and addition of tables to the schema. For example, tables storing individual patient data in the original LOVD schema, only store referral numbers which correspond to particular NGS analyses in our version of LOVD (as per the requirements of Sect. 2 regarding PII data). Further, our schema extends LOVD with additional tables, e.g., for recording data from ClinVar, data gathered from clinical analysis report forms, filled out by clinicians during variant interpretation, etc.
The Variant Annotation component offers a functionality to browse the annotated data through a basic user interface that retrieves data from the aforementioned database and displays them in a tabular form. This interface allows the user to identify any variants of interest that should reported in the context of the investigation of the case of a particular patient. Finally, the users are also able to report newly identified links between variants and phenotypes, diseases, treatments, etc. These special-purpose contributions are very important since they may be related to particularities of the population being investigated by the lab that owns the platform and may be a very valuable addition for investigating research questions by medical researchers (see Sect. 3.3).
3.3 Knowledge Base
Our platform offers a Knowledge Base component which consists of the Results Repository along with a graphical UI-enabled analytics query engine. This knowledge base is intended for medical researchers and, hence, should not require familiarity with any database and/or respective query language. To facilitate this requirement, our platform uses metabaseFootnote 11 running on top of mariaDB.
Metabase is an open source tool that allows for querying databases through user friendly UIs and without requiring users to be familiar with database query languages, or the underlying database schemas. It offers automatic explorations of the tables of databases it connects to, and allows for performing and saving queries on their data. It is particularly tailored to performing statistical queries, offering a variety of visualization options for the results, such as bar charts, pie charts, etc. Metabase supports connecting to a variety of different database management systems, such as MySQL, PostgreSQL, MongoDB, and others.
In the case of our platform’s knowledge base, we use MariaDB as the underlying RDBMS. The core of this database is a collection of clinical variant interpretation data collected in a single table, alongside their connection to external exam referral data links (see Sect. 2). The clinical interpretation data stored originate from the Variant Annotation system of the platform, in particular, data from clinical interpretation result forms provided by the tool. These data concern the characterization of variants detected using the platform’s workflow (i.e., pathogenic, uncertain/unknown significance), the genes on which they were found, alongside data pertaining to the particular analysis (i.e., the frequency and coverage of the variants).
Registered users of the platform may access the knowledge base through a web interface provided by metabase. The web interface facilitates access to the underlying database tables through an intuitive UI, through which users can ask simple questions by applying filters on values of the selected table’s columns, and summarizations (i.e., aggregation queries and/or grouping) on the data by clicking on buttons provided for each of these functions. Further options are provided with regards to the display format of the results. By default these are shown in table format, but users may choose more appropriate visualizations, such as bar charts, or chronological time series, which are additionally customizable in terms of display (user can change display colors, types of axes, texts in legends, etc.).
Researchers querying the data are given the option to save each performed query. Saved queries, alongside their resulting visualizations, can be added to dashboards, which are readily available to users when accessing metabase. Various dashboards can focus on particular aspects of the data (e.g., statistical data of variants based on each gene they were found on), allowing for a fine grained organization and real time monitoring of the summary statistics recorded in the knowledge base. Finally, the various dashboards and saved questions make up a powerful tool that enables the sharing of results among different users of the platform.
4 Scalability Experiments and Discussion
In order to test the scalability of the platform, we collected execution time measurements from two different pipelines, with a different number of pipeline instances (runs) executing concurrently. The experiment was carried out on an installation of the system with 26 nodes. Each node has sufficient resources to execute two tasks (pipeline steps) where each task is allocated 8 CPU threads and 16 Gb RAM. Regarding storage, 8 nodes with a total of 36 Gb RAM are used to serve 6 Tb of disk space, split between transient NFS space and longer-term CEPH S3 space.
These resources suffice to execute the number of concurrent runs we show in Table 1 without scheduling and without deploying multiple runs on the same node. These execution times show that there is relatively small deviation between execution times and, consequently, it does not make sense to invest in migrating still-executing tasks to nodes where processing has finished and, in general, in more dynamic scheduling.
We also observe that processing times scale well, and bigger batches do not require linearly longer execution times. As discussed in the introduction, processing takes place in batches of roughly identical runs as soon as FASTQ inputs become available from an NGS engine. This motivates our approach of parallelizing the execution of multiple, independent of each other, runs. Furthermore, bioinformatics pipelines are computationally challenging and, subsequently, most of the execution time is consumed in actual computation rather than network and disk transfers. Having relatively small network and disk overhead means that we expect (and actually observe in Table 1) a sub-linear relationship between the number of runs in a batch and execution time for the batch.
However, some non-constant overhead and some noticeable execution time deviation is still visible in the results. This is due to the fact that input files are large and in our experiments (as foreseen by the use case) all runs are loaded at the same time, so that network and hard disk access becomes a temporary bottleneck at the beginning of the execution of each batch. The phenomenon dampens after the first step, as it is highly unlikely that all runs terminate their steps at exactly the same time. This observation motivated our devoting to storage the considerable resources mentioned above.
5 Conclusions and Future Work
We presented the architecture and implementation of the end-to-end precision medicine platform developed for the Greek National Precision Medicine Network for Oncology. The architecture describes the platform as an ecosystem of Cloud-native applications offering services over a backbone that executes variant calling workflows. The platform is implemented using the Cromwell orchestrator and the Kubernetes container deployment, scaling, and management system. Our tests confirm that our architecture and our choices on which systems to integrate in order to materialize the architecture are sound, and fit well with our use cases and with the nature of bioinformatics pipelines.
Besides the focus on scalable Cloud deployment, another innovation is using an internal Gitlab instance as the provider not only of container images, but also of identity services and access right management. This maximally exploits the flexibility of the Gitlab role-based access control system and its UI for managing users and roles.
Future work includes defining a digital artifact that combines reference to specific container image tags, reference data versions, and outputs. Such an artifact will constitute a fully specified experiment that can be reliably reproduced on any instance of our architecture.
Further work will look into interesting integrations with client applications and external resources. This includes developing higher-level analytics interfaces, such as in R or as Python Notebooks. Another useful integration is with clinical databases that follow standard schemas such as the OHDSI Common Data Model,Footnote 12 facilitating the processing of medical research queries and analyses.
Notes
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
An exhaustive list of annotation databases used with VEP’s default configuration can be found here:
https://www.ensembl.org/info/docs/tools/vep/script/VEP_script_documentation.pdf.
- 10.
This plugin retrieves LOVD variation data from http://www.lovd.nl.
- 11.
- 12.
References
Fjukstad, B., Bongo, L.A.: A review of scalable bioinformatics pipelines. Data Sci. Eng. 2, 245–251 (2017)
Fokkema, I.F., Taschner, P.E., Schaafsma, G.C., Celli, J., Laros, J.F., den Dunnen, J.T.: LOVD v.2.0: the next generation in gene variant databases. Hum. Mutat. 32(5), 557–563 (2011)
McLaren, W., et al.: The ensembl variant effect predictor. Genome Biol. 17(1), 1–14 (2016)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this paper

Cite this paper
Mouchakis, G. et al. (2021). A Cloud-Native NGS Data Processing and Annotation Platform. In: Rezig, E.K., et al. Heterogeneous Data Management, Polystores, and Analytics for Healthcare. DMAH Poly 2021 2021. Lecture Notes in Computer Science(), vol 12921. Springer, Cham. https://doi.org/10.1007/978-3-030-93663-1_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-93663-1_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-93662-4
Online ISBN: 978-3-030-93663-1
eBook Packages: Computer ScienceComputer Science (R0)