
Abstract
In this chapter, we start with the review on three classes of methodologies for oncology dose-escalation trial design: the 3+3, the statistical model-based approach including Continuous Reassessment Method (CRM) and Bayesian Logistic Regression Model (BLRM), and the toxicity interval-based algorithms such as Bayesian Optimal Interval Design (BOIN) and Toxicity Probability Interval method (TPI) and their respective variations. The focus of this chapter is to give a comprehensive outline of the various statistical extensions of these methods to address the statistical challenges caused by the prolonged safety evaluation window, or equivalently, the fast enrollment rate. They include, in CRM and BLRM class, the weighted likelihood function method (TITE-CRM), TITE-CRM aided by suspension rule or Bayesian predictive risk for toxicity to avoid aggressive dose escalation, the TITE-CRM that leverages drug cycle information, adaptive time-to-event toxicity distribution, and three-parameter logistic regression extension on the basis of BLRM. In the toxicity interval-based class, we review R-TPI method for the Toxicity Probability Interval method, TITE-BOIN which imputes the unobserved DLT, and BOIN12 which models the long-term toxicity and efficacy concurrently. The methods under discussion can play a valuable role in improving the accuracy of optimal dose identification without sacrificing patient safety or significantly prolonging the trial duration.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Oncology
- Kaplan-Meier
- MCMC
- R-TPI
- Rolling-TPI
- Long-term dose-limiting toxicity
- DTL
- Bayesian optimal interval
- BOIN
- Modified Toxicity probability interval
- mTPI
- Escalation with overdose control
- EWOC
- First-in-human
- FIH
- Non-tolerated dose
- NTD
- DLT
- BOIN12
- Likelihood function method
- Maximumly tolerated dose
- MTD
- Phase 2 trial
- RD2P
- Continuous reassessment method
- CRM
- TITE-CRM
- TITE-BOIN
- R-TPI
- Bayesian logistic regression model
- BLRM
- Logistic regression model
- Toxicity interval-based algorithms
- Toxicity probability interval method
- TPI
- Bayesian optimal interval design
- BOIN
1 Introduction
In the pharmaceutical industry, identifying the proper dose of experimental drugs is a critical mission in early phase development. In the field of oncology, the 1960s–1970s witnessed the advent of chemo/radiotherapy for cancer treatment. In these settings, the correlation between the dose of chemotherapy drugs and efficacy is established within a certain dose range. However, dose is a double-edged sword, that is, when the dose is too low, there is little chance for patients to derive treatment benefit while enduring possible toxicity. On the other hand, the unnecessarily high dose increases the risks of adverse events that might offset the improvement of the quality of life as a result of tumor response. Dose-limiting toxicity, DLT, is defined as the type of adverse events that prohibit further dose escalation in the hope for better efficacy.
In a traditional oncology setting, it is generally accepted that the chances for both DLT and tumor response increase concurrently as the dose escalates. A desirable dose that can be used in the latter development stage, therefore, should be at the level that strikes a proper balance between the possibility of achieving efficacy and the level of toxicity that can be managed. Such a dose level, which is defined as maximumly tolerated dose (MTD) or recommended dose for phase 2 trial (RD2P), typically has a DLT rate ranging from 20 to 30%, depending on the specific disease condition and drugs’ mechanism of action [18].
The first-in-human (FIH) oncology trial is typically a dose-escalation study with the primary objective of identifying MTD or RD2P. The oldest yet still the most widely used approach is a rule-based algorithm such as the 3+3 method [17]. Briefly, the patients will be enrolled to a specific dose level in a cohort with a fixed size (typically N = 3). If none of them experiences any DLT, the next cohort of three subjects will be enrolled to the next higher level. If one subject has at least one DLT, the current dose will be expanded to another three subjects to further characterize the safety profile with the emphasis on DLT. If two or more subjects have DLT among six subjects, the dose will be declared as a non-tolerated dose (NTD). The dose that is one level below NTD, if already has six subjects tested for DLT, will be declared as MTD.
Apparently, the 3+3 method suffers several shortcomings. In theory, it can only target MTD with a DLT rate between 17 and 33% without adequate precision due to its simple rule-based nature. Secondarily, empirical experience shows that the 3+3 approach tends to prematurely stop a trial by identifying as MTD the dose level that is lower than a potentially efficacious and tolerable level. As a result, this design leads to a majority of the trial participants being treated at the suboptimal level and not getting the clinical benefit they otherwise could.
It has been reported that one of the main reasons for the failures in late-phase clinical development is improper dose selection during early phase trials. The methodology such as the 3+3 design, which lacks statistical rigor, is arguably to blame. In 1990, O’Quigley et al. [14] proposed a statistical model-based dose-finding algorithm called Continual Reassessment Method (CRM), which later adopted the full Bayesian solution. It updates the parametric model for dose-toxicity curves based on prior knowledge and the accumulative data in real time. For a first-in-human trial with sparse data and rapid decision-making, this approach is conceptually appealing and in practice demonstrates ability superior to 3+3 in identifying dose levels that have a better chance to succeed in the later development stage. Within the Bayesian framework inspired by CRM, a more modified version of model-based methods, such as Escalation with Overdose Control (EWOC) and Bayesian Logistic Regression Model (BLRM), have also been put forward and widely applied in the pharmaceutical industry [13].
On the other hand, it has been debated whether it is either necessary or feasible to characterize the full dose-toxicity model using sparse phase I trial data. Both CRM and BLRM require Bayesian modeling of the toxicity data at all dose levels, increasing the operational complexity. More importantly, they are not as simple and transparent as the 3+3 design which the clinical team can readily understand and deploy.
Therefore, simplified versions of model-based methods, sometimes dubbed as statistical model-assisted methods, have also been proposed, as exemplified by the Bayesian Optimal Interval (BOIN) design and the Modified Toxicity probability Interval (mTPI) design [8, 11]. These models do not attempt to characterize the whole dose-toxicity relationship within the dose range being tested; instead, they base the dose recommendation on the frequentist or Bayesian posterior probability of observed toxicity at individual dose level in relation to a prespecified target DLT interval. By doing away with modeling all the observed toxicity data all at once, these interval-based designs provide transparent decision rules that are uniformly applicable to all the dose levels, which is based on the exhaustive enumeration of foreseeable cohort size and DLT number. Essentially, BOIN and mTPI methods provide nearly as transparent and simple implementation as the 3+3 design, with a performance at least comparable to the more complicated CRM or BLRM model.
Among many challenges Phase I dose-finding trials face, delayed or long-term dose-limiting toxicity (DLT) is the one that greatly increases the trial duration. In the first-in-human trial, subjects are tested at a new drug/dose in a very small cohort size, for the sake of caution, typically not exceeding three, before the next group of subjects can be dosed. In order for experimental drugs to be studied in an affordable sample size (N = 30 ~ 40) with a reasonable time span, the current dose level needs to be cleared of DLT quickly before the next dose level can be tested. Fortunately, the traditional concept of DLT, conceived during the early days of chemotherapy, presumes cytotoxicity-related DLT develops shortly after the first dosing within the first cycle (28 days). These features make the phase I dose-escalation trial what they look like today.
As more and more molecularly targeted therapies enter the pipeline and market, however, they demonstrate diverse mechanisms of action (MOA) that could impact the onset of DLT. For example, immuno-oncology therapies such as PD-1 checkpoint pathway inhibitors are known for their delayed immuno-response related toxicities and efficacy [12]. Since all the current patients need to clear the DLT window before any new patients can enter the trial, a long DLT observation window will lead to a prolonged trial duration. As an example, a simulation study showed that it will take 4–8 years to complete a dose-escalation trial with 24–48 patients if the DLT window is as long as 6 months [3]. Similarly, even if the DLT observation period itself is not exceedingly long, a relatively rapid enrollment, in case of the high willingness of patient participation, may cause a backlog and long waiting list of enrollment and eventually turn away patients who urgently need the opportunity coming with the potential new therapies.
In these two scenarios, it is beneficial to allow new patients to start treatment while the ones before them are still in the DLT observation period. For an early phase trial with small sample size, however, it is not efficient nor ethical to disregard even the partial information without the ultimate DLT outcome yet. Currently, there are numerous approaches to allow new patients to be enrolled in trial while taking account into the incomplete information carried by patients who have not yet complete DLT window. In this chapter, we will use three sections to discuss the current algorithms to handle the late-onset DLT problems in dose-escalation trials. This section is the summary of the background for the dose-escalation trial. The second section will summarize the basic types of dose-escalation algorithms, and the third section will review the extensions of these basic methods to the case of late-onset toxicity or fast patient enrollment.
2 Dose-Escalation Algorithm
2.1 The 3+3 method
Firstly, escalate the dose from the lowest level to the highest level in cohort size of three subjects.
-
(1)
If no DLT is encountered among the three subjects, escalate to the next higher dose level.
-
(2)
In the case of one DLT, three more subjects will be enrolled to the same dose level.
-
(3)
In the case of two or more DLT, the next cohort of three subjects will be dosed at one level lower.
-
(4)
Eventually, if two or more DLT are observed among six subjects treated at one dose, this dose will be declared as a non-tolerable dose (NTD). The dose that is one level below NTD, if already being tested in six subjects, will be declared as MTD.
2.2 Model-Based Method
2.2.1 Continual Reassessment Method
First of all, a guessed DLT probability (π(θ)d) for all the dose levels will be solicited from the consultation with the clinical team based on the best available knowledge, such as clinical data from the similar compound, preclinical PK, and toxicity data, etc. The parametric dose-toxicity relationship is expressed by the following one-parameter power model:
where a suggested prior for log(θ) is a normal distribution with mean 0 and variance 1.342. If the prior median of θ is 1, C d is the prior median at dose d.
With the one-parameter exponential function as likelihood function and log normal as the prior distribution for θ, one can derive the posterior distribution of θ via Bayesian theorem:
The next dose level, recommended for the incoming new cohort of patients, will be the one whose posterior point estimate of DLT rate is the closest to the MTD level with prespecified DLT rate [14].
2.2.2 Bayesian Logistic Regression Model
Unlike the one-parameter exponential model for CRM, Neuenschwander et al recommend a two-parameter logistic curve to model the dose-toxicity relationship. This curve has quite a few resemblances in the field of biology and medicine, thus has good acceptance among clinicians and translational scientists.
The logistic model, coupled with the Bernoulli distribution of DLT status, forms the likelihood function for DLT rate. The prior distribution of alpha and beta is specified by lognormal distribution as follows:
where the mean of the logistic parameter can be derived from historical data of the same or similar compound while the variance can be calibrated based on the level of certainty on this prior knowledge [13].
Another feature of BLRM framework, besides making the parametric inference on the dose-toxicity relationship, is to take into consideration the uncertainty of the point estimate of the posterior distribution, which is updated by the upcoming toxicity data based on the prior distribution. The rationale is that various DLT rates can be considered equivalent if they fall into a probability interval that is close or distant enough from MTD with the prespecified DLT probability. Briefly, the MCMC draws from posterior distribution are tabulated based on their chance of falling into the probability regions such as “too low/under dose”, “about right/on target” and “too high/overly toxic”. The dose level that maximizes the on-target probability, while maintaining the risk of overdose below a prespecified value such as 25%, will be recommended to the next cohort of patients. This approach has been shown to avoid aggressive escalation encountered in CRM method.
2.3 Toxicity Interval-Based Method
2.3.1 Modified Toxicity Probability Interval (mTPI)
“All models are wrong, some are useful.”
If the main purpose of a dose-escalation algorithm is to identify the one singular dose level that achieves the proper balance between efficacy and safety, some might argue, the attempt to characterize the whole dose-toxicity curve, which could be complex and parameter-rich, might seem to be an overkill. In this spirit, Ji et al proposed a probability interval-based method which only focuses on the toxicity estimation for the current dose level, without borrowing information from other dose levels such as CRM and BLRM [8].
This simple approach is fundamentally Bayesian. With a flat prior beta (1,1), the posterior distribution of DLT rate for the current dose can be expressed as follows:
where n i is the number of patients enrolled at dose level I, and r i is the number of patients who experience DLT.
Like BLRM approach, the rate of DLT can be split into three regions: low/under-dose, medium/on-target and high/overdose, which correspond to three different decisions: escalate, retainment and de-escalate, respectively. The chance of true DLT falling into these regions can be modeled by the posterior distribution of the DLT rate, which is often a bell-shaped beta distribution. The region with the highest Unit Probability Mass (UPM), which is specified in the following formula:
will be the recommended decision for the next cohort of patients.
The implementation of this rule causes some unease in practice. For example, when three out of six patients experience DLT, the escalation region will have the highest UPM, thus becoming the recommended decision of the next dose, when most clinicians would probably agree that this kind of safety profile might warrant de-escalation.
To fine-tune mTPI, mTPI-2, the modified version of the original method has been proposed by the same group [5]. Instead of relying on the overall UPM for the whole decision region (low, medium and overdose), a series of sub-regions are constructed within each decision region using the length for the narrowest interval of the three, which typically is the on-target region. Then the maximum UPM for the resulting sub-intervals from the three regions will be compared, and the region with the highest maximum sub-region UPM will be selected as the recommended action. This change enables dose de-escalation in the case of 3 DLT out of 6 subjects.
2.3.2 Bayesian Optimal Interval Design (BOIN)
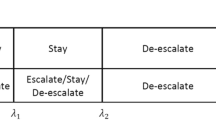
BOIN is another popular interval-based method that, similar to mTPI-2, makes dose recommendation based on the local point estimator of the toxicity at an individual dose level [10]. It first specifies three toxicity boundaries: Φ, Φ1 and Φ2, where Φ is the target DLT rate for MTD, Φ1 is the highest DLT considered to be suboptimal, and Φ2 is the lowest DLT rate deemed too toxic. Conceptually, the implementation of BOIN is even simpler than mTPI-2. It directly compares the observed toxicity rate to Φ1 and Φ2 , then makes dose recommendation as follows:
-
(1)
Escalate the dose when DLT rate is lower than Φ1
-
(2)
De-escalate when DLT rate is higher than Φ2
-
(3)
Otherwise have the next cohort of patients remain on the sample dose level
Φ, as the target MTD level, is solicited from the clinical team through consultation. The selection of Φ1 and Φ2 can be optimized by minimizing the selection error rate through the following formulation:
where λ1j and λ2j are the joint error rates when it comes to making decision in relation to lower and higher bounds of the target DLT interval. It can be shown that Φ1 = 0.6Φ and Φ2= 1.4Φ provides satisfactory operating characteristics in most clinical scenarios.
3 Time-to-Event Consideration
As discussed in the previous section, both scenarios including long DLT follow-up window/normal enrollment time and normal DLT window/fast enrollment rate may lead to a significant patient backlog. This could result in excessively long trial duration and ethical issues such as delaying patients with terminal illness the access to potential life-saving experimental drugs. To solve this problem, numerous extensions have been built on the previously described frameworks, allowing for the continuous enrollment of new patients before all the current patients have completed DLT evaluation period. We will summarize these developments in this section.
3.1 The 3+3 Method
The rolling-six method has been proposed by Skolnik et al as an extension to the 3+3 rule to accommodate the need to keep enrollment going while the patients of the current cohort are still under DLT evaluation [16]. Instead of suspending enrollment after every three subjects in the 3+3 method, this rolling-six design allows six patients to be under concurrent evaluation before halting enrollment.
Briefly, if the number of the patients who are at the current dose level reaches three, the fourth patient will
-
(1)
Be escalated to the next higher level of the current dose if all three subjects’ DLT window are cleared
-
(2)
Stay at the current dose level if at least one subject among the three has not completed their DLT window or one DLT has been reported from these three subjects
-
(3)
Be de-escalated to the next lower level of the current dose if two or more DLT had been reported.
The dosing decision of the fifth and the sixth subject will be the same as above. Extensive simulations results demonstrate that the expansion of three-patients cohort to the “rolling-six” cohort lowers the duration of the dose-escalation trial without exposing patients to excessive toxicity.
The detailed decision rule is summarized as follows (Table 1):
3.2 CRM/BLRM
3.2.1 Weighted Likelihood Function Method (TITE-CRM)
The essence of model-based Bayesian framework is to construct posterior distribution of toxicity profile by combing the prior distribution and observed data. In the case of the CRM, the likelihood function of binary DLT data is shown by the following:
A natural challenge, therefore, is how to deal with the partial toxicity data when concurrently enrolling new patients, while the current patients have not yet finished the whole DLT evaluation window. Entirely discarding the data points due to the lack of the final toxicity call would be inefficient. Before reaching the end of DLT window, a DLT-free subject with a long follow-up already carries more information than the one who just starts the treatment. This difference should be reflected in the data likelihood function when it comes to model update, which is particularly important to the situation of data scarcity in phase I dose-escalation trial.
One solution, as Cheung et al proposed in 2000, is to have the information of unfinished patients contribute less to the posterior distribution than the patients who have the known DLT outcome [3]. This is achieved by penalizing the contribution of an unfinished patient with a weighted likelihood function as follows:
Cheung et al. showed that a simple linear form of weight function from 0 to 1, in which the information carried by uncompleted DLT-free subject is proportional to the ratio of his/her follow-up time to the length of DLT window, is adequate to provide the satisfactory estimate of MTD while reducing the whole trial duration. The weight function can be expressed as follows:
This weight function assumes a uniform distribution for time-to-DLT. They had also shown that the more complicated forms of the time-to-DLT distribution, such as logistic and Weibull distribution, have similar performance in improving MTD identification and shortening trial duration.
Cheung et al. ’s method is called time-to-event continuous reassessment method (TITE-CRM) because the time-to-DLT event is taken into account in the update of the posterior distribution.
3.2.2 TITE-CRM with Suspension Rule
TITE-CRM, in theory, allows for continuous patient accrual without any suspension, for the model already takes full advantage of data that, even when they are incomplete, are available in real time. The real-world clinical practice, however, shows that TITE-CRM could lead to overly aggressive escalation behavior. In order to mitigate this risk, Polley introduced a principled approach to halting accrual for new subjects when the current ones are not adequately followed up [15].
Two user-defined threshold values, m and c, are solicited from clinicians for this purpose. First of all, m is the maximum waiting time a physician is willing to place a prospective subject on the waiting list. c is a threshold to measure the extent of patient safety being evaluated. If the total follow-up time for the patients on the current dose level, defined as V, exceeds threshold c, then it means that the current safety assessment is adequate and the new subjects can start treatment right away without any delay. Otherwise, the clinical team will assign the prospective patient a waiting time that is proportional to the inadequacy of current safety follow-up, setting a cap at m, the maximum waiting time that the clinician team can tolerate. This rule can be expressed by the following formula:
where S is the waiting time.
Simulation study shows that this mitigation improves the overall trial safety without scarifying the accuracy of the MTD identification.
3.2.3 TITE-CRM with Predictive Risk
In parallel, a more computationally intensive approach had been proposed by Bekele et al. [1]. Instead of assuming DLT occurs at a constant rate during the entire span of clinical observation, they use sequential ordinal modeling to describe the relationship between the dose and time-to-toxicity with the likelihood function as follows:
This method is basically Bayesian. It calculates the predictive toxicity probability from the posterior distribution. If the predictive toxicity for the prospective patient is too high, too low, on-target, or on-target with a high level of uncertainty, the decision will be to de-escalate, escalate, stay on the same dose or stop accrual to collect more safety information from the ongoing patients, respectively.
The appeal of this approach is that the decision to suspend accrual can be made quantitatively with the predictive toxicity risk. Due to its complexity in rule-setting and derivation of the posterior distribution, however, this method is not used as widely as TITE-CRM and its other variations.
3.2.4 TITE-CRM with Cycle Information
It can be argued that the uniform distribution may be too simplistic to model the true nature of time-to-toxicity distribution, to which the aggressive dose-escalation behavior of TITE-CRM may attribute. Huang et al. propose to leverage the cyclic nature of cancer drug administration to model the time-to-DLT distribution [6]. Due to the cumulative effect of drug exposure, the patients, even when they are not followed up long enough at the current cycle, may carry a large amount of safety information if they are already at a later cycle without experiencing any DLT in the previous cycles. As result, the weight, which will be used to adjust the contribution of incomplete observation to the posterior DLT distribution, is an adaptive function that combines the DLT probability distribution of the previous cycle with the proportion of local safety follow-up time to cycle length, as follows:
Then the implantation of the weighted likelihood function, the update of prior distribution will follow in the same manner as the standard TITIE-CRM.
3.2.5 TITE-CRM with Adaptive Time-to-DLT Distribution
Another line of effort recognized that the distribution of time-to-toxicity, similar to the dose-toxicity relationship, can be adaptively learned from the real data. Braun proposed that the probability of DLT, rather than being assumed to have constant rate across the evaluation window, can be modeled by a beta distribution Beta (1, θ) where θ can vary with the dose and determine whether DLT is early or late-onset event. The objective of this approach is to let the learning of time-to-event distribution follow a data-driven mode without the strong assumption for the constant rate [2].
In Braun’s method, the initial uniform distribution of time-to-DLT over DLT assessment window [0, T] is generalized to a beta distribution Beta (1, θ), which is an adaptive weight function with unknown parameter θ:
where T is DLT window and u is the incomplete follow-up time.
This becomes uniform distribution when θ =1, as the initial TITE-CRM paper adopted. To model the DLT kinetics as close to reality as possible, θ is allowed to vary with dose as follows:
The likelihood function involving λ and β is:
In the following computation, λ, with the prior N (0, σ2), can be inferred from the posterior DLT distribution along with β, which characterizes the dose-toxicity relationship in the CRM and the TITE-CRM.
Adaptively training the weight function and time-to-event distribution based on real toxicity data seems to be data-driven and less arbitrary. However, as the author suggested, this approach may not manifest its full potential in a phase I setting with a very small sample size. Furthermore, estimating additional parameters might have a statistical cost that, when the performance gain is arguably marginal, is not justifiable.
3.2.6 BLRM Adaptation
In the early phase dose-escalation trial for oncology, it is often time quite common to consider the first cycle as DLT evaluation window, when DLT is projected to occur rather soon after the first dose. This practice may negate the need to account for the time to toxicity in case of long DLT assessment period. When one has a good rationale to extend the DLT window beyond the first cycle, nonetheless, it turns out not to be trivial to explicitly define the length of DLT window. Zheng et al. proposed a three-parameter logistic regression model, built on BLRM framework advocated by Neuenschwander et al, to model patients’ different extent of drug exposure during the whole duration, beyond an arbitrarily determined DLT window [22].
BLRM is based upon the assumption that only the tangible variable that impacts DLT rate is the dose, which can be modeled by two parameters: the DLT rate at reference dose (α) and the slope of dose-toxicity curve (β). The approach of Zheng et al. extends BLRM to an additional parameter, the ratio of treatment time of a patient to a reference time window, which can be adopted from a commonly used DLT window. The joint likelihood function based on three-parameter logistic model is described as follows:
This function will become identical to BLRM formation when the time-to-DLT is capped at T 0, making it a fixed DLT window design.
The rest of the computation can follow the routine of BLRM, though what Zheng et al. actually used in their paper is to go after the dose level with the posterior DLT mean closest to the target MTD.
3.3 Model-Assisted Method
3.3.1 R-TPI
The m-TPI2 version of time-to-DLT adjustment is called R-TPI (Rolling-TPI) [4]. Interestingly, this modification, in order to maintain the simplicity in its original formulation, does not require statistically modeling the partial information carried by the patients who have not completed the full DLT evaluation. Instead, R-TPI operates similarly as the rolling-six design.
It first makes dose recommendations based on the status of completed subjects alone, without considering the pending ones. Then, by assuming the safest case scenario (no DLT for pending patients) and the most toxic scenario (all the pending patients will develop DLT eventually by the end of DLT evaluation period), the algorithm checks whether the initial decision is altered by these hypotheticals. If yes, it means that the pending result for the incomplete patients would be a game-changer for dose decision and cautions must be taken; thus the initial dose recommendation will be moderated in terms of its aggressiveness, or the trial will require more pending patients to complete their DLT observation period before the new patient can start the treatment, in order to garner more safety information.
Specifically, the study statistician will work with the clinical team to determine a trial parameter C, which is the maximum pending patients the team can tolerate before enrolling any new patients from safety perspective. Therefore, if the number of pending patients exceeds parameter C, study team would have no option but to halt the patient accrual. On the other hand, if all patients (n d) in the current dose level complete the required observation window, the decision rule will follow as a routine m-TPI2 approach.
The situation becomes trickier if the number of incomplete patients is between 0 and C, where the following rule will be followed if that is the case:
-
(1)
If the m-TPI2 decision is de-escalation after excluding the pending patients (m d), it means that the safety profile for the current level is a bit precarious even solely based on the completed subjects (n d). Then m-TPI2 calculation will be repeated assuming all the pending patients (m d) are DLT-free eventually. The following are two possible outcomes:
-
(a)
If the result remains de-escalation, the final decision will be de-escalation, reflecting the fact that even the safest assumption for the pending subjects (m d) would not neutralize the overly toxic signal from the patients with known outcomes.
-
(b)
If the recommended change is staying on the same dose (S), it implies moderate but volatile toxicity signal based on the complete patients (n d). Then R-TPI will check the number of patients who are enrolled to the current dose (k d) in the last batch. If it is below a certain threshold (say, three), which should be prespecified by the study team and simulation exercises, the new patient will be enrolled to the current dose, that is, to increase the sample size thus reducing the uncertainty by honoring the recommended action: stay. If k d is greater than the prespecified threshold, on the other hand, it means that the current dose level already has an adequate sample size; the only way to increase the information content will be to halt new patient accrual, letting more pending patients reach their endpoint.
-
(a)
-
(2)
If the m-TPI2 decision is escalating after excluding the pending patients (md), it means that the current dose level is confidently safe even solely based on the completed subjects (n d). Then m-TPI2 calculation will be repeated assuming all the pending patients (m d) without unknown outcome will develop DLT eventually.
-
(a)
If the re-calculated decision remains escalation, then the final decision will be to escalate, reflecting the fact that even the most toxic assumption for the pending subjects (m d) would not change the conclusion of dose being safe based on the patients with the known outcomes.
-
(b)
If the recommended change is staying on the same dose, it implies a moderate toxicity signal based on the complete patients (n d), which has a high degree of uncertainty. Then the R-TPI will check the number of patients who are enrolled to the current dose (k d) in the last batch. If it is below a certain threshold (say, three), new patients will be added to the current dose, that is, to increase the sample size and reduce the uncertainty, by honoring the recommended action: stay. If k d is greater than the prespecified threshold, it means that the current dose level already has an adequate sample size; the only way to increase the information content will be to halt the patient accrual, letting more pending patients reach their endpoint.
Similar to m-TPI2, the decision rule for R-TPI can also be pre-calculated in the protocol. Its strength of transparency is not lost (Table 2).
-
(a)
3.3.2 TITE-BOIN
The time-to-event version of BOIN method takes a different approach from R-TPI. When the new patients are waiting while some patients on the current dose level are still pending without final DLT results, the algorithm goes ahead to impute the DLT result for these pending patients, so that the waiting patients can enter the study in a timely manner, based on both the observed and imputed DLT results [20].
It can be shown that the point estimator of DLT rate at the current dose level, based on both the observed and unknown data, can be given as follows:
where O is the set of patients with known outcome of DLT (observed) and M is the set of patients whose DLT status is still pending. With c being the total number of pending patients, STFT is the sum of total follow-up time for the c patients divided by the length of DLT follow-up window, representing the ratio of information carried by those pending patients up to time t i. This representation assumes the uniform distribution of DLT rate across the DLT evaluation window, which has been proven robust in previous literatures. The only unknown entity in the right side of the equation is p, which can be estimated from the posterior beta distribution for DLT rate based on a vague prior and the patients who cleared DLT window. The p hat on the left will be compared with the toxicity interval in the regular BOIN to facilitate the dose recommendation.
One common strength of R-TPI and TITE-BOIN is that they both produce a transparent decision table in protocol before the first patient is accrued. For TITIE-BOIN at each dose level, the sample size, the number of DLT, the number of pending patients, and the threshold value of STFT are enumerated with the corresponding four different decision-makings for the incoming patients: de-escalate, stay, suspend accrual, and escalation, as follows (Table 3):
3.3.3 BOIN12
During a long DLT period, which we have discussed so far, it is possible for efficacy signal to emerge alongside with toxicity. An efficient design, therefore, is called for to select the dose with optimal risk-benefit trade-off. Sometimes, it is desirable to model both efficacy and toxicity simultaneously even when DLT window is not very long. For example, CAR-T cell therapy can induce quick and robust efficacy response and potentially severe toxicity at the same time.
Lin et al. proposed that the efficacy/toxicity balance can be quantitated by the following 2 × 2 table [9].

where u 1 − u 4 represent the utility scores which can be solicited from consultation with the clinical team. Typically, the optimal situation, in which tumor response is achieved in absence of toxicity, can be rewarded 100 (u 1) points with magnitude of 0–100, while toxicity without efficacy, the most undesirable scenario, has score of 0. Different scores between 0 and 100 can be assigned to u2 and u3 based on medical consideration, such as value of tumor response, the clinical sequelae of DLT, etc.
Corresponding to the four scenarios laid out above, the number of patients who have clinical outcomes (efficacy/toxicity) can be denoted as Y(d) = (y 1(d), y 2(d), y 3(d), y 4(4)), which can be modeled by multinomial distribution. In order to simplify the computation, however, the authors proposed an “quasi-beta distribution” method to solve this complex situation with binomial approximation.
In a regular binomial setting for toxicity alone, the number of DLT at dose d follows a binomial distribution B (p, n). Similarly, the equivalent of the binomial DLT count in “quasi-likelihood” theory here is x(d), the weighted utility score which can be normalized as follows:
x(d) follows a “quasi-binomial” distribution Bq (u(d), n(d)) where the expected u(d) is as follows:
and n(d) is the total patients treated at dose level d.
The likelihood function, as shown below, has a similar form as binomial probability:
In Bayesian framework, if we assign probability of the utility, u(d), to a beta distribution Beta (α, β) (α = 1 and β = 1 renders a flat prior), its posterior distribution can be formulated as follows:
In BOIN12 algorithm, the dose recommendation is based on the posterior inference on the probability of the utility, as opposed to DLT rate used in standard BOIN method. Its step-by-step guideline can be laid out as follows:
-
1.
Define the DLT rate boundary for de-escalation and escalation (λ e , λ d) in the same fashion as regular BOIN method and start to treat patients at the lowest dose level.
-
2.
Upon observing the DLT rate at dose level d, follow the rules below:
-
a.
If the observed DLT rate is greater than the upper boundary λ d, de-escalate to the next lower level d − 1.
-
b.
If DLT rate is on the target range (between λ e and λ d), and there are adequate number of patients at the current level (no less than a prespecified number, say, 6 or 9, etc), the dose level d or (d − 1) will be recommended for the new patients, depending on which level has the higher posterior probability of the drug utility, Pr(u(d) > u b |D(d)). Here dose level d+1 is not considered for potential better utility because of relatively strong confidence that the current level is not underdose.
-
c.
If the observed DLT rate is below the lower bound λ e, or within the target range (between λ e and λ d) but with a high degree of uncertainty (the number of treated patients at the current dose is less than a prespecified threshold value), even the dose level d+1 which is one level higher than the current dose, in addition to the current level d and its adjacent lower level d−1, can be explored for their posterior probability of the utility score. Among these three levels, the one with the highest posterior probability of functional utility will be recommended to the new patients.
-
a.
-
3.
Once the maximum sample size is reached, DLT level at each dose level will be estimated isotonically and the one whose DLT rate is closest to the target DLT rate will be declared as MTD. The final recommended dose for phase 2 (RDP2) should be the dose level with highest estimated utility score while not exceeding MTD level.
BOIN12 method retains the strength of BOIN and mTPI-2 that is the transparency to pre-tabulate all the decision-making points prior to the start of a trial. The following is example of decision table (Table 4).
3.3.4 Imputation of Unobserved DLT Data
The idea of using imputed DLT outcome from the pending subjects to guide dose recommendation, as has been done in TITE-BOIN method, has seen its application in other literatures [7, 11, 21]. We will have two examples as follows.
Liu et al. showed that the imputed DLT status from pending patients, along with the observed DLT count, can be fit into regular CRM model, leading to the update of the posterior DLT rate and dose recommendation for the incoming patients based on the posterior mean [11]. This process is an iterative process consisting two fundamental steps: (1) imputation of missing DLT value, (2) posterior estimation of the CRM parameter, as summarized below:
-
1.
The time to DLT for subject i is modeled by a piece-wise exponential model as follows:
$$ L\left(\mathbf{\mathcal{Y}}|\boldsymbol{\lambda} \right)=\prod_{i=1}^n\prod_{k=1}^K{\lambda}_k^{\delta_{i,k}}\exp \left\{-{\mathcal{Y}}_i{\lambda}_k{s}_{ik}\right\} $$
where y is DLT status, s ik is the length of k sub-interval of DLT evaluation window, λ k is the constant hazard rate for kth time interval. In Bayesian framework, λ k is assumed to follow a prior Gamma distribution:
where the value C can be calibrated to render the prior vague (C = 2).
This leads to the posterior distribution, which is conditional on the observed data and model parameters including the power parameter α of the CRM and the DLT hazard rate λ, as follows:
The missing DLT data can be imputed by drawing posterior samples from the distribution above.
-
2.
The observed and imputed DLT data can be used to update the posterior distribution of the CRM model, from which α can be sampled. The DLT hazard rate λ k can be sampled from the conjugate Gamma posterior distribution as follows:
$$ {\lambda}_k\mid \mathbf{\mathcal{Y}}\sim {\boldsymbol{G}}_{\boldsymbol{a}}\left(\frac{{\overset{\sim }{\lambda}}_k}{C}+\sum_{i=1}^n{\delta}_{ik},\frac{1}{C}+\sum_{i=1}^n{y}_i{s}_{ik}\right) $$ -
3.
The drawn samples of α and λ k will be fed back into step 1, updating imputation of the missing DLT data. Then α and λ k will be sampled again from the posterior distribution of the CRM model based on the renewed imputed data. This iterative process, which is also called Bayesian data augmentation method, will go on until Markov chain sampling achieves convergence.
-
4.
The final posterior sampling of α will determine the point estimator of the posterior DLT rate, which will determine the dose recommendation.
This approach is evidently complex. So far there is no definite evidence showing it outperforms other simpler approaches such as R-TPI.
In a related paper published by the same group, this iterative data augmentation process is implemented by EM (Estimation-Maximization) algorithm [21]. In the Estimation step, the missing DLT outcome of y i can be substituted with its expectation in the form of
Then in the M step, the MLE of the CRM power parameter α and the DLT hazard rate λk by the following likelihood function:
K = 1, …, K, an estimate analogous to Kaplan-Meier’s estimator and
respectively.
Furthermore, this EM-CRM framework also allows multiple dose-toxicity “skeletons” to be selected, and the Bayesian model selection and averaging will be employed to give the best estimate for the CRM power parameter α, which is the basis for DLT rate estimation and subsequent dose recommendation.
3.4 Use Kaplan-Meier Method to Derive Fractional DLT for Pending Subjects
Finally, the missing DLT status of pending patients can be replaced by fraction of 1, depending on the proportion of follow-up time to the full DLT observation window. This fractional value can be used in either rule-based method such as the 3+3 after rounding, or model-based method such as CRM/BLRM , given that the specification of likelihood function can take fraction as the input value [19].
When a subject completes the DLT window, his DLT status y i will be either 0 or 1, depending on whether they experience DLT. When a patient’s final DLT status is still pending upon a new patient enters, however, their fractional DLT values can be computed as follows:
where S is Kaplan-Meier estimator of survival function at u i or τ. The KM survival estimator is expressed as:
With the missing DLT data replaced by a fractional DLT, they can serve as input for any dose-escalation method mentioned above.
4 Summary
In this chapter, we started with the review on three classes of methodologies for oncology dose-escalation trial design: the 3+3, the statistical model-based approach including Continuous Reassessment Method (CRM) and Bayesian Logistic Regression Model (BLRM), and the toxicity interval-based algorithms such as Bayesian Optimal Interval Design (BOIN) and Toxicity Probability Interval method (TPI) and their respective variations. The focus of this chapter is to give a comprehensive outline of the various statistical extensions of these methods to address the statistical challenges caused by the prolonged safety evaluation window, or equivalently, the fast enrollment rate. They include, in CRM and BLRM class, the weighted likelihood function method (TITE-CRM), TITE-CRM aided by suspension rule or Bayesian predictive risk for toxicity to avoid aggressive dose escalation, the TITE-CRM that leverages drug cycle information, adaptive time-to-event toxicity distribution, and three-parameter logistic regression extension on the basis of BLRM. In the toxicity interval-based class, we review R-TPI method for Toxicity Probability Interval method, TITE-BOIN which imputes the unobserved DLT and BOIN12 which models the long-term toxicity and efficacy concurrently. The methods under discussion can play a valuable role in improving the accuracy of optimal dose identification without sacrificing patient safety or significantly prolonging the trial duration.
References
Bekele, B., Ji, Y., Shen, Y., Thall, P.: Monitoring late-onset toxicities in phase I trials using predicted risks. Biostatistics. 9(3), 442–457 (2008)
Braun, T.: Generalizing the TITE-CRM to adapt for early- and late-onset toxicities. Stat Med. 25(12), 2071–2083 (2006)
Cheung, Y., Chappell, R.: Sequential designs for phase I clinical trials with late-onset toxicities. Biometrics. 56(4), 1177–1182 (2000)
Guo, W., Ji, Y., Li, D.: R-TPI: rolling toxicity probability interval design to shorten the duration and maintain safety of phase I trials. J Biopharm Stat. 29(3), 411–424 (2019)
Guo, W., Wang, S.J., Yang, S., Lynn, H., Ji, Y.: A Bayesian interval dose-finding design addressing Ockham’s razor: mTPI-2. Contemp Clin Trials. 58, 23–33 (2017)
Huang, B., Kuan, P.: Time-to-event continual reassessment method incorporating treatment cycle information with application to an oncology phase I trial. Biom J. 56(6), 933–946 (2014)
Jin, I., Liu, S., Thall, P., Yuan, Y.: Using data augmentation to facilitate conduct of phase I-II clinical trials with delayed outcomes. J Am Stat Assoc. 109(506), 525–536 (2014)
Ji, Y., Liu, P., Li, Y., Bekele, B.N.: A modified toxicity probability interval method for dose-finding trials. Clin Trials. 7(6), 653–663 (2010)
Lin, R., Zhou, Y., Yan, F., Li, D., Yuan, Y.: Personal communication (2020)
Liu, S., Yuan, Y.: Bayesian optimal interval designs for phase I clinical trials. J. R. Stat. Soc. Ser. C. 64, 507–523 (2015)
Liu, S., Yin, G., Yuan, Y.: Bayesian data augmentation dose finding with continual reassessment method and delayed toxicity. Ann Appl Stat. 7(4), 1837–2457 (2013)
Shah, N.J., et al.: Delayed toxicities with anti-PD-1 and anti-PDL-1 immune checkpoint inhibitors (ICIs). J. Clin. Oncol. 36(15 suppl) (2018)
Neuenschwander, B., Branson, M., Gsponer, T.: Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med. 27(13), 2420–2439 (2008)
O’Quigley, J., Pepe, M., Fisher, L.: Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics. 46(1), 33–48 (1990)
Polley, M.: Practical modifications to the time-to-event continual reassessment method for phase I cancer trials with fast patient accrual and late-onset toxicities. Stat Med. 30(17), 2130–2143 (2011)
Skolnik, J., Barrett, J., Jayaraman, B., Patel, D., Adamson, P.: Shortening the timeline of pediatric phase I trials: the rolling six design. J Clin Oncol. 26(2), 190–195 (2008)
Storer, B.E.: Design and analysis of phase I clinical trials. Biometrics. 45(3), 925–937 (1989)
Ting, N., Chen, D.G., Ho, S., Cappelleri, J.C.: Phase II Clinical Development of New Drugs. Springer, Cham (2017)
Yin, G., Zheng, S., Xu, J.: Fractional dose-finding methods with late-onset toxicity in phase I clinical trials. J Biopharm Stat. 23(4), 856–870 (2013)
Yuan, Y., Lin, R., Li, D., Nie, L., Warren, K.: Time-to-event bayesian optimal interval design to accelerate phase I trials. Clin Cancer Res. 24(20), 4921–4930 (2018)
Yuan, Y., Yin, G.: Robust EM continual reassessment method in oncology dose finding. J Am Stat Assoc. 106(495), 818–831 (2011)
Zheng, W., Zhao, Y., Lu, Y., Miao, H., Liu, H.: A Bayesian three-parameter logistic model for early- and late-onset DLTs in oncology phase I studies. J Biopharm Stat. 26(2), 339–351 (2016)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Wei, X., Liu, R. (2022). Review of Statistical Treatment for Oncology Dose-Escalation Trial with Prolonged Evaluation Window or Fast Enrollment. In: Lio, Y., Chen, DG., Ng, H.K.T., Tsai, TR. (eds) Bayesian Inference and Computation in Reliability and Survival Analysis. Emerging Topics in Statistics and Biostatistics . Springer, Cham. https://doi.org/10.1007/978-3-030-88658-5_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-88658-5_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-88657-8
Online ISBN: 978-3-030-88658-5
eBook Packages: Mathematics and StatisticsMathematics and Statistics (R0)