
Abstract
The lung microbiome in adult asthma is the product of a long period of dynamic shaping by microorganisms and the host from infancy onwards. The use of next-generation sequencing technologies has facilitated new, detailed studies of the lung and GI microbiomes and their relation to immunologic and molecular features of asthma. In this chapter we evaluate the studies that have examined the microbiome of the normal and asthmatic adult airways, how the development of the gastrointestinal and upper airway microbiomes and their interactions with the host in infancy and childhood shape asthma in early life, the relationships between asthma phenotypes and the airway microbiome, and emerging understanding of the fungal mycobiome in asthma and the potential effects of medications on the microbiome. The studies done to date provide a descriptive platform and challenge us to more longitudinal, larger, and mechanistic studies to determine the clinical utility of the lung microbiome in the diagnosis and treatment of adult asthma.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
Introduction
Asthma continues to be one of the most common chronic diseases worldwide. It affects almost 300 million people and has an increasing prevalence in developed countries, including the U.S. As important as it is in childhood, where it affects up to 10% of children, it is also of major importance to the adult population. Many children with asthma will continue to have asthma their entire lives; others will experience some temporary abatement followed by recrudescence. Asthma that occurs de novo in adults, without any evidence of airway hyperreactivity or narrowing in childhood, is increasingly recognized. Indeed, asthma in the elderly has been recognized for decades, and the prevalence of asthma in this population also is increasing. The mechanisms proposed by which people develop asthma or are predisposed to its development are numerous. From a century ago when the origin of asthma was considered to be allergic in nature, we now understand increasingly that asthma is a rich interplay of genetics, inflammation, and environmental exposures. Of the latter, allergens, pollutants, and viral infection long have been identified, but increasingly, both the microbiome of the lungs and that of the gastrointestinal system (“gut microbiome”) are increasingly understood to have some role, though the mechanisms by which each might do so, and the interplay between them, and of each with other environmental and inflammatory cues, are yet to be understood.
This goals of this chapter are the following: (1) to synthesize recent evidence on the airway microbiome and its potential role in chronic asthma and asthma phenotypes; (2) to summarize evidence on the role of environmental microbial exposures and the developing upper airway and gut microbiomes in infancy on early-life asthma; and (3) to highlight the scientific challenges, critical research questions, and promising avenues to advance understanding of the microbiome’s impact on asthma. Current knowledge on asthma biology and clinical characteristics also will be discussed, for the purpose of and tailored to these goals.
The Microbiome of the Airways in Asthma
Overview
There is now recognition gained over the past decade that the airways are not sterile in the normal state, and that indeed there exists an ecology of bacteria, likely together with fungi and viruses, in the airways. In this chapter the airway microbiome is defined as the composition of the bacterial and fungal organisms from trachea to alveoli as identified by an array of their genomes from next-generation sequencing. However, sampling the lower airways poses challenges (as discussed below). Studies focused on understanding the role of “airway” microbiota on asthma inception in early life predominantly have evaluated the upper respiratory tract (e.g. nasopharyngeal sampling) rather than the lower airways. Differences in the microbiome of the upper and lower respiratory tract both have been associated with asthma and are discussed separately in this chapter.
Studying the airway microbiome in asthma is motivated by the following important questions: First, do differences or changes in the microbiome have a role in modulating airway inflammation? Second, do the changes in airway ecology and the microbiome noted in asthma track with any of the defined asthma phenotypes in clinically important ways? Indeed, a dysbiotic airway microbiome may be correlated to or even essential to a select phenotype. Third, which comes first – a dysbiotic airway microbiome followed by airway inflammation, or airway inflammation (be it eosinophilic, neutrophilic, both or neither) that leads over time to altered ecological niches that support only select airway microbiota? And finally, if we manage through some manipulation (be it drugs, antibiotics, probiotics or something else) to nudge the airway ecology back towards a more “normal” state, will we in turn downregulate or modulate airway inflammation and perhaps clinical asthma control?
The relationships between the airway microbiome and asthma inflammatory phenotypes are complex. Defining the relationships between the airway microbiome and these phenotypes, however, might inform better our understanding of the underlying pathophysiology of each phenotype. Further, markers within the microbiome, be it the abundance of particular taxa, a diversity index, or ratio of select taxa, may serve as biomarkers that add to inflammatory, genetic, and clinical biomarkers to define phenotypes and guide the use of therapies. The past decade has seen the utility of inflammatory biomarkers in the definition of phenotypes such as the usefulness of blood and sputum eosinophil counts as a marker for the type-2 (T2) high phenotype that predicts the response to therapies directed against interleukin (IL)-5 or the IL-4Rα receptor. Yet even here, patients who meet the criteria for this phenotype have, at best, a 50% response to these agents. The door is open to the potential use of the microbiome to shape and sharpen our understanding of asthma endotypes.
Further, a deeper understanding of the microbiome in chronic asthma may lead to therapies directed against select microbiota. As we will discuss later in this chapter, first steps have already been taken with the use of antibiotics such as azithromycin. Better and more targeted therapies, perhaps including probiotics (live microorganisms that are intended to have health benefits), prebiotics (nondigestible food components that selectively stimulate the growth or activity of desirable microorganisms) or bacterial products, may develop into adjunct therapies for select asthma phenotypes.
Defining the Airway Microbiome in Asthma
“The healthy lower airways are sterile” is a dogma taught over the last century of medicine. Indeed, the lungs were initially not considered important enough to study in the first phases of the Human Microbiome Project [1]. In retrospect, the flaw in such dogma is obvious: the lung is exposed to bacteria from early infancy from its continuous exposure to 8000 liters daily of inspired air and to secretions from the nose, oropharynx, and the gastrointestinal tract [2,3,4], and it would be remarkable indeed if the lung were not to be colonized by at least some microbiota in all that time. But the view of the sterility of the lung was ordered by the technology of its time: from the earliest days of microbiology, we could only consider those organisms that could be grown in culture, and it was uncommon indeed for any bacteria to be cultured from samples collected from the lung in its normal state. While organisms might be introduced to the lung from elsewhere, the innate defenses of the lung, including anti-bacterial defensive proteins and immune cells, and the “mucociliary elevator” that lift particles and contaminants including bacteria from peripheral airways to glottis, balanced this immigration exactly. Perturbations of this balance led to shorter-term infections such as bacterial, viral, or fungal pneumonia, and longer-term infections that characterize the progression of diseases such as cystic fibrosis [5,6,7,8,9]. But the mucociliary elevator and innate defense of the normal lung served to maintain sterility.
This dogma profoundly changed with the willingness of investigators to sample the lung more directly by bronchoscopy and by advent of next-generation sequencing. As described in earlier chapters, sequencing that exploited variations in the 16S rRNA gene, a highly conserved locus of the bacterial genome, permitted identification of nearly all bacterial organisms to an increasingly precisely identified level of homology in a given ecological space, regardless of their ability to grow in any external culture system. Culture-independent profiling based on sequence polymorphisms in the 16S rRNA gene, and sequences unique to the fungal ribosome, permit identification of these microbiota and examination of the relationships of a microbial community to disease phenotypes [10]. Such a community would encompass both those organisms that were either transitory in passage down or back up the airways, or were more permanent survivors in a dedicated ecological niche. This community could be described by taxonomy (the identification and compositional abundance of microbes), the diversity of the microbial community both within each airway niche or between groups of patients (e.g. by asthma phenotype), and the functionality of these organisms based on readouts of their predicted gene functions or products. Such information provides insight into the interactions between microbiota in the same ecological space, and their ability to provoke (or not) the host-defense systems. Understanding the bacterial ecology of the airway then could be related to the changes within that airway specific to asthma: inflammation, changes in defensive mechanisms, and changes in airway structure.
How to Study the Airway Microbiome in Asthma
Before embarking on a review of the studies to date, one must consider how best to sample the airways, when such sampling should be done, and which patient populations should be studied. Chapters 1, 2 and 3 discussed study design and sampling considerations, including the methods for collection, processing, and subsequent data analysis (the bioinformatics “‘pipeline”), with an eye towards enhancing reproducibility. This comes from hard-won experience in studies of other diseases and fields. We note the specific issues with sample collection with regard to asthma in this section.
The Problems of Sampling
The first issue to be overcome is that of sampling: what is the best way to examine the airway microbiome in asthma? Differences in sampling methods, including the compartments sampled, may induce variability of results and both limit comparisons between studies and limit our ability to apply these findings to patient care.
The choice of which compartment to sample is the first question. Asthma is a disease of the large airways, and one would start with examining these airways – but which airways? The large airways can be seen as encompassing two regions: the trachea and main-stem bronchi, which are in direct communication with the oropharynx [11], and the lower-order conducting airways (generations 3 through about 10), which while connected to the trachea and to the peripheral airways may present as a unique microbial niche [12]. There are potential differences between these spaces, central, conducting, and peripheral, and each may be influenced by the microbiome of the adjoining space(s), the rates of microbial migration into and out of the space, elimination by any host defense factors, and microbial reproduction.
The next question is how to sample these spaces. Samples can be collected by bronchoscopy; an unprotected brush inserted through the channel of the scope will collect whatever material that has contaminated the channel to that point in time, including any oral and upper airway microbiota. A “protected” brush inside a catheter with a gelatin plug at its tip may fare better, but then special care is required to remove contaminants from around the protective sleeve. Brushing collects material, including epithelial cells and mucins, at the airway surface. The latter may have trapped (again, the “ciliary elevator”) bacteria brought from the more peripheral airways. Thus, the endobronchial sample is an amalgam of what is there at that point in time, plus whatever has been lifted from below. An endobronchial washing with saline would invariably collect material from the peripheral airways unless a balloon is used to block this; this is cumbersome and requires significant expertise. Bronchial biopsies would include surface bacteria and also bacteria in the submucosa. In a comparison of paired endobronchial washes and biopsies obtained in a small cohort of patients with severe asthma, Millares et al. [13] found that the two types of samples had modestly different relative abundance, beta-diversity, and predicted functional capabilities based on an analytical method (PICRUSt). Finally, as one readily appreciates upon viewing data from any study using small brushes, there is a low biomass inherent both to the low absolute numbers of bacteria and the small sample mass both in the airway and on the brush. In sampling the central airways then, endobronchial brushings are generally used, but investigators must keep in mind these limitations.
What about the peripheral airways in asthma? Over the last two decades evidence has suggested the presence of small- and peripheral-airways disease in asthma [14,15,16,17]. Might then sampling of this space be worthwhile? This could be done by bronchoalveolar lavage (BAL); this fluid would represent all airways distal to the tip of a wedged bronchoscope, including the alveoli, and of course any organisms already in the channel of the bronchoscope. Such samples are then heterogeneous, and worse, of very low biomass, often an order of magnitude lower than that seen for the central airway samples in terms of bacterial burden [18].
The low biomass of either central or peripheral airway samples creates a significant additional technical burden, that of distinguishing what is in the lung from what is contaminating the sample (upper airway or from the scope channel) along with the background of any 16S rRNA that might be present in reagents and the various kits used for initial isolation [19, 20]. Both organizational (e.g., scope cleaning, testing of reagents) and computational methods can be used to ameliorate partially this problem [21] but investigations of the airway are challenged in a way that studies with much higher biomass, such as the GI (gut) microbiome, are not.
When to Study the Airway Microbiome
Many of the adult studies done to date examine associations between clinical and biological parameters of asthma with the microbiome collected at a single time point. These cross-sectional studies are invaluable snapshots, particularly when compared to either normal subjects or to patients with other airways diseases such as COPD or cystic fibrosis. One recent review tabulates the findings of 31 cross-sectional studies done between 2010 and early 2019 in pediatric and adult asthma; such tabulation clearly shows the substantial variance in key findings [22]. The major limitation of any cross-sectional study is, of course, the lack of data at a future point in time, and (usually) the inability to collect the same or similar data, particularly biological specimens, from previous points. Thus, cross-sectional studies are invaluable to delineate associations but less so to understand mechanisms.
A particular challenge for any asthma longitudinal study is the inability to collect samples of the lower airways repeatedly over time by bronchoscopy. While the risks of bronchoscopy are very low for asthma patients in a research setting [23, 24], relatively few research participants will consent to more than a single invasive procedure, and the costs associated with repeated bronchoscopy are prohibitive in any large trial. Serial sampling then might be best done via assessment of sputum. One early study demonstrated that the sputum microbiome differed in patients with severe or mild asthma compared to normal subjects [25]. But examination of these samples presupposes that the sputum microbiome is representative of that of the lower airway. Evidence that such a supposition is true (or true enough to be useful in investigation) comes from a direct comparison of sputum and lower airway microbiota in the AsthmaNet Microbiome study, a cooperative, multicenter observational trial that was sponsored by the National Heart Lung and Blood Institute (NHLBI) [26]. This study examined paired samples consisting of protected endobronchial brushings, induced sputum, oral washings, and nasal brushings in a cohort of patients with mild asthma. This study was careful to account for potential causes of contamination. As might be expected, although compositionally similar to the endobronchial microbiota of the lower airway, the microbiota in induced sputum were distinct and reflected enrichment of oral bacteria [26]. Patients with asthma or atopy were more likely to have bacterial taxonomy in induced sputum that reflected the lower airway and were more distinct from that seen in the oral cavity. This study suggests that within limits, repeated survey of the lower airway microbiome over time in patients with asthma (but not necessarily healthy subjects) may be approximated using induced sputum.
Considering Environmental Influences on Asthma and the Human Microbiome
Urbanization with attendant air and traffic pollution clearly are associated with increased asthma prevalence. While asthma prevalence has increased over the decades in highly developed countries, low- and middle-income countries now are seeing an increase, particularly among urban populations [27, 28]. The biologic, social, and environmental factors are complex and as yet incompletely understood but revolve around a rich interplay of air quality, pollution, diet, exercise or the lack of it, use of antibiotics, reduced exposures to “rural” allergens and increased exposures to city allergens, and changes in the patterns and types of childhood infection. With these in mind, it is clear that the rural environment is associated with a lower prevalence of asthma [27, 29,30,31], a protection that is lost in rural to urban migration [32]. With the difficulties of quantifying exposures, lifestyle, and even whether a given residence is urban or rural, it is challenging to relate changes in the environment to changes in the microbiome that in turn might influence asthma.
One interesting approach to address the potential protective effect of the rural environment has been to examine the role of the farm microbiome and its microbial products on the susceptibility to asthma and atopy in children. Early-life farm exposures have been shown to reduce the risk of asthma in children [33,34,35,36,37,38]. In a study of 196 children with asthma with and without atopy, increased asthma severity was associated with an increased concentration of allergenic fungal species, high total fungal concentrations, and high bacterial richness in house dust [39, 40]. Differences in the prevalence of asthma and atopy are seen in children raised in different farming environments, such as between children of Amish (traditional farming) versus Hutterite (modern farming) heritage [38]. The farm environment can be carried indoors to mix with other influences including pet dander, food, dust mites, and other eukaryotic small organisms. Indoor microbiota are different in farm environments and are associated with asthma and atopy prevalence, and depending on context may be either protective or exacerbating [41, 42]. Here again, whether the indoor and farm microbiota elicit changes in the lower airway microbiome is not known. Indeed, in the study of Hrusch et al., that examined house dust differences in Amish versus Hutterite heritage, intranasal instillation of house dust extracts from either location to ovalbumin-sensitized and challenged mice elicited opposite effects: the house dust from Hutterite farms augmented whereas the Amish dust extracts inhibited the airway responsiveness, eosinophilia, and IgE levels induced by allergen [35]. But the differences in microbiota and other components in these two extracts were not defined, and so it is not clear what component of the dust is responsible. This one genre of work then illustrates some of the complexities of relating the larger environment to changes in the microbiome of a locale, and to determine whether a change in that microbiome, or its subsequent effect on the host, is responsible for asthma.
We often think of “the environment” as a macro or global or regional event, but in ecological terms the environment is everything about us from the great outdoors to our most personal indoor settings. For many, the indoor or “built” environment is key: most people in the developed world spend the majority of their time indoors in which the microbial community, both that within the home and that brought to the indoors, may be shared [43, 44]. Indeed, humans live within a personal, aerosol “bio-cloud” of their own microbiome that settles about them that can be used to identify individuals [45, 46], and in modern life it is straightforward to see that one’s aerosolized organisms (from oral pharynx, nose, skin but perhaps also exhaled from the lung) could be transferred to and then inhaled by another person. Indeed, this is a mechanism by which infection (bacterial or viral) may be transmitted. The proximity of people in the indoor environment, be it home, office, school, or day-care center, permits more efficient “sharing” and transfer of these microbiota. As one example, day care attendance early in life is associated with decreased asthma prevalence at elementary school age and adolescence [47, 48], though there was no measurement or association of the environmental or child-carried microbiome in these studies.
Endotoxin exposure is a risk for wheezing but not for asthma [49, 50], an apparent contradiction that has not yet been resolved. Indeed, indoor microbial exposure to endotoxin and fungal antigens might reduce the risk of asthma in certain settings, as demonstrated in the Prevention and Incidence of Asthma and Mite Allergy birth cohort study of children with atopic mothers in New Zealand [51]. Counterbalancing these studies, both specific bacterial products (e.g., endotoxin) and fungal products (e.g., chitin, glucans) in indoor environments are associated with increased asthma prevalence in children [52,53,54,55]. Perhaps the organisms in the indoor environment themselves may change asthma susceptibility. One small study demonstrated that the microbiota in home dust differed in dwellings that housed low-income asthmatic children versus non-asthmatic children; in the former group the sampled house dust contained an increased abundance of Proteobacteria and Cyanobacteria [56]. One potentially important component of house dust can be pet dander, and it is clear that the indoor microbiota of homes with pets differ from homes that are pet-free [57,58,59,60]. Allergen presence and dog dander within a home are correlated with changes in the indoor microbiome in house dust [61]. Such differences in the indoor microbiome may in part account for the known association of dogs with protection from allergic sensitization and asthma in early life [62,63,64]. No studies to date have connected the taxonomy of house dust microbiota to that of the lower airway microbiome or has related house dust exposure to the changes in the airway microbiome in patients with asthma. Further, the specific biogeography within an indoor environment, ventilation, and the microbiomes of the occupants of a home all might contribute to the “bio-cloud” that we inhale, and thereby may modulate in some way our airway microbiome. Larger studies that associate these factors to risks of asthma, both in children and in adults, are needed.
The impact of seasonality in the larger outdoor environment (and perhaps also the indoor environment) on asthma has long been recognized in many regions of the world due to the variations in humidity, base temperature, and rapid changes in temperature, and high concentrations of air pollutants and allergens [65,66,67,68,69,70,71,72,73,74,75]. These are, of course, conditioned by individual clinical characteristics, phenotypes, and other causal and exacerbating factors. One potential mechanism among many by which these factors may elicit changes in asthma control may be by changing the airway microbiome. Studies that relate seasonality to the “bio-cloud” and thence to the lung microbiome clearly are needed.
The Airway Microbiome in Health
Having some understanding of the limitations of sampling and study design, and of where and how people with asthma might live, we can examine the airway microbiome in asthma. To do so, we first discuss briefly what is known about the “normal” microbiome in health. One of the earliest papers to examine the normal lower airways community was that of Charlson et al. who sampled multiple sites in the pharynx, central airway by protected endobronchial brushing, and peripheral airways and alveoli by lavage, in six healthy subjects [11]. They demonstrated bacterial communities in the lower airways, both central and peripheral, that were low in biomass and that were indistinguishable from the pharyngeal flora. These bacteria were represented by five major phyla, including Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria. Their data strongly suggested that the lower airway microbiome was a subset of that of the upper airway and occurred due to colonization from aspirated microorganisms. This work represented the first baseline data for the microbiome of lower healthy airways. The lungs, indeed, were not sterile. Subsequent studies that compare a disease-associated airway microbiome to normal generally have observed similar findings. For example, Morris et al. [76] examined the upper and lower airway microbiomes in 64 healthy subjects in a multisite study sponsored by the NHLBI Lung HIV Microbiome Project (LHMP); of these, 45 were nonsmokers and 19 were current smokers. Many of the microbiota identified in the lung were also noted in the mouth, but several, including Haemophilus and Enterobacteriaceae, were in higher proportion in the lower airways than upper, suggesting that the lung microbiome did not derive entirely from the mouth and pharynx. Further, while the upper microbiome differed between smoking and nonsmoking subjects, the lung microbiome did not. Another interesting observation was that while patients from eight different sites were included in their study, there were no significant differences in the diversity indices compared across the clinical centers, suggesting a remarkable uniformity in the healthy lung microbiome. Similar results with regard to the lack of difference in the lung microbiome between healthy nonsmokers and healthy smokers without lung disease were noted in bronchial wash samples [77] and bronchoalveolar lavage fluid [78]. Additional studies have examined the geography of the microbial communities of the pharynx (oral wash), nose (swab), stomach (gastric aspirate) and lung (BAL) in various combinations. One such study in 28 healthy subjects showed that both the oral and gastric microbiomes were both different and richer than that seen in the lower, peripheral airways [79]. Marked subject-to-subject variation was noted, a finding that has been seen in other studies of different microbiomes. Dickson et al. [12] examined 15 healthy subjects by bronchoscopy, sampling (in order) from the peripheral lung by BAL to conducting airways at several locations and then central airways by protected brushes. They demonstrated that spatial variation in microbiota within an individual was significantly less than variation across individuals, and that community richness decreased as samples went from trachea to conducting airway to peripheral airway. Another study of 86 normal subjects from the LHMP examined oral washes and bronchoalveolar lavage and demonstrated that the lower, peripheral airways as sampled by BAL did not mirror completely the oropharynx [80]. In a more recent study of 124 healthy subjects in which sputum was collected as part of a study examining the airway microbiome in patients with COPD, Firmicutes, Bacteroidetes, and Actinobacteria were the major phyla constituting 88% of the total reads in these healthy subjects; Streptococcus, Veillonella, Prevotella, Actinomyces, and Rothia were the dominant genera [81]. The genus Haemophilus, in contrast, formed only 3% of the healthy microbiome. These findings were increasingly confirmed in subsequent studies comparing the normal versus asthmatic airway microbiomes in studies cited in the following sections. Similar findings have been reported in which a normal population is recruited as a control for COPD studies with reference to the lower airway microbiome (as examples, see [77, 78]).
It becomes clear then that the adult normal airways have a bacterial microbiome that is small in biomass, well defined, and derived in part, but only in part, from that of the upper airway. The major phyla that are reproducibly found include Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria. While there can be significant variation between normal, healthy subjects, there is less (but not zero) variation between regions of the lung. How is it different in individuals with asthma, when does it become different, and might a dysbiotic microbiome have a role in asthma and airway inflammation? In the next section we discuss first the evidence linking early-life exposures and the developing microbiomes of the upper airways as well as of the gut, to asthma development in childhood.
Early-Life Asthma and the Microbiome
Extensive evidence has highlighted the concept of a critical window in early life during which the developing immune system is shaped by exposures that influence subsequent risk for allergic diseases including asthma [82]. Such exposures can be categorized broadly as follows: (1) characteristics of the external environment that impact opportunity for microbial contact (e.g. farm vs. nonfarm household); (2) lifestyle practices that shape the establishment of microbiota and maturation of the microbiome (e.g. breastfeeding, pet ownership); (3) medical events and treatments that may support or interrupt microbiota establishment, its ecological succession and interactions with host immunity (e.g. respiratory virus infections, antibiotics, probiotics). Numerous studies have been published on these areas over the last several decades. Reported links to various types of exposures, coupled with mechanistic studies implicating the role of microbiota, have underscored the multifactorial nature of early-life interactions that shape risk for asthma. This section will highlight select studies that have contributed to this evidence. These include recent investigations that have applied advanced analytical methods to examine the microbiome coupled, in some cases, with mechanism-directed experiments. In doing so, such studies demonstrate the power of combining cross-disciplinary approaches to advance insights into the complex host–microbiota interactions that shape asthma pathogenesis.
The Early Microbiome in Asthma and the External Environment
As mentioned earlier, differences in the prevalence of allergic conditions between persons living in different environments or geographic regions have long been recognized, well before the current era of next-generation sequencing methods to survey microbial content. Epidemiologic comparisons between different populations of similar genetic ancestry have reported significant differences in the rates of allergy and asthma, observations that persist in more recent reports [35, 36, 38, 83, 84]. These include studies comparing individuals living in the Karelia region of Finland versus Russia [84], in farming versus nonfarming households of Germany and other parts of Europe [36, 83], and between Amish and Hutterite communities in North America [38, 85]. These and other similar data [86] support the now well-accepted notion that characteristics of the home environment can affect risk for childhood allergy or asthma.
Differences in hygiene levels and related living practices impact the frequency and nature of contact with microbes. Thus, many studies have examined markers of microbial load or the types of microbes found in household dust samples [36, 55, 87], finding differences associated with atopy or asthma. In general, a higher microbial load or diversity of microbes found in household samples is associated with a lower risk or prevalence of atopy or asthma. However, the specific microbes associated with these outcomes vary by study and location. This highlights not only geographical differences, but also the likely importance of the sum effects of a microbial community on microbiome–host interactions. A number of factors affect microbial content within homes, which include pets that are exposed to the outdoors and the flow or tracking of outside air or soil into homes [61, 64, 87]. As noted in the section concerning the indoor environment and the microbiome, recent studies also have shown interactions between household bacterial and allergen load that significantly modify their associations with asthma [88, 89], adding further complexity to how environmental exposures shape immune responses that lead to asthma.
To apply this knowledge in a way that may guide environmental interventions, a recent study aimed to quantify the “farm home microbiota” effect by developing an index (“FaRMI”) derived from the relative abundances of bacteria/archaea measured in farm-home floor dust [90]. Relative abundance data from samples collected in a rural Finnish birth cohort were used to derive the index, which was then applied to samples from a nonfarm/suburban Finnish birth cohort. The overall microbial composition of samples was distinct between the two cohorts. Compared to suburban homes, rural home dust was characterized by higher bacterial richness and enriched in members of specific bacterial orders (Bacteroidales, Clostridiales, and Lactobacillales). No differences in fungal richness were identified. Among suburban homes a higher FaRMI in dust samples was negatively associated with asthma. Similar relationships were observed when FaRMI was derived and used to analyze samples from an independent German birth cohort. Moreover, the asthma-protective effect observed with a high FaRMI was independent of atopic sensitization, suggestive of microbiota-specific contribution. Further research is still needed to understand how environmental exposure to particular consortia of bacteria elicit immune-protective effects against asthma in early life.
Other recent studies have shed insight into this by phenotyping innate and adaptive immune responses between groups at differential risk for asthma, such as among Amish versus Hutterite children. The prevalence of asthma and atopy is strikingly lower among the Amish, and significantly higher levels of endotoxin were measured in Amish house dust [38, 85]. Major differences also have been observed in the proportions and functional markers of innate immune cells in blood and in T-cell phenotypes [35]. As mentioned earlier from this study, using a mouse model of allergic asthma, intranasal administration of Amish house dust extracts prevented airway hyperreactivity and lung eosinophilia, contrasting from the effects observed with Hutterite house dust extract [38]. The protective effects were abrogated in mice deficient for MyD88 and Trif, two important molecules at intersecting innate immune-signaling pathways. Although no information is currently available on whether there are differences in the dust-associated microbial or allergen content, results from the in vivo models indicate that specific components in the Amish house dust are at least partly responsible for the observed protection against allergic airway inflammation. Similar observations were made in another study in which dust from dog-owning homes was administered to mice using models of asthma; this resulted in attenuation of allergic airway inflammation, with Lactobacillus johnsonii identified as one responsible species [91].
The GI Microbiome and Early-Life Asthma
The establishment of gut microbiota is essential for normal immune development and discussion of this is important as it pertains to asthma risk in childhood. The gut microbiome is established in the same way the lung microbiome is starting shortly after birth, by aspiration or swallowing of oral microorganisms. Transfer of bacteria from the gut to the lung also may be indirect, as GI bacteria may be taken up into macrophages and dendritic cells that then may migrate to the lung [92]. The consequences of lacking microbiota, as seen in germ-free mice who are born and raised under sterile conditions, include underdeveloped gut mucosa-associated lymphoid tissue and dysfunctional immune responses [93]. That the gut microbiome plays a key role in the pathogenesis of early-onset asthma is supported also by the evidence that certain interventions, which affect maturation of the gut microbiome, are associated with atopy and asthma in pre-school and school-age children [94]. These include perinatal factors such as mode of delivery (Caesarean section) and exposure to antibiotics, while breastfeeding is associated with decreased risk [95,96,97]. These factors have been shown to affect the trajectory of gut microbiota development over the first two years of life [98]. Bokulich et al. [98] found that Cesarean section led to depleted Bacteroidetes populations in infants, altering establishment by maternal-derived bacteria that otherwise would occur via vaginal delivery. Antibiotic administration significantly suppressed Clostridiales, including Lachnospiraceae, which include species that produce butyrate and other short-chain fatty acids that regulate host immunity. As alluded to, the mechanisms through which gut commensal bacteria shape local and peripheral immune responses involve a multitude of pathways [99, 100]. The role of vitamin D in immune function [101] and the effects of short-chain fatty acids (SCFA) [97, 102] and biogenic amines, both of which are produced by certain gut bacteria [103,104,105], all may play a role in asthma pathogenesis.
A number of recent studies from large birth cohorts have investigated more specifically the relationships between infant gut microbiota composition and markers of atopy, asthma incidence or prevalence in later childhood [106,107,108,109,110]. Earlier culture-based studies provided the initial evidence that fecal prevalence of specific bacterial species differed between children who did or did not go on to develop atopy. For example, Kalliomaki et al. [111] observed a reduced ratio of Bifidobacteria to Clostridia isolated by culture, and in parallel found an overall difference in stool bacterial fatty acid profiles at age 3 weeks between infants who had evidence of atopy versus those who did not at age 12 months. Such differences observed from samples collected in very early life (within the first 1–3 months) may seem surprising, but similar observations have been made from other birth cohorts. For example, in a study of 319 infants enrolled in the Canadian Healthy Infant Longitudinal Development (CHILD) Study, Arrieta et al. [112] performed 16S rRNA gene sequencing of fecal samples collected at age 3 months and found decreased fecal prevalence of four bacterial genera members (Lachnospira, Veillonella, Faecalibacterium, Rothia) in children who were at increased risk for asthma. Infants at increased risk also had reduced fecal LPS concentration, as suggested by in silico analysis of predicted bacterial gene functions, and also reduced fecal levels of the SCFA acetate. Intriguingly, inoculation of germ-free mice with primary isolates from these four bacterial taxa ameliorated airway inflammation in adult progeny, demonstrating a causal role for members of these bacterial genera in allergic airway disease.
The exact “critical window” of microbiota–host interactions that sets a risk trajectory towards early atopic asthma remains to be firmly established. Other birth cohort studies have examined the gut microbiome at later time points (up to 1 year), finding relationships to subsequent asthma or atopy risk even in samples collected later in the first year of life [109, 110]. Among 690 infants in the Copenhagen Birth Cohort [110] (fecal samples analyzed at ages one week, one month and one year), the 1-year samples were compositionally distinct from the early time points. Yet, of the two clusters defining microbiota differences in the 1-year samples, this distinction was most apparent among infants born to an asthmatic mother. Moreover, it was within this group of at-risk infants (i.e. asthmatic mothers) that associations with asthma at age 5 were characterized by differences in the relative abundance of Veillonella, Lachnospiraceae incertae sedis, Bifidobacterium, Alistipes and Ruminococcus, and other bacteria. No significant links to later asthma with the 1-year gut microbiota composition were observed among the infants born to non-asthmatic mothers. The noted interaction between gut microbial community “type” and a family history of asthma highlights the intersecting factors that add complexity to understanding mechanisms that result in increased asthma risk.
Beyond bacteria, the role of fungal communities (also referred to as mycobiota) is of tremendous interest. However, human investigations focusing on this and in the context of pediatric asthma remain sparse. Given the dynamics of the gut microbiome in early life, along with established knowledge regarding immune responses to fungi, it is reasonable to suspect fungi play a role in asthma. Direct interactions between fungi and bacteria also may be important, but much remains unknown about inter-kingdom interactions and their role in human diseases. However, data from mouse models have shown, broadly, that fungi play a role in type 2 inflammatory responses. For example, oral treatment of mice with antifungal drugs results in restructuring of their gut mycobiota (reduced Candida, increased Aspergillus and other fungal species) and in a mouse model of asthma, led to increased allergic lung inflammation characterized by eosinophil infiltration, and increased type 2 immune responses measured in blood [113]. Mice whose intestinal tracts were newly colonized with Candida albicans displayed not only fungal-specific Th17 responses, but also increased susceptibility to allergic airway inflammation [114]. Studies from human birth cohorts have reported differences in the relative abundances of specific fungi as a feature of atopy- and asthma-associated gut dysbiosis [109, 112]. In a U.S. cohort [109] clustering of bacterial and fungal community data revealed a cluster characterized by low relative abundance of Bifidobacteria, Akkermansia, and Faecalibacterium and a higher relative abundance of Candida and Rhodotorula fungi. Infants in this cluster had the highest risk of atopic sensitization to aeroallergens at age 2.
The Nasopharyngeal Microbiome and Early-Life Asthma
To date, much of the literature studying the role of the microbiome in childhood asthma has focused on the environment and trajectories of gut microbiota establishment and succession. There has been recent interest as well in the upper respiratory tract (URT) microbiome, in particular the nasopharyngeal (NP) compartment. Several types of sampling approaches have been applied to collect samples from the URT, including nasal swab, nasal aspirate, and hypopharyngeal swabs via the oral cavity [115,116,117,118,119,120,121]. Despite differences in sample collection, studies utilizing any of these specimen types have identified links between URT bacterial microbiota and the development of childhood asthma. Like the gut, the composition of nasopharyngeal bacteria is dynamic in the first few weeks to months of life, even in healthy infants [115, 117]. Similar dynamism has been described for bacteria profiled from hypopharyngeal aspirate samples taken in the first 3 months of life [116].
In a study of 112 infants sampled frequently in the first year of life, factors differentially associated with NP bacterial composition included mode of delivery, infant feeding, crowding, and recent antibiotic use [115]. In contrast to the lower respiratory tract, Corynebacterium and Dolosigranulum are more prevalent members of the NP microbiome, and evidence suggests they are associated with healthy states. Bosch et al. [115] observed that children experiencing more respiratory tract infections (RTIs) in the first year of life already displayed an aberrant microbiota developmental trajectory at age one month, compared to children experiencing fewer or no RTIs. The observed alterations involved decreased stability of the NP microbial community over time, reduction in Corynebacterium and Dolosigranulum, and early enrichment in Moraxella.
Viruses as a cause of RTIs (e.g. rhinovirus and respiratory syncytial virus; RV and RSV) are an important risk factor for childhood asthma [122]. Thus, studies examining longitudinal relationships between viral RTIs, changes in URT microbiota (nasal or hypopharyngeal) and asthma outcomes, are of great interest to elucidate the potential role that airway bacteria may play in modulating asthma risk. In a multicenter cohort of infants hospitalized with RSV-induced bronchiolitis, delayed clearance of RSV (defined by the same RSV subtype identified three weeks later) was associated with a Haemophilus-dominant NP microbiome at the time of initial hospitalization [119]. This suggests that an individual’s existing NP microbiota pattern may play a role in determining the severity or outcome of viral RTIs. In a study of 234 infants from an Australian birth cohort, Teo et al. observed that NP bacterial profiles defined by predominance of Moraxella, Streptococcus, Haemophilus were significantly associated with acute viral RTIs [121]. Intriguingly, shifts in NP bacterial composition were detected in samples obtained preceding RTIs. Moreover, the consequences of having an RTI-associated NP bacterial profile (i.e. defined by Moraxella, Streptococcus or Haemophilus) differed by atopic status. Atopic children were more likely to have a “persistent wheeze” phenotype by age 5 in contrast to non-atopic children with the same NP microbiota profile. These observations suggest that allergic state modifies the outcome of viral RTIs coupled to altered NP bacterial composition.
Mechanistic links between dysbiosis of the upper airway microbiome and childhood asthma are not fully understood. In the COPSAC birth cohort, higher relative abundances of Veillonella and Prevotella in hypopharyngeal aspirates collected at age one month were associated with asthma by age 6 years and associated with reduced TNF-α and IL-1β and increased CCL2 and CCL17 in nasal epithelial lining fluid, markers of both Type 1 and Type 2-related immune responses [123]. Another recent study from this cohort observed associations between airway bacterial richness at age 1 week and allergic rhinitis at age 6 years, which was mediated by an epigenetic signature correlating with expression of genes for lysosome and bacterial invasion of epithelial cell pathways [124]. More research is needed to dissect causal relationships between altered airway microbiota, acute viral RTIs, and the associated immune responses to understand how these factors intersect and temporally influence asthma risk.
Lastly, whether differences in the NP microbiome may modulate asthma outcomes in older children has been examined in several recent studies. Zhou et al. analyzed nasal blow samples from 214 children (mean age 8 years) to determine if bacterial composition changed at the onset of loss of asthma control and whether particular microbiota characteristics associated with the number of these events over the course of one year [120]. Children whose nasal microbiota was dominated by Corynebacterium and Dolosigranulum experienced the lowest number of events. Furthermore, shifts to a Moraxella-dominated nasal microbiota detected at the onset of the event was associated with greater likelihood of progressing to a severe asthma exacerbation. Similar observations related to Moraxella-dominant nasal microbiota and risk of exacerbations were seen in a study of 413 children between the ages of 6 and 17 years [125]. Lung function and bronchial hyper-reactivity have also been associated with greater NP relative abundance of Streptococcus and Staphylococcus, respectively, among older children with asthma [118].
The Microbiome in Adult Asthma
Exploration of the lower airway microbiome in adults with asthma follows from the studies outlined above in early childhood asthma. Organisms introduced early in life might provoke inflammation and injury, or limit the responses to that inflammation, such that over time repeated microbial exposure or periodic insult could lead to an on-going inflammatory state that would be locked-in by early adult life. This dysbiotic theory could include not just bacteria but also viral infection, and could explain how repeated infection, overt or sub-clinical, would serve as an effector that over time could lead stimulate worsening inflammation and airway damage in adult asthma. This could be combined with the insults from other environmental stimuli such as allergens and pollutants, and indeed such multiple stimuli would work in concert or synergistically. Alternately, inflammation and injury from these other sources could over time alter the ecological space in the lower airway in a way that ordinary, commensal organisms seen in very low biomass could no longer survive, to be replaced (or augmented and supplemented) by new phyla and genera. This lung disease theory would suggest that for the most part, the microbiome was more of a reactor or even an innocent bystander, being acted on rather than acting to change the airway micro-environment. Of course, both mechanisms could be operative as a synergistic theory of on-going and mutually reinforcing airway injury, such that the microbiome is both effector and reactor. Finally, there are clearly a certain percentage of adults who lack any evidence of asthma in childhood, who lack atopy, who nevertheless have asthma [126,127,128]. Asthma in these patients is clearly heterogenous in nature and due to several phenotypes [128, 129]. A de novo dysbiosis of the lower airway microbiome might explain how these patients developed airway inflammation and clinical symptoms, and as in children, it is also possible that the microbiome might be a reactor or bystander. With this in mind, over the past decade there have been a number of studies that have examined the association of the airway microbiome and asthma in adults.
One early study used endobronchial brushes in a small cohort of subjects to demonstrate an increased relative abundance of Proteobacteria, particularly Haemophilus, and a decreased abundance of Prevotella in adult patients with either asthma or COPD compared to control subjects [130]. Millares et al. [13] examined 13 patients with severe asthma; this study did not include a control cohort, but did demonstrate a significant abundance of Streptococcus and Prevotella in bronchial biopsies. Goleva et al. [131] demonstrated that the microbiome present in bronchoalveolar lavage fluid of control subjects and subjects with either corticosteroid “resistant” or “sensitive” asthma differed modestly in relative abundance of selected genera, though there were no differences at the phylum level between asthmatic patients and normal subjects. Further, the overall bacterial burden was low. Another small study demonstrated in a cohort of 10 control subjects and 10 subjects with mild asthma that three major phyla, Firmicutes, Actinobacteria, and Proteobacteria, accounted for over 90% of total 16S rRNA sequences profiled from the sputum supernatants of subjects with mild asthma. Here again, Proteobacteria were significantly enriched compared to microbial communities in the sputum of control subjects [132]. These data suggested that the lower airway microbiome could indeed differ in asthma. However, differences in sample collection and sample location within the lung, varying phenotypes of asthmatic subjects, and differing use of medications, particularly inhaled and oral corticosteroids, were likely significant confounders.
Larger studies in time confirmed first that the airway microbiome of patients with asthma were different, if modestly so, from that of the normal airway microbiome, and began to address the more obvious confounders. Using brush samples previously collected in the Macrolides in Asthma (MIA) study, Huang et al. [133] demonstrated higher 16S rRNA amplicon concentrations and diversity in endobronchial brushings obtained from asthmatic patients versus healthy controls that correlated with bronchial hyperresponsiveness. This study was large, 65 adults with sub-optimally controlled, mild to moderate asthma, and contained a small control cohort. Many but not all subjects had evidence of 16S rRNA in their endobronchial brushes, and not all of these could be amplified. Of the 42 asthmatic and 5 control subjects with sufficient product, a clear difference in bacterial burden could be identified. While the normal airway was not sterile, the asthmatic airway had a greater bacterial burden. Going beyond a taxonomic approach, the study demonstrated that the degree of bronchial hyperreactivity to methacholine correlated not to the relative abundance of any genus or phylum, but rather to the overall community diversity of the bacterial population. This approach emphasized that the overall ecological community perhaps matters more than simply an “over” or “under” abundance of a single bacteria. This work was among the first to demonstrate a relation of bacterial burden and community diversity to a physiologic parameter important to asthma.
A follow-on study then examined patients with more severe asthma, collected from the Bronchoscopic Exploratory Research Study of Biomarkers in Corticosteroid-refractory Asthma (BOBCAT) study [134]. The ability to examine larger populations was facilitated by the ability to “tag on” to previous sample collections, illustrating the importance of biospecimen collections in asthma studies, while also imposing limitations as to how samples may have been collected, processed and stored. In this study, patients with severe asthma had differences in bacterial composition of endobronchial brushes based on body mass index, assessment in asthma symptom control, the number of sputum neutrophils, and the number of eosinophils in bronchial biopsies. These bacterial communities did not diverge from normal in the same way: for example, microbial communities associated with poor symptom control and sputum neutrophils were predominant for Proteobacteria, whereas patients with a higher body mass index had an enrichment in airway microbiota for Bacteroidetes and Firmicutes. While it was difficult with a smaller cohort to examine differences in phenotypes, expression of several Th-17 genes in airway epithelial cells was associated with Proteobacteria dominance in the microbiome. Finally, the airway dysbiosis in patients with severe asthma appeared to differ from that noted in subjects with milder asthma who were using inhaled corticosteroids.
Other asthma cohorts also focused on relating asthma clinical parameters to the microbiome. Denner et al. [18] related the abundance of select taxa to corticosteroid use and to pulmonary function, specifically FEV1. Using endobronchial brushings, they found that Lactobacillus, Pseudomonas, and Rickettsia were significantly enriched in samples from asthmatic patients, whereas Prevotella, Streptococcus, and Veillonella were enriched in brush samples from control subjects. In this regard their control data agreed with that of Charlson and other studies with a normal cohort, increasing our confidence that such organisms may be considered, in at least most healthy people, as commensal. Further, Denner et al. found that Pseudomonas was in greater abundance in patients receiving oral corticosteroids and with a lower FEV1 – that is to say, in patients with more severe asthma. Zhang et al. [25] using induced sputum also were able to demonstrate an increased prevalence of Pseudomonas in patients with severe asthma compared to controls. Li et al. [135] demonstrated a higher abundance of Pseudomonadaceae in severe asthmatic subjects compared to those with milder disease and with control subjects. Other studies using sputum or brushes have not replicated the increased relative abundance of Pseudomonas but have instead observed enrichment of genera such as Neisseria and Moraxella [130, 132, 136, 137], illustrating the importance of method, sample collection, and patient population. Further, not every study finds a difference between healthy subjects and patients with asthma, particularly if the disease is mild enough that patients are not being treated with inhaled corticosteroids [138]. Indeed, in studies in which patients with differing disease severity are included, those with mild disease generally have few differences from normal [18, 137], though one recent paper has demonstrated association of both sputum and oral microbiota to immunologic features such as atopy status and the presence/absence of a T2 phenotype in subjects with mild asthma [136]. It is clear that patients with more severe asthma, particularly those with frequent exacerbations or requiring the use of high-dose inhaled or oral corticosteroids, have a more disordered airway microbiome.
Among the ecological markers of a microbial community is the diversity of the community, both within a defined group of subjects (alpha-diversity) and between groups (beta-diversity). In many chronic illnesses in which the microbiome has been examined (as one example, Crohn’s disease [139]), alpha-diversity is decreased in patients with illness compared to control – that is to say, there are fewer different types of bacteria in the ecological space, reflecting an ecological collapse that may be related to the disease state and the consequences of the two-way response of host and microbial community. Studies to date in asthma have been mixed with regard to changes in diversity. Two fairly early studies demonstrated a higher diversity in asthmatic patients compared to healthy controls [132, 133], whereas other studies found no significant changes between these groups [135, 138, 140]. The Denner study noted decreased diversity in asthma patients versus healthy subjects as measured by the Shannon alpha-diversity index [18], while the AsthmaNet microbiome study found that diversity as measured by Faith’s phylogenetic diversity index, a phylogeny-based measure of biodiversity, was increased in asthmatic patients versus healthy subjects [137]. Other recent studies have demonstrated lower alpha-diversity indices in patients with severe asthma versus milder disease; these studies have been larger and have had a greater proportion of asthma patients with severe disease [18, 26, 141]. The weight of the evidence currently suggests that in asthma, as in other chronic illness, microbial diversity decreases, though this may be difficult to demonstrate in patients with mild airways disease. Moreover, there are many ways of examining and comparing microbial diversity, and the inconsistencies between the above noted studies may reflect this. These measures represent today our best efforts to understand what shapes differences between groups of patients.
Longitudinal Studies
The study of the airway microbiome in adult asthma has been limited by the lack of longitudinal studies. Several studies have been done in children with asthma via sampling of the nasopharynx, which as previously noted may not reflect the lower airways. These studies have identified links between URT bacterial microbiota and the subsequent development of childhood asthma [115,116,117,118,119,120,121]. Similar studies have not been done in adults to date; we therefore do not know the time-associated changes in the asthmatic microbiome. Indeed, we do not even know the dynamic changes that occur in the normal lower airway microbiome and how these might be influenced by the environment and diet. It is well established that the GI microbiome is highly variable in early life and is great influenced on a daily basis by diet, environment, and the use of antibiotics [142,143,144,145]. Tantalizing clues summarized in following sections suggest that the environment and antibiotics may alter the airway microbiome as well, and that management of such alterations might well improve asthma control. To date, however, the longitudinal human studies, greatly needed, are lacking.
In summary, the airway adult microbiome is different in patients with asthma. After consideration of all the differences in study methods, sampling locations, sequencing methods, and varying patient populations and disease severity, both the taxonomy and diversity of the asthmatic adult microbiome differs somewhat from health and changes more as disease state worsens. What we cannot demonstrate yet is a distinct profile that uniquely and near-irrevocably makes clear that, given this microbiome, this patient must have asthma. Simply put: there are no differences in kind, only in degree.
The GI Microbiome in Adult Asthma
As previously discussed, the GI microbiome clearly has a modulatory and very likely contributory role in the development of early-life, childhood asthma. It is becoming clearer that the GI microbiome may also have an on-going role in adult asthma, by introduction of organisms to the lung via aspiration, by production of immunomodulatory factors, by alterations in the function of immune cells, all of which then lead to changes in airway inflammation. The relationships between the GI microbiota, their products and immunomodulatory signals, and the lung (i.e. the gut-lung axis) could extend, regulate and exacerbate T2-high responses in asthma, and may also have a role in Th17-driven asthma [146, 147]. Metabolites from GI microbiota also could influence T cell plasticity and function and dendritic cell function.
Current estimates suggest that the adult gut contains approximately 1014 bacteria, perhaps 7 to 8 log-fold more than the lung; two-thirds of these are specific to an individual [148, 149]. Four phyla, Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria predominate in the GI microbiome as in the lung. Genera such as Clostridium, Faecalibacterium, Ruminococcus, Roseburia, Eubacterium, Bifidobacterium, Prevotella, and Bacteroides are dominant in the normal intestinal microbiome [149]; many of these are found in low abundance if at all in the normal lung microbiome. Under normal circumstances the predominant GI bacteria both prevent the growth and aggression of harmful and pathogenic microbiota and participate in a number of beneficial immune modulating functions [149, 150]. As one example, they aid in digesting food products by fermenting complex carbohydrates that then produces SCFA that regulate inflammation and allergic responses [151,152,153]. A dysbiotic GI microbiome with a disrupted ecology not only leads to intestinal inflammation and dysfunction but also to worsened allergic inflammation and responses elsewhere [151]. A number of factors, including age, diet and fiber, childbirth, antibiotic ingestion, and intestinal disease all can lead to a GI microbiome that is temporarily or permanently dysbiotic [149, 151].
Our understanding of the relative contributions of the GI and lung microbiomes in adult asthma is evolving. The gut-lung axis is best considered as a transfer of metabolites, immune cells and immunomodulatory signalers from the gut to the lung (though reverse transfer could also occur), such that changes in gut microbial ecology or a gut dysbiosis could influence adult respiratory diseases including asthma [154,155,156]. A recent review examines the potential immune regulators that may be generated by a microbiome [153]. Many studies that examine this axis focus on acute infection models (e.g., influenza, pneumonia, mycobacteria) [152, 157,158,159,160], and the literature regarding early-life interactions was summarized above. Both the GI and lung microbiomes may be modulated by allergens (inhaled and/or swallowed) that both directly alter the respective barrier function in each system and elicit immune cell activation. House dust mite antigen, long known to be an allergic irritant in the lungs and indeed used in mouse models of T2-mediated allergic lung inflammation, also impairs the barrier function of the intestine upon ingestion [161]; likewise, particulate matter found in air pollution not only can elicit airway inflammation but also colonic epithelial inflammation [162]. A “leaky” barrier may allow ingress of bacteria; this has been demonstrated in clinical situations such as the adult respiratory distress syndrome [163] but such transfer has not been demonstrated in chronic inflammatory lung diseases. Intestinal epithelial cells and immune cells may assimilate signals directly from the directly abutting or nearby GI microbiome in ways that both shape a local response and a response at distal sites, including the lung [164]. As one example relevant to asthma, certain Bacteroides species that can synthesize polysaccharide A (PSA), introduced into germ-free mice, elicit a higher number of circulating IL-10 producing CD4+ T cells and Th1 cells compared to non-PSA synthesizing species [165]; this might drive, for example, T2-low asthma. In a mouse model of allergic airways disease that results from administration of antibiotics and disruption of the GI microbiome followed by ovalbumin sensitization and challenge, introduction of Candida albicans to the gut elicits a greater Th2-mediated inflammatory airway response [166]. In a study of young and old mice that mimics the effect of aging, older mice challenged with house dust mite allergen had greater airway inflammation, and had a different GI microbial structure with a decrease in the ratio of Bacteroidetes to Firmicutes, compared to similarly-challenged young mice [167]. The combination of GI microbiome manipulations and allergen airway challenge, or lung infection challenge, in animal models is a particularly useful model that may allow us to understand the interplay of the gut-lung axis on airway inflammation, particularly in longitudinal models.
Studies of the gut-lung axis in adults humans with asthma are few to date. A recent interesting pilot study has demonstrated differences in the gut bacterial community structure in a group of adults with mild-to-moderate asthma compared to control subjects without known lung disease [168]. The gut microbiome within each subject was stable in the absence of changes in asthma status, while there was a strong association between FEV1 and differences in bacterial composition at the phylum level with changes in both Bacteroidetes and Firmicutes and a lower B/F ratio in asthmatic subjects. Future studies will need to examine both gut microbial parameters and circulating mediators that may be secreted by GI immune cells provoked by the gut microbiome, and then sample (by bronchoscopy if possible) changes in local lung inflammation, particularly T cell phenotypes and the presence of cytokines released by different T-helper cell populations. These more mechanistic clinical studies then may help delineate the role of the gut-lung axis in asthma.
Asthma Phenotypes and the Airway Microbiome
Of particular importance among adult asthma microbiome studies done to date have been those relating the microbiome to biological markers of a particular phenotype.
Key Asthma Phenotypes and Endotypes
A complete review of the state of knowledge of asthma phenotypes is beyond the scope of this chapter, and readers are referred to excellent, recent reviews of this topic [169,170,171]. Heterogeneity in asthma and airway inflammation was understood over a century ago [172], and it is clear today that asthma is a heterogenous and complex disease with genetic, environmental, immunological, and behavioral inputs that cannot be explained by one single pathophysiologic mechanism. Over the past two decades, our understanding of asthma has been guided by the T2-inflammation hypothesis that provides an organizing immunologic and molecular framework for the fundamental and well-known associations of atopy, early life exposures, and eosinophilic airway inflammation. Both CD4+ helper T lymphocytes (Th2) and innate lymphoid cells (ILC2) [173] generated as a response to type-2 inflammation release signaling cytokines (such as IL-4, IL-5, and IL-13) and chemokines that drive a particular type of airway inflammation notable for eosinophilic infiltration of the mucosa and submucosa [174, 175]. This idea is further refined by the recognition of the endotype, that is, a condition or phenotype that is defined by a distinct functional or pathobiological mechanism [176], that might be defined (at least somewhat) by statistical clustering or big-data approaches, the use of molecular or genetic signatures, and the response to biological therapies, showing an end-result, real-world usefulness of the defined endotype [177]. The T2-high endotype, driven by eosinophilic inflammation that is signaled by T2-related cells that are in turn induced by epithelial cell, viral, and allergic/atopic stimulation [178], and is responsive to anti-IL-4 and anti-IL-5 therapies, is perhaps the best understood of the asthma syndromes and accounts for perhaps one-fourth of all patients with asthma. The T2-low phenotypes, best understood as “not T2-high”, clearly are heterogenous, varied, and not well separated from each other. A neutrophil-dominant asthma phenotype characterized by high proportion of sputum neutrophils, perhaps driven by cytokines such as IL-17 and IL-22, and mixed neutrophil-eosinophil phenotypes, are part of this T2-low paradigm [179,180,181,182,183]. Clearly microbial (bacterial or fungal) products, or the organisms themselves, could be part of the signaling processes in either T2-high or T2-low inflammation.
The T2 Phenotypes and the Microbiome
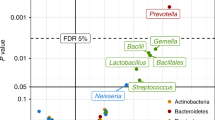
Given the state of asthma phenotypes today, the incorporation of key microbial markers might help improve our ability to define and apply these phenotypes to patient care. Studies to date have attempted to examine certain phenotype markers, generally dividing their cohort based on one or a couple of key clinical or biomarkers, and then describing differences in the microbiome. Again, these are generally cross-sectional studies with small cohorts compared to the typical sizes of phenotype-driven biological drug clinical trials in asthma, so the ability to refine these findings into actionable hypotheses are modest. One early study was that of Li et al. [135] in which the sputum microbiome was examined in a cohort of mild and severe asthma patients divided, for phenotype purposes, based on a sputum eosinophil count >3% and sputum neutrophil count >61%. Select bacterial families, including Actinomycetaceae and Enterobacteriaceae, were more abundant in patients with the eosinophilic inflammatory phenotype. In a study of 23 patients with corticosteroid-free (and thus mild) asthma compared to 10 healthy controls by bronchoscopy, asthma patients with low endobronchial eosinophils had decreased alpha-diversity and increased beta-diversity compared to both those asthma patients with high eosinophils and the healthy controls [184]. Several genera were significantly depleted (e.g., Aeribacillus, Halomonas, and Sphingomonas) or enriched (e.g., Actinomyces, Bacteroides, and Neisseria) in eosinophil-low versus eosinophil-high patients. A recent related study from China examined “non-eosinophilic” versus eosinophilic asthma and demonstrated decreased alpha-diversity in the former [141]. The first AsthmaNet Microbiome study [137] found that asthma patients defined as T2-high on the basis of gene expression in epithelial cells collected on endobronchial brushings had a significantly lower bacterial burden compared to patients defined as T2-low on the absence of this expression (Fig. 5.1). In contrast, the BOBCAT trial failed to demonstrate an association between T2-high related genes expressed in epithelial cells and either microbial taxa or diversity [134]. On balance, patients with T2-high asthma may have select changes in their airway microbiome that might be useful in defining further the phenotype, though further explorations between the microbiome and gene signatures in larger studies would be welcome.

The relationship of asthma, phenotype and atopy with bacterial load from the Asthma Microbiome I study. Figure (a) demonstrates the distribution of patients with asthma and atopy (AA), patients with asthma but no atopy (ANA), and healthy controls based on a gene scoring system using endobronchial brush samples. Using this, for the patients who are considered to be of the T2-high asthma phenotype (above the dashed line in (a) and to the left in (b), bacterial load is substantially lower than patients who are T2-low phenotype. With further validation, one could foresee that bacterial load could be used to help predict a correct phenotype for a patient with asthma: if high, the patient may have T2-low asthma. From reference (137) with permission
Patients with T2-low, neutrophilic asthma, defined in part as a ≥ 60% proportion of neutrophils in sputum, constitute a separate asthma phenotype. Defined generally by the absence of T2-high markers and by the presence of a higher proportion of sputum neutrophils, this phenotype incorporates about 60 to 75% of patients with asthma and is clearly heterogeneous [129, 185, 186]. This phenotype is said to be associated with more severe asthma and a poor response to corticosteroid therapy [187,188,189] (though the astute asthma clinician can readily find both milder cases in this phenotype and patients with T2-high asthma who are dependent on oral corticosteroids). Neutrophilic T2-low asthma may include patients with a “Th-17” phenotype, as higher concentrations of cytokines from Th-17 lymphocytes, such as IL-17A, IL-22, Il-23, TNF-α, and IL-8, can drive neutrophilic-predominant asthma [129, 179,180,181,182,183, 186, 188, 190,191,192,193], though a recent review questions the directionality of the association and suggests that IL-17 could be protective in asthma [194]. Examination of the airway microbiome in these patients generally demonstrates lower bacterial diversity and higher dissimilarity compared to those with eosinophilic asthma [146, 184, 187]. An early study from Wood et al. [195] examined airway neutrophilia in patients with asthma; patients with higher sputum neutrophil counts also had a higher load of potentially pathogenic bacteria as defined by culture. Likewise, in pre-school children with persistent wheezing who underwent bronchoscopy and BAL, those with peripheral airway neutrophilia (81% of all children), and a majority of these had elevated bacterial counts [196]. These studies suggested that neutrophilia and infection were linked in at least some cases of asthma but of course could not suggest directionality. In more recent studies employing NGS, such associations have been demonstrated in more detail. One study examined patients with either severe asthma or moderate to severe COPD seen at the time of exacerbation and separated into clusters based on factor analysis of sputum mediators. In both diseases, patients with neutrophilic predominance by sputum and the presence of mediators such as Il-1β, IL-6, and TNF-α had increased proportions of Proteobacteria, whereas patients with eosinophilic predominance and the presence of IL-5, IL-13, and CCL26 had increased proportions of Bacteroidetes [197]. The study derived from the BOBCAT cohort demonstrated a positive correlation between Th17-associated genes in airway epithelium and several microbial taxa, particularly the increased abundance of Proteobacteria and that of families such as Pasteurellaceae, Enterobacteriaceae, and Bacillaceae [134]. Taylor et al. [187] showed that select genera, such as Gemella, Rothia, and Streptococcus, were decreased, as was alpha-diversity, in patients with neutrophilic versus eosinophilic asthma. In these patients, there was an inverse correlation between phylogenetic diversity and the proportion of sputum neutrophils. Yang et al. [146] demonstrated that patients with neutrophilic asthma had a higher bacterial burden that had less community richness and diversity, and had a taxonomic distribution that was distinct with an increased relative abundance of both Haemophilus and Moraxella, compared to patients with nonneutrophilic asthma. The patients with neutrophilic asthma also had higher concentrations of mediators such as IL-6, IL-8, IL-17A, and TNF-α that could drive increased airway neutrophilia.
These associations support the idea, yet to be proven conclusively, that patients with neutrophil-predominant, T2-low asthma have a dysbiotic microbiome that may drive the airway neutrophilia and contribute to the pathogenesis of this asthma phenotype. Infection and allergic inflammation may coexist, of course, and there are patients with both neutrophilic and eosinophilic asthma – a phenotype that is labeled as a “mixed” phenotype [128, 170, 181, 198]. The bacterial load, as defined using 16S rRNA gene copy numbers, is similar in the sputum of patients labeled with the mixed phenotype compared to the neutrophilic phenotype, and both are higher than that seen in patients who are “paucigranulocytic,” with neither neutrophils nor eosinophils in sputum [146]. One interesting potential mechanistic explanation for this may be that allergic inflammation may promote bacterial persistence. Evidence for this comes from work by Essilfie et al. [199] in which mice were infected with live or killed H. influenzae and then sensitized and challenged with ovalbumin or placebo in a standard allergic inflammation model. This combination led to “persistent” airway inflammation, present 26 and 31 days after infection, while in an OVA-model alone inflammation typically has resolved. H. influenzae load was greater in the OVA-treated mice, and conversely, H. influenzae treatment suppressed some features of eosinophilic airways disease. More intriguingly, chronic H. influenzae infection combined with allergen challenge induced clear steroid resistance. A more recent study employing a similar mouse model with H. influenzae and OVA challenge demonstrated a similar induction of corticosteroid resistance which was accompanied by defects in regulatory T cell (Treg) associated immunosuppression, airway remodeling, and goblet cell hyperplasia [200]. Taken together, these data clearly suggested that the combination of infection and allergen challenge promoted more chronic infection, changed the nature of the allergic airway inflammation, and elicited glucocorticoid resistance. The “mixed” phenotype that combines features of T2-high and T2-low (or perhaps better said, T2-high and T17-driven) then could be influenced in part by the lung microbiome.
One recent paper of potential interest to the asthma microbiome community examined the ability to stratify risk for COPD exacerbations by assessing the ratio of the relative abundance of Gammaproteobacteria and Firmicutes in serial sputum samples collected at the time of exacerbation. This “G/F ratio” revealed three separate groups of patients by cluster analysis; one group designated “HG” with a predominance of Gammaproteobacteria had a G/F ratio that correlated positively with increases in select inflammatory markers such as C-reactive protein and IL-1β over baseline, and negatively with FEV1 [201]. This study shows the potential of using the microbiome as a diagnostic tool to identify patients who then might be treated appropriately (or at least, differently). Such studies are very much needed in asthma.
Taken together, measures of airway bacterial burden, diversity or specific compositional features may help sharpen the distinction between T2-high and T2-low asthma phenotypes. However, the specific use of microbial markers in this regard, and their combination with inflammatory markers such as blood eosinophils, FeNO, and sputum cell counts or mediators, has not yet been formally done. Further, a dysbiotic microbiome may contribute to inflammatory changes and perhaps clinical outcomes, particularly with regard to the T2-low phenotype variant that is neutrophilic asthma. Larger, longitudinal studies that incorporate appropriate markers and microbiome analysis will be needed, as will more mechanistic studies in appropriate animal models. In particular, the question of whether dysbiosis drives neutrophilic asthma or whether neutrophilic asthma creates an ecological niche in which select microbiota can thrive, needs to be addressed.
The Obesity-Asthma Phenotype and the Microbiome
One T2-low phenotype that at least somewhat separates from other phenotypes, particularly for severe and exacerbation-prone asthma, is that associated with obesity. Several epidemiological studies have suggested that obesity predisposes to asthma [202,203,204,205]. The effects of obesity on asthma prevalence [206,207,208], severity [209, 210] and response to treatment [208], including bariatric surgery [211], are complex. Obesity influences several asthma phenotypes [169, 208, 212] and response to asthma controller therapies [213,214,215]. Whether obesity causes a distinct asthma phenotype or whether it simply worsens pre-existing disease, either by changes in lung mechanics or by a change in airway inflammation, has been controversial, but growing evidence suggests that inflammation and oxidative stress may link obesity with asthma [215,216,217,218,219,220,221]. The adipocyte secreted hormones adiponectin (in its high molecular weight form, HMW-APN) and leptin are key regulators in long-term body weight, energy homeostasis, and fatty acid oxidation. Adiponectin-deficient mice have increased inflammatory cell infiltration in airways after allergen challenge [222]; a similar effect is seen after ozone exposure that is reversed with adiponectin expression [223, 224]. Leptin is pro-inflammatory, and increases in leptin levels are associated with airway hyperreactivity and pro-allergic responses [225, 226]. Higher leptin and lower APN levels are seen in adult asthmatics [227]. Taken together, there is a clear association of adipokines, particularly leptin and APN, with asthma. Obesity associated with asthma declares later in life in adults, particularly in women [205, 208, 228], associates with asthma symptoms and exacerbations, with low expression of T2-high associated biomarkers such as blood eosinophils, serum IgE, and FeNO [210, 211, 229,230,231], and with systemic inflammatory markers such as IL-6 [232, 233] and C-reactive protein [233]. These latter markers and the absence of the T2-high markers suggest a T2-low phenotype.
Few studies have specifically examined the lung microbiome in obesity in general, or specifically obesity and asthma. The previously noted BOBCAT study showed a significant association of obesity (BMI > 30) with a distinct lung microbiome with a higher relative abundance of Bacteroidetes, Firmicutes, Prevotella, and certain Clostridium species, and lower abundance of Proteobacteria [134]. Endobronchial specimens of the obese subjects showed fewer mucosal and submucosal eosinophils compared to the lean subjects that paralleled the changes in the microbiome. Regrettably, larger studies have not yet specifically addressed the airway microbiome in obesity. One recent study examined the fungal microbiome (next section) and included a follow-up analysis of the bacterial microbiome described by Denner et al. [18]. While this analysis did not identify any significant association between BMI and any bacterial taxa, there were positive relationships between predicted functional bacterial pathways, such as galactose metabolism and linoleic acid metabolism, and BMI [234].
The data to date then suggest a potential role for the airway microbiome in obesity-associated asthma. Larger studies that delineate more precisely the relationship between the microbiome and adult patients with the obesity-associated T2-low asthma phenotype would be welcome. Especially important would be longitudinal studies that relate successful weight loss, either medical or surgical, with a change in the lung microbiome and concomitant improvement in asthma control.
Fungal Microbiome in Asthma
The microbiome of any ecological space in the human body will include more than just bacteria. Just as we expect to inhale and aspirate bacteria on a near-continuous basis, we may expect constant exposure of the upper and lower airways to fungi and the development of a fungal mycobiome within the larger microbiome. Fungi are eukaryotes, and the genera most commonly associated with allergy in humans are Alternaria, Aspergillus, Cladosporium, and Penicillium. Fungi produce spores, or conidia, that can remain dormant until they are ready to germinate. Mesophilic fungi such as Alternaria and Cladosporium grow best at temperatures between 20 and 30 °C and thus ordinarily do not germinate in the body; they instead cause respiratory allergies. Thermotolerant fungi such as Aspergillus, Candida, and Penicillium grow well at 37 °C and thus may elicit both allergy and grow well in the lungs, causing infection. This makes fungi unique compared to other allergens (e.g., pollens) and eukaryotes (e.g., house dust mites) that do not grow within the host. Inhalation of fungal spores, their fragments or their secreted products then may elicit lung disease. Larger spores such as Alternaria generally deposit in the upper airways whereas smaller spores such as Aspergillus may reach the small airways. In either case, germination, survival and growth then contribute to the mycobiome.
Fungi may also be found in the GI microbiome and are delivered by oral secretions or by food, and are generally considered to be transient and not colonizing [235]. Dysbiosis of GI fungi may have a role in early-life development of asthma and may be co-associated with bacterial dysbiosis [109, 112]. Mice treated with oral antibiotics that remove bacteria, particularly Lactobacillus, may have a fungal overgrowth that then can elicit an exaggerated response following airway antigen challenge via M2-macrophage polarization [236]. Fungus-free mice in which commensal or dysbiotic fungi are introduced into the GI tract can develop Th2-mediated for the former, and Th17-mediated for the latter immune responses in allergic airway inflammation [237]. In this way, a dysbiotic GI fungal community can influence distant lung immune responses.
Fungi are ubiquitous outdoors [238]; of those that may be important to asthma pathogenesis, Cladosporium and Alternaria are prominent [239]. Fungi can be found indoors in homes and offices in appreciable numbers. Older homes and homes with a “damp” indoor environment have a higher fungal burden, particularly for Aspergillus and Penicillium [240,241,242]. Each of these can be associated with asthma. Dust collected from homes can be rich in fungi and has a distinct fungal microbiome; this richness correlated with the age of the home and the relative humidity as well as with dog ownership [60], may be influenced but by outdoor air, and in contrast to the bacterial microbiome, less influenced by human occupants [60, 243, 244].
As with bacteria, at one point the lung was not considered to harbor fungi under normal conditions, as fungi could not be cultured [245]. It has long been appreciated that fungal sensitization is seen in at least some patients with asthma [246] and that there is a clear association between fungal allergen sensitivity and the presence of asthma and other respiratory diseases [247], particularly with Alternaria and Aspergillus [248,249,250,251]. At one extreme end this is manifested as allergic bronchopulmonary aspergillosis (ABPA), which may be considered as an allergic response to the fungus in the airway. ABPA presents as severe or poorly controlled asthma with a high serum IgE concentration, persistent eosinophilia, and bronchiectasis [252, 253]. These patients will have an elevated Aspergillus-specific IgE, detectable Aspergillus-specific IgG, and a positive skin-prick test for Aspergillus [254, 255]. There are patients with severe asthma who will meet only some of these criteria, or who are sensitized to a fungus other than Aspergillus; such patients may be sensitized (demonstrated by skin-prick testing or by blood allergen tests) to several genera of “environmental” fungi such as Cladosporium, Alternaria, Penicillium, Candida, and Trichophyton that are common in air and soil [256]. The terms “fungal asthma” or “severe asthma with fungal sensitization” (SAFS) have been used in these cases that excludes ABPA [257]. While much attention is correctly paid to indoor mold and fungus exposure [241], particularly during infancy and early-life development [240, 258, 259], airborne outdoor fungi also can trigger asthma exacerbations in children and adolescents, particularly those already sensitized to Cladosporium [74].
Fungal products can promote allergic responses by virtue of being allergens and pathogen-associated molecular pattern (PAMP) molecules. These responses link a dysbiotic mycobiome to the inflammatory changes in asthma. Recent reviews of these products and their mechanisms have been published [260,261,262,263].
A dysbiotic lung mycobiome may be present in human asthma. As sequencing of the ITS1 (internal transcribed spacer 1) region of 18S-rRNA has become available and as reference libraries to interpret this sequencing, and to exclude human and other eukaryotic 18S, have become more complete, one can investigate the presence of the mycobiome in the lung. Van Woerden et al. examined the presence of sequenced fungi in sputum from patients with asthma and normal subjects; the former had a doubled-incidence of mold in the home. Grifola sordulenta, Malassezia pachydermatis, Psathyrella candolleana, Termitomyces clypeatus had a higher relative abundance in the sputum of asthma patients, while Cladosporium cladosporioides, Eremothecium sinecaudum, Systenostrema alba, and Vanderwaltozyma polyspora had a higher relative abundance in the sputum of control subjects. Malassezia pachydermatis is associated with atopic dermatitis [264], suggesting a role in atopy. Bronchoalveolar lavage done in 15 children with severe asthma with and without known fungal sensitization showed an increased abundance of fungal genera such as Rhodosporidium, Pneumocystis, Leucosporidium, and Rhodotorula compared to that seen in 11 normal children [265]. Interestingly, they did not detect an increased prevalence of environmental fungi such as Aspergillus and Alternaria. Bronchoalveolar lavage done in young to middle-aged adults with ABPA, SAFS, asthma without evidence of fungal sensitization, and control subjects noted that the healthy subjects had a low fungal burden with an abundance of Malasezziales [266]. In contrast, asthmatic patients with or without fungal sensitization or ABPA had an increased burden of A. fumigatus complex. The load of this fungus differed little between patients with a current history of itraconazole therapy versus no therapy; whereas patients with past history had a higher load. Both total fungal burden and the load of A. fumigatus complex were higher in those asthma patients receiving oral or inhaled corticosteroids. This study made clear the potential role and burden of Aspergillus in patients with severe asthma, even those without known fungal sensitization or ABPA.
A new study has now examined the potential interactions of the fungal and bacterial microbiomes in asthma with an emphasis on the T2-high phenotype. Sharma et al. [234] analyzed ITS1 sequences in endobronchial brushes and BAL samples from 39 asthmatic subjects separated by T2 and atopy status and 19 control subjects previously reported for their bacterial microbiome [18]. Asthma subjects with markers for T2-high disease had a lower fungal alpha-diversity than T2-low subjects and control subjects, and beta-diversity also differed based on T2 status in endobronchial samples. As with the bacterial microbiome, there is a fungal mycobiome in normal, healthy subjects that is small in biomass. In BAL fluid, the relative abundance of Trichoderma, Alternaria, Cladosporium, and Fusarium species were significantly enriched in asthmatic patients, while Blumeria species, Mycosphaerella species, and different Fusarium species were enriched in healthy control subjects. Differences in endobronchial brushes were more modest, with an increased relative abundance of Penicillium in asthmatic subjects and those with atopy, and Trichoderma was increased in T2-high asthmatic patients. Seven key fungal genera – Alternaria, Aspergillus, Cladosporium, Fusarium, Penicillium, Trichoderma, and Mycosphaerella – were significantly associated with asthma, T2 inflammation, and atopy. The authors then examined co-occurrence networks to examine differential associations between fungal and bacterial taxa. Samples from endobronchial brushes had a greater density of connections that maintained highly connected sets of taxa compared to BAL, and in each sample set, asthmatic patients had greater connections compared to controls. Examples of these are given in Fig. 5.2. From these networks, select fungal genera were associated with select bacterial genera. Although the keystone fungal taxa remained similar between BAL fluid and EB samples, co-occurring bacterial taxa were distinct between the two regions in asthmatic patients. Further, using a random forest model, the authors could identify top discriminatory fungal taxa, particularly Alternaria, Cladosporium, Mycosphaerella, and Aspergillus, that could classify asthmatic and healthy subjects with up to 72% accuracy. This study makes clear that fungal load, in addition to bacterial load here and in the study by Durack et al. [137], and the presence of select fungal and bacterial taxa can help to differentiate T2-high from T2-low asthma. These studies need repetition in larger and more varied patients, but the promise of being able to apply the lung microbiome as part of the delineation of asthma phenotypes and endotypes is becoming increasingly clear.

Significant co-occurrence relationships of fungi and bacteria between different modules in patients with asthma using samples collected from BAL fluid (a) or endobronchial brushes (b). These networks represent statistically significant correlations; a connection stands for a strong (Spearman ρ < 0.6) and significant (P < 0.01) correlation. Fungal nodes are labeled in black inside the network, and bacterial nodes are labeled outside the network in different colors depending on the module to which they belong. Nodes are colored by modules or communities (group of taxa) based on Louvain community detection algorithm. The size of the node is controlled by the number of connections). The edge width is proportional to the weight of correlation. The level of modularity ranges from 8 to 16 for all networks. For genera with more than 1 differentially abundant taxa, the ESV number is shown as a subscript. Co-occurrence relationships could be used to determine which bacteria and fungi might be predicted to be present in the airway microbiome when one or more are detected in a clinical sample, and could be used (for example) as an aid to determining a correct phenotype for a patient with asthma. From reference (234) with permission
Specific Anti-Fungal Therapies in Asthma
In patients with asthma with either ABPA or fungal sensitization, antifungal therapy with itraconazole or similar azoles can be a useful adjunct [267,268,269], and this forms part of the evidence base for the potential role of the mycobiome in asthma, even as Aspergillus, as previously noted, may not be present or even in substantial prevalence in some patients with fungal asthma. Several trials have looked at the utility of these therapies in severe asthma in the absence of clear evidence of ABPA. Denning et al. [257] conducted a randomized, placebo-controlled trial of itraconazole, an agent with a wide spectrum of anti-fungal activity, in 58 adult patients with severe asthma and SAFS. These patients had been receiving or had recently received oral corticosteroids, were skin-prick test positive for one of several fungi, were negative for Aspergillus precipitins (IgG), and had a circulating IgE concentration of less than 1000 IU/ml. Other asthma therapies were optimized before enrollment. Patients receiving anti-fungal therapy had a significant improvement in asthma quality of life scores and a decrease in total IgE concentrations. Interestingly, relapse in symptoms after discontinuation of itraconazole was common. Other azole agents also have noted improvement in quality of life scores for patients with SAFS [270, 271]. A more recent study examined another potent anti-fungal agent, amphotericin B, as nebulized therapy in 21 patients with severe asthma who were labeled either as SAFS or had a diagnosis of ABPA. In contrast to the Denning study, the response rate as measured by quality of life improvement was less than 15% and substantial side-effects, including bronchospasm were noted [272].
Effects of Asthma Medications on the Airway Microbiome
As we begin to consider longitudinal studies of the airway microbiome in asthma in both children and adults, two immediate questions arise: first, how might asthma-related medications change the microbiome for better or for worse, and second, might we introduce medications that would change the microbiome and thereby improve asthma control?
Corticosteroid Therapy and the Airway Microbiome
Inhaled corticosteroids are a mainstay of asthma controller therapy, and oral corticosteroids are near-universally employed in the treatment of severe acute exacerbations. While these agents have no antimicrobial activity per se, inhaled steroid therapy often ameliorates underlying airway inflammation, and this may alter the local microbiome. Early studies of the microbiome in asthma were small and generally included patients receiving inhaled corticosteroids, and therefore it was not possible to separate a steroid-specific effect. Goleva et al. noted no differences between corticosteroid-resistant and corticosteroid-sensitive patients with asthma at the phyla level regarding diversity, richness and taxonomy, but select genera, including Haemophilus, were increased in the steroid-resistant group [131]. Denner et al. demonstrated increased Proteobacteria and decreased Bacteroidetes, and increased Pseudomonas and decreased Prevotella and Veillonella, in patients received inhaled corticosteroids versus those without such therapy [18]. From these studies it becomes clear that the airway microbiome is perhaps different in patients taking inhaled corticosteroids as a controller therapy for asthma. The question then becomes, does the addition of such therapy change diversity or taxonomy? Or is the necessity for use of corticosteroids a marker for more severe underlying disease that is itself responsible for the changes, or do the corticosteroids elicit a host response that then alters the microbiome? In the AsthmaNet Microbiome Study [137], a subset of recruited patients with steroid-naïve asthma received inhaled fluticasone or placebo for 6 weeks, with bronchoscopy and sputum collection before and after therapy. Patients who responded to steroid therapy with an improvement in methacholine responsiveness (ICS responders) were compared to nonresponders; the former had a baseline bacterial microbiome more similar to that of healthy controls with an enrichment of Streptococcaceae, Fusobacteriaceae, and Sphingomonodaceae, whereas nonresponders at baseline were enriched in Microbacteriaceae, Pasteurellaceae, and asthma-associated Haemophilus genera. Microbiome analysis of endobronchial brush samples was limited by insufficient 16S rRNA amplicon in paired samples (pre- and post-intervention) from some subjects. At the taxon level ICS treatment resulted in increased relative abundance of Microbacteriaceae and Neisseria and Moraxella species, and depletion of a specific Fusobacterium, which was not observed with the placebo treatment [137]. Parallel observations including differential changes between the two treatment groups, before and after the interventions, were observed in a subsequent analysis of induced sputum from participants in the same study [136]. Two points can be made here based on the studies conducted to date. First, inhaled corticosteroid therapy in patients with mild asthma and who respond to such therapy clearly have some changes in airway taxonomy. Second, much larger trials are needed if we are to determine better the changes in the microbiome after initiation of corticosteroid therapy, and the interplay of such a treatment effect compared to changes in host inflammation elicited by corticosteroid therapy.
Corticosteroids are potent inhibitors of pro-inflammatory molecules and cells, and it is tempting to suggest that glucocorticoids would also alter expression and production of innate immune responses that generally prevent infection. This in fact does not occur and indeed, corticosteroids generally fail to inhibit the expression of many of the genes involved in innate immunity [273]. In airways, the epithelium produces a number of innate immunity-related proteins that destroy or suppress microorganisms, including complement, collectins, lysozyme, lactoferrin, secretory leukocyte protease inhibitor, and defensins. Defensins, small cationic proteins that are expressed either constitutively or can be induced by various pathogens [274], are regulated by Toll-like receptors (TLR) such as TLR2 [275, 276]. Corticosteroids may enhance Toll-like receptor (TLR) 2 expression, the absence of which leads to an inability to clear organisms such as Mycoplasma and an inability to induce human β-defensin [277, 278]. Another anti-microbial protein is CCL20, also known as MIP-3a, that is similar to defensins. CCL20 is expressed in the airway epithelium and can be induced by bacteria, and is regulated by several TLRs [274, 279, 280]. In cultured airway epithelial cells, treatment with corticosteroids enhances the production of CCL20 [281, 282]. Treatment of airway epithelial cells with budesonide but not fluticasone elicits increased expression of both CCL20 and lactotransferrin [283]. One older study demonstrates that corticosteroids increase secretory leukocyte protease inhibitor transcripts in airway epithelial cells [284].
Counterbalancing the idea that corticosteroid therapy may be benign or even beneficial with reference to the lung microbiome in asthma are data from a number of clinical trials in COPD that inhaled corticosteroid treatment can increase the risk of pneumonia [285,286,287,288,289,290]. In the few studies to date that report asthma patients separately from COPD with regard to pneumonia incidence, little increased pneumonia risk is seen with the use of inhaled corticosteroids in asthma: for example, the START trial (Steroid Treatment As Regular Therapy), with over 7200 patients demonstrated no increased risk of pneumonia in patients treated with budesonide compared to placebo [291]. Two smaller randomized trials of either fluticasone [292] or budesonide demonstrated similar findings [293]. A recent meta-analysis of the risk for pneumonia in patients with asthma has reported little if any increased risk [294]. The mechanisms by which corticosteroids then alter the airway microbiome are not yet clear, but data to date suggest that by preserving and perhaps even enhancing select innate immunity responses that are critical to fighting bacterial colonization, corticosteroids may change the microbiome in some way that is beneficial in asthma.
Beta-Adrenergic Agonist Therapy and the Airway Microbiome
Inhaled beta-adrenergic agonists, short-acting or long-acting, are a mainstay of therapy in asthma. To date, there are no reports of the effect of these drugs specifically on the lung microbiome. In any study or experiments in which these agents are co-administered with inhaled corticosteroids, one cannot exclude a modulating effect., perhaps by interactions on genes targeted by the glucocorticoid receptor [295, 296].
Macrolide Antibiotic Therapy and the Airway Microbiome
Antibiotics are commonly given to patients with asthma exacerbations, even as there is little evidence of their efficacy [297]. Recognition that at least some patients were colonized with bacteria such as Chlamydophila pneumoniae or Mycoplasma pneumoniae that contributed to their asthma symptoms [298,299,300,301,302,303] led to the idea that perhaps antibiotic therapy directed against these organisms would be useful therapy. The most studied antibiotic in asthma to date has been the macrolides.
Macrolide antibiotics, from the parent erythromycin to currently available agents including azithromycin and clarithromycin, are one of the most widely used antibiotic classes and have a role in the treatment of other obstructive airways diseases such as COPD, cystic fibrosis, non-CF bronchiectasis, and bronchiolitis [304,305,306,307,308,309,310]. As noted above, chronic asthma may be triggered repeatedly by viral respiratory infection and by select bacteria such as Chlamydophila pneumoniae and Mycoplasma pneumoniae, and this led to a suggestion that macrolide therapy might be useful in asthma. Further, macrolides have potential immunomodulatory and antiviral properties [311, 312], perhaps via suppression of NFκB [313,314,315,316], that extend beyond their role as direct antibacterial agents. Examples that are relevant to the airways and asthma include the ability of azithromycin therapy to maintain airway epithelial barrier integrity in culture models of Pseudomonas infection [317], which as noted earlier in this chapter may be found in increased relative abundance in asthmatic airway microbiome, suppress biofilm formation by Pseudomonas aeruginosa [318], and blocks quorum sensing by P. aeruginosa [319]. Macrolides inhibit mucin formation in cultured, differentiated airway epithelial cells induced by Fusobacterium nucleatum that is independent of any anti-bacterial activity [320]. Azithromycin can inhibit epithelial cell apoptosis and the epithelial-to-mesenchymal transition that occurs with ovalbumin challenge in mouse models [321, 322]. Despite the risks of anti-macrolide resistance that could develop from long-term use [323], these potential nonbactericidal mechanisms combined with the potential for direct action have led to exploration of macrolide therapy as a potential adjunct asthma therapy.
Given the scientific rationale for their use, their efficacy in clinical asthma trials has been variable and until recently disappointing. From the 1950s to 1970s, studies suggested that the macrolide troleandomycin could be a steroid-sparing agent in patients with severe asthma receiving parental corticosteroids, perhaps by blocking metabolism of methylprednisolone [324,325,326,327]. One trial demonstrated that azithromycin was unlikely to be a steroid-sparing agent in children with moderate to severe asthma who were receiving high dose inhaled corticosteroids; however, this trial had difficulty recruiting participants and was prematurely terminated for futility [328].
As “atypical” bacteria were recognized to be present in at least some children or adults with asthma, consideration was given to the antibiotic effect of macrolides. Here, results were inconsistent: for example, Kraft et al. [301] demonstrated that clarithromycin treatment increased FEV1 in patients who had evidence of either M. pneumoniae or C. pneumoniae infection based on PCR analysis of upper and lower airway samples. Against this, a report from the Asthma Clinical Research Network demonstrated that clarithromycin therapy did not improve asthma outcomes in those patients with suboptimally controlled asthma [329]. This trial was stratified based on PCR evidence for the same two microorganisms, and neither group improved. These trials required evidence of bacterial infection by either culture or PCR demonstration, and both were insensitive compared to next-generation sequencing. Similar trials examined different macrolide antibiotics generally over six to 26 weeks with assessment by clinical markers, and these trials in adults had varied results [330,331,332,333,334,335,336,337]. As recently as 2015, a Cochrane systematic review was inconclusive as to the clinical use of macrolides in asthma [338].
More recent studies took advantage of our improving understanding of asthma phenotypes. The recognition of neutrophilic asthma as an asthma phenotype, the suggestion that this phenotype was associated with increased bacterial load and potential pathogens such as Haemophilus influenzae and Moraxella catarrhalis and with neutrophil-associated cytokines such as IL-8 [195], and the understanding that neutrophilic inflammation was crucial to the pathogenesis of panbronchiolitis in which macrolide antibiotics had a demonstrated role [310, 339], all suggested a role for macrolides in neutrophilic asthma. Simpson et al. [333] demonstrated that clarithromycin therapy decreases both the number of neutrophils and concentrations of IL-8 in the sputum of patients with severe asthma. A randomized clinical trial (AZISAST ) compared the effectiveness of azithromycin for prevention of exacerbations in severe asthma among patients treated daily for 6 months. The primary endpoints, that of the rate of severe exacerbations and lower respiratory tract infection, were not met overall, but in pre-defined patients with non-eosinophilic asthma (blood eosinophils <200/μl and fractional excretion of nitric oxide below the lower limit of normal), patients with at least one primary endpoint event decreased from 62% in the placebo group to 33% in the treated group, a relative risk reduction of 54% [331]. To the extent that neutrophilic, non-eosinophilic asthma might be driven by the microbiome, these data suggested that macrolide therapy could be useful in treatment.
As next-generation sequencing became available, the question of whether macrolides could change the airway microbiome directly and thus change asthma control could be asked. One very early small trial examined the lower airway microbiome sampled by bronchial washing for and six weeks after treatment with daily azithromycin; therapy was associated with decreased richness and reductions in Pseudomonas, Hemophilus, and Staphylococcus [340]. In a follow-on to the AZISAST study [331], Santiago et al. [341] examined the oropharyngeal microbial community in 13 patients with moderate to severe asthma (8 receiving azithromycin, 5 receiving placebo) at baseline, during and after 6 months treatment with either azithromycin or placebo. They found that the overall composition of the oral microbiome in these patients differed little to that of the healthy population. Treatment over 6 months with azithromycin increased the relative abundance of Streptococcus salivarius and decreased that of Leptotrichia wadei. The authors noted that they used oropharyngeal samples out of concern that collection of lower airway samples might induce asthma, a concern not borne out in other studies. Another recent study that demonstrated the utility of azithromycin as adjunct therapy for both T2-high and T2-low severe, uncontrolled asthma was the AMAZES trial, a placebo-controlled, randomized clinical trial in which 48 week therapy with thrice-weekly azithromycin reduced asthma exacerbations and improved quality of life [342]. Subsequent analysis of sputum samples from a subset of these patients demonstrated that azithromycin treatment did not alter bacterial load, nor alter the relative abundance of select pathogens such as Moraxella and Pseudomonas, but did decrease Faith’s phylogenetic diversity index and decreased the relative abundance of Haemophilus [343]. One concern in this study was a noted increase in several macrolide resistance genes as noted by PCR analysis of sputum. Taken together, the microbiome analysis performed from the AZISAST and AMAZES studies begins to make clear that whatever the effects of azithromycin are on asthma, its effects on the microbiome appear modest. This raises the question as to whether the immunomodulatory effects of this antibiotic are more important.
Other clinical trials outside of asthma have examined the effect of macrolides on the airway microbiome. Azithromycin therapy during the treatment of respiratory syncytial virus bronchiolitis infection in infants decreased the abundance of Moraxella in nasal lavage samples collected pre and post treatment [344]. Other bacterial taxa were unchanged by treatment, and the lower abundance in Moraxella was associated with less respiratory wheezing in the ensuing twelve months. Likewise, treatment with erythromycin for 48 weeks in 84 adults enrolled in the Bronchiectasis and Low-dose Erythromycin Study (BLESS) demonstrated lower abundance of Actinomyces and Streptococcus but an increased abundance of Haemophilus in oropharyngeal swabs [345]. In COPD, azithromycin therapy has been considered as among first-line therapies for treatment of exacerbations [346, 347], and as a long-term adjunct treatment of GOLD class 4 COPD [348,349,350]. One study examined the lower airway microbiome by BAL in 20 patients with COPD before and eight weeks after treatment with either daily azithromycin or placebo. In this trial, azithromycin treatment did not alter bacterial burden but did decrease the relative abundance of a number of taxa, alpha-diversity, and concentrations of chemokines and cytokines such as CXCL1, TNF-α, and IL-13 [351]. As in the asthma studies, the effects of macrolide therapy on the microbiome were modest.
In summary, it becomes clear that macrolide therapy has complex effects on the lung, including on the lung microbiome. These changes may be due both to the direct, antimicrobial effect on the bacteria and by more indirect effects as an immunomodulator, with the latter perhaps being the more important. Resolving these questions may provide a better rationale for the use of macrolide therapy in asthma.
Other Antibiotics and the Airway Microbiome
Lastly, older studies have examined the use of penicillin-based antibiotics in patients hospitalized for asthma exacerbations and found no evidence for efficacy [352, 353]. To date, there are no NGS-based studies that examine the role of penicillin-based antibiotics, or of other antibiotics commonly employed in outpatients with respiratory symptoms (e.g., sulfonamides, cephalosporins, tetracycline) on the airway microbiome in asthmatic patients.
Probiotics and the Airway Microbiome
The GI microbiome has become an important and increasingly studied etiological factor in a number of immunological, neurological, and malignant diseases. As noted earlier, there is increasing recognition of how the gut-lung axis could influence T cell plasticity and function and dendritic cell function. While much attention has been focused on the gut-lung axis in early life, one recent study has demonstrated differences in the gut bacterial community structure in a group of adults with asthma compared to control subjects without known lung disease [168]. Differences in specific gut bacterial communities at the phylum level (Bacteroidetes and Firmicutes) associated with FEV1, and select OTUs were either decreased (Bacteroides, Enterobacteriaceae) or increased (Bifidobacterium, and Lachnospiraceae) in subjects with asthma. Cluster analysis done by variation in gut bacterial community structure demonstrated three different clusters in asthma patients; one such cluster had greater airway hyperresponsiveness and bronchodilator reversibility. Thus, the GI microbiome could be used as part of a phenotyping assessment of asthma patients.
For these reasons, modulating the adult GI microbiome by probiotic therapy with live microorganisms that alter these functions in favor of asthma control and better health, is an attractive consideration, and one that might avoid many of the problems associated with antibiotic use.
Lactobacillus is one probiotic that has received considerable attention. A low relative abundance of Lactobacillus has been shown to be associated with early development of allergy in infants [354,355,356]. Oral supplementation with different Lactobacillus species, particularly L. reuteri, alleviated airway inflammation, decreased IgE production, and decreased production of T2-associated cytokines induced by house dust mite allergen in a mouse model [357]. Likewise, oral gavage with L. paracasei L9 attenuated airway hyperresponsiveness, eosinophil infiltration in airways, and decreased serum IgE after exposure to particulate matter 2.5 (PM2.5) in mice sensitized and challenged with ovalbumin [358]. Treatment of ovalbumin sensitized and challenged mice with Lactobacillus rhamnosus GG (LGG) by gavage improved both airway inflammation and airway remodeling as evidenced by reduced collagen deposition and expression of markers such as T-bet, GATA3, and Foxp3 [359]. These animal studies clearly establish a potential use for probiotic therapy in airway inflammation.
Trials in which infants at high risk for atopic disease (usually by virtue of having one or two atopic parents) have received probiotic therapy, usually Lactobacillus rhamnosus GG (LGG) by oral supplementation, have had varying results. One early trial in Finland suggested a benefit by decreasing subsequent atopic disease, a benefit that extended to age 5 [360,361,362]. However, a different trial done at about the same time demonstrated no clinical benefit from LGG when given to pregnant women with a family history of atopic dermatitis in primary prevention of atopic dermatitis to their offspring [363]. A trial in which preschool children with allergic asthma or rhinitis consumed fermented milk containing L. casei demonstrated no improvement in asthma control [364]. Likewise, in the Trial of Infant Probiotic Supplementation (TIPS), oral administration of LGG supplementation for six months in 92 high-risk infants on the subsequent incidence of eczema (atopic dermatitis), with development of asthma examined as a secondary end-point of the study, failed to demonstrate a benefit for either disease [365]. In a follow-on study from the TIPS trial, Durack et al. [366] examined gut microbiota maturation in these infants, and demonstrated that LGG supplementation in infants with a “meconium” dysbiosis of their GI microbiome had increased diversification of their microbiota and increased production of anti-inflammatory lipids during LGG treatment. However, within six months of cessation of supplementation, all benefits had been lost. Thus it is not clear today whether probiotic intervention in early life conveys a lasting benefit in asthma and other allergic diseases.
In contrast, trials in children and adolescents with asthma suggest some benefit with probiotic therapy. An early trial of L. gasseri supplementation in children aged 6 to 12 years with asthma and allergic rhinitis over two months demonstrated improved pulmonary function compared to placebo [367]. A more recent placebo-controlled, randomized clinical trial examined supplementation of L. paracasei, L. fermentum, and their combination on asthma control over three months in 160 adolescents aged 6 to 18 years with asthma. Children continued usual care and asthma medications in this time. The children receiving Lactobacillus therapy in any combination had lower asthma severity and improved control compared to those children receiving placebo [368]. A small pilot trial from Brazil in 30 children aged 6 to 17 years with asthma received a probiotic containing L. reuteri or placebo; children receiving the probiotic over 60 days had an improvement in symptoms and asthma control test scores [369]. A recent meta-analysis of 17 randomized controlled trials in 5264 children suggested that LGG supplementation elicited a reduction in the occurrence of asthma but not other atopic diseases in post-natal periods [370].
One promising probiotic agent is OM-85 Bronchovaxom (OM-85 BV), a low-endotoxin alkaline extract of 21 strains of five pathogenic bacteria that on oral administration as a prophylactic agent reduces the frequency and duration of respiratory tract infection in children [371,372,373,374]. Used in pre-treatment, OM-85 BV prevents airway hyperreactivity and inflammation in Leishmania major antigen sensitized and challenged mice by increasing the number of regulatory T cells in the airway [375]. The recent EOLIA trial compared the effect of sublingual administration of a similar polyvalent mechanical bacterial lysate tablet versus placebo for three months in 152 children aged 6 to 16 years with asthma, a history of asthma exacerbations, and sensitivity to dust mite allergen. While the major endpoint of a change in ACT score was not met, the number of asthma exacerbations decreased in children treated with the bacterial lysate [376]. Data regarding OM-85 BV in adults is sparse to date. The upcoming PrecISE precision intervention trial for severe asthma sponsored by NHBLI will examine the potential utility of OM-85 BV in two patient cohorts, adolescent and adult respectively, with severe asthma in an adaptive trial design.Footnote 1
No other probiotic trials for adults with asthma are published to date.
Effects of Asthma Exacerbations on the Airway Microbiome
Asthma exacerbations can be triggered by a variety of environmental exposures; one major trigger is respiratory tract viral infection [377,378,379]. Children with asthma may have exacerbations triggered by bacterial pathogens such as H. influenzae, M. pneumoniae, M. catarrhalis, or C. pneumoniae, as noted previously, in addition to viruses. In contrast, respiratory bacterial infection is not commonly associated with asthma exacerbations in adults; this is a noticeable distinction between asthma and COPD [380]. While there are scant data to date on the lung microbiome in adults before and after exacerbation, the AZISAST and AMAZES trials with azithromycin suggest that modification of the lung microbiome may change the frequency of and susceptibility to exacerbations [331, 342, 343, 381], though the changes in the microbiome in the AMAZES trial were, as noted previously, modest and suggested that azithromycin may exert some of its protective effects via nonbacterial mechanisms.
There is some indirect evidence that viral infection may increase susceptibility to subsequent bacterial growth, and that this may be relevant to asthma. Viruses such as rhinovirus, influenza, and respiratory syncytial virus (RSV) can increase airway epithelial cell barrier permeability via disruption of tight junctions, alter expression of adhesion molecules that then may promote bacterial invasion, impair cilia function, decrease the expression or effectiveness of host defense proteins such as the Toll-like receptors and secretory IgA, and increase mucous production [382,383,384,385,386,387,388]. Indeed, it is clear that the epithelium is the principal site of first attack for most viral infections of the airway [389]. Any viral infection would also alter the local environment in terms of nutrient availability, oxidative status, and presence of mediators that may change bacterial virulence and survival [385]. Whether any of these events are important to asthma exacerbations in terms of susceptibility, risk or duration, or in a subsequent alteration of the airway milieu that in turn may promote a longer-term dysbiotic microbiome, is unknown.
Future Directions
We have learned much about the lung microbiome in health and in obstructive lung disease over the past decade. Next-generation sequencing, bioinformatics, mechanistic studies of the microbiome in mice, and the ability to gather increasingly large cohorts of human participants in clinical trials have increasingly demonstrated that the lung microbiome in asthma is dysbiotic and may well have a role in pathogenesis and disease progression in select asthma phenotypes. To date, much of this research, while increasingly elegant in research methods has been descriptive and based on single time-point observations.
There is a clear and compelling need to apply an ecological framework to understanding the pathogenesis of asthma and for longitudinal studies that are large and well organized with the following characteristics: collection of multiple microbiological specimens from the lung, upper respiratory tract and gut; additional biospecimens and genetic samples to work out biomarkers of disease and phenotype to deeply characterize host response factors, and collection of individual-specific environmental and behavioral data (Fig. 5.3). Such studies are needed involving multiple, diverse populations around the world, and at present are particularly lacking in the study of adult asthma. The single largest impediment to such studies is that no agency, government or private, has been willing to date to pay for the twenty-first century equivalent of a “Framingham” style asthma study. Such studies must surmount the substantial costs of collection and analysis and the significant burdens placed on the participants. Further, these studies must assume the task of accounting for differing environments, both indoor and outdoor, that may alter the microbiome (gut or lung) and the host defenses, seasonality, and the interactions of nonmicrobial allergens with the microbiome and with host responses. The human subjects of course have a varying genome and differences in the expression of each of the “-omic” areas that might be studied, as well as interactions between these -omics. In the end, these studies would need to provide specific etiologic, mechanistic, and associative insights about the microbiome and asthma so as to inform experimental cell-based and animal models of host-microbiome interactions, which then could come back to appropriate clinical trials.

(a) Ecological interactions shape both host biology and their microbiomes. The human body is a complex ecosystem that experiences concurrent microbial and nonmicrobial exposures from external/built environments, other animals, diet, medication, occupational exposures, and pollutants. The human ecosystem is also impacted by concomitant inflammatory and/or immune disorders An ecological framework is necessary to advance mechanistic insights into these multidirectional interactions and how they shape asthma. Adapted from reference (396) with permission. (b) The longitudinal nature of ecological interactions from prior to birth to adulthood that shape asthma risk, development, and clinical outcomes, with reference to the microbiomes and the relationships of microbiota to the immune system and key environmental influences. Adapted from reference (397) with permission
If the microbiome has a role in asthma, it must do so by inducing a response in the human host. The large studies will need to correlate key features of the microbiome, and features that go beyond simple taxonomy or diversity, with host clinical, cellular, and molecular disease markers. As one example, endobronchial brushings collected from patients with and without malignant lung nodules, sequenced for both bacterial (16S rRNA) and host transcriptome profiles, demonstrated that patient transcriptomic signatures relevant to lung cancer pathogenesis were associated with increased relative abundance for Streptococcus and Veillonella in the brush samples [390]. Such a study done in patients with asthma of a carefully curated phenotype (e.g., the T2-low, Th-17 high phenotype that appears to be most correlated with a high bacterial burden, as previously discussed), combined with a multiple -omics strategy could examine, for example, single nucleotide polymorphisms and micro-RNA on the host side and metagenomic analysis on the microbiome side, to test whether similar associations exist. Select differences then could be tested in newer dedicated cell- and organ-cultures, including cultures that use engineered substrates populated with cells collected from airways or derived from stem cells, and “lung-on-a-chip” microsystems [391].
Animal models in which a human microbiome is transplanted into germ-free (GF) (“axenic”) mice also provide a model in which causal relationships can be explored between the microbiome and a host. There is recognition that while the GF mouse, lacking its own microbiome, represents a “blank slate” [392,393,394,395] upon which human microbiota can be specifically tested, alone, in select combinations, or as a collected ecology from human subjects, to provoke host responses, particularly when combined with airway allergen challenge. There are clear differences between human and mouse immunology that must be taken into account in explaining the results of such experiments. Further, facilities to generate and care for these mice are frightfully expensive – these mice are literally worth their weight in gold. Such limitations for now limit these experiments.
Perhaps the greatest impediment to date has been one that is now finally disappearing – lung biologists now agree that the lung is not sterile. This recognition must translate to a recognition that large clinical trials should be sampling the lung and gut microbiomes as part of their design. Even if not analyzed at the time for the potential role in modulating the response to a pharmacological therapy, the use of these samples (as in the BOBCAT trial, [134]) in post-hoc explorations not only offer the potential to relate the microbiome to the studied phenotype or group of patients but also, by adding larger numbers of patients, provide important corroboration to our understanding.
Among the key questions to be addressed in the future is whether intervening in the adult microbiome in asthma matters. If the “critical window” in which the lung or gut microbiome (with other factors) influences the host immunology to develop an asthma phenotype is open only during early life, then modification of the microbiome in adults may be well beyond the time in which such modifications can have any useful effect. That the use of simple antibiotics such as azithromycin can modulate asthma clinical control suggests that this extreme and perhaps dour view of the microbiome is unwarranted, and that the microbiome is at least to some degree “plastic” and changeable. Modification of the microbiome need not be direct to be useful; changing the host immune system (e.g., even with inhaled corticosteroids) in ways that in turn modify the airway microbiome may elicit a useful downstream effect. Modification of the microbiome need not work in every adult with asthma to be useful: the asthma community is correctly vested in anti-IL-5 and anti-IL-4/IL-13 biologic therapy for the relatively modest number of patients with severe, T2-high asthma (15 to 30% of all asthma patients are T2-high, and 10 to 20% have severe asthma, so in the end we are addressing 2 to 6% of the asthma population). A microbiome-based therapy that addresses a phenotype presently unreachable, such as the “neutrophilic” phenotype that is T2-low and perhaps Th-17-high, would be a substantial advance.
The ultimate goal in understanding the lung microbiome in asthma is to uncover epidemiologic, diagnostic or therapeutic features that will improve patient care. The microbiome may aid us in delineating phenotypes and understanding markers of disease risk and progression. Not only might we find therapies that may modify the microbiome (lung or gut or both) that improve clinical outcomes, as importantly, we might uncover beneficial or deleterious effects of current therapies. All these will shape our understanding of adult asthma and lead to more personalized care. Understanding the microbiome in asthma then is much like understanding the immunology, cell biology and epidemiology of this disease: a necessary part that will shape how we care for patients in the future.
Notes
- 1.
Information about the PrecISE trial can be found at https://preciseasthma.org/preciseweb/
References
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804–10.
Bisgaard H, Hermansen MN, Buchvald F, Loland L, Halkjaer LB, Bonnelykke K, et al. Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med. 2007;357(15):1487–95.
Madan JC, Koestler DC, Stanton BA, Davidson L, Moulton LA, Housman ML, et al. Serial analysis of the gut and respiratory microbiome in cystic fibrosis in infancy: interaction between intestinal and respiratory tracts and impact of nutritional exposures. MBio. 2012;3(4).
Huxley EJ, Viroslav J, Gray WR, Pierce AK. Pharyngeal aspiration in normal adults and patients with depressed consciousness. Am J Med. 1978;64(4):564–8.
Labelle AJ, Arnold H, Reichley RM, Micek ST, Kollef MH. A comparison of culture-positive and culture-negative health-care-associated pneumonia. Chest. 2010;137(5):1130–7.
Garau J, Baquero F, Pérez-Trallero E, Pérez JL, Martín-Sánchez AM, García-Rey C, et al. Factors impacting on length of stay and mortality of community-acquired pneumonia. Clin Microbiol Infect. 2008;14(4):322–9.
Kosmidis C, Denning DW. The clinical spectrum of pulmonary aspergillosis. Thorax. 2015;70(3):270–7.
O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373(9678):1891–904.
Bell SC, Mall MA, Gutierrez H, Macek M, Madge S, Davies JC, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med. 2020;8(1):65–124.
Neelakanta G, Sultana H. The use of metagenomic approaches to analyze changes in microbial communities. Microbiology insights. 2013;6:37–48.
Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184(8):957–63.
Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle GB, et al. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc. 2015;12(6):821–30.
Millares L, Bermudo G, Perez-Brocal V, Domingo C, Garcia-Nunez M, Pomares X, et al. The respiratory microbiome in bronchial mucosa and secretions from severe IgE-mediated asthma patients. BMC Microbiol. 2017;17(1):20.
Alfieri V, Aiello M, Pisi R, Tzani P, Mariani E, Marangio E, et al. Small airway dysfunction is associated to excessive bronchoconstriction in asthmatic patients. Respir Res. 2014;15(1):86.
Balzar S, Chu HW, Strand M, Wenzel S. Relationship of small airway chymase-positive mast cells and lung function in severe asthma. Am J Respir Crit Care Med. 2005;171(5):431–9.
Balzar S, Wenzel SE, Chu HW. Transbronchial biopsy as a tool to evaluate small airways in asthma. Eur Respir J. 2002;20(2):254–9.
Hamid Q, Song Y, Kotsimbos TC, Minshall E, Bai TR, Hegele RG, et al. Inflammation of small airways in asthma. J Allergy Clin Immunol. 1997;100(1):44–51.
Denner DR, Sangwan N, Becker JB, Hogarth DK, Oldham J, Castillo J, et al. Corticosteroid therapy and airflow obstruction influence the bronchial microbiome, which is distinct from that of bronchoalveolar lavage in asthmatic airways. J Allergy Clin Immunol. 2016;137(5):1398–405. e3
Cox MJ, Cookson WO, Moffatt MF. Sequencing the human microbiome in health and disease. Hum Mol Genet. 2013;22(R1):R88–94.
Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87.
Carney SM, Clemente JC, Cox MJ, Dickson RP, Huang YJ, Kitsios GD, et al. Methods in lung microbiome research. Am J Respir Cell Mol Biol. 2020;62(3):283–99.
Abdel-Aziz MI, Vijverberg SJH, Neerincx AH, Kraneveld AD. Maitland-van der Zee AH. The crosstalk between microbiome and asthma: exploring associations and challenges. Clin Exp Allergy. 2019;49(8):1067–86.
Moore WC, Evans MD, Bleecker ER, Busse WW, Calhoun WJ, Castro M, et al. Safety of investigative bronchoscopy in the severe asthma research program. J Allergy Clin Immunol. 2011;128(2):328–36. e3
Elston WJ, Whittaker AJ, Khan LN, Flood-Page P, Ramsay C, Jeffery PK, et al. Safety of research bronchoscopy, biopsy and bronchoalveolar lavage in asthma. Eur Respir J. 2004;24(3):375–7.
Zhang Q, Cox M, Liang Z, Brinkmann F, Cardenas PA, Duff R, et al. Airway microbiota in severe asthma and relationship to asthma severity and phenotypes. PLoS One. 2016;11(4):e0152724.
Durack J, Huang YJ, Nariya S, Christian LS, Mark Ansel K, Beigelman A, et al. Bacterial biogeography of adult airways in atopic asthma. Microbiome. 2018;6(1):104.
Rodriguez A, Brickley E, Rodrigues L, Normansell RA, Barreto M, Cooper PJ. Urbanisation and asthma in low-income and middle-income countries: a systematic review of the urban-rural differences in asthma prevalence. Thorax. 2019;74(11):1020–30.
Neffen H, Chahuan M, Hernandez DD, Vallejo-Perez E, Bolivar F, Sanchez MH, et al. Key factors associated with uncontrolled asthma – the asthma control in Latin America Study. J Asthma. 2020;57(2):113–22.
Nicolaou N, Siddique N, Custovic A. Allergic disease in urban and rural populations: increasing prevalence with increasing urbanization. Allergy. 2005;60(11):1357–60.
Platts-Mills TA, Cooper PJ. Differences in asthma between rural and urban communities in South Africa and other developing countries. J Allergy Clin Immunol. 2010;125(1):106–7.
Sole D, Cassol VE, Silva AR, Teche SP, Rizzato TM, Bandim LC, et al. Prevalence of symptoms of asthma, rhinitis, and atopic eczema among adolescents living in urban and rural areas in different regions of Brazil. Allergol Immunopathol. 2007;35(6):248–53.
Rodriguez A, Vaca MG, Chico ME, Rodrigues LC, Barreto ML, Cooper PJ. Rural to urban migration is associated with increased prevalence of childhood wheeze in a Latin-American city. BMJ Open Respir Res. 2017;4(1):e000205.
Ruokolainen L, Paalanen L, Karkman A, Laatikainen T, von Hertzen L, Vlasoff T, et al. Significant disparities in allergy prevalence and microbiota between the young people in Finnish and Russian Karelia. Clin Exp Allergy. 2017;47(5):665–74.
Feng M, Yang Z, Pan L, Lai X, Xian M, Huang X, et al. Associations of early life exposures and environmental factors with asthma among children in rural and urban areas of Guangdong. China Chest. 2016;149(4):1030–41.
Hrusch CL, Stein MM, Gozdz J, Holbreich M, von Mutius E, Vercelli D, et al. T-cell phenotypes are associated with serum IgE levels in Amish and Hutterite children. J Allergy Clin Immunol. 2019;144(5):1391–401. e10
Ege MJ, Mayer M, Normand AC, Genuneit J, Cookson WO, Braun-Fahrlander C, et al. Exposure to environmental microorganisms and childhood asthma. N Engl J Med. 2011;364(8):701–9.
Ege MJ, Strachan DP, Cookson WO, Moffatt MF, Gut I, Lathrop M, et al. Gene-environment interaction for childhood asthma and exposure to farming in Central Europe. J Allergy Clin Immunol. 2011;127(1):138–44, 44 e1–4.
Stein MM, Hrusch CL, Gozdz J, Igartua C, Pivniouk V, Murray SE, et al. Innate immunity and asthma risk in Amish and Hutterite farm children. N Engl J Med. 2016;375(5):411–21.
Dannemiller KC, Gent JF, Leaderer BP, Peccia J. Indoor microbial communities: influence on asthma severity in atopic and nonatopic children. J Allergy Clin Immunol. 2016;138(1):76–83. e1
Dannemiller KC, Gent JF, Leaderer BP, Peccia J. Influence of housing characteristics on bacterial and fungal communities in homes of asthmatic children. Indoor Air. 2016;26(2):179–92.
Dorado Garcia A, Lidwien AMS, Bossers A, Inge MW, Schmitt H, Markus JE, et al. Indoor airborne microbiota composition associated with asthma and atopy in rural children. Eur Respir J. 2018;52.
Riedler J, Braun-Fahrlander C, Eder W, Schreuer M, Waser M, Maisch S, et al. Exposure to farming in early life and development of asthma and allergy: a cross-sectional survey. Lancet. 2001;358(9288):1129–33.
Adams RI, Bateman AC, Bik HM, Meadow JF. Microbiota of the indoor environment: a meta-analysis. Microbiome. 2015;3:49.
Dannemiller KC, Weschler CJ, Peccia J. Fungal and bacterial growth in floor dust at elevated relative humidity levels. Indoor Air. 2017;27(2):354–63.
Meadow JF, Altrichter AE, Bateman AC, Stenson J, Brown GZ, Green JL, et al. Humans differ in their personal microbial cloud. PeerJ. 2015;3:e1258.
Adams RI, Bhangar S, Pasut W, Arens EA, Taylor JW, Lindow SE, et al. Chamber bioaerosol study: outdoor air and human occupants as sources of indoor airborne microbes. PLoS One. 2015;10(5):e0128022.
Ball TM, Castro-Rodriguez JA, Griffith KA, Holberg CJ, Martinez FD, Wright AL. Siblings, day-care attendance, and the risk of asthma and wheezing during childhood. N Engl J Med. 2000;343(8):538–43.
Kramer U, Heinrich J, Wjst M, Wichmann HE. Age of entry to day nursery and allergy in later childhood. Lancet. 1999;353(9151):450–4.
Mendy A, Gasana J, Vieira ER, Forno E, Patel J, Kadam P, et al. Endotoxin exposure and childhood wheeze and asthma: a meta-analysis of observational studies. J Asthma. 2011;48(7):685–93.
Thorne PS, Mendy A, Metwali N, Salo P, Co C, Jaramillo R, et al. Endotoxin exposure: predictors and prevalence of associated asthma outcomes in the United States. Am J Respir Crit Care Med. 2015;192(11):1287–97.
Douwes J, van Strien R, Doekes G, Smit J, Kerkhof M, Gerritsen J, et al. Does early indoor microbial exposure reduce the risk of asthma? The prevention and incidence of asthma and mite allergy birth cohort study. J Allergy Clin Immunol. 2006;117(5):1067–73.
Michel O, Kips J, Duchateau J, Vertongen F, Robert L, Collet H, et al. Severity of asthma is related to endotoxin in house dust. Am J Respir Crit Care Med. 1996;154(6 Pt 1):1641–6.
Goldman DL, Vicencio AG. The chitin connection. mBio. 2012;3(2).
Douwes J, Zuidhof A, Doekes G, van der Zee SC, Wouters I, Boezen MH, et al. (1-->3)-beta-D-glucan and endotoxin in house dust and peak flow variability in children. Am J Respir Crit Care Med. 2000;162(4 Pt 1):1348–54.
Braun-Fahrlander C, Riedler J, Herz U, Eder W, Waser M, Grize L, et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N Engl J Med. 2002;347(12):869–77.
Ciaccio CE, Barnes C, Kennedy K, Chan M, Portnoy J, Rosenwasser L. Home dust microbiota is disordered in homes of low-income asthmatic children. J Asthma. 2015;52(9):873–80.
Barberan A, Dunn RR, Reich BJ, Pacifici K, Laber EB, Menninger HL, et al. The ecology of microscopic life in household dust. Proc Biol Sci. 2015;282(1814).
Maier RM, Palmer MW, Andersen GL, Halonen MJ, Josephson KC, Maier RS, et al. Environmental determinants of and impact on childhood asthma by the bacterial community in household dust. Appl Environ Microbiol. 2010;76(8):2663–7.
Sitarik AR, Havstad S, Levin AM, Lynch SV, Fujimura KE, Ownby DR, et al. Dog introduction alters the home dust microbiota. Indoor Air. 2018;28(4):539–47.
Kettleson EM, Adhikari A, Vesper S, Coombs K, Indugula R, Reponen T. Key determinants of the fungal and bacterial microbiomes in homes. Environ Res. 2015;138:130–5.
Richardson M, Gottel N, Gilbert JA, Gordon J, Gandhi P, Reboulet R, et al. Concurrent measurement of microbiome and allergens in the air of bedrooms of allergy disease patients in the Chicago area. Microbiome. 2019;7(1):82.
Ownby DR, Johnson CC, Peterson EL. Exposure to dogs and cats in the first year of life and risk of allergic sensitization at 6 to 7 years of age. JAMA. 2002;288(8):963–72.
Gao X, Yin M, Yang P, Li X, Di L, Wang W, et al. Effect of exposure to cats and dogs on the risk of asthma and allergic rhinitis: a systematic review and meta-analysis. Am J Rhinol Allergy. 2020;34(5):703–14.
Fujimura KE, Johnson CC, Ownby DR, Cox MJ, Brodie EL, Havstad SL, et al. Man’s best friend? The effect of pet ownership on house dust microbial communities. J Allergy Clin Immunol. 2010;126(2):410–2, 2 e1–3.
Silverman RA, Stevenson L, Hastings HM. Age-related seasonal patterns of emergency department visits for acute asthma in an urban environment. Ann Emerg Med. 2003;42(4):577–86.
Chen CH, Xirasagar S, Lin HC. Seasonality in adult asthma admissions, air pollutant levels, and climate: a population-based study. J Asthma. 2006;43(4):287–92.
Tsai SS, Cheng MH, Chiu HF, Wu TN, Yang CY. Air pollution and hospital admissions for asthma in a tropical city: Kaohsiung. Taiwan Inhalation toxicology. 2006;18(8):549–54.
Zhang Y, Peng L, Kan H, Xu J, Chen R, Liu Y, et al. Effects of meteorological factors on daily hospital admissions for asthma in adults: a time-series analysis. PLoS One. 2014;9(7):e102475.
Kim KH, Jahan SA, Kabir E. A review on human health perspective of air pollution with respect to allergies and asthma. Environ Int. 2013;59:41–52.
Cohen HA, Blau H, Hoshen M, Batat E, Balicer RD. Seasonality of asthma: a retrospective population study. Pediatrics. 2014;133(4):e923–32.
Soyiri IN, Reidpath DD, Sarran C. Forecasting asthma-related hospital admissions in London using negative binomial models. Chron Respir Dis. 2013;10(2):85–94.
Wisniewski JA, McLaughlin AP, Stenger PJ, Patrie J, Brown MA, El-Dahr JM, et al. A comparison of seasonal trends in asthma exacerbations among children from geographic regions with different climates. Allergy Asthma Proc. 2016;37(6):475–81.
Boulet LP, Cartier A, Thomson NC, Roberts RS, Dolovich J, Hargreave FE. Asthma and increases in nonallergic bronchial responsiveness from seasonal pollen exposure. J Allergy Clin Immunol. 1983;71(4):399–406.
Tham R, Vicendese D, Dharmage SC, Hyndman RJ, Newbigin E, Lewis E, et al. Associations between outdoor fungal spores and childhood and adolescent asthma hospitalizations. J Allergy Clin Immunol. 2017;139(4):1140–7. e4
Atkinson RW, Kang S, Anderson HR, Mills IC, Walton HA. Epidemiological time series studies of PM2.5 and daily mortality and hospital admissions: a systematic review and meta-analysis. Thorax. 2014;69(7):660–5.
Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187(10):1067–75.
Einarsson GG, Comer DM, McIlreavey L, Parkhill J, Ennis M, Tunney MM, et al. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax. 2016;71(9):795–803.
Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One. 2011;6(2):e16384.
Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio. 2015;6(2):e00037.
Beck JM, Schloss PD, Venkataraman A, Twigg H 3rd, Jablonski KA, Bushman FD, et al. Multicenter comparison of lung and oral microbiomes of HIV-infected and HIV-uninfected individuals. Am J Respir Crit Care Med. 2015;192(11):1335–44.
Haldar K, George L, Wang Z, Mistry V, Ramsheh MY, Free RC, et al. The sputum microbiome is distinct between COPD and health, independent of smoking history. Respir Res. 2020;21(1):183.
Dominguez-Bello MG, Godoy-Vitorino F, Knight R, Blaser MJ. Role of the microbiome in human development. Gut. 2019;68(6):1108–14.
von Mutius E, Martinez FD, Fritzsch C, Nicolai T, Roell G, Thiemann HH. Prevalence of asthma and atopy in two areas of West and East Germany. Am J Respir Crit Care Med. 1994;149(2 Pt 1):358–64.
Laatikainen T, von Hertzen L, Koskinen JP, Makela MJ, Jousilahti P, Kosunen TU, et al. Allergy gap between Finnish and Russian Karelia on increase. Allergy. 2011;66(7):886–92.
Holbreich M, Genuneit J, Weber J, Braun-Fahrlander C, Waser M, von Mutius E. Amish children living in northern Indiana have a very low prevalence of allergic sensitization. J Allergy Clin Immunol. 2012;129(6):1671–3.
Vedanthan PK, Mahesh PA, Vedanthan R, Holla AD, Liu AH. Effect of animal contact and microbial exposures on the prevalence of atopy and asthma in urban vs rural children in India. Ann Allergy Asthma Immunol. 2006;96(4):571–8.
Pakarinen J, Hyvarinen A, Salkinoja-Salonen M, Laitinen S, Nevalainen A, Makela MJ, et al. Predominance of Gram-positive bacteria in house dust in the low-allergy risk Russian Karelia. Environ Microbiol. 2008;10(12):3317–25.
Lynch SV, Wood RA, Boushey H, Bacharier LB, Bloomberg GR, Kattan M, et al. Effects of early-life exposure to allergens and bacteria on recurrent wheeze and atopy in urban children. J Allergy Clin Immunol. 2014;134(3):593–601. e12
Mendy A, Wilkerson J, Salo PM, Cohn RD, Zeldin DC, Thorne PS. Exposure and sensitization to pets modify endotoxin association with asthma and wheeze. J Allergy Clin Immunol Pract. 2018;6(6):2006–13. e4
Kirjavainen PV, Karvonen AM, Adams RI, Taubel M, Roponen M, Tuoresmaki P, et al. Farm-like indoor microbiota in non-farm homes protects children from asthma development. Nat Med. 2019;25(7):1089–95.
Fujimura KE, Demoor T, Rauch M, Faruqi AA, Jang S, Johnson CC, et al. House dust exposure mediates gut microbiome lactobacillus enrichment and airway immune defense against allergens and virus infection. Proc Natl Acad Sci U S A. 2014;111(2):805–10.
Bingula R, Filaire M, Radosevic-Robin N, Bey M, Berthon JY, Bernalier-Donadille A, et al. Desired turbulence? gut-lung axis, immunity, and lung cancer. J Oncol. 2017;2017:5035371.
Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–23.
Huang YJ, Marsland BJ, Bunyavanich S, O’Mahony L, Leung DY, Muraro A, et al. The microbiome in allergic disease: current understanding and future opportunities-2017 PRACTALL document of the American Academy of Allergy, Asthma & Immunology and the European Academy of Allergy and Clinical Immunology. J Allergy Clin Immunol. 2017;139(4):1099–110.
Silvers KM, Frampton CM, Wickens K, Pattemore PK, Ingham T, Fishwick D, et al. Breastfeeding protects against current asthma up to 6 years of age. J Pediatr. 2012;160(6):991–6. e1
Raciborski F, Tomaszewska A, Komorowski J, Samel-Kowalik P, Bialoszewski AZ, Walkiewicz A, et al. The relationship between antibiotic therapy in early childhood and the symptoms of allergy in children aged 6–8 years – the questionnaire study results. Int J Occup Med Environ Health. 2012;25(4):470–80.
Roduit C, Scholtens S, de Jongste JC, Wijga AH, Gerritsen J, Postma DS, et al. Asthma at 8 years of age in children born by caesarean section. Thorax. 2009;64(2):107–13.
Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med. 2016;8(343):343ra82.
McLoughlin RM, Mills KH. Influence of gastrointestinal commensal bacteria on the immune responses that mediate allergy and asthma. J Allergy Clin Immunol. 2011;127(5):1097–107. quiz 108–9.
Marsland BJ, Trompette A, Gollwitzer ES. The gut-lung Axis in respiratory disease. Ann Am Thorac Soc. 2015;12(Suppl 2):S150–6.
Ly NP, Litonjua A, Gold DR, Celedon JC. Gut microbiota, probiotics, and vitamin D: interrelated exposures influencing allergy, asthma, and obesity? J Allergy Clin Immunol. 2011;127(5):1087–94. quiz 95–6
Cait A, Hughes MR, Antignano F, Cait J, Dimitriu PA, Maas KR, et al. Microbiome-driven allergic lung inflammation is ameliorated by short-chain fatty acids. Mucosal Immunol. 2018;11(3):785–95.
Pugin B, Barcik W, Westermann P, Heider A, Wawrzyniak M, Hellings P, et al. A wide diversity of bacteria from the human gut produces and degrades biogenic amines. Microb Ecol Health Dis. 2017;28(1):1353881.
Barcik W, Pugin B, Westermann P, Perez NR, Ferstl R, Wawrzyniak M, et al. Histamine-secreting microbes are increased in the gut of adult asthma patients. J Allergy Clin Immunol. 2016;138(5):1491–4. e7
Barcik W, Pugin B, Bresco MS, Westermann P, Rinaldi A, Groeger D, et al. Bacterial secretion of histamine within the gut influences immune responses within the lung. Allergy. 2019;74(5):899–909.
Bisgaard H, Li N, Bonnelykke K, Chawes BL, Skov T, Paludan-Muller G, et al. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J Allergy Clin Immunol. 2011;128(3):646–52 e1–5.
Abrahamsson TR, Jakobsson HE, Andersson AF, Bjorksten B, Engstrand L, Jenmalm MC. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin Exp Allergy. 2014;44(6):842–50.
Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med. 2015;7(307):307ra152.
Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med. 2016;22(10):1187–91.
Stokholm J, Blaser MJ, Thorsen J, Rasmussen MA, Waage J, Vinding RK, et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat Commun. 2018;9(1):141.
Kalliomaki M, Kirjavainen P, Eerola E, Kero P, Salminen S, Isolauri E. Distinct patterns of neonatal gut microflora in infants in whom atopy was and was not developing. J Allergy Clin Immunol. 2001;107(1):129–34.
Arrieta MC, Arevalo A, Stiemsma L, Dimitriu P, Chico ME, Loor S, et al. Associations between infant fungal and bacterial dysbiosis and childhood atopic wheeze in a nonindustrialized setting. J Allergy Clin Immunol. 2018;142(2):424–34. e10
Wheeler ML, Limon JJ, Bar AS, Leal CA, Gargus M, Tang J, et al. Immunological consequences of intestinal fungal Dysbiosis. Cell Host Microbe. 2016;19(6):865–73.
Shao TY, Ang WXG, Jiang TT, Huang FS, Andersen H, Kinder JM, et al. Commensal Candida albicans positively calibrates systemic Th17 immunological responses. Cell Host Microbe. 2019;25(3):404–17. e6
Bosch A, de Steenhuijsen Piters WAA, van Houten MA, Chu M, Biesbroek G, Kool J, et al. Maturation of the infant respiratory microbiota, Environmental drivers, and health consequences. A prospective cohort study. Am J Respir Crit Care Med. 2017;196(12):1582–90.
Mortensen MS, Brejnrod AD, Roggenbuck M, Abu Al-Soud W, Balle C, Krogfelt KA, et al. The developing hypopharyngeal microbiota in early life. Microbiome. 2016;4(1):70.
Bosch A, Levin E, van Houten MA, Hasrat R, Kalkman G, Biesbroek G, et al. Development of upper respiratory tract microbiota in infancy is affected by mode of delivery. EBioMedicine. 2016;9:336–45.
Kim BS, Lee E, Lee MJ, Kang MJ, Yoon J, Cho HJ, et al. Different functional genes of upper airway microbiome associated with natural course of childhood asthma. Allergy. 2018;73(3):644–52.
Mansbach JM, Hasegawa K, Piedra PA, Avadhanula V, Petrosino JF, Sullivan AF, et al. Haemophilus-dominant nasopharyngeal microbiota is associated with delayed clearance of respiratory syncytial virus in infants hospitalized for bronchiolitis. J Infect Dis. 2019;219(11):1804–8.
Zhou Y, Jackson D, Bacharier LB, Mauger D, Boushey H, Castro M, et al. The upper-airway microbiota and loss of asthma control among asthmatic children. Nat Commun. 2019;10(1):5714.
Teo SM, Tang HHF, Mok D, Judd LM, Watts SC, Pham K, et al. Airway microbiota dynamics uncover a critical window for interplay of pathogenic bacteria and allergy in childhood respiratory disease. Cell Host Microbe. 2018;24(3):341–52. e5
Beigelman A, Bacharier LB. Infection-induced wheezing in young children. J Allergy Clin Immunol. 2014;133(2):603–4.
Thorsen J, Rasmussen MA, Waage J, Mortensen M, Brejnrod A, Bonnelykke K, et al. Infant airway microbiota and topical immune perturbations in the origins of childhood asthma. Nat Commun. 2019;10(1):5001.
Morin A, McKennan CG, Pedersen CT, Stokholm J, Chawes BL, Malby Schoos AM, et al. Epigenetic landscape links upper airway microbiota in infancy with allergic rhinitis at 6 years of age. J Allergy Clin Immunol. 2020.
McCauley K, Durack J, Valladares R, Fadrosh DW, Lin DL, Calatroni A, et al. Distinct nasal airway bacterial microbiotas differentially relate to exacerbation in pediatric patients with asthma. J Allergy Clin Immunol. 2019;144(5):1187–97.
Masoli M, Fabian D, Holt S, Beasley R. Global initiative for Asthma P. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59(5):469–78.
Sood A, Qualls C, Schuyler M, Arynchyn A, Alvarado JH, Smith LJ, et al. Adult-onset asthma becomes the dominant phenotype among women by age 40 years. The longitudinal CARDIA study. Ann Am Thorac Soc. 2013;10(3):188–97.
Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181(4):315–23.
Samitas K, Zervas E, Gaga M. T2-low asthma: current approach to diagnosis and therapy. Curr Opin Pulm Med. 2017;23(1):48–55.
Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578.
Goleva E, Jackson LP, Harris JK, Robertson CE, Sutherland ER, Hall CF, et al. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am J Respir Crit Care Med. 2013;188(10):1193–201.
Marri PR, Stern DA, Wright AL, Billheimer D, Martinez FD. Asthma-associated differences in microbial composition of induced sputum. J Allergy Clin Immunol. 2013;131(2):346–52 e1–3.
Huang YJ, Nelson CE, Brodie EL, Desantis TZ, Baek MS, Liu J, et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol. 2011;127(2):372–81 e1–3.
Huang YJ, Nariya S, Harris JM, Lynch SV, Choy DF, Arron JR, et al. The airway microbiome in patients with severe asthma: associations with disease features and severity. J Allergy Clin Immunol. 2015;136(4):874–84.
Li N, Qiu R, Yang Z, Li J, Chung KF, Zhong N, et al. Sputum microbiota in severe asthma patients: relationship to eosinophilic inflammation. Respir Med. 2017;131:192–8.
Durack J, Christian LS, Nariya S, Gonzalez J, Bhakta NR, Ansel KM, et al. Distinct associations of sputum and oral microbiota with atopic, immunologic, and clinical features in mild asthma. J Allergy Clin Immunol. 2020;146(5):1016–26.
Durack J, Lynch SV, Nariya S, Bhakta NR, Beigelman A, Castro M, et al. Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J Allergy Clin Immunol. 2017;140(1):63–75.
Jung JW, Choi JC, Shin JW, Kim JY, Park IW, Choi BW, et al. Lung microbiome analysis in steroid-Nasmall yi, Ukrainianve asthma patients by using whole sputum. Tuberc Respir Dis (Seoul). 2016;79(3):165–78.
Colquhoun C, Duncan M, Grant G. Inflammatory bowel diseases: host-microbial-environmental interactions in dysbiosis. Diseases. 2020;8(2).
Lee JJ, Kim SH, Lee MJ, Kim BK, Song WJ, Park HW, et al. Different upper airway microbiome and their functional genes associated with asthma in young adults and elderly individuals. Allergy. 2019;74(4):709–19.
Pang Z, Wang G, Gibson P, Guan X, Zhang W, Zheng R, et al. Airway microbiome in different inflammatory phenotypes of asthma: a cross-sectional study in Northeast China. Int J Med Sci. 2019;16(3):477–85.
Sheflin AM, Melby CL, Carbonero F, Weir TL. Linking dietary patterns with gut microbial composition and function. Gut Microbes. 2017;8(2):113–29.
Laforest-Lapointe I, Arrieta MC. Patterns of early-life Gut microbial colonization during human immune development: an ecological perspective. Front Immunol. 2017;8:788.
Ianiro G, Tilg H, Gasbarrini A. Antibiotics as deep modulators of gut microbiota: between good and evil. Gut. 2016;65(11):1906–15.
Zarrinpar A, Chaix A, Xu ZZ, Chang MW, Marotz CA, Saghatelian A, et al. Antibiotic-induced microbiome depletion alters metabolic homeostasis by affecting gut signaling and colonic metabolism. Nat Commun. 2018;9(1):2872.
Yang X, Li H, Ma Q, Zhang Q, Wang C. Neutrophilic asthma is associated with increased airway bacterial burden and disordered community composition. Biomed Res Int. 2018;2018:9230234.
Yang X, Jiang Y, Wang C. Does IL-17 respond to the disordered lung microbiome and contribute to the neutrophilic phenotype in asthma? Mediat Inflamm. 2016;2016:6470364.
Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J. 2017;474(11):1823–36.
Mathieu E, Escribano-Vazquez U, Descamps D, Cherbuy C, Langella P, Riffault S, et al. Paradigms of lung microbiota functions in health and disease, particularly, in asthma. Front Physiol. 2018;9:1168.
Ghosh AR. Appraisal of microbial evolution to commensalism and pathogenicity in humans. Clin Med Insights Gastroenterol. 2013;6:1–12.
Pascal M, Perez-Gordo M, Caballero T, Escribese MM, Lopez Longo MN, Luengo O, et al. Microbiome and allergic diseases. Front Immunol. 2018;9:1584.
Samuelson DR, Welsh DA, Shellito JE. Regulation of lung immunity and host defense by the intestinal microbiota. Front Microbiol. 2015;6:1085.
Kim CH. Immune regulation by microbiome metabolites. Immunology. 2018;154(2):220–9.
Trivedi R, Barve K. Gut microbiome a promising target for management of respiratory diseases. Biochem J. 2020;477(14):2679–96.
Dumas A, Bernard L, Poquet Y, Lugo-Villarino G, Neyrolles O. The role of the lung microbiota and the gut-lung axis in respiratory infectious diseases. Cell Microbiol. 2018;20(12):e12966.
Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15(1):55–63.
Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo F, Roelofs JJ, de Boer JD, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut 2016;65(4):575–83.
Galvao I, Tavares LP, Correa RO, Fachi JL, Rocha VM, Rungue M, et al. The metabolic sensor GPR43 receptor plays a role in the control of Klebsiella pneumoniae infection in the lung. Front Immunol. 2018;9:142.
Chen CJ, Wu GH, Kuo RL, Shih SR. Role of the intestinal microbiota in the immunomodulation of influenza virus infection. Microbes Infect. 2017;19(12):570–9.
Hu Y, Feng Y, Wu J, Liu F, Zhang Z, Hao Y, et al. The gut microbiome signatures discriminate healthy from pulmonary tuberculosis patients. Front Cell Infect Microbiol. 2019;9:90.
Tulic MK, Vivinus-Nebot M, Rekima A, Rabelo Medeiros S, Bonnart C, Shi H, et al. Presence of commensal house dust mite allergen in human gastrointestinal tract: a potential contributor to intestinal barrier dysfunction. Gut. 2016;65(5):757–66.
Vignal C, Pichavant M, Alleman LY, Djouina M, Dingreville F, Perdrix E, et al. Effects of urban coarse particles inhalation on oxidative and inflammatory parameters in the mouse lung and colon. Part Fibre Toxicol. 2017;14(1):46.
Dickson RP, Singer BH, Newstead MW, Falkowski NR, Erb-Downward JR, Standiford TJ, et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol. 2016;1(10):16113.
Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20(2):159–66.
Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122(1):107–18.
Noverr MC, Falkowski NR, McDonald RA, McKenzie AN, Huffnagle GB. Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infect Immun. 2005;73(1):30–8.
Vital M, Harkema JR, Rizzo M, Tiedje J, Brandenberger C. Alterations of the murine gut microbiome with age and allergic airway disease. J Immunol Res. 2015;2015:892568.
Begley L, Madapoosi S, Opron K, Ndum O, Baptist A, Rysso K, et al. Gut microbiota relationships to lung function and adult asthma phenotype: a pilot study. BMJ Open Respir Res. 2018;5(1):e000324.
Ray A, Oriss TB, Wenzel SE. Emerging molecular phenotypes of asthma. Am J Physiol Lung Cell Mol Physiol. 2015;308(2):L130–40.
Ilmarinen P, Tuomisto LE, Kankaanranta H. Phenotypes, risk factors, and mechanisms of adult-onset asthma. Mediat Inflamm. 2015;2015:514868.
Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet. 2008;372(9643):1107–19.
Huber HL, Koessler KK. The pathology of bronchial asthma. Arch Intern Med. 1922;30(6):689–760.
Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. 2018;174(5):1054–66.
Coyle AJ, Le Gros G, Bertrand C, Tsuyuki S, Heusser CH, Kopf M, et al. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am J Respir Cell Mol Biol. 1995;13(1):54–9.
Anderson GP, Coyle AJ. TH2 and ‘TH2-like’ cells in allergy and asthma: pharmacological perspectives. Trends Pharmacol Sci. 1994;15(9):324–32.
Russell CD, Baillie JK. Treatable traits and therapeutic targets: goals for systems biology in infectious disease. Curr Opin Syst Biol. 2017;2:140–6.
Wenzel SE. Complex phenotypes in asthma: current definitions. Pulm Pharmacol Ther. 2013;26(6):710–5.
Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity. 2015;43(1):29–40.
Wenzel SE, Szefler SJ, Leung DY, Sloan SI, Rex MD, Martin RJ. Bronchoscopic evaluation of severe asthma. Persistent inflammation associated with high dose glucocorticoids. Am J Respir Crit Care Med. 1997;156(3 Pt 1):737–43.
Hastie AT, Moore WC, Li H, Rector BM, Ortega VE, Pascual RM, et al. Biomarker surrogates do not accurately predict sputum eosinophil and neutrophil percentages in asthmatic subjects. J Allergy Clin Immunol. 2013;132(1):72–80.
Hastie AT, Moore WC, Meyers DA, Vestal PL, Li H, Peters SP, et al. Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J Allergy Clin Immunol. 2010;125(5):1028–36. e13
Linden A. Role of interleukin-17 and the neutrophil in asthma. Int Arch Allergy Immunol. 2001;126(3):179–84.
Green RH, Brightling CE, Woltmann G, Parker D, Wardlaw AJ, Pavord ID. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax. 2002;57(10):875–9.
Sverrild A, Kiilerich P, Brejnrod A, Pedersen R, Porsbjerg C, Bergqvist A, et al. Eosinophilic airway inflammation in asthmatic patients is associated with an altered airway microbiome. J Allergy Clin Immunol. 2017;140(2):407–17. e11
Fitzpatrick AM, Chipps BE, Holguin F, Woodruff PG. T2-“low” asthma: overview and management strategies. J Allergy Clin Immunol Pract. 2020;8(2):452–63.
Sze E, Bhalla A, Nair P. Mechanisms and therapeutic strategies for non-T2 asthma. Allergy. 2020;75(2):311–25.
Taylor SL, Leong LEX, Choo JM, Wesselingh S, Yang IA, Upham JW, et al. Inflammatory phenotypes in patients with severe asthma are associated with distinct airway microbiology. J Allergy Clin Immunol. 2018;141(1):94–103.e15.
McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181(6):4089–97.
Al-Ramli W, Préfontaine D, Chouiali F, Martin JG, Olivenstein R, Lemière C, et al. TH17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol. 2009;123(5):1185–7.
Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. 2015;7(301):301ra129.
Nakagome K, Matsushita S, Nagata M. Neutrophilic inflammation in severe asthma. Int Arch Allergy Immunol. 2012;158(Suppl 1):96–102.
Chesne J, Braza F, Mahay G, Brouard S, Aronica M, Magnan A. IL-17 in severe asthma. Where do we stand? Am J Respir Crit Care Med. 2014;190(10):1094–101.
Bullone M, Carriero V, Bertolini F, Folino A, Mannelli A, Di Stefano A, et al. Elevated serum IgE, oral corticosteroid dependence and IL-17/22 expression in highly neutrophilic asthma. Eur Respir J. 2019;54(5).
Hynes GM, Hinks TSC. The role of interleukin-17 in asthma: a protective response? ERJ Open Res. 2020;6(2).
Wood LG, Simpson JL, Hansbro PM, Gibson PG. Potentially pathogenic bacteria cultured from the sputum of stable asthmatics are associated with increased 8-isoprostane and airway neutrophilia. Free Radic Res. 2010;44(2):146–54.
Schwerk N, Brinkmann F, Soudah B, Kabesch M, Hansen G. Wheeze in preschool age is associated with pulmonary bacterial infection and resolves after antibiotic therapy. PLoS One. 2011;6(11):e27913.
Ghebre MA, Pang PH, Diver S, Desai D, Bafadhel M, Haldar K, et al. Biological exacerbation clusters demonstrate asthma and chronic obstructive pulmonary disease overlap with distinct mediator and microbiome profiles. J Allergy Clin Immunol. 2018;141(6):2027–36. e12
Demarche S, Schleich F, Henket M, Paulus V, Van Hees T, Louis R. Detailed analysis of sputum and systemic inflammation in asthma phenotypes: are paucigranulocytic asthmatics really non-inflammatory? BMC Pulm Med. 2016;16:46.
Essilfie AT, Simpson JL, Dunkley ML, Morgan LC, Oliver BG, Gibson PG, et al. Combined Haemophilus influenzae respiratory infection and allergic airways disease drives chronic infection and features of neutrophilic asthma. Thorax. 2012;67(7):588–99.
Yang X, Wang Y, Zhao S, Wang R, Wang C. Long-term exposure to low-dose Haemophilus influenzae during allergic airway disease drives a steroid-resistant neutrophilic inflammation and promotes airway remodeling. Oncotarget. 2018;9(38):24898–913.
Haldar K, Bafadhel M, Lau K, Berg A, Kwambana B, Kebadze T, et al. Microbiome balance in sputum determined by PCR stratifies COPD exacerbations and shows potential for selective use of antibiotics. PLoS One. 2017;12(8):e0182833.
Ford ES. The epidemiology of obesity and asthma. J Allergy Clin Immunol. 2005;115(5):897–909. quiz 10
Taylor B, Mannino D, Brown C, Crocker D, Twum-Baah N, Holguin F. Body mass index and asthma severity in the National Asthma Survey. Thorax. 2008;63(1):14–20.
Sideleva O, Dixon AE. The many faces of asthma in obesity. J Cell Biochem. 2014;115(3):421–6.
Rasmussen F, Hancox RJ. Mechanisms of obesity in asthma. Curr Opin Allergy Clin Immunol. 2014;14(1):35–43.
Sood A, Cui X, Qualls C, Beckett WS, Gross MD, Steffes MW, et al. Association between asthma and serum adiponectin concentration in women. Thorax. 2008;63(10):877–82.
Sood A, Qualls C, Schuyler M, Thyagarajan B, Steffes MW, Smith LJ, et al. Low serum adiponectin predicts future risk for asthma in women. Am J Respir Crit Care Med. 2012;186(1):41–7.
Dixon AE, Shade DM, Cohen RI, Skloot GS, Holbrook JT, Smith LJ, et al. Effect of obesity on clinical presentation and response to treatment in asthma. J Asthma. 2006;43(7):553–8.
Sood A, Dominic E, Qualls C, Steffes MW, Thyagarajan B, Smith LJ, et al. Serum adiponectin is associated with adverse outcomes of asthma in men but not in women. Front Pharmacol. 2011;2:55.
Raviv S, Dixon AE, Kalhan R, Shade D, Smith LJ. Effect of obesity on asthma phenotype is dependent upon asthma severity. J Asthma. 2011;48(1):98–104.
Dixon AE, Pratley RE, Forgione PM, Kaminsky DA, Whittaker-Leclair LA, Griffes LA, et al. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol. 2011;128(3):508–15 e1–2.
Sutherland ER, Lehman EB, Teodorescu M, Wechsler ME, National Heart L. Blood Institute’s Asthma Clinical Research N. Body mass index and phenotype in subjects with mild-to-moderate persistent asthma. J Allergy Clin Immunol. 2009;123(6):1328–34. e1
Camargo CA Jr, Boulet LP, Sutherland ER, Busse WW, Yancey SW, Emmett AH, et al. Body mass index and response to asthma therapy: fluticasone propionate/salmeterol versus montelukast. J Asthma. 2010;47(1):76–82.
Sutherland ER, Camargo CA Jr, Busse WW, Meltzer EO, Ortega HG, Yancey SW, et al. Comparative effect of body mass index on response to asthma controller therapy. Allergy Asthma Proc. 2010;31(1):20–5.
Telenga ED, Tideman SW, Kerstjens HA, Hacken NH, Timens W, Postma DS, et al. Obesity in asthma: more neutrophilic inflammation as a possible explanation for a reduced treatment response. Allergy. 2012;67(8):1060–8.
King GG, Brown NJ, Diba C, Thorpe CW, Munoz P, Marks GB, et al. The effects of body weight on airway calibre. Eur Respir J. 2005;25(5):896–901.
Farah CS, Kermode JA, Downie SR, Brown NJ, Hardaker KM, Berend N, et al. Obesity is a determinant of asthma control independent of inflammation and lung mechanics. Chest. 2011;140(3):659–66.
Manna P, Jain SK. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: causes and therapeutic strategies. Metab Syndr Relat Disord. 2015;13(10):423–44.
Leiria LO, Martins MA, Saad MJ. Obesity and asthma: beyond T(H)2 inflammation. Metab Clin Exp. 2015;64(2):172–81.
Todd DC, Armstrong S, D’Silva L, Allen CJ, Hargreave FE, Parameswaran K. Effect of obesity on airway inflammation: a cross-sectional analysis of body mass index and sputum cell counts. Clin Exp Allergy. 2007;37(7):1049–54.
van Veen IH, Ten Brinke A, Sterk PJ, Rabe KF, Bel EH. Airway inflammation in obese and nonobese patients with difficult-to-treat asthma. Allergy. 2008;63(5):570–4.
Medoff BD, Okamoto Y, Leyton P, Weng M, Sandall BP, Raher MJ, et al. Adiponectin deficiency increases allergic airway inflammation and pulmonary vascular remodeling. Am J Respir Cell Mol Biol. 2009;41(4):397–406.
Verbout NG, Benedito L, Williams AS, Kasahara DI, Wurmbrand AP, Si H, et al. Impact of adiponectin overexpression on allergic airways responses in mice. J Allergy. 2013;2013:349520.
Kasahara DI, Kim HY, Williams AS, Verbout NG, Tran J, Si H, et al. Pulmonary inflammation induced by subacute ozone is augmented in adiponectin-deficient mice: role of IL-17A. J Immunol. 2012;188(9):4558–67.
Sideleva O, Suratt BT, Black KE, Tharp WG, Pratley RE, Forgione P, et al. Obesity and asthma: an inflammatory disease of adipose tissue not the airway. Am J Respir Crit Care Med. 2012;186(7):598–605.
Zheng H, Zhang X, Castillo EF, Luo Y, Liu M, Yang XO. Leptin enhances TH2 and ILC2 responses in allergic airway disease. J Biol Chem. 2016;291(42):22043–52.
Aydin M, Koca C, Ozol D, Uysal S, Yildirim Z, Kavakli HS, et al. Interaction of metabolic syndrome with asthma in postmenopausal women: role of adipokines. Inflammation. 2013;36(6):1232–8.
Muc M, Mota-Pinto A, Padez C. Association between obesity and asthma – epidemiology, pathophysiology and clinical profile. Nutr Res Rev. 2016:1–8.
Aujla SJ, Ross KR, Chmiel JF, Holguin F. Airway molecular phenotypes in pediatric asthma. Curr Opin Allergy Clin Immunol. 2011;11(2):122–6.
Lang JE, Hossain J, Dixon AE, Shade D, Wise RA, Peters SP, et al. Does age impact the obese asthma phenotype? Longitudinal asthma control, airway function, and airflow perception among mild persistent asthmatics. Chest. 2011;140(6):1524–33.
Chapman TJ, Georas SN. Regulatory tone and mucosal immunity in asthma. Int Immunopharmacol. 2014;23(1):330–6.
White SR, Laxman B, Naureckas ET, Hogarth DK, Solway J, Sperling AI, et al. Evidence for an IL-6–high asthma phenotype in asthmatic patients of African ancestry. J Allergy Clin Immunol. 2019;144(1):304–6.e4.
Peters MC, McGrath KW, Hawkins GA, Hastie AT, Levy BD, Israel E, et al. Plasma interleukin-6 concentrations, metabolic dysfunction, and asthma severity: a cross-sectional analysis of two cohorts. Lancet Respir Med. 2016;4(7):574–84.
Sharma A, Laxman B, Naureckas ET, Hogarth DK, Sperling AI, Solway J, et al. Associations between fungal and bacterial microbiota of airways and asthma endotypes. J Allergy Clin Immunol. 2019;144(5):1214–27. e7.
Auchtung TA, Fofanova TY, Stewart CJ, Nash AK, Wong MC, Gesell JR, et al. Investigating colonization of the healthy adult gastrointestinal tract by fungi. mSphere. 2018;3(2).
Kim YG, Udayanga KG, Totsuka N, Weinberg JB, Nunez G, Shibuya A. Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE(2). Cell Host Microbe. 2014;15(1):95–102.
Li X, Leonardi I, Semon A, Doron I, Gao IH, Putzel GG, et al. Response to fungal dysbiosis by gut-resident CX3CR1(+) mononuclear phagocytes aggravates allergic airway disease. Cell Host Microbe. 2018;24(6):847–56. e4
Peay KG, Kennedy PG, Talbot JM. Dimensions of biodiversity in the Earth mycobiome. Nat Rev Microbiol. 2016;14(7):434–47.
Hollins PD, Kettlewell PS, Atkinson MD, Stephenson DB, Corden JM, Millington WM, et al. Relationships between airborne fungal spore concentration of Cladosporium and the summer climate at two sites in Britain. Int J Biometeorol. 2004;48(3):137–41.
Tischer CG, Hohmann C, Thiering E, Herbarth O, Muller A, Henderson J, et al. Meta-analysis of mould and dampness exposure on asthma and allergy in eight European birth cohorts: an ENRIECO initiative. Allergy. 2011;66(12):1570–9.
Sharpe RA, Bearman N, Thornton CR, Husk K, Osborne NJ. Indoor fungal diversity and asthma: a meta-analysis and systematic review of risk factors. J Allergy Clin Immunol. 2015;135(1):110–22.
Salo PM, Yin M, Arbes SJ Jr, Cohn RD, Sever M, Muilenberg M, et al. Dustborne Alternaria alternata antigens in US homes: results from the National Survey of Lead and Allergens in Housing. J Allergy Clin Immunol. 2005;116(3):623–9.
Adams RI, Miletto M, Lindow SE, Taylor JW, Bruns TD. Airborne bacterial communities in residences: similarities and differences with fungi. PLoS One. 2014;9(3):e91283.
Mendell MJ, Mirer AG, Cheung K, Tong M, Douwes J. Respiratory and allergic health effects of dampness, mold, and dampness-related agents: a review of the epidemiologic evidence. Environ Health Perspect. 2011;119(6):748–56.
Pashley CH, Fairs A, Morley JP, Tailor S, Agbetile J, Bafadhel M, et al. Routine processing procedures for isolating filamentous fungi from respiratory sputum samples may underestimate fungal prevalence. Med Mycol. 2012;50(4):433–8.
Mak G, Porter PC, Bandi V, Kheradmand F, Corry DB. Tracheobronchial mycosis in a retrospective case-series study of five status asthmaticus patients. Clin Immunol. 2013;146(2):77–83.
Gergen PJ, Turkeltaub PC. The association of individual allergen reactivity with respiratory disease in a national sample: data from the second National Health and Nutrition Examination Survey, 1976–80 (NHANES II). J Allergy Clin Immunol. 1992;90(4 Pt 1):579–88.
Halonen M, Stern DA, Wright AL, Taussig LM, Martinez FD. Alternaria as a major allergen for asthma in children raised in a desert environment. Am J Respir Crit Care Med. 1997;155(4):1356–61.
Salo PM, Arbes SJ Jr, Sever M, Jaramillo R, Cohn RD, London SJ, et al. Exposure to Alternaria alternata in US homes is associated with asthma symptoms. J Allergy Clin Immunol. 2006;118(4):892–8.
Downs SH, Mitakakis TZ, Marks GB, Car NG, Belousova EG, Leuppi JD, et al. Clinical importance of Alternaria exposure in children. Am J Respir Crit Care Med. 2001;164(3):455–9.
Black PN, Udy AA, Brodie SM. Sensitivity to fungal allergens is a risk factor for life-threatening asthma. Allergy. 2000;55(5):501–4.
Agarwal R, Chakrabarti A. Allergic bronchopulmonary aspergillosis in asthma: epidemiological, clinical and therapeutic issues. Future Microbiol. 2013;8(11):1463–74.
Patterson TF, Thompson GR 3rd, Denning DW, Fishman JA, Hadley S, Herbrecht R, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;63(4):e1–e60.
Agarwal R, Chakrabarti A, Shah A, Gupta D, Meis JF, Guleria R, et al. Allergic bronchopulmonary aspergillosis: review of literature and proposal of new diagnostic and classification criteria. Clin Exp Allergy. 2013;43(8):850–73.
Agarwal R, Maskey D, Aggarwal AN, Saikia B, Garg M, Gupta D, et al. Diagnostic performance of various tests and criteria employed in allergic bronchopulmonary aspergillosis: a latent class analysis. PLoS One. 2013;8(4):e61105.
Denning DW, O’Driscoll BR, Hogaboam CM, Bowyer P, Niven RM. The link between fungi and severe asthma: a summary of the evidence. Eur Respir J. 2006;27(3):615–26.
Denning DW, O’Driscoll BR, Powell G, Chew F, Atherton GT, Vyas A, et al. Randomized controlled trial of oral antifungal treatment for severe asthma with fungal sensitization: the Fungal Asthma Sensitization Trial (FAST) study. Am J Respir Crit Care Med. 2009;179(1):11–8.
Thacher JD, Gruzieva O, Pershagen G, Melen E, Lorentzen JC, Kull I, et al. Mold and dampness exposure and allergic outcomes from birth to adolescence: data from the BAMSE cohort. Allergy. 2017;72(6):967–74.
Reponen T, Vesper S, Levin L, Johansson E, Ryan P, Burkle J, et al. High environmental relative moldiness index during infancy as a predictor of asthma at 7 years of age. Ann Allergy Asthma Immunol. 2011;107(2):120–6.
Williams PB, Barnes CS, Portnoy JM, Environmental AW. Innate and adaptive immune response to fungal products and allergens. J Allergy Clin Immunol Pract. 2016;4(3):386–95.
Bartemes KR, Kita H. Innate and adaptive immune responses to fungi in the airway. J Allergy Clin Immunol. 2018;142(2):353–63.
Iliev ID, Leonardi I. Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat Rev Immunol. 2017;17(10):635–46.
Li E, Landers CT, Tung HY, Knight JM, Marshall Z, Luong AU, et al. Fungi in mucoobstructive airway diseases. Ann Am Thorac Soc. 2018;15(Suppl 3):S198–204.
Gaitanis G, Magiatis P, Hantschke M, Bassukas ID, Velegraki A. The Malassezia genus in skin and systemic diseases. Clin Microbiol Rev. 2012;25(1):106–41.
Goldman DL, Chen Z, Shankar V, Tyberg M, Vicencio A, Burk R. Lower airway microbiota and mycobiota in children with severe asthma. J Allergy Clin Immunol. 2018;141(2):808–11. e7
Fraczek MG, Chishimba L, Niven RM, Bromley M, Simpson A, Smyth L, et al. Corticosteroid treatment is associated with increased filamentous fungal burden in allergic fungal disease. J Allergy Clin Immunol. 2018;142(2):407–14.
Stevens DA, Schwartz HJ, Lee JY, Moskovitz BL, Jerome DC, Catanzaro A, et al. A randomized trial of itraconazole in allergic bronchopulmonary aspergillosis. N Engl J Med. 2000;342(11):756–62.
Wark PA, Hensley MJ, Saltos N, Boyle MJ, Toneguzzi RC, Epid GD, et al. Anti-inflammatory effect of itraconazole in stable allergic bronchopulmonary aspergillosis: a randomized controlled trial. J Allergy Clin Immunol. 2003;111(5):952–7.
Patel AR, Patel AR, Singh S, Singh S, Khawaja I. Treating allergic bronchopulmonary aspergillosis: a review. Cureus. 2019;11(4):e4538.
Chishimba L, Niven RM, Cooley J, Denning DW. Voriconazole and posaconazole improve asthma severity in allergic bronchopulmonary aspergillosis and severe asthma with fungal sensitization. J Asthma. 2012;49(4):423–33.
Agbetile J, Bourne M, Fairs A, Hargadon B, Desai D, Broad C, et al. Effectiveness of voriconazole in the treatment of Aspergillus fumigatus-associated asthma (EVITA3 study). J Allergy Clin Immunol. 2014;134(1):33–9.
Chishimba L, Langridge P, Powell G, Niven RM, Denning DW. Efficacy and safety of nebulised amphotericin B (NAB) in severe asthma with fungal sensitisation (SAFS) and allergic bronchopulmonary aspergillosis (ABPA). J Asthma. 2015;52(3):289–95.
Schleimer RP. Glucocorticoids suppress inflammation but spare innate immune responses in airway epithelium. Proc Am Thorac Soc. 2004;1(3):222–30.
Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol. 2011;45(2):189–201.
Hertz CJ, Wu Q, Porter EM, Zhang YJ, Weismuller KH, Godowski PJ, et al. Activation of Toll-like receptor 2 on human tracheobronchial epithelial cells induces the antimicrobial peptide human beta defensin-2. J Immunol. 2003;171(12):6820–6.
Wang X, Zhang Z, Louboutin JP, Moser C, Weiner DJ, Wilson JM. Airway epithelia regulate expression of human beta-defensin 2 through Toll-like receptor 2. FASEB J. 2003;17(12):1727–9.
Homma T, Kato A, Hashimoto N, Batchelor J, Yoshikawa M, Imai S, et al. Corticosteroid and cytokines synergistically enhance toll-like receptor 2 expression in respiratory epithelial cells. Am J Respir Cell Mol Biol. 2004;31(4):463–9.
Wu Q, Jiang D, Minor MN, Martin RJ, Chu HW. In vivo function of airway epithelial TLR2 in host defense against bacterial infection. Am J Physiol Lung Cell Mol Physiol. 2011;300(4):L579–86.
Sim SH, Liu Y, Wang D, Novem V, Sivalingam SP, Thong TW, et al. Innate immune responses of pulmonary epithelial cells to Burkholderia pseudomallei infection. PLoS One. 2009;4(10):e7308.
Starner TD, Barker CK, Jia HP, Kang Y, McCray PB Jr. CCL20 is an inducible product of human airway epithelia with innate immune properties. Am J Respir Cell Mol Biol. 2003;29(5):627–33.
Persson C, Uller L. Glucocorticoids induce the production of the chemoattractant CCL20 in airway epithelium. Eur Respir J. 2015;45(3):859–60.
Zijlstra GJ, Fattahi F, Rozeveld D, Jonker MR, Kliphuis NM, van den Berge M, et al. Glucocorticoids induce the production of the chemoattractant CCL20 in airway epithelium. Eur Respir J. 2014;44(2):361–70.
van den Berge M, Jonker MR, Miller-Larsson A, Postma DS, Heijink IH. Effects of fluticasone propionate and budesonide on the expression of immune defense genes in bronchial epithelial cells. Pulm Pharmacol Ther. 2018;50:47–56.
Abbinante-Nissen JM, Simpson LG, Leikauf GD. Corticosteroids increase secretory leukocyte protease inhibitor transcript levels in airway epithelial cells. Am J Phys. 1995;268(4 Pt 1):L601–6.
Wedzicha JA, Calverley PM, Seemungal TA, Hagan G, Ansari Z, Stockley RA, et al. The prevention of chronic obstructive pulmonary disease exacerbations by salmeterol/fluticasone propionate or tiotropium bromide. Am J Respir Crit Care Med. 2008;177(1):19–26.
Calverley PM, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356(8):775–89.
Singh S, Amin AV, Loke YK. Long-term use of inhaled corticosteroids and the risk of pneumonia in chronic obstructive pulmonary disease: a meta-analysis. Arch Intern Med. 2009;169(3):219–29.
Morjaria JB, Rigby A, Morice AH. Inhaled corticosteroid use and the risk of pneumonia and COPD exacerbations in the UPLIFT study. Lung. 2017;195(3):281–8.
Hollis S, Jorup C, Lythgoe D, Martensson G, Regnell P, Eckerwall G. Risk of pneumonia with budesonide-containing treatments in COPD: an individual patient-level pooled analysis of interventional studies. Int J Chron Obstruct Pulmon Dis. 2017;12:1071–84.
Wang CY, Lai CC, Yang WC, Lin CC, Chen L, Wang HC, et al. The association between inhaled corticosteroid and pneumonia in COPD patients: the improvement of patients’ life quality with COPD in Taiwan (IMPACT) study. Int J Chron Obstruct Pulmon Dis. 2016;11:2775–83.
Sheffer AL, Silverman M, Woolcock AJ, Diaz PV, Lindberg B, Lindmark B. Long-term safety of once-daily budesonide in patients with early-onset mild persistent asthma: results of the inhaled steroid treatment as regular therapy in early asthma (START) study. Ann Allergy Asthma Immunol. 2005;94(1):48–54.
Woodcock A, Bateman ED, Busse WW, Lotvall J, Snowise NG, Forth R, et al. Efficacy in asthma of once-daily treatment with fluticasone furoate: a randomized, placebo-controlled trial. Respir Res. 2011;12:132.
Noonan M, Rosenwasser LJ, Martin P, O’Brien CD, O’Dowd L. Efficacy and safety of budesonide and formoterol in one pressurised metered-dose inhaler in adults and adolescents with moderate to severe asthma: a randomised clinical trial. Drugs. 2006;66(17):2235–54.
Htun ZM, Aldawudi I, Katwal PC, Jirjees S, Khan S. Inhaled corticosteroids as an associated risk factor for asthmatic pneumonia: a literature review. Cureus. 2020;12(6):e8717.
Rider CF, Altonsy MO, Mostafa MM, Shah SV, Sasse S, Manson ML, et al. Long-acting beta2-adrenoceptor agonists enhance glucocorticoid receptor (GR)-mediated transcription by gene-specific mechanisms rather than generic effects via GR. Mol Pharmacol. 2018;94(3):1031–46.
Marchini G, Carnevali S, Facchinetti F. Formoterol counteracts the inhibitory effect of cigarette smoke on glucocorticoid-induced leucine zipper (GILZ) transactivation in human bronchial smooth muscle cells. Eur J Pharmacol. 2019;850:8–14.
Normansell R, Sayer B, Waterson S, Dennett EJ, Del Forno M, Dunleavy A. Antibiotics for exacerbations of asthma. Cochrane Database Syst Rev. 2018;6:CD002741.
Johnston SL, Martin RJ. Chlamydophila pneumoniae and Mycoplasma pneumoniae: a role in asthma pathogenesis? Am J Respir Crit Care Med. 2005;172(9):1078–89.
Sutherland ER, Martin RJ. Asthma and atypical bacterial infection. Chest. 2007;132(6):1962–6.
Blasi F, Johnston SL. The role of antibiotics in asthma. Int J Antimicrob Agents. 2007;29(5):485–93.
Kraft M, Cassell GH, Pak J, Martin RJ. Mycoplasma pneumoniae and Chlamydia pneumoniae in asthma: effect of clarithromycin. Chest. 2002;121(6):1782–8.
Gil JC, Cedillo RL, Mayagoitia BG, Paz MD. Isolation of Mycoplasma pneumoniae from asthmatic patients. Ann Allergy. 1993;70(1):23–5.
Biscardi S, Lorrot M, Marc E, Moulin F, Boutonnat-Faucher B, Heilbronner C, et al. Mycoplasma pneumoniae and asthma in children. Clin Infect Dis. 2004;38(10):1341–6.
Wong C, Jayaram L, Karalus N, Eaton T, Tong C, Hockey H, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet. 2012;380(9842):660–7.
Altenburg J, de Graaff CS, Stienstra Y, Sloos JH, van Haren EH, Koppers RJ, et al. Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: the BAT randomized controlled trial. JAMA. 2013;309(12):1251–9.
Albert RK, Connett J, Bailey WC, Casaburi R, Cooper JA Jr, Criner GJ, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365(8):689–98.
Equi A, Balfour-Lynn IM, Bush A, Rosenthal M. Long term azithromycin in children with cystic fibrosis: a randomised, placebo-controlled crossover trial. Lancet. 2002;360(9338):978–84.
Yadav H, Peters SG, Keogh KA, Hogan WJ, Erwin PJ, West CP, et al. Azithromycin for the treatment of obliterative bronchiolitis after hematopoietic stem cell transplantation: a systematic review and meta-analysis. Biol Blood Marrow Transplant. 2016;22(12):2264–9.
Li H, Zhou Y, Fan F, Zhang Y, Li X, Yu H, et al. Effect of azithromycin on patients with diffuse panbronchiolitis: retrospective study of 51 cases. Intern Med. 2011;50(16):1663–9.
Lin X, Lu J, Yang M, Dong BR, Wu HM. Macrolides for diffuse panbronchiolitis. Cochrane Database Syst Rev. 2015;1:CD007716.
Kanoh S, Rubin BK. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin Microbiol Rev. 2010;23(3):590–615.
Parnham MJ, Erakovic Haber V, Giamarellos-Bourboulis EJ, Perletti G, Verleden GM, Vos R. Azithromycin: mechanisms of action and their relevance for clinical applications. Pharmacol Ther. 2014;143(2):225–45.
Aghai ZH, Kode A, Saslow JG, Nakhla T, Farhath S, Stahl GE, et al. Azithromycin suppresses activation of nuclear factor-kappa B and synthesis of pro-inflammatory cytokines in tracheal aspirate cells from premature infants. Pediatr Res. 2007;62(4):483–8.
Cheung PS, Si EC, Hosseini K. Anti-inflammatory activity of azithromycin as measured by its NF-kappaB, inhibitory activity. Ocul Immunol Inflamm. 2010;18(1):32–7.
Kobayashi Y, Wada H, Rossios C, Takagi D, Higaki M, Mikura S, et al. A novel macrolide solithromycin exerts superior anti-inflammatory effect via NF-kappaB inhibition. J Pharmacol Exp Ther. 2013;345(1):76–84.
Matsumura Y, Mitani A, Suga T, Kamiya Y, Kikuchi T, Tanaka S, et al. Azithromycin may inhibit interleukin-8 through suppression of Rac1 and a nuclear factor-kappa B pathway in KB cells stimulated with lipopolysaccharide. J Periodontol. 2011;82(11):1623–31.
Halldorsson S, Gudjonsson T, Gottfredsson M, Singh PK, Gudmundsson GH, Baldursson O. Azithromycin maintains airway epithelial integrity during Pseudomonas aeruginosa infection. Am J Respir Cell Mol Biol. 2010;42(1):62–8.
Mulet X, Macia MD, Mena A, Juan C, Perez JL, Oliver A. Azithromycin in Pseudomonas aeruginosa biofilms: bactericidal activity and selection of nfxB mutants. Antimicrob Agents Chemother. 2009;53(4):1552–60.
Hoffmann N, Lee B, Hentzer M, Rasmussen TB, Song Z, Johansen HK, et al. Azithromycin blocks quorum sensing and alginate polymer formation and increases the sensitivity to serum and stationary-growth-phase killing of Pseudomonas aeruginosa and attenuates chronic P. aeruginosa lung infection in Cftr(-/-) mice. Antimicrob Agents Chemother. 2007;51(10):3677–87.
Nagaoka K, Yanagihara K, Harada Y, Yamada K, Migiyama Y, Morinaga Y, et al. Macrolides inhibit Fusobacterium nucleatum-induced MUC5AC production in human airway epithelial cells. Antimicrob Agents Chemother. 2013;57(4):1844–9.
Liu Y, Pu Y, Li D, Zhou L, Wan L. Azithromycin ameliorates airway remodeling via inhibiting airway epithelium apoptosis. Life Sci. 2017;170:1–8.
Pu Y, Liu Y, Liao S, Miao S, Zhou L, Wan L. Azithromycin ameliorates OVA-induced airway remodeling in Balb/c mice via suppression of epithelial-to-mesenchymal transition. Int Immunopharmacol. 2018;58:87–93.
Serisier DJ. Risks of population antimicrobial resistance associated with chronic macrolide use for inflammatory airway diseases. Lancet Respir Med. 2013;1(3):262–74.
Kaplan MA, Goldin M. The use of triacetyloleandomycin in chronic infectious asthma. Antibiot Annu. 1958;6:273–6.
Itkin IH, Menzel ML. The use of macrolide antibiotic substances in the treatment of asthma. J Allergy. 1970;45(3):146–62.
Zeiger RS, Schatz M, Sperling W, Simon RA, Stevenson DD. Efficacy of troleandomycin in outpatients with severe, corticosteroid-dependent asthma. J Allergy Clin Immunol. 1980;66(6):438–46.
Szefler SJ, Rose JQ, Ellis EF, Spector SL, Green AW, Jusko WJ. The effect of troleandomycin on methylprednisolone elimination. J Allergy Clin Immunol. 1980;66(6):447–51.
Strunk RC, Bacharier LB, Phillips BR, Szefler SJ, Zeiger RS, Chinchilli VM, et al. Azithromycin or montelukast as inhaled corticosteroid-sparing agents in moderate-to-severe childhood asthma study. J Allergy Clin Immunol. 2008;122(6):1138–44. e4
Sutherland ER, King TS, Icitovic N, Ameredes BT, Bleecker E, Boushey HA, et al. A trial of clarithromycin for the treatment of suboptimally controlled asthma. J Allergy Clin Immunol. 2010;126(4):747–53.
Cameron EJ, Chaudhuri R, Mair F, McSharry C, Greenlaw N, Weir CJ, et al. Randomised controlled trial of azithromycin in smokers with asthma. Eur Respir J. 2013;42(5):1412–5.
Brusselle GG, Vanderstichele C, Jordens P, Deman R, Slabbynck H, Ringoet V, et al. Azithromycin for prevention of exacerbations in severe asthma (AZISAST): a multicentre randomised double-blind placebo-controlled trial. Thorax. 2013;68(4):322–9.
Hahn DL, Grasmick M, Hetzel S, Yale S, Group AS. Azithromycin for bronchial asthma in adults: an effectiveness trial. JABFM. 2012;25(4):442–59.
Simpson JL, Powell H, Boyle MJ, Scott RJ, Gibson PG. Clarithromycin targets neutrophilic airway inflammation in refractory asthma. Am J Respir Crit Care Med. 2008;177(2):148–55.
Hahn DL, Plane MB, Mahdi OS, Byrne GI. Secondary outcomes of a pilot randomized trial of azithromycin treatment for asthma. PLoS Clin Trials. 2006;1(2):e11.
Kostadima E, Tsiodras S, Alexopoulos EI, Kaditis AG, Mavrou I, Georgatou N, et al. Clarithromycin reduces the severity of bronchial hyperresponsiveness in patients with asthma. Eur Respir J. 2004;23(5):714–7.
Black PN, Blasi F, Jenkins CR, Scicchitano R, Mills GD, Rubinfeld AR, et al. Trial of roxithromycin in subjects with asthma and serological evidence of infection with Chlamydia pneumoniae. Am J Respir Crit Care Med. 2001;164(4):536–41.
Amayasu H, Yoshida S, Ebana S, Yamamoto Y, Nishikawa T, Shoji T, et al. Clarithromycin suppresses bronchial hyperresponsiveness associated with eosinophilic inflammation in patients with asthma. Ann Allergy Asthma Immunol. 2000;84(6):594–8.
Kew KM, Undela K, Kotortsi I, Ferrara G. Macrolides for chronic asthma. Cochrane Database Syst Rev. 2015;9:CD002997.
Kadota J, Mukae H, Ishii H, Nagata T, Kaida H, Tomono K, et al. Long-term efficacy and safety of clarithromycin treatment in patients with diffuse panbronchiolitis. Respir Med. 2003;97(7):844–50.
Slater M, Rivett DW, Williams L, Martin M, Harrison T, Sayers I, et al. The impact of azithromycin therapy on the airway microbiota in asthma. Thorax. 2014;69(7):673–4.
Lopes Dos Santos Santiago G, Brusselle G, Dauwe K, Deschaght P, Verhofstede C, Vaneechoutte D, et al. Influence of chronic azithromycin treatment on the composition of the oropharyngeal microbial community in patients with severe asthma. BMC Microbiol. 2017;17(1):109.
Gibson PG, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, et al. Efficacy of azithromycin in severe asthma from the AMAZES randomised trial. ERJ Open Res. 2019;5(4).
Taylor SL, Leong LEX, Mobegi FM, Choo JM, Wesselingh S, Yang IA, et al. Long-Term Azithromycin Reduces Haemophilus influenzae and Increases Antibiotic Resistance in Severe Asthma. Am J Respir Crit Care Med. 2019;200(3):309–17.
Zhou Y, Bacharier LB, Isaacson-Schmid M, Baty J, Schechtman KB, Sajol G, et al. Azithromycin therapy during respiratory syncytial virus bronchiolitis: upper airway microbiome alterations and subsequent recurrent wheeze. J Allergy Clin Immunol. 2016;138(4):1215–9.e5.
Choo JM, Abell GCJ, Thomson R, Morgan L, Waterer G, Gordon DL, et al. Impact of long-term erythromycin therapy on the oropharyngeal microbiome and resistance gene reservoir in non-cystic fibrosis bronchiectasis. mSphere. 2018;3(2).
Milstone AP. Use of azithromycin in the treatment of acute exacerbations of COPD. Int J Chron Obstruct Pulmon Dis. 2008;3(4):515–20.
Vermeersch K, Gabrovska M, Aumann J, Demedts IK, Corhay JL, Marchand E, et al. Azithromycin during acute chronic obstructive pulmonary disease exacerbations requiring hospitalization (BACE). A multicenter, randomized, double-blind, placebo-controlled trial predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med. 2019;200(7):857–68.
Simpson JL, Powell H, Baines KJ, Milne D, Coxson HO, Hansbro PM, et al. The effect of azithromycin in adults with stable neutrophilic COPD: a double blind randomised, placebo controlled trial. PLoS One. 2014;9(8):e105609.
Uzun S, Djamin RS, Kluytmans JA, Mulder PG, van’t Veer NE, Ermens AA, et al. Azithromycin maintenance treatment in patients with frequent exacerbations of chronic obstructive pulmonary disease (COLUMBUS): a randomised, double-blind, placebo-controlled trial Prophylactic antibiotic therapy for chronic obstructive pulmonary disease (COPD) Azithromycin for prevention of exacerbations of COPD. Lancet Respir Med. 2014;2(5):361–8.
Vermeersch K, Belmans A, Bogaerts K, Gyselinck I, Cardinaels N, Gabrovska M, et al. Treatment failure and hospital readmissions in severe COPD exacerbations treated with azithromycin versus placebo – a post-hoc analysis of the BACE randomized controlled trial. Respir Res. 2019;20(1):237.
Segal LN, Clemente JC, Wu BG, Wikoff WR, Gao Z, Li Y, et al. Randomised, double-blind, placebo-controlled trial with azithromycin selects for anti-inflammatory microbial metabolites in the emphysematous lung. Thorax. 2017;72(1):13–22.
Graham VA, Milton AF, Knowles GK, Davies RJ. Routine antibiotics in hospital management of acute asthma. Lancet. 1982;1(8269):418–20.
Shapiro GG, Eggleston PA, Pierson WE, Ray CG, Bierman CW. Double-blind study of the effectiveness of a broad spectrum antibiotic in status asthmaticus. Pediatrics. 1974;53(6):867–72.
Johansson MA, Sjogren YM, Persson JO, Nilsson C, Sverremark-Ekstrom E. Early colonization with a group of Lactobacilli decreases the risk for allergy at five years of age despite allergic heredity. PLoS One. 2011;6(8):e23031.
Bjorksten B, Sepp E, Julge K, Voor T, Mikelsaar M. Allergy development and the intestinal microflora during the first year of life. J Allergy Clin Immunol. 2001;108(4):516–20.
Sjogren YM, Jenmalm MC, Bottcher MF, Bjorksten B, Sverremark-Ekstrom E. Altered early infant gut microbiota in children developing allergy up to 5 years of age. Clin Exp Allergy. 2009;39(4):518–26.
Li L, Fang Z, Liu X, Hu W, Lu W, Lee YK, et al. Lactobacillus reuteri attenuated allergic inflammation induced by HDM in the mouse and modulated gut microbes. PLoS One. 2020;15(4):e0231865.
Wang X, Hui Y, Zhao L, Hao Y, Guo H, Ren F. Oral administration of Lactobacillus paracasei L9 attenuates PM2.5-induced enhancement of airway hyperresponsiveness and allergic airway response in murine model of asthma. PLoS One. 2017;12(2):e0171721.
Wu CT, Lin FH, Lee YT, Ku MS, Lue KH. Effect of Lactobacillus rhamnosus GG immunopathologic changes in chronic mouse asthma model. J Microbiol Immunol Infect. 2019;52(6):911–9.
Kalliomaki M, Salminen S, Arvilommi H, Kero P, Koskinen P, Isolauri E. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357(9262):1076–9.
Kalliomaki M, Salminen S, Poussa T, Arvilommi H, Isolauri E. Probiotics and prevention of atopic disease: 4-year follow-up of a randomised placebo-controlled trial. Lancet. 2003;361(9372):1869–71.
Kalliomaki M, Salminen S, Poussa T, Isolauri E. Probiotics during the first 7 years of life: a cumulative risk reduction of eczema in a randomized, placebo-controlled trial. J Allergy Clin Immunol. 2007;119(4):1019–21.
Kopp MV, Hennemuth I, Heinzmann A, Urbanek R. Randomized, double-blind, placebo-controlled trial of probiotics for primary prevention: no clinical effects of Lactobacillus GG supplementation. Pediatrics. 2008;121(4):e850–6.
Giovannini M, Agostoni C, Riva E, Salvini F, Ruscitto A, Zuccotti GV, et al. A randomized prospective double blind controlled trial on effects of long-term consumption of fermented milk containing Lactobacillus casei in pre-school children with allergic asthma and/or rhinitis. Pediatr Res. 2007;62(2):215–20.
Cabana MD, McKean M, Caughey AB, Fong L, Lynch S, Wong A, et al. Early probiotic supplementation for eczema and asthma prevention: a randomized controlled trial. Pediatrics. 2017;140(3).
Durack J, Kimes NE, Lin DL, Rauch M, McKean M, McCauley K, et al. Delayed gut microbiota development in high-risk for asthma infants is temporarily modifiable by lactobacillus supplementation. Nat Commun. 2018;9(1):707.
Chen YS, Jan RL, Lin YL, Chen HH, Wang JY. Randomized placebo-controlled trial of lactobacillus on asthmatic children with allergic rhinitis. Pediatr Pulmonol. 2010;45(11):1111–20.
Huang CF, Chie WC, Wang IJ. Efficacy of lactobacillus administration in school-age children with asthma: a randomized, placebo-controlled trial. Nutrients. 2018;10(11).
Moura JCV, Moura ICG, Gaspar GR, Mendes GMS, Faria BAV, Jentzsch NS, et al. The use of probiotics as a supplementary therapy in the treatment of patients with asthma: a pilot study and implications. Clinics (Sao Paulo). 2019;74:e950.
Du X, Wang L, Wu S, Yuan L, Tang S, Xiang Y, et al. Efficacy of probiotic supplementary therapy for asthma, allergic rhinitis, and wheeze: a meta-analysis of randomized controlled trials. Allergy Asthma Proc. 2019;40(4):250–60.
Steurer-Stey C, Lagler L, Straub DA, Steurer J, Bachmann LM. Oral purified bacterial extracts in acute respiratory tract infections in childhood: a systematic quantitative review. Eur J Pediatr. 2007;166(4):365–76.
Schaad UB, Mutterlein R, Goffin H, Group BV-CS. Immunostimulation with OM-85 in children with recurrent infections of the upper respiratory tract: a double-blind, placebo-controlled multicenter study. Chest. 2002;122(6):2042–9.
Gutierrez-Tarango MD, Berber A. Safety and efficacy of two courses of OM-85 BV in the prevention of respiratory tract infections in children during 12 months. Chest. 2001;119(6):1742–8.
Schaad UB. OM-85 BV, an immunostimulant in pediatric recurrent respiratory tract infections: a systematic review. World J Pediatr. 2010;6(1):5–12.
Navarro S, Cossalter G, Chiavaroli C, Kanda A, Fleury S, Lazzari A, et al. The oral administration of bacterial extracts prevents asthma via the recruitment of regulatory T cells to the airways. Mucosal Immunol. 2011;4(1):53–65.
Emeryk A, Bartkowiak-Emeryk M, Raus Z, Braido F, Ferlazzo G, Melioli G. Mechanical bacterial lysate administration prevents exacerbation in allergic asthmatic children-the EOLIA study. Pediatr Allergy Immunol. 2018;29(4):394–401.
Friedlander SL, Busse WW. The role of rhinovirus in asthma exacerbations. J Allergy Clin Immunol. 2005;116(2):267–73.
Mikhail I, Grayson MH. Asthma and viral infections: an intricate relationship. Ann Allergy Asthma Immunol. 2019;123(4):352–8.
Ramsahai JM, Hansbro PM, Wark PAB. Mechanisms and Management of Asthma Exacerbations. Am J Respir Crit Care Med. 2019;199(4):423–32.
Huang YJ, Erb-Downward JR, Dickson RP, Curtis JL, Huffnagle GB, Han MK. Understanding the role of the microbiome in chronic obstructive pulmonary disease: principles, challenges, and future directions. Transl Res. 2017;179:71–83.
Gibson PG, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (AMAZES): a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10095):659–68.
Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med. 2008;178(12):1271–81.
Smith CM, Kulkarni H, Radhakrishnan P, Rutman A, Bankart MJ, Williams G, et al. Ciliary dyskinesia is an early feature of respiratory syncytial virus infection. Eur Respir J. 2014;43(2):485–96.
Frey A, Lunding LP, Ehlers JC, Weckmann M, Zissler UM, Wegmann M. More than just a barrier: the immune functions of the airway epithelium in asthma pathogenesis. Front Immunol. 2020;11:761.
Denney L, Ho LP. The role of respiratory epithelium in host defence against influenza virus infection. Biom J. 2018;41(4):218–33.
Ritchie AI, Jackson DJ, Edwards MR, Johnston SL. Airway epithelial orchestration of innate immune function in response to virus infection. A focus on asthma. Ann Am Thorac Soc. 2016;(13 Suppl 1):S55–63.
Jiao J, Wang C, Zhang L. Epithelial physical barrier defects in chronic rhinosinusitis. Expert Rev Clin Immunol. 2019;15(6):679–88.
Looi K, Buckley AG, Rigby PJ, Garratt LW, Iosifidis T, Zosky GR, et al. Effects of human rhinovirus on epithelial barrier integrity and function in children with asthma. Clin Exp Allergy. 2018;48(5):513–24.
Lejeune S, Deschildre A, Le Rouzic O, Engelmann I, Dessein R, Pichavant M, et al. Childhood asthma heterogeneity at the era of precision medicine: modulating the immune response or the microbiota for the management of asthma attack. Biochem Pharmacol. 2020;179:114046.
Tsay JJ, Wu BG, Badri MH, Clemente JC, Shen N, Meyn P, et al. Airway microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am J Respir Crit Care Med. 2018;198(9):1188–98.
Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. 2010;328(5986):1662–8.
Herbst T, Sichelstiel A, Schar C, Yadava K, Burki K, Cahenzli J, et al. Dysregulation of allergic airway inflammation in the absence of microbial colonization. Am J Respir Crit Care Med. 2011;184(2):198–205.
Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD, et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med. 2014;20(6):642–7.
Singanayagam A, Ritchie AI, Johnston SL. Role of microbiome in the pathophysiology and disease course of asthma. Curr Opin Pulm Med. 2017;23(1):41–7.
Di Gangi A, Di Cicco ME, Comberiati P, Peroni DG. Go with your gut: the shaping of T-cell response by Gut microbiota in allergic asthma. Front Immunol. 2020;11:1485.
Kozik A, Huang YJ. Ecological interactions in asthma: from environment to microbiota and immune responses. Curr Opin Pulm Med. 2020;26(1):27–32.
Kozik AJ, Huang YJ. The microbiome in asthma: role in pathogenesis, phenotype, and response to treatment. Ann Allergy Asthma Immunol. 2019;122(3):270–5.
Acknowledgements
We acknowledge the many investigators, research staff, and the many patients with and without asthma around the world, who have participated in the studies contributing knowledge to the topics discussed in this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
White, S.R., Huang, Y.J. (2022). The Role of the Microbiome in Asthma Inception and Phenotype. In: Huang, Y.J., Garantziotis, S. (eds) The Microbiome in Respiratory Disease. Respiratory Medicine. Humana, Cham. https://doi.org/10.1007/978-3-030-87104-8_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-87104-8_5
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-87103-1
Online ISBN: 978-3-030-87104-8
eBook Packages: MedicineMedicine (R0)