
Abstract
Reactive oxygen species (ROS) transform cell responses through miscellaneous processes. These act as signalling molecules when present in low concentrations or damage cell machinery when present in high levels. ROS-mediated mitochondrial damage leads to lesser oxidative phosphorylation and enhanced cell death. Aberrant cell death and redox signalling are implicated in numerous pathological conditions such as cardiac, neurodegenerative, and metabolic diseases. Cells have adopted an auto-degradation process as a cytoprotective strategy by inhibition of aberrant cell death and oxidation stress along with activation of autophagy. Autophagy, a pervasive degradation or turnover homeostatic mechanism of cell organelles and components, involves differentiation and ageing. Any dysfunction of autophagy can lead to anomalous mitochondrial function and ROS. Redox signalling cascade is related to the etiology of ageing, and the cellular machinery that regulates redox, autophagy and ageing has been elucidated. Redox signalling orchestrates autophagy and ageing processes leading to altered transduction of redox homeostasis. This chapter discusses ageing process, generation of ROS, redox signalling, autophagy types and mechanisms, the interplay between redox signalling, autophagy and ageing.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Ageing
Ageing can be defined as an intrinsic, universal process experienced by all living beings and depicted as the steady accretion of molecular and cellular damage (Alonso-Fernández and De la Fuente 2011; Carmona and Michan 2016). Ageing is a heterochronic and heterogeneous process. The heterochronic nature of ageing can be defined as the asynchrony by which different tissues and cells age within an organism (Alonso-Fernández and De la Fuente 2011; Peng et al. 2014). Several prominent deleterious changes may occur because of ageing including telomere shortening, DNA damage, oxidative stress, and inflammatory senescence-associated secretory phenotype (SASP) (Cheon et al. 2019). Hence, ageing is one of the perfect examples of homeostasis deterioration being related to impaired biological systems (Abbas et al. 2017). As a heterogeneous process, ageing may occur across diverse organisms at various rates; even among members of the same species, ageing occurs at variable rates (Alonso-Fernández and De la Fuente 2011; Carmona and Michan 2016; Urtamo et al. 2019).
1.1 Ageing Process
Growing old is a process called quasi-programmed i.e., a plan that is sustained and not once switched off. It is a developmental program with aimless continuation even after the completion of assigned tasks, as predicted by evolutionary theory. Ageing is not caused by damage, in fact, damage occurs due to ageing (Blagosklonny 2008). The damage driven by ageing results in the deterioration of an organism’s body function, causing several diseases and gradually lessens the stress resistance (Davalli et al. 2016). The biogerontologists demonstrated that both environmental conditions and genetics contribute to the onset of ageing, which can be explained by the fact that 75% of individuals get older due to environmental factors including behavioural patterns, whereas 25% of individuals are accounted by genetics (Fernández-Ballesteros et al. 2013; Labat-Robert and Robert 2015). Therefore, other than genes, ageing is ascribed to socio-environmental conditions as well as behavioural and personal events (Theurey and Pizzo 2018).
1.2 Healthy Ageing
The principal challenge is to figure out the basis of healthy ageing. The study of desirable phenotypes of ageing that are healthy ageing and longevity has been mentioned as ‘positive biology’ (Farrelly 2012; Brooks-Wilson 2013). However, healthy ageing can be defined as warding off the decline of cellular and molecular processes for the longest period of the lifespan. Not surprisingly, increased longevity and healthy ageing have a strong association. The main difference between healthy ageing and longevity is that the former emphasizes health span and the latter is aimed at life span. Hence, both are closely related to each other because a person, who exceptionally has a long lifespan, tends to be healthy for much of his life (Fig. 7.1). This statement can be justified by the fact that certain factors such as stress resistance, protection against age-related diseases, and cellular homeostasis can be promoted by dietary, genetic, and/or pharmacological interventions which ultimately tend to enhance the lifespan and vice versa (Carmona and Michan 2016). The extrinsic skin age process is called photo-ageing, and the skin being an outermost organ is affected not only by ultraviolet radiation but also by other environmental factors including tobacco smoking and air pollution (Gu et al. 2020).

Factors contributing to ageing. Increased ROS/RNS (reactive nitrogen species) generation damage protein, mitochondria and this damage propagate to neighboring organelles (lysosomes), cells and other macromolecules (DNA). Dietary habits, sedentary lifestyle and environmental factors along with declined autophagy further aggravate the ageing process
1.3 Cellular Ageing
Cellular factors that influence ageing are strongly connected with progressively compromised autophagy (a process that causes degradation of cell debris) (Cheon et al. 2019). ROS along with genetic factors are the major cause of intrinsic ageing (Abbas et al. 2017). In diverse eukaryotic species, autophagy is associated with a vital supervisory position for ageing as it can eliminate impaired molecules and cell organelles (Wang and Xu 2020). The removal of defective mitochondria, a significantly selective pathway of autophagy, is known as mitophagy (Poljšak et al. 2012; Couve et al. 2013; Markaki et al. 2017).
Ageing symptoms can be mitigated by cellular antioxidant systems and restoration or induction of autophagy (Wong et al. 2020). Over the last 30 years, main anti-ageing cures; consumption of polyamine-rich antioxidant, autophagic inducers, and caloric control were found to be effective. Given the ever-increasing ageing population and human lifespan along with the occurrence of cardiovascular disease, it is mandatory to figure out the fundamental autophagy mechanisms. From an autophagic approach, two processes leading to ageing occur simultaneously i.e., mitophagy and augmentation in ROS levels (Ren and Zhang 2018; Barbosa et al. 2019).
2 Oxidative Stress and Ageing
How ageing is influenced by reactive oxygen species (ROS) is a mystery in biology (Ewald 2018). The free radicals or ROS are responsible for the development of ageing, as illustrated by theories on ageing that is from programmed cell death to ‘natural inevitable cell damage’. The report for ageing research would carefully and accurately be described as “It is the free radicals and oxygen is poisonous when it is present as reactive specie”. The remarkably common concept regarding ageing is the free radical concept of age established in earlier work (Gerschman 1954). Oxidative toxicity is instigated by free radicals as these radicals trigger the destruction of cell machinery and tissues. However, the production of free radicals generally arises through metabolic reactions. Thus, the concept inspired of Harman’s theory that comprehensive elimination of the so-called detrimental compounds could lessen such impairments, subsequently, slow the ageing progression (Abbas et al. 2017; Pomatto and Davies 2018).
Oxidative damage can be defined as free radical’s accumulation due to the over-production of free radicals that cannot be processed progressively or due to fewer antioxidants bioavailability (Weidinger and Kozlov 2015; Simioni et al. 2018). The decline of antioxidant defending ability as compared to ROS leads to oxidation tension. The surplus ROS can primarily promote inflammation and increased concomitant cytokine synthesis, which can additionally activate the formation of ROS (Pisoschi and Pop 2015; Luo et al. 2020a, b).
Oxidative stress promotes the development of ageing as well as several long-lasting and deteriorating ailments such as inflammation, arthritis, cancer, autoimmune disorders, cardiac disease and neuropathies (Chandrasekaran et al. 2017). Oxidative stress disrupts bio-signalling, due to which different pathophysiological events could occur and successive modifications at different life phases, specifically in the older stage (Szentesi et al. 2019). When acceptable equilibrium among antioxidant-oxidant processes is maintained, then the modifying influence on ROS synthesis and deactivation occurs continuously in normal as well as pathological settings (Davalli et al. 2016).
Since ROS are responsible for molecular damage, certain mechanisms have been evolved by the organisms for the protection against abnormally increased ROS. Both low and acute ROS exposure can trigger protective mechanisms (Ewald 2018). Recently a novel idea termed “hormesis” has been introduced, conferring that the cellular response for a more deleterious condition may be corrected by small dosages of a stressor. This could enhance cellular fitness and lifespan (Barbosa et al. 2019). This acute or low ROS exposure in model organisms can enhance lifespans such as in rodents, nematodes, flies, and yeast. Low or acute ROS levels can function as a secondary messenger at the physiological level to modify bio-signalling and play a significant role in an adjustment under oxidative stress (Ewald 2018). In this context, the beneficial effect of low levels of ROS is significant as it triggers the homeostatic responses; however, severe impairment or ageing could be due to its unequal accumulation. ROS performs several cellular activities such as inflammation, cell signal transduction, differentiation, cell survival, cell death, immune response, and gene transcription. Therefore, the balance between antioxidants (AOs) and oxidation species is important for a biological role such as adaptation, growth, and regulation (Warraich et al. 2020).
2.1 Synthesis of ROS
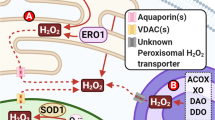
Reactive oxygen–nitrogen species (RONS) including potent nitrogen and oxygen species are normally generated as by-products during the cellular redox process. Such by-products may be non-radical as well as radical dynamic compounds (Pham-Huy et al. 2008; Powers et al. 2011). Numerous exogenous RONS sources are transition or heavy metals, drugs, radiation, alcohol, air pollution and tobacco. The endogenous RONS sources are lipoxygenase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, angiotensin II and myeloperoxidase (MPO). The main intracellular ROS source is enzymes (Liguori et al. 2018). ROS are produced within several cellular compartments. These compartments include mitochondria during oxidative phosphorylation, NADPH oxidase at the plasma membrane, cyclooxygenases, oxidase, cytoplasm, and lipid oxidation inside the peroxisomes (Campisi et al. 2019). Although all these sources contribute to producing an overall oxidative burden during ageing, maximum production of ROS occurs during oxidative phosphorylation. In addition to these sources, other major endogenous oxidant sources are monoamine oxidase, nitric oxide synthase, cytochrome p450, mitochondrial respiratory chain (RC), several oxidoreductases such as xanthine oxidase and enzymes that activate xenobiotic such as NADPH oxidases by causing inflammatory and infection responses (Warraich et al. 2020).
Oxidant generation from all these sources can vary with pathophysiological conditions and increases with age. However, contact with endogenous sources of oxidants is much more extensive as the internal oxidants are produced continuously during the lifetime (Warraich et al. 2020). The radicals and non-radical compounds that can trigger any deleterious response due to free oxygen are identified as ROS and comprise of superoxide anion (O−), H2O2 (hydrogen peroxide), hydroperoxyl (HO2), hydroxidochlorine (HClO), dioxygen (O=O) alkoxyl radical (R–O⋅), and hydroxyl radical (HO⋅). ROS include derivatives of oxygen and are either non-radical or radical oxidizing agents and/or are easily convertible to radicals (Lambert and Brand 2009; Cui et al. 2012; Genova and Lenaz 2015).
The sequential oxidation reduction reactions of mitochondria generate ATP along with free radicals. Other sources of free radicals are vigorous physical activity and infectious conditions that involve activation of phagocytes (Pham-Huy et al. 2008; Pisoschi and Pop 2015). Free radicals in bio-systems were observed in the 1950s and since then it was believed that these radicals contribute largely to diverse pathological processes and ageing (Lushchak 2015). The interaction of oxygen with certain molecules produces radicals with an outmost shell having unpaired electrons such as O2 i.e., diatomic oxygen. These potent radicals in a cell are generated by the removal or gain of a single electron and behave as reductants or oxidants (Bailey 2019). The same spin quantum number is carried by electrons when all electrons are not located in the same π* anti-bonding orbital. This parallel spin is responsible for their less reactivity with non-radical species. Conversion of O2 (molecular oxygen) into extra active singlet oxygen O2 (dioxygen) is likely to happen by the supply of energy to reverse the rotation of one electron. Therefore, both electrons either occupy the same π* orbital by pairing or they may remain in two discrete orbitals. The spin constraint can be controlled by the addition of an electron to oxygen. Non-radicals are synthesized during stress conditions in the body and the normal aerobic metabolism such as H2O2 (Genova and Lenaz 2015; Liguori et al. 2018). Numerous forms of ROS possess distinctive characteristics. hiROS (highly reactive oxygen species) damage biomolecules while loROS (less reactive oxygen species) impair cell signalling mechanism (Scialò et al. 2017). The term “ROS physioma” is used for the hiROS family and includes superoxide anion, hydroxyl radical and hydrogen peroxide. Superoxide radicals participate in the generation of other ROS such as H2O2 and HO⋅ (Zarkovic 2020).
The manifestation of cellular degenerative characteristics of Friedreich’s ataxia (FRDA) is due to mitochondrial ROS generation and is enhanced by the deficient mitochondrial frataxin. The increased level of ROS in FRDA cells is due to the defect produced in complex IV by defective synthesis of heme and the damaged cytochrome c as well as heme in complex IV and III. Thus, heme defect in FRDA results in limited oxidase and cytochrome c action as well as increased mitochondrial ROS, all take place due to defective iron-sulphur centres. Consequently, shortage of functional heme, the presence of defective heme and eventually increased ROS synthesis happens (Napoli et al. 2006).
Several diseases have a strong association with ageing, which are progressed due to the stimulation of a protein pore mPTP (mitochondrial permeability transition pore) on the internal membrane where the development of the voltage-gated channel occurs, stimulated by calcium overloaded mitochondria and ROS. This initiates the production of more ROS by the complete opening of mPTP, and it may also cause matrix DNA to be released into inter-membrane space for the hydrolytic reaction. This results in cell DNA depletion thus contributing to the activation of the ageing (Rottenberg and Hoek 2017). It is assumed that the opening of mPTP is increased by ageing and vice versa. ROS also initiates the stimulation of mPTP. The activated mPTP causes damage to mitochondria during oxidative reactions, hence further worsening the clinical features of FRDA. Many deleterious properties of dysfunctional mitochondria and ROS towards ageing and longevity are interceded by activated mPTP (Panel et al. 2018).
3 Ageing and Mitochondria
In human mtDNA (mitochondrial DNA), 16,659 base pairs are present. Mammalian cells contain several copies of mitochondrial DNA. Human mtDNA codes for rRNA, tRNA, and thirteen different proteins which are crucial for the functional and structural stability of mitochondria (Dröse and Brandt 2012). Frequent studies indicate that the mutations in mitochondrial DNA may affect the production of ATP. Neurodegenerative diseases and early phases of ageing are associated with disturbance in the integrity of mitochondrial DNA. In mitochondria, various metabolic pathways including TCA (tri-carboxylic acid) cycle, beta-oxidation of fatty acids, amino acids oxidation and one-carbon cycle takes place. Mitochondria are responsible for generating 90% of the cell’s energy (Warraich et al. 2020).
Ageing at the molecular level is caused by a lifelong accumulation of numerous damages, many of which have yet to be completely elucidated (Table 7.1). Functional abnormalities in mitochondria are considered indicators of ageing (Bolduc et al. 2019). Over time, the mitochondrial DNA destruction and production of ROS occurs which ultimately leads to the cell’s inefficiency to recognize the vital function of mitochondria. This information contributed to the formation of MFRTA (mitochondrial free radical theory of ageing) theory although it is controversial to some extent (Son and Lee 2019). Many researchers have updated, extended, and questioned it. Nonetheless, two basic statements were not revised. First; the antioxidant/oxidant imbalance ensues with ageing, which causes the accumulation of damaged macromolecules. Second, oxidative damage causes the degenerative phenotype of ageing. A few studies questioned the second statement, but the first statement is well known that in the elderly, damaged macromolecules accumulate due to the antioxidant/oxidant imbalance (Luo et al. 2020a, b).
MFRTA theory (Lara et al. 2018) is currently termed as OST (oxidative stress theory). Several studies indicate that during ageing, alteration in mitochondria and mtDNA, accelerated structural disintegration, declined phosphorylation during aerobic metabolism, improved synthesis of ROS, and harmful impact on nucleic acid, fats, and protein occur (Haas 2019). An estimated mitochondrial mutation rate is ten times greater than nuclear DNA. The mtDNA has lesser repairing ability and deficiency of histones which influence cancer and ageing (Kammeyer and Luiten 2015).
As concluded from the above review of literature, the mammalian mitochondria are the central places for ROS production and thus the mtDNA is more vulnerable to harm due to their existence adjacent to the ROS production site. It has been demonstrated that mitochondrial DNA contains a large amount of 8-hydroxy-20-deoxyguanosine (a by-product of oxidation) than DNA present in the nucleus (Cui et al. 2012). The decreased mitochondrial function correlates with the emergence of several ailments. However, the general idea is that ROS causes the accumulation of damaged macromolecules which leads to ageing. The number of impaired mitochondria is increased during ageing, and it produces less ATP and more ROS. In cells, the harmful effect due to DNA damage occurs by blocking replication, breaks in DNA strands, transcription, and rearrangement of chromosomes (Warraich et al. 2020).
With the increase in age, the progressive oxidative reactions induce the phenomenon of strand breaks in DNA and mutations in somatic mitochondrial DNA. The RC complex damage occurs by the mutations in mitochondrial DNA which results in a cycle having more alternations in mtDNA and increased synthesis of ROS (Bonomini et al. 2015). Mitochondrial damage and energy crisis due to ROS and oxidation stress cause neurodegenerative disorders and trigger ageing. Destruction of mtDNA is linked with ageing, but it is not clear that which one is directly affecting vascular ageing; mitochondrial DNA damage or mitochondrial dysfunction (Foote et al. 2018; Stefanatos and Sanz 2018).
3.1 ROS Production in Mitochondria
Chance and colleagues documented the ROS production in the respiratory chain for the first time in 1966 (Chance et al. 1979). Within the chain, prominent sources of ROS production are the first and third complexes when a direct reaction between electrons derived from NADH, ubiquinone and oxygen or other electron acceptors occurs, resulting in the generation of free radicals. Thus, during the respiration process, ROS are the normal side products but their reaction with fats, proteins and nucleic acids deteriorates these molecules (Marchi et al. 2012). The isolated mitochondria generate a significant proportion of ROS by two approaches, principally through (i) first complex in a certain condition such as reduced coenzyme Q and high proton motive force (Fp) as there is no ATP synthesis; (ii) when a higher ratio of reduced and oxidized NAD present in the mitochondrial matrix. When active production of ATP occurs by mitochondria, less amount of O2 (singlet oxygen) is produced, consequently, lower NADH/NAD+ and Fp ratio are present. ROS generation in isolated mitochondria takes place at eleven sites, although only three sites are more relevant for in vivo ROS production: complex I (NADH dehydrogenase), complex II (succinate dehydrogenase), and complex III (cytochrome bc1 complex). However, the recent investigation indicated that at complex II, the substantial formation of O2 (singlet oxygen) occurs (Eleutherio et al. 2018).
According to Brand (2016), hydrogen-free radicals are formed in mammalian mitochondria at eleven different sites. Each site is closely related to substrate catabolism and RC and has its unique properties. Sequential action of electrons transport through mitochondrial chain complexes produces a difference of chemical concentration and electric charges. Eventually, proton transport takes place from the matrix to the inter-membrane part. During this transmission, the direct electron transfer to oxygen chiefly at I, II, and III complexes results in the production of superoxide (one electron transport) or hydrogen peroxide (two electrons transport) (Eleutherio et al. 2018).
Surprisingly, ROS generation does not occur at complex IV (cytochrome c oxidase), although it causes reduction of oxygen to water, elaborating that ROS production and electron leakage in RC can be inhibited. ROS generation in RC complexes is significant in the regulation of different biochemical processes such as cellular differentiation, and deterioration in ischemia/reoxygenation injury (Scialò et al. 2017), whereas the ROS also mediates the progression of some cancer types. The ROS formed from the third complex are shared between the mitochondrial matrix and IMS. While ROS generated by complex 1 are transferred to the matrix only, thereby illustrating the adoption of different distribution mechanisms for both the complexes (Reczek and Chandel 2015; Stefanatos and Sanz 2018).
When the electron transfer takes place at complex I (from NADH to CoQ), the generation of ROS may occur at two places, IQ (CoQ attachment position) and IF (FMN position) within the matrix. ROS generation in complex II takes place on IIF position, having an association with succinate dehydrogenase. The small amount of ROS production by complex III is negligible in comparison to ROS produced from CI. The ROS formed by CIII is transported into IMS and matrix. Within IMS and matrix, ROS are converted into H2O2, reactions catalyzed by enzymes (superoxide dismutase) SOD2 (matrix) and SOD1 (IMS). This H2O2 has a significant function in physiological mechanisms. Superoxide presumably functions at the site of its synthesis is short-lived and has less membrane-penetrability in comparison to the H2O2 that is uncharged, extra stable and can pass through aquaporin channels, thus a better multipurpose signalling particle. Though analogous to superoxide molecules, substantial destruction of molecules by H2O2 takes place in the presence of free Fe2+. The presence of this cation results in the production of highly active OH-radicals as shown in Fig. 7.2 (Warraich et al. 2020).

Mitochondrial redox mediated signaling. ROS in mitochondria is produced from the escape of electrons to form superoxide (O2−) in complex I and complex III of the electron transport chain. O2 − is converted to H2O2 by SOD1 and SOD2 in intermembrane space and matrix. H2O2 is reduced to water by Gpx. Both O2− and H2O2 are mtROS that act as signaling molecules triggering metabolic processes in the cytoplasm and nucleus. SOD: Superoxide dismutase, Gpx: Glutathione peroxidase, NO: Nitric oxide, PTM: Post-transcriptional modifications
The generation of a progenitor ROS i.e., superoxide anion occurs at almost nine mitochondrial sites (Andreyev et al. 2005). Moreover, NADPH oxidases also produce superoxide, which catalyses the controlled formation of O2 (singlet oxygen) by coupling the electrons derived from NADPH to oxygen. The topological significance of sites generating ROS and variation among various ROS peculiarly superoxide and hydrogen peroxide should be appreciated. For example, superoxide in matrix generating oxidation–reduction bio-signalling at IQ site may vary from the superoxide at complex IIIQo that sends such bio-signalling into the cytosol. Likewise, hydrogen peroxide producing redox signalling from both these will be different (Warraich et al. 2020).
A momentous correlation between ROS production rate, RC complexes action, and mitochondrial membrane potential (ΔΨm) exists. If increased production of ROS occurs by the dissipation of mitochondrial membrane potential and respiration is inhibited, then the rate of free radical formation can be reduced by uncoupling which activates the drop in ΔΨm (Angelova and Abramov 2018). Even though in RC a large amount of ROS generation occurs under resting situation, O2 generation also takes place by the proteins in the matrix including enzymes of the citric acid cycle (pyruvate, alpha-ketoglutarate dehydrogenase, aconitase) and by different complexes. Studies have confirmed that the function of RC is disturbed during ageing and the reason might be the weakened antioxidant defence and increased accumulation of ROS from impaired metabolic process. Though, mitohormesis induction at a young age to avoid age-associated illnesses and management of RC activity in old age for enhancement of lifespan using therapeutics can be a bright scenario to continue (Bouska et al. 2019).
4 Redox Signalling
The molecular basis of the ageing process in humans is a complicated unanswered question, although numerous studies support the concept of the altered mitochondrial function being the major regulatory point during ageing (Jang et al. 2018). Throughout the history of scientific research, ROS and redox reactions have been declared potent and harmful agents. However, recently it has been suggested that ROS are involved in the regulation of bio-signalling. Explicit properties of ROS are modified mostly by covalently modified cysteine in specific redox-sensing target proteins. Oxidized cysteine residues then lead to alterable enzymatic modifications. Thus, ROS regulate an array of biological phenomenon such as the generation of inflammatory and growth factor responses. Any disturbance in redox signalling can lead to pathological manifestations (Finkel 2011). Redox biology has emerged from pathological findings and deals with the role of oxidative species in signalling pathways. The earliest studies from the 1930s by McCord and Fridovich on the discovery of superoxide dismutase also described the association between signalling and redox reactions. Initially, most of the researchers focused on damage of biomolecules by free radicals but later on, it was realized that peroxides such as H2O2 and lipid oxidation electrophiles are helpers in signal transduction and regulators of some transcription factors (Fig. 7.2). The new field of redox signalling emerged from the combination of signal transduction and redox biology. It is well established that redox signals are changed in ageing, but these signals are important in maintaining normal homeostasis (Forman 2016).
4.1 Dual Role of ROS
ROS are toxic products of aerobic metabolism that play a dual role. These are required for many signalling reactions such as programmed cell death. It is plausible to believe that ROS have benefits in bio-system viability and maintenance of specific intracellular ROS level is essential to sustain life (Mittler 2017). ROS perform an important function in the production of blood cells, differentiation and the equilibrium between dormancy and production of relevant stem cells. Suitable ROS levels can be an ideal therapeutic target for the treatment of blood cancers (Samimi et al. 2020).
The most interesting biological paradox in recent scientific history is the fact that ROS are toxic yet signal supporting molecules. Redox mechanisms based on the scavenging chemistry of oxidants have been evolved that facilitate the wellbeing of the living organism during the ageing process (D’Autréaux and Toledano 2007).
4.2 Redox Signalling
Signalling systems in living organisms are dependent upon chemicals with free energy, redox atmosphere and ion channels across the membranes. Thiol-sulfhydryl redox systems are controlled in dynamic yet non-equilibrium settings, are constantly oxidized and differ in redox potential within cellular organelles or intracellular compartments. Redox signalling uses “sulphur switch” which means cysteine amino acids that undergo reversible oxidation reactions, covalent incorporation of nitric oxide, reversible binding of glutathione, and the addition of acyl or sulfhydryl groups (Paulsen and Carroll 2013). These redox signalling systems control delivery, biological affinity, movement and sensitivity of signalling proteins, thereby influencing the rate and activities of redox systems. Thio-sulfhydryl reservoirs are oxidized with increasing age, lifestyle, environmental pollution and diseases such as diabetes, obesity and neurodegenerative diseases (Jones 2010).
Foyer and Noctor (2016) stated that considering the limited data available on ROS mediated signalling, some knowledge can be extracted about the stress-triggered signalling process. ROS transmit information in response to internal or external stimuli. It is speculated that cells can rely on ROS to monitor metabolic flux and ROS may become the symbol of sustainability in the context of ever-changing demanding situations. Previously, it was stated by Kamata and Hirata (1999) that biochemical signalling involves the binding of growth factor to receptors to activate tyrosine kinase that stimulates MAP (mitogen-activated protein), PLC-gamma (phospholipase C-gamma) and PI3K (phosphatidylinositol-3 kinase). The message is conveyed to the nucleus for gene expression after activation of many transcription factors. ROS such as H2O2 activates tyrosine kinases, triggering a cascade of molecules like MAP kinases and PLC-gamma (Zhang et al. 2016). This alters calcium levels, leading to stimulate or block many transcription factors. Redox molecules either stimulate or suppress some transcriptional factors, applying to double-check regulation of cell signalling. ROS also acts as a secondary messenger in response to the extracellular stimulus by crosstalk between signalling systems and redox systems. Cell death also has redox regulation by oxidation and reduction reactions. Some death signals generate ROS and which in turn activates death machinery. It is confirmed that a cell’s destiny is ultimately decided by crosstalk between bio-signalling pathways and redox species (Redza-Dutordoir and Averill-Bates 2016).
ROS can affect cellular proliferation, apoptosis, necrosis, gene expression and signalling processes especially involving mitogen-activating protein kinases cascades (Sies and Jones 2020). Within the human redox pool, damage caused by ROS is mended and key players of redox atmosphere are enzymes and antioxidants that establish a reduced environment by continuous addition of energy (Genestra 2007). The ability of living matter to extract energy from the environment, transform it to another form and use it for growth and reproduction is the basic attribute of life. During aerobic metabolism in mitochondria, oxidative phosphorylation generates ROS as a by-product that can damage cell machinery, though recently these have been recognized as signal molecules. ROS-controlled bio-signalling processes play important role in homeostasis (Shadel and Horvath 2015).
5 Autophagy
Participation of the lysosomal system to degrade intracellular organelles and macromolecules through a process named autophagy and of extracellular components and membrane through a process known as heterophagy or endocytosis is well-known for more than half a century (Kaushik and Cuervo 2018). The autophagy term is derivative of a Greek word with the meaning “to eat itself”. Autophagy is a catabolic and well-preserved process that has evolved in all eukaryotic cells during evolution and is a method for transportation of extra and intracellular components from cells to lysosomes, where their degradation and subsequent recycling take place. This may occur because of natural or stress response. The autophagy process was first observed in 1962 by T. Ashford and K. Porter in the cells of the rat liver after receiving glucagon. The name was given in 1963 soon after its discovery by a Belgian biochemist Christian de Duve (Fîlfan et al. 2017; Arensman and Eng 2018; Barbosa et al. 2019). Subsequently, recognition of genes linked to autophagy and autophagy morphology in yeast cells was described in the 1990s by Japanese researcher Yoshinori Ohsumi (Wang and Xu 2020).
Autophagy allows cell survival by inducing mobilization of endogenous macromolecules during periods of nutrient deprivation. It plays a paramount role in a stressful environment by maintaining the homeostasis in cells and metabolic stability during routine degradation, synthesis, and successive replacement of cytoplasmic components (Tan et al. 2014; Arensman and Eng 2018).
5.1 Lysosomal System in Autophagy
The contribution of autophagy, in almost all cases, in diverse physiological functions is attributed to its two major functions: as a procedure to eliminate undesirable cell components or as a source of energy. The lysosomal system has the unique ability to degrade and sequester the complete cellular portion, and this ability confers a paramount involvement of autophagy in certain circumstances where widespread cellular transformation is required for example embryogenesis, cell differentiation as well as degradation that occurs during some types of apoptosis (Buratta et al. 2020).
Lysosome, cell’s recycling centre is fully devoted to the degradation of various macromolecules both from the intracellular and from the extracellular environment. The lysosomal membrane contains permeases for recycling vital building blocks that are produced from the degraded products (e.g. fatty acids, sugars, cholesterol, and amino acids, etc.) in the cytosol. The lumen of the lysosomes also contains about 40 types of hydrolytic enzymes such as glycosidase, lipases, proteases, phosphatases and nucleases. The degradation of soluble individual proteins, cell organelles as well as particulate structures occurs through lysosomes. This characteristic of the lysosomal system makes it specifically relevant under circumstances when the formation of irreversible aggregates and oligomers starts from damaged proteins (Cuervo 2008; Hubbard et al. 2012). Moreover, the degradation of cytosolic components by lysosomes occurs non-specifically; however, it may also be distinguished between the targets to be degraded with the participation of a degradation label like a chaperone and a complicated process to create a way for the targeted proteins to cross the membrane of lysosomes through a specific translocation complex (Yang et al. 2019).
Autophagy also plays a role in attained (acquired) and inborn (inherited) immunity through digestion, sampling, and by presenting the peptides from both their cellular milieu as well as from the invasive pathogens that get an entry into the cells (Cuervo 2008). Generally, autophagy can target either the degradation of selective cargo or the bulk. However, many studies have elaborated that the process of autophagy specifically targets its cargo for degradation. Examples are the specifically chosen degradation of mitochondria (mitophagy), protein masses (aggrephagy), ribosomes (ribophagy), and lysosomes (lysophagy) (Barbosa et al. 2019; Wong et al. 2020). Several inducers start the process of autophagy such as various stressors, while one of the major stimuli is a nutrient restriction which can rapidly activate the autophagy process along with protein synthesis inhibition (Boya et al. 2013). Several genes linked to autophagy (Atg) that encode proteins involved in autophagy have been reported (Shibutani et al., 2015; Luo et al. 2020a, b). The complexes of ATG proteins primarily have several categories (Table 7.2).
5.2 Autophagy Mechanism
There are three steps of autophagy: initiation, formation, and degradation.
Start-up stage: Firstly, the autophagy regulation is mediated by environmental signals including the level of nutrients and exogenous stress agents such as heat or hypoxia over a multifaceted system of signalling pathways and proteins. The serine-threonine protein kinases are the key proteins in the initiation phase and are characterized by the mammalian target of rapamycin (mTOR). Two mechanisms proceed in different modes for the modulation of autophagy i.e., mechanistic target of rapamycin (mTOR) and nutrient-sensing pathways that include adenosine monophosphate-activated kinase (AMPK). Activation of AMPK induced autophagy during nutrient-depleted conditions drives a high AMP/ATP ratio. Consequently, phosphorylation and stimulation of ULK1, a serine/threonine-protein kinase, occurs that is an activator of autophagy. Conversely, an elevated level of amino acids is detected by mTORC1 a nutrient-sensing complex at the lysosomal membrane containing mTOR, endorses biomass production and cell proliferation actively and suppresses the autophagy process by repressing ULK1 (Gu et al. 2020; Wong et al. 2020).
Formation stage: The next stage requires the participation of the ATG proteins group to control the double-membrane vesicle generation for ingesting the cytoplasmic material (Madeo et al. 2015). The ingestion occurs by the formation of phagophore (Gu et al. 2020). The membrane used for the formation of phagophore originates from various locations including endosomes, mitochondria, endoplasmic reticulum, golgi complex, and plasma membrane (Lapierre et al. 2015). Moreover, certain parts of the plasma membrane having marked ATG16L1 may be delivered to the core of autophagic machinery by recycling endosomes (Madeo et al. 2015). Phagophore causes encapsulation of misfolded proteins or dysfunctional organelles, followed by the formation of autophagosome by extension and edge gradual fusion of phagophore. Beclin-1/Atg6 and Vps34 (vesicular protein sorting 34) are two distinct class III phosphatidylinositol 3-kinase (PI3-kinase) complexes in mammalian systems, involved in the phagophore extension (Itakura et al. 2008). Inhibition of phosphatidylinositol 3-kinases (PI3K) by 3-Methyladenine (3-MA) causes inhibition of autophagy due to blockage of autophagosome formation (Gu et al. 2020; Wang and Xu 2020). A previous study elaborated that the elongation of the phagophore includes ATG6 (Beclin-1), through contact with various binding partners; it likewise comprises a setting for incorporation of autophagy and programmed cell death processes. Many ATG16L1-interacting ATG proteins, including ATG12 and ATG5, participate in the stabilization of incipient phagophores (Madeo et al. 2015).
Degradation stage: Finally, at the third and last step of autophagy, phagophores enclosing the degraded cellular material form vesicular autophagosomes. Autophagosomes at that point merge with lysosomes forming autolysosomes for digestion through the transmission of microtubules cytoskeletal network system. The endomembrane of the autophagosome and components encapsulated by it are degraded by hydrolase present in the autolysosome. The permeases then release the degraded products into the cytoplasm and are reused (Madeo et al. 2015; Gu et al. 2020). The autolysosome formation should be precisely processed as incomplete autolysosome processing can instead result in the production of the residual body having indigestible material. The accumulated unprocessed autophagic vacuoles cause various age-related diseases including neurodegenerative disorders. This suggests that in aged individuals the cell’s potential to effectively manage and accomplish the autophagy is progressively diminished. SNARE (soluble NSF [N-ethyl-maleimide-sensitive Rab7, Dynein, and HSPB1 (heat shock 27 kDa protein 1) are involved in these processes (Lapierre et al. 2015; Wang and Xu. 2020).
5.3 Types of Autophagy
Three principal types of autophagy (Fig. 7.3) can be differentiated based on the procedure through which cytoplasmic components are carried to the lysosomes and the volume of sequestered substrates (Morgunova et al. 2016). Macroautophagy is commonly described as autophagy, the second type is conserved from yeast to mammals, named microautophagy while the third type has only been explained in mammals, termed as chaperone-mediated autophagy (CMA) (Cuervo et al. 2005).

Types of autophagy. Autophagy leads to the degradation of cargo and discharge of breakdown products into the cytosol for reuse. Macroautophagy depends on autophagosomes formation in the cytosol, to remove and transport materials to the lysosome. Autophagy gene Beclin 1, an ortholog of Atg6/vacuolar protein sorting (Vps)-30 protein plays role in positioning proteins before autophagial degradation. CMA (chaperone-mediated autophagy) transports unfolded proteins directly across the lysosomal membrane. Substrate proteins with specific KFERQ sequences are recognized by HSC70 and are transported into lysosomes through binding with LAMP2A protein. Microautophagy comprises the uptake of materials through invagination of the lysosomal membrane. ESCRT, cytosolic proteins together with numerous supporting proteins facilitate membrane bending/budding forming multivesicular bodies. Multivesicular bodies ultimately fuse with the lysosome for the degradation of materials. HSC70: heat shock protein 70 complex, LAMP2A: lysosome-associated membrane protein type 2A, ESCRT: endosomal sorting complexes required for transport
5.3.1 Macroautophagy
It is a well-studied and stress-induced type of autophagy where degradation occurs through autophagosome having a vacuole with bilayer and autophagolysosome, a lysosome having single-membrane. A group of proteins is involved for the completion of various steps in macroautophagy such as restraining membrane formation, length extension, growth, the merging of lysosomes, and breakdown (Lőrincz and Juhász 2020). These are generically named Atg proteins and were elucidated for the first time in yeast. Overexpression and Knockdowns of the genes in different organisms encoding these proteins have greatly increased the knowledge about the involvement of macroautophagy in enormous pathological as well as normal physiological processes. Macroautophagy helps a cell to renew the non-nuclear intracellular materials, destroy the worn-out structures and produce the new ones by utilizing the building blocks generated during “digestion” (Cuervo et al. 2005; Morgunova et al. 2016).
5.3.2 Microautophagy
In mammalian cells, microautophagy has been reported to take place in late endosomes instead of lysosomes, hence named endosomal microautophagy (Sahu et al. 2011). It comprises the transport of components from the cytosol to the endosome with the formation of endosomal membrane invaginations, and this process is facilitated by the ESCRT (endosomal sorting complexes required for transport). Hence, in microautophagy, invaginations are formed by the lysosome, and macromolecules as well as small structures are sequestered without the formation of autophagosomes as in macroautophagy (Macian 2019). Microautophagy is also named basal autophagy because in cells this process is maintained at a constant level. When a cell experiences deficiency of energy it utilizes the process of microautophagy (Morgunova et al. 2016).
5.3.3 Chaperone-mediated autophagy (CMA)
In CMA rearrangement of the lysosomal membrane is not required; instead, chaperone proteins are involved in the transportation of “faulty” proteins to the lysosome. Specific “chaperones” recognition and binding to the target substrate molecules result in their degradation. The specific sequences of amino acid on degraded substrates are required by these “molecular chaperones” (Morgunova et al. 2016; Luo et al. 2020a, b).
Many scientific reports have defined CMA as an extremely degradative and regulated process that involves the participation of HSC70 (heat shock protein 70 complex). Additionally, receptor lysosome-associated membrane protein type 2A (LAMP2A) undergoes multimerization.
The degradation of proteins by CMA must have a KFERQ motif in amino acid sequence, which is essential for the binding of chaperone HSC70. For lysosomal docking, binding of LAMP2A (twelve amino-acid tails in the cytosol) to the HSC70 complex and substrate is required. Moreover, multimerization of LAMP2A is mandatory for substrate transfer inside the lysosome. The multimeric complex releases cytosolic HSC70, after which an HSP90 chaperone present at the membrane of lysosome lumen associates with LAMP2A to stabilize it all through the substrate transfer process. Finally, to end the process of translocation, there is a requirement for a luminal chaperone HSC70 and once inside, the lysosomal enzymes degraded the targeted protein (Barbosa et al. 2019).
CMA is very sensitive and mainly liable for protein breakdown only (Luo et al. 2020a, b). Hence, transportation of only soluble cytosolic proteins occurs this way and for entry to the lysosomes, unfolding of the proteins is necessary (Massey et al. 2006; Kaushik and Cuervo 2018).
5.4 Ageing and Autophagy
Researchers have focused to explore the role of lysosomal and autophagy in ageing. Current studies have indicated that autophagy might have an impact on ageing, stress induced by oxidation, inflammation, and astrocytes functionality (Wang and Xu 2020). Almost all cells and tissues experience reduced autophagic activity as an organism ages, and it was supposed to largely contribute to the commencement of several detrimental age-associated ailments and various aspects of the age-related phenotypes (Cuervo 2008). Decreased autophagy is linked with enhanced ageing, while stimulation of autophagy may induce effective age control potentials (Madeo et al. 2010). In yeast, during nutrient deprivation, autophagy is crucial for survival as it provides energy and new nutrients by enabling the recycling of macromolecules (Rubinsztein et al. 2011). Logically, an indication that ageing is accelerated by autophagosomes formation deficiency proposed that health span should be prolonged by the enhancement of autophagic activity, specifically if there was insufficient normal autophagy to respond against cell impairment associated with ageing. Genetic manipulations in various species are specially developed to enhance autophagy capable of extending longevity (Madeo et al. 2015).
One of the earliest proofs that elevated autophagy have a paramount role in enhancing lifespan originates from the inspiring opinion that in C. elegans, autophagy is caused by the failure of insulin-like growth factor pathway and autophagy inhibition is due to essential Atg genes mutation, which hampers the improvement of lifespan (Meléndez et al. 2003). Pyo and collaborators conducted an experimental work to evaluate the association between increased lifespan and genetic overexpression of one Atg. The authors observed a rise in the process of autophagy and anti-ageing features by overexpressing the Atg5 gene in mice as compared to the wild-type mice (Pyo et al. 2013).
Another study was performed in Ana María Cuervo’s laboratory that explained the significance of autophagy in ageing. A double transgenic mouse model was generated in aged mice within which for CMA, expression of the lysosome receptor could be modulated. It was concluded that ageing characteristics can be limited at the organ and cellular levels by the enhancement of this receptor (Zhang and Cuervo 2008). Furthermore, expression of the Atg5 showed increased insulin sensitivity, improved resistance to age-related obesity, showing an enhanced metabolism in aged individuals. However, several studies failed to verify that longevity can be achieved by the upregulation of one autophagic factor. Moreover, enhanced lifespan has been shown by many KO (knock-out) mouse models; however, the connection with ageing and the molecular mechanisms behind it are not yet well-defined (Barbosa et al. 2019).
Similar characteristics are shared by an animal model named senescence-accelerated mouse prone 8 (SAMP8) that are non-genetically altered strains of mice (Ma et al. 2011). During ageing, certain remarkable changes related to autophagy were detected in the SAMP8 mice brain, such as reduced autophagy activity and accumulation of ubiquitin-positive proteins (Tan et al. 2014). Dysfunctional management of bio-signalling pathways including AMPK and mTOR resulting in cellular vulnerabilities that are conducive for the progression of neurodegeneration, cancer, and metabolic disease as well as impaired autophagy which ultimately contributes to ageing. However, a reduction in the function of mTORC1 is enough to enhance the life duration in worms, mold, mice, and flies (Wong et al. 2020).
Overexpression of an autophagy stimulator viz. HLH-30 was identified in C. elegans to increase the lifetime and is homologous to the transcription factor EB (TFEB) in mammals (Lapierre et al. 2013). An in vitro ageing model in humans indicated that over-expression of certain factors of the autophagy machinery including LC3/ATG8 and ATG12 contributed to the maintenance of mitochondria and long life. This suggests advantageous effects of pro-autophagic protein expression either in humans or within other mammalian cells. Moreover, within the nervous system, initiation of autophagy is of great importance for example in D. melanogaster, overexpression of brain-specific LC3/ATG8 was linked to prolonging age (Simonsen et al. 2008) and the absence of polyglutamine stretch of huntingtin in the brain exhibited a rise in autophagy which was linked to enhance longevity (Zheng et al. 2010).
The over-expression of Atg8 leads to longevity in muscle and neurons of adult Drosophila flies. Similarly, neuron-specific autophagy in adult Drosophila is induced in both non-cell and cell autonomously by the overexpression of Atg1, hence also results in extension of lifespan. Concordant with the biological significance of all findings, numerous relevant genes such as Atg5, Atg6, Atg1, Atg7, and Atg8 represent decreased appearance in flies with ageing and in human as well as mice muscle, ATG7 and LC3 proteins levels also decline with age. Generally, age-related and genetic loss of adequate lysosomal and autophagic function associate with the progress of various metabolic as well as anti-amnesic diseases. Examples are the mutations that result in loss of function of enormous genes relevant to autophagy such as Becn1/VPS30/ATG6, Atg5, and Atg7 decrease autophagy and higher accumulation of aggregated, disordered proteins in Alzheimer’s disease (Ab and MAPT/tau), Huntington disease (HTT/huntingtin), and Parkinson’s disease (SNCA/a-synuclein) (Lapierre et al. 2015).
Most ageing studies in humans are conducted using postmortem tissues, biopsies, peripheral blood samples and different types of cells that can be propagated by employing an artificial culturing medium in vitro. In all these cases, individual environmental factors/lifestyle is difficult to control between people, which leads to considerable experimental variability. Moreover, the literature reports additional ageing models that are used for better elucidation about the multi-factorial process of ageing in different species. Some emerging models include the longest-lived (30 years) rodent mole nude rat, the longest-lived mammal named the bowhead whale (200 years), killifish, the shortest-lived (~four months) vertebrate, and bivalve mollusks (life span up to 500 years). Diverse experimental approaches in all of these species have been applied to figure out the biological signals of ageing that range from detection of age-related biomarkers, regulatory proteins, genes/polymorphisms, compounds/metabolites, hormones, and different diets and decreased intake of calorie to intermittent fasting, which may defer the start of age-associated diseases and/or confer resistance as well as modify the process of ageing and longevity to manage with the environmental challenges (Carmona and Michan 2016). Autophagy activation can delay ageing, but at the same time, several studies have highlighted the opposite view. Hence, further work is warranted to assess the exact relationship between ageing and autophagy processes (Gu et al. 2020).
5.4.1 Role of Macroautophagy and CMA in Ageing
Alteration in macroautophagy and CMA happens with ageing; consequently, the contribution of autophagy to longevity has been supported by both vertebrate and invertebrate transgenic models (Hubbard et al. 2012). Generally, CMA and macroautophagy activities decline during ageing (Salminen and Kaarniranta 2009). However, there is a lack of evidence that how microautophagy activity might be affected during the process of ageing (Macian 2019). The dramatic increase in the function of both macroautophagy and CMA has been observed during stress, which facilitates cells to adjust to the environment (Morgunova et al. 2016).
Two well-characterized types of extralysosomal ‘waste’ are indigestible oxidized protein and senescent mitochondria (Cuervo et al. 2005). The organellar homeostasis is critically regulated by autophagy specifically that of mitochondria (Rubinsztein et al. 2011). Macroautophagy degrades all cellular structures, but mitochondrial digestion known as mitophagy is of peculiarly great significance because the long-term existence of the cell is dependent on the quality control of such organelles. Macroautophagy is strongly connected with the biogenesis of mitochondria; in few circumstances, the fundamental constituents from the “old” mitochondria are utilized by the cell for the formation of new mitochondria (Yen and Klionsky 2008; Morgunova et al. 2016).
The aged post-mitotic cells contain many mitochondria that are structurally deteriorated and enlarged, exhibiting swelling and disintegration of cristae, usually causing the formation of amorphous material. The mitochondria that are excessively enlarged are frequently named ‘giant’. The basic mechanism elaborating age-associated changes in mitochondria is still under discussion. Initial deterioration of mitochondria can be ascribed to the damage by ROS along with an inefficient performance of mitochondrial repair systems such as mtDNA repair as well as Lon and AAA proteases. Defective mitochondria should be degraded and autophagocytosed but the accumulated damaged mitochondria with age either escape macroautophagy or they require replicative advantage over normal mitochondria (Cuervo et al. 2005).
Dysfunctional mitochondrion that has lost membrane potential is more susceptible to release ROS along with toxic apoptotic mediators (Fig. 7.4). These are eliminated selectively by autophagy through ubiquitin Ser65 phosphorylation reactions carried out by PINK1 kinase (PTEN induced kinase 1), following attachment of ubiquitin proteins by the E3 ligase Parkin, compared to ‘‘healthy’’ mitochondrion. The autophagy uses PINK1 kinase and E3 ligase Parkin as markers to distinguish between damaged and healthy mitochondria, promote binding of dysfunctional mitochondria to phagophore by recruiting the mitochondrial autophagy receptors P62/SQSTM1 and NDP52 (Rubinsztein et al. 2011; Gu et al. 2020).

Effect of ageing on autophagy. Activated autophagy causes the elimination of damaged mitochondria by using PINK1 kinase and E3 ligase Parkin markers, while reduced autophagy results in the accumulation of waste product, making the process of ageing more lethal and causing several other destructive ailments. PINK1 kinase: PTEN induced kinase 1
It is strongly suggested that the working of macroautophagy, especially defective mitochondrial breakdown, is impaired due to ageing. Cell organelles undergo a complex and inducible macroautophagy process that includes Beclin1 and other Atg genes activation. The stimulation of macroautophagy occurs in response to hormone treatments and during various stress conditions. Interestingly, both the repression of growth hormone-insulin-like growth factor (GH- IGF-1) axis and caloric restriction can reverse age-associated variations and cause activation of macroautophagy. The increased stress resistance is a special feature of hermetic lifetime extension in C. elegans and long-lived mouse models (Cypser et al. 2006; Murakami 2006; Salminen and Kaarniranta 2009).
It has been observed that during ageing, housekeeping processes become compromised as the rate of protein turnover determines the quality of the cellular housekeeping mechanisms. During ageing, the accumulation of waste material in cells after mitosis indicates the inefficiency of physiological housekeep such as the proteolytic system. The proteasomal degradation activity is decreased in ageing (Salminen and Kaarniranta 2009), and the mechanism involves quality control checks, regulation of translation, protein folding, and breakdown by cell machinery. UPS (ubiquitin–proteasome system) and autophagy-lysosome system are involved in it. However, the nature of cross-talk between autophagy and proteasome is still not clear (Sun-Wang et al. 2020).
Indeed, autophagy performs a paramount role in proteostasis regulation, a mechanism which has been recently identified as one of the fundamental age causing mechanism (Macian 2019). Proteins are the components that enable or directly perform several functions of cells, taken together they constitute the “proteome”. The term proteostasis or protein stability refers to the capacity of cells to protect protein function and structure against ambient stressors such as changes in oxidative stress, pH, temperature, radiation, and ageing (Ruan et al. 2020; Sabath et al. 2020). Vulnerability in proteostasis correlates with changes in longevity and ageing rates among various species. Different researches identified that long-lived organisms are highly resistant to numerous environmental stressors and protein unfolding, thereby have maintained endogenous enzymatic activities in comparison to the species that are short-lived having less robust/effective proteostasis (Treaster et al. 2014). The proteostasis network present in the cells is well elaborated that involves autophagy, chaperones, protein synthesis, the ubiquitin–proteasome pathway, and the unfolded-protein response. These network constituents altogether are designed for the maintenance of recycling of long-lived products, counteracting protein misfolding, protein turnover and clearing up unfolded proteins. Studies revealed that proteostasis networks with age can be compromised resulting in the accumulation of protein and the aggregation of damaged and/or unfolded proteins (Carmona and Michan 2016).
One of the main functions performed by autophagy is proteostasis. CMA is engaged in the elimination of potent oxidized proteins by degradation in lysosomes (Barbosa et al. 2019). Cuervo and Dice (2000) demonstrated that during ageing the activity of CMA reduces in rat liver. The impaired transport of defective proteins into lysosomes as well as their binding to the membrane of lysosomes was observed with ageing. Interestingly, the results of various studies concluded that the progressive age-related decreased expression of the receptor protein in CMA uptake and LAMP-2A protein results in the reduced efficiency of CMA. Contrarily, certain targeting protein expression in CMA i.e., HSC70 protein was generally unaffected by ageing. These studies were further extended by the Cuervo laboratory, indicating that in transgenic mice the inhibition of age-associated deterioration of LAMP-2A protein can sustain a functional and an active CMA till older age. Moreover, the CMA conservation was significantly linked with improved liver function and decreased accumulation of defective proteins (Zhang and Cuervo 2008). During ageing, the decline of the LAMP-2A mechanism in lysosomes appears to be post-transcriptional as the LAMP-2A efficiency during transcription is not affected by ageing. This identifies that the function and assembly of the LAMP-2A complex in lysosomal membranes were affected by ageing; hence, impaired CMA properties occur during ageing. Moreover, HSP90 protein levels, a key constituent of LAMP-2A complex assembly, also decrease in the liver of ageing rats (Salminen and Kaarniranta 2009).
6 Conclusion
The interplay between redox signalling, autophagy and ageing is although quite dynamic, yet many aspects are still unknown. This highlights the probable challenge to identify specified pathways, the bulk of stimuli and metabolites. It is plausible that ageing process comprises multiple vicious cycles of ROS generation, redox signalling, and autophagial degradation of mitochondria. Defective and effective autophagy may decline or accelerate normal physiological conditions. Much of the data presented here are from those model organisms in which morbidity, mortality, and ageing may vary from what impacts human life and health. Nevertheless, it is expected that the biological progressions allied with the long life of model organisms may defend against ailments that afflict people.
Now the next puzzle is what best should be adopted to improve the healthy ageing process without any side effects. Being aged is a reality that everyone accepts. We want to live a healthy life i.e., healthy ageing. But unfortunately, it is not quite possible because in most cases ageing causes detrimental ailments. There are numerous factors including external as well as internal that collectively contribute to make ageing a detrimental process. Among these, increased oxidative stress and the decreased autophagy process are significant as both are interconnected with each other. During ageing, if the autophagy activity suppresses due to certain factors, it can neither destroy ROS-producing mitochondria nor maintains the homeostasis process, hence making the process of ageing even worse. Therefore, it is fascinating to speculate, although it is not proven experimentally, that supplements such as inducers that enhance the autophagy activity as well as certain dietary antioxidants may be beneficial to maintain the oxidant-antioxidant balance inside the body, promote health and postpone our inevitable fate.
References
Abbas G, Salman A, Rahman SU, Ateeq MK, Usman M, Sajid S et al (2017) Ageing mechanisms: linking oxidative stress, obesity and inflammation. Matrix Sci Med 1:30–33. https://doi.org/10.26480/msm.01.2017.30.33
Alonso-Fernández P, De la Fuente M (2011) Role of the immune system in ageing and longevity. Curr Aging Sci 4:78–100. https://doi.org/10.2174/1874609811104020078
Andreyev AY, Kushnareva YE, Starkov AA (2005) Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) 70:200–214. https://doi.org/10.1007/s10541-005-0102-7
Angelova PR, Abramov AY (2018) Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett 592:692–702. https://doi.org/10.1002/1873-3468.12964
Arensman MD, Eng CH (2018) Self-digestion for lifespan extension: enhanced autophagy delays aging. Mol Cell 71:485–486. https://doi.org/10.1016/j.molcel.2018.08.002
Bailey DM (2019) Oxygen, evolution and redox signalling in the human brain; quantum in the quotidian. J Physiol 597:15–28. https://doi.org/10.1113/JP276814
Barbosa MC, Grosso RA, Fader CM (2019) Hallmarks of aging: an autophagic perspective. Front Endocrinol (Lausanne) 9:790. https://doi.org/10.3389/fendo.2018.00790
Blagosklonny MV (2008) Aging: ROS or TOR. Cell Cycle 7:3344–3354. https://doi.org/10.4161/cc.7.21.6965
Bolduc JA, Collins JA, Loeser RF (2019) Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med 132:73–82. https://doi.org/10.1016/j.freeradbiomed.2018.08.038
Bonomini F, Rodella LF, Rezzani R (2015) Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis 6:109–120. https://doi.org/10.14336/AD.2014.0305
Bouska M, Huang K, Kang P, Bai H (2019) Organelle aging: Lessons from model organisms. J Genet Genomics 46:171–185. https://doi.org/10.1016/j.jgg.2019.03.011
Buratta S, Tancini B, Sagini K, Delo F, Chiaradia E, Urbanelli L et al (2020) Lysosomal exocytosis, xosome release and secretory autophagy: the autophagic- and endo-lysosomal systems go extracellular. Int J Mol Sci 21:2576. https://doi.org/10.3390/ijms21072576
Boya P, Reggiori F, Codogno P (2013) Emerging regulation and functions of autophagy. Nat Cell Biol 15:713–720. https://doi.org/10.1038/ncb2788
Brand MD (2016) Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 100:14–31. https://doi.org/10.1016/j.freeradbiomed.2016.04.001
Brooks-Wilson AR (2013) Genetics of healthy aging and longevity. Hum Genet 132:1323–1338. https://doi.org/10.1007/s00439-013-1342-z
Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E (2019) From discoveries in ageing research to therapeutics for healthy ageing. Nature 571:183–192. https://doi.org/10.1038/s41586-019-1365-2
Carmona JJ, Michan S (2016) Biology of healthy aging and longevity. Rev Invest Clin 68:7–16. PMID: 27028172
Chance B, Sies H, Boveris A (1979) Hydroperoxide metabolism in mammalian organs. Physiol Rev 59:527–605. https://doi.org/10.1152/physrev.1979.59.3.527
Chandrasekaran A, Idelchik MDPS, Melendez JA (2017) Redox control of senescence and age-related disease. Redox Biol 11:91–102. https://doi.org/10.1016/j.redox.2016.11.005
Cheon SY, Kim H, Rubinsztein DC, Lee JE (2019) Autophagy, cellular aging and age-related human diseases. Exp Neurobiol 28:643–657. https://doi.org/10.5607/en.2019.28.6.643
Couve E, Osorio R, Schmachtenberg O (2013) The amazing odontoblast: activity, autophagy, and aging. J Dent Res 92:765–772. https://doi.org/10.1177/0022034513495874
Cuervo AM (2008) Autophagy and aging: keeping that old broom working. Trends Genet 24:604–612. https://doi.org/10.1016/j.tig.2008.10.002
Cuervo AM, Dice JF (2000) Age-related decline in chaperone-mediated autophagy. J Biol Chem 275:31505–31513. https://doi.org/10.1074/jbc.M002102200
Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A (2005) Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 1:131–140. https://doi.org/10.4161/auto.1.3.2017
Cui H, Kong Y, Zhang H (2012) Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct 2012:646354. https://doi.org/10.1155/2012/646354
Cypser JR, Tedesco P, Johnson TE (2006) Hormesis and aging in Caenorhabditis elegans. Exp Gerontol 41:935–939. https://doi.org/10.1016/j.exger.2006.09.004
D’Autréaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8:813–824. https://doi.org/10.1038/nrm2256
Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D (2016) ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid Med Cell Longev 2016:3565127. https://doi.org/10.1155/2016/3565127
Dröse S, Brandt U (2012) Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol 748:145–169. https://doi.org/10.1007/978-1-4614-3573-0_6
Eleutherio E, Brasil AA, França MB, de Almeida DSG, Rona GB, Magalhães RSS (2018) Oxidative stress and aging: learning from yeast lessons. Fungal Biol 122:514–525. https://doi.org/10.1016/j.funbio.2017.12.003
Ewald CY (2018) Redox signaling of NADPH oxidases regulates oxidative stress responses, immunity and aging. Antioxidants (Basel) 7:130. https://doi.org/10.3390/antiox7100130
Farrelly C (2012) ‘Positive biology’ as a new paradigm for the medical sciences. Focusing on people who live long, happy, healthy lives might hold the key to improving human well-being. EMBO Rep 13:186–188. https://doi.org/10.1038/embor.2011.256
Fernández-Ballesteros R, Robine JM, Walker A, Kalache A (2013) Active aging: a global goal. Curr Gerontol Geriatr Res 2013:298012. https://doi.org/10.1155/2013/298012
Fîlfan M, Sandu RE, Zăvăleanu AD, GreşiŢă A, Glăvan DG, Olaru DG, Popa-Wagner A (2017) Autophagy in aging and disease. Rom J Morphol Embryol 58:27–31. PMID: 28523294
Finkel T (2011) Signal transduction by reactive oxygen species. J Cell Biol 194:7–15. https://doi.org/10.1083/jcb.201102095
Foote K, Reinhold J, Yu EPK, Figg NL, Finigan A, Murphy MP et al (2018) Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice. Aging Cell 17:e12773. https://doi.org/10.1111/acel.12773
Forman HJ (2016) Redox signaling: an evolution from free radicals to ageing. Free Radic Biol Med 97:398–407. https://doi.org/10.1016/j.freeradbiomed.2016.07.003
Foyer CH, Noctor G (2016) Stress-triggered redox signalling: what’s in pROSpect? Plant Cell Environ 39:951–964. https://doi.org/10.1111/pce.12621
Genestra M (2007) Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell Signal 19:1807–1819. https://doi.org/10.1016/j.cellsig.2007.04.009
Genova ML, Lenaz G (2015) The interplay between respiratory supercomplexes and ROS in aging. Antioxid Redox Signal 23:208–238. https://doi.org/10.1089/ars.2014.6214
Gerschman R (1954) Oxygen poisoning and x-irradiation: a mechanism in common. Glutathione 288–291. https://doi.org/10.1016/B978-1-4832-2900-3.50030-4
Gu Y, Han J, Jiang C, Zhang Y (2020) Biomarkers, oxidative stress and autophagy in skin aging. Ageing Res Rev 59:101036. https://doi.org/10.1016/j.arr.2020.101036
Haas RH (2019) Mitochondrial dysfunction in aging and diseases of aging. Biology (Basel) 8:48. https://doi.org/10.3390/biology8020048
Hubbard VM, Valdor R, Macian F, Cuervo AM (2012) Selective autophagy in the maintenance of cellular homeostasis in aging organisms. Biogerontology 13:21–35. https://doi.org/10.1007/s10522-011-9331-x
Itakura E, Kishi C, Inoue K, Mizushima N (2008) Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19:5360–5372. https://doi.org/10.1091/mbc.e08-01-0080
Jang JY, Blum A, Liu J, Finkel T (2018) The role of mitochondria in aging. J Clin Invest 128:3662–3670. https://doi.org/10.1172/JCI120842
Jones DP (2010) Redox sensing: orthogonal control in cell cycle and apoptosis signalling. J Intern Med 268:432–448. https://doi.org/10.1111/j.1365-2796.2010.02268.x
Kamata H, Hirata H (1999) Redox regulation of cellular signalling. Cell Signal 11:1–14. https://doi.org/10.1016/s0898-6568(98)00037-0
Kammeyer A, Luiten RM (2015) Oxidation events and skin aging. Ageing Res Rev 21:16–29. https://doi.org/10.1016/j.arr.2015.01.001
Kaushik S, Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19:365–381. https://doi.org/10.1038/s41580-018-0001-6
Labat-Robert J, Robert L (2015) Longevity and aging. Mechanisms and perspectives. Pathol Biol (Paris) 63:272–276. https://doi.org/10.1016/j.patbio.2015.08.001
Lambert AJ, Brand MD (2009) Reactive oxygen species production by mitochondria. Methods Mol Biol 554:165–181. https://doi.org/10.1007/978-1-59745-521-3_11
Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu CC, Visvikis O, Chang JT et al (2013) The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun 4:2267. https://doi.org/10.1038/ncomms3267
Lapierre LR, Kumsta C, Sandri M, Ballabio A, Hansen M (2015) Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 11:867–880. https://doi.org/10.1080/15548627.2015.1034410
Lara RC, Araújo GR, Mello AP (2018) Oxidative stress: in vitro comparative evaluation of the resveratrol modulator capacity in neuro 2-A lines and human leukocyte cells. Curr Trends Metabolomics 2018:1–8
Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D et al (2018) Oxidative stress, aging, and diseases. Clin Interv Aging 13:757–772. https://doi.org/10.2147/CIA.S158513
Lőrincz P, Juhász G (2020) Autophagosome-lysosome fusion. J Mol Biol 432:2462–2482. https://doi.org/10.1016/j.jmb.2019.10.028
Luo J, Mills K, le Cessie S, Noordam R, van Heemst D (2020a) Ageing, age-related diseases and oxidative stress: what to do next? Ageing Res Rev 57:100982. https://doi.org/10.1016/j.arr.2019.100982
Luo F, Sandhu AF, Rungratanawanich W, Williams GE, Akbar M, Zhou S et al (2020b) Melatonin and autophagy in aging-related neurodegenerative diseases. Int J Mol Sci 21:7174. https://doi.org/10.3390/ijms21197174
Lushchak VI (2015) Free radicals, reactive oxygen species, oxidative stresses and their classifications. Ukr Biochem J 87:11–18. https://doi.org/10.15407/ubj87.06.011
Ma Q, Qiang J, Gu P, Wang Y, Geng Y, Wang M (2011) Age-related autophagy alterations in the brain of senescence accelerated mouse prone 8 (SAMP8) mice. Exp Gerontol 46:533–541. https://doi.org/10.1016/j.exger.2011.02.006
Macian F (2019) Autophagy in T cell function and aging. Front Cell Dev Biol 7:213. https://doi.org/10.3389/fcell.2019.00213
Madeo F, Tavernarakis N, Kroemer G (2010) Can autophagy promote longevity? Nat Cell Biol 12:842–846. https://doi.org/10.1038/ncb0910-842
Madeo F, Zimmermann A, Maiuri MC, Kroemer G (2015) Essential role for autophagy in life span extension. J Clin Invest 125:85–93. https://doi.org/10.1172/JCI73946
Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P (2012) Mitochondria-ROS crosstalk in the control of cell death and aging. J Signal Transduct 2012:329635. https://doi.org/10.1155/2012/329635
Markaki M, Metaxakis A, Tavernarakis N (2017) The role of autophagy in aging: molecular mechanisms. In: Autophagy: cancer, other pathologies, inflammation, immunity, infection, and aging. Academic Press, pp 123–138
Massey AC, Kiffin R, Cuervo AM (2006) Autophagic defects in aging: looking for an “emergency exit”? Cell Cycle 5:1292–1296. https://doi.org/10.4161/cc.5.12.2865
Meléndez A, Tallóczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B (2003) Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301:1387–1391. https://doi.org/10.1126/science.1087782
Mittler R (2017) ROS are good. Trends Plant Sci 22:11–19. https://doi.org/10.1016/j.tplants.2016.08.002
Morgunova GV, Klebanov AA, Khokhlov AN (2016) Some remarks on the relationship between autophagy, cell aging, and cell proliferation restriction. Mosc Univ Biol Sci Bull 71:207–211. https://doi.org/10.3103/S0096392516040088
Murakami S (2006) Stress resistance in long-lived mouse models. Exp Gerontol 41:1014–1019. https://doi.org/10.1016/j.exger.2006.06.061
Napoli E, Taroni F, Cortopassi GA (2006) Frataxin, iron-sulfur clusters, heme, ROS, and aging. Antioxid Redox Signal 8:506–516. https://doi.org/10.1089/ars.2006.8.506
Panel M, Ghaleh B, Morin D (2018) Mitochondria and aging: a role for the mitochondrial transition pore? Aging Cell 17:e12793. https://doi.org/10.1111/acel.12793
Paulsen CE, Carroll KS (2013) Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev 113:4633–4679. https://doi.org/10.1021/cr300163e
Peng C, Wang X, Chen J, Jiao R, Wang L, Li YM et al (2014) Biology of ageing and role of dietary antioxidants. Biomed Res Int 2014:831841. https://doi.org/10.1155/2014/831841
Pham-Huy LA, He H, Pham-Huy C (2008) Free radicals, antioxidants in disease and health. Int J Biomed Sci 4:89–96. PMID: 23675073
Pisoschi AM, Pop A (2015) The role of antioxidants in the chemistry of oxidative stress: a review. Eur J Med Chem 97:55–74. https://doi.org/10.1016/j.ejmech.2015.04.040
Poljšak B, Dahmane RG, Godić A (2012) Intrinsic skin aging: the role of oxidative stress. Acta Dermatovenerol Alp Pannonica Adriat 21:33–36. https://doi.org/10.2478/v10162-012-0009-0
Pomatto LCD, Davies KJA (2018) Adaptive homeostasis and the free radical theory of ageing. Free Radic Biol Med 124:420–430. https://doi.org/10.1016/j.freeradbiomed.2018.06.016
Powers SK, Ji LL, Kavazis AN, Jackson MJ (2011) Reactive oxygen species: impact on skeletal muscle. Compr Physiol 1:941–969. https://doi.org/10.1002/cphy.c100054
Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI et al (2013) Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 4:2300. https://doi.org/10.1038/ncomms3300
Reczek CR, Chandel NS (2015) ROS-dependent signal transduction. Curr Opin Cell Biol 33:8–13. https://doi.org/10.1016/j.ceb.2014.09.010
Redza-Dutordoir M, Averill-Bates DA (2016) Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 1863:2977–2992. https://doi.org/10.1016/j.bbamcr.2016.09.012
Ren J, Zhang Y (2018) Targeting autophagy in aging and aging-related cardiovascular diseases. Trends Pharmacol Sci 39:1064–1076. https://doi.org/10.1016/j.tips.2018.10.005
Rottenberg H, Hoek JB (2017) The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 16:943–955. https://doi.org/10.1111/acel.12650
Ruan L, Wang Y, Zhang X, Tomaszewski A, McNamara JT, Li R (2020) Mitochondria-associated proteostasis. Annu Rev Biophys 49:41–67. https://doi.org/10.1146/annurev-biophys-121219-081604
Rubinsztein DC, Mariño G, Kroemer G (2011) Autophagy and aging. Cell 146:682–695. https://doi.org/10.1016/j.cell.2011.07.030
Sabath N, Levy-Adam F, Younis A, Rozales K, Meller A, Hadar S et al (2020) Cellular proteostasis decline in human senescence. Proc Natl Acad Sci U S A 117:31902–31913. https://doi.org/10.1073/pnas.2018138117
Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A et al (2011) Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20:131–139. https://doi.org/10.1016/j.devcel.2010.12.003
Salminen A, Kaarniranta K (2009) Regulation of the aging process by autophagy. Trends Mol Med 15:217–224. https://doi.org/10.1016/j.molmed.2009.03.004
Samimi A, Khodayar MJ, Alidadi H, Khodadi E (2020) The dual role of ROS in hematological malignancies: stem cell protection and cancer cell metastasis. Stem Cell Rev Rep 16:262–275. https://doi.org/10.1007/s12015-019-09949-5
Scialò F, Fernández-Ayala DJ, Sanz A (2017) Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front Physiol 8:428. https://doi.org/10.3389/fphys.2017.00428
Shadel GS, Horvath TL (2015) Mitochondrial ROS signaling in organismal homeostasis. Cell 163:560–569. https://doi.org/10.1016/j.cell.2015.10.001
Shibutani ST, Saitoh T, Nowag H, Münz C, Yoshimori T (2015) Autophagy and autophagy-related proteins in the immune system. Nat Immunol 16:1014–1024. https://doi.org/10.1038/ni.3273
Sies H, Jones DP (2020) Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 21:363–383. https://doi.org/10.1038/s41580-020-0230-3
Simioni C, Zauli G, Martelli AM, Vitale M, Sacchetti G, Gonelli A, Neri LM (2018) Oxidative stress: role of physical exercise and antioxidant nutraceuticals in adulthood and aging. Oncotarget 9:17181–17198. https://doi.org/10.18632/oncotarget.24729
Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4:176–184. https://doi.org/10.4161/auto.5269
Son JM, Lee C (2019) Mitochondria: multifaceted regulators of aging. BMB Rep 52:13–23. https://doi.org/10.5483/BMBRep.2019.52.1.300
Stefanatos R, Sanz A (2018) The role of mitochondrial ROS in the aging brain. FEBS Lett 592:743–758. https://doi.org/10.1002/1873-3468.12902
Sun-Wang JL, Ivanova S, Zorzano A (2020) The dialogue between the ubiquitin-proteasome system and autophagy: implications in ageing. Ageing Res Rev 64:101203. https://doi.org/10.1016/j.arr.2020.101203
Szentesi P, Csernoch L, Dux L, Keller-Pintér A (2019) Changes in redox signaling in the skeletal muscle with aging. Oxid Med Cell Longev 2019:4617801. https://doi.org/10.1155/2019/4617801
Tan CC, Yu JT, Tan MS, Jiang T, Zhu XC, Tan L (2014) Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol Aging 35:941–957. https://doi.org/10.1016/j.neurobiolaging.2013.11.019
Theurey P, Pizzo P (2018) The aging mitochondria. Genes (Basel) 9(1):22. https://doi.org/10.3390/genes9010022
Treaster SB, Ridgway ID, Richardson CA, Gaspar MB, Chaudhuri AR, Austad SN (2014) Superior proteome stability in the longest lived animal. Age (Dordrecht) 36:9597. https://doi.org/10.1007/s11357-013-9597-9
Urtamo A, Jyväkorpi SK, Strandberg TE (2019) Definitions of successful ageing: a brief review of a multidimensional concept. Acta Biomed 90:359–363. https://doi.org/10.23750/abm.v90i2.8376
Wang JL, Xu CJ (2020) Astrocytes autophagy in aging and neurodegenerative disorders. Biomed Pharmacother 122:109691. https://doi.org/10.1016/j.biopha.2019.109691
Warraich UE, Hussain F, Kayani HUR (2020) Aging—oxidative stress, antioxidants and computational modeling. Heliyon 6:e04107. https://doi.org/10.1016/j.heliyon.2020.e04107
Weidinger A, Kozlov AV (2015) Biological Activities of reactive oxygen and nitrogen species: oxidative stress versus signal transduction. Biomolecules 5:472–484. https://doi.org/10.3390/biom5020472
Wong SQ, Kumar AV, Mills J, Lapierre LR (2020) Autophagy in aging and longevity. Hum Genet 139:277–290. https://doi.org/10.1007/s00439-019-02031-7
Yang Q, Wang R, Zhu L (2019) Chaperone-mediated autophagy. Adv Exp Med Biol 1206:435–452. https://doi.org/10.1007/978-981-15-0602-4_20
Yen WL, Klionsky DJ (2008) How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 23:248–262. https://doi.org/10.1152/physiol.00013.2008
Zarkovic N (2020) Roles and functions of ROS and RNS in cellular physiology and pathology. Cells 9:767. https://doi.org/10.3390/cells9030767
Zhang C, Cuervo AM (2008) Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 14:959–965. https://doi.org/10.1038/nm.1851
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y et al (2016) ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev 2016:4350965. https://doi.org/10.1155/2016/4350965
Zheng S, Clabough EB, Sarkar S, Futter M, Rubinsztein DC, Zeitlin SO (2010) Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet 6:e1000838. https://doi.org/10.1371/journal.pgen.1000838
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Ethics declarations
Conflict of Interest:
All authors declare they have no conflict of interest.
Ethical Approval:
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hussain, F., Warraich, UEA., Jamil, A. (2022). Redox Signalling, Autophagy and Ageing. In: Çakatay, U. (eds) Redox Signaling and Biomarkers in Ageing. Healthy Ageing and Longevity, vol 15. Springer, Cham. https://doi.org/10.1007/978-3-030-84965-8_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-84965-8_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-84964-1
Online ISBN: 978-3-030-84965-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)