
Abstract
Our understanding of the process of autophagy and its role in health and diseases has grown remarkably in the last two decades. Early work established autophagy as a general bulk recycling process which involves the sequestration and transport of intracellular material to the lysosome for degradation. Currently, autophagy is viewed as a nexus of metabolic and proteostatic signalling that can determine key physiological decisions from cell fate to organismal lifespan. Here, we review the latest literature on the role of autophagy and lysosomes in stress response and longevity. We highlight the connections between autophagy and metabolic processes, the network associated with its regulation, and the links between autophagic dysfunction, neurodegenerative diseases, and aging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autophagy is a conserved intracellular process that encapsulates and delivers macromolecular cargoes such as proteins and organelles to lysosomes for subsequent degradation (reviewed in Galluzzi et al. 2017a). Three forms of autophagy have been characterized based on distinct mechanisms of cargo delivery to lysosomes: (1) Macroautophagy, whereby double-membraned vesicles called autophagosomes engulf autophagic cargo and directs them to lysosomes upon fusion, (2) Chaperone-mediated autophagy (CMA), where cargo is translocated into the lysosomal lumen by recognition of their KFERQ-like domain by cytosolic chaperones such as heat shock cognate 70 kDa proteins, and (3) Microautophagy, by which cargo enters lysosomes through membrane invaginations.
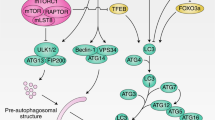
Overall, autophagy can direct either the degradation of bulk or selective cargo. Examples of the latter include the selective breakdown of protein aggregates (aggrephagy), mitochondria (mitophagy), lysosomes (lysophagy), and ribosomes (ribophagy). Notably, the most studied form of autophagy, macroautophagy (hereafter simply referred to as autophagy), is regulated by a core autophagic machinery of key autophagy-related (ATG) proteins (reviewed in Nakatogawa et al. 2009). The process is initiated by the Unc-51-like kinase 1 (ULK1)/ATG13/ATG101/focal adhesion kinase family-interacting protein of 200 kDa (FIP200) initiation complex in response to low cellular nutrient/energy levels, which recruits the Beclin1/ATG14/VPS15/VPS34 Class III PI3K complex to generate phosphatidylinositol 3-phosphate (PI3P) for nucleation and formation of the cup-shaped, double-membraned pre-autophagosomal phagophore at the autophagic cargo site. Subsequent phagophore expansion requires the ATG5/ATG12/ATG16L complex and the proteins ATG8 family of microtubule-associated protein light chain 3 (LC3) and gamma-aminobutyric acid receptor-associated (GABARAP). At this stage, the ATG5/ATG12/ATG16L complex promotes the conjugation of ATG4-cleaved LC3/GABARAP (LC3-I) to phosphatidylethanolamine (PE) to form LC3-II, which integrates into phagophore membranes. The phagophore expands until it eventually engulfs the autophagic cargo in an autophagosome. In selective autophagy, the delivery of specific autophagic cargoes into LC3-containing phagophores/autophagosomes is mediated through the interaction of cargo-bound autophagy receptors with LC3 via their LC3-interacting motifs (reviewed in Deng et al. 2017). A well-studied example is SQSTM1/P62, a selective autophagy receptor which binds to ubiquitinated protein aggregates and delivers them for aggrephagy clearance. While P62 mediates several types of “phagies”, other autophagy receptors which mediate organelle-specific selective autophagy have been uncovered. Notably, age-related conditions such as neurodegenerative diseases are known to be caused by or correlated with mutations and dysregulation of ATG proteins, selective autophagy, and their receptors (reviewed in Deng et al. 2017; Menzies et al. 2017). To circumvent such diseases of aging, efforts to pharmacologically modulate autophagy are at the forefront of multiple research programs in academia and the pharmaceutical industry (reviewed in Galluzzi et al. 2017b). In this review, we highlight the latest links between autophagy and metabolism, the recent elucidation of transcriptional regulatory mechanisms governing autophagy and the emerging evidence of the impact of dysfunctional autophagy has on neurodegenerative diseases and aging.
Links between autophagy and metabolism
Nutrient sensing governing autophagy induction
The regulation of autophagy is mediated by environmental cues such as nutrient levels and external stressors like hypoxia or heat through a complex network of proteins and signalling pathways (reviewed in Lahiri et al. 2019). Nutrient-sensing pathways such as adenosine monophosphate–activated kinase (AMPK) and mechanistic target of rapamycin (mTOR) act in an opposing manner to modulate autophagy (reviewed in He and Klionsky 2009). Nutrient-depleted conditions driving a high AMP/ATP ratio induce autophagy by activating AMPK (Bujak Adam et al. 2015; Wang et al. 2017a), which in turn phosphorylates and activates ULK1, the serine/threonine protein kinase activator of autophagy (Egan et al. 2011). Conversely, detection of elevated amino acids by mTORC1 (reviewed in Perera and Zoncu 2016; Saxton and Sabatini 2017), the nutrient-sensing complex containing mTOR at the lysosomal membrane, actively promotes cell proliferation, biomass production and represses autophagy by suppressing ULK1. Under nutrient-deprived conditions or by pharmacological inhibition, mTOR inactivation relieves ULK1 suppression and consequentially induces autophagy (Diaz-Troya et al. 2008). While amino acid sensing activation of mTOR is well established, glucose and cholesterol have also been recently shown to modulate lysosomal-targeted mTOR activation (Castellano et al. 2017; Wolfson et al. 2017). These findings suggest a more systematic integration of metabolism by mTOR which is counter-balanced by cellular energy state sensing via AMPK to properly drive the autophagy process and satisfy homeostatic demands.
Dysfunctional regulation of the mTOR and AMPK signalling pathways can contribute to organismal aging by impairing autophagy and by giving rise to cellular vulnerabilities conducive for the development of cancer, metabolic disease, and neurodegeneration (reviewed in Kaushik and Cuervo 2018; Laplante and Sabatini 2012; Valvezan and Manning 2019; Zhou et al. 2017). Reducing mTORC1 function is sufficient to extend lifespan in yeast, worms, flies, and mice (Vellai et al. 2003; Simonsen et al. 2008; Harrison et al. 2009; reviewed in Kapahi et al. 2010; Lawrence and Zoncu 2019). As mTORC1 activity is modulated at the lysosomal membrane (Lawrence and Zoncu 2019), the lysosome has emerged as a key organelle integrating decisions pertaining to metabolic fate and macromolecule recycling. Proper lysosomal function, which is essential for efficient degradation of autophagic cargoes, was also shown to decline with age, particularly in key organs such as liver, brain, and muscle (Lawrence and Zoncu 2019). Many lysosomal storage diseases (LSDs) share similar phenotypes with aging tissues, such as aberrant mTOR activity and compromised autophagic flux (Platt et al. 2012). Considering the central role of the lysosome in modulating growth, metabolism and somatic maintenance, the process of aging is likely comprised of LSD-like demonstrations that prevent cells from properly degrading macromolecules and enacting proper nutrient signalling (Lloyd-Evans and Haslett 2016).
One of the benefits of functional autophagy on metabolic homeostasis is the ability of this process to provide cells with a source of precursors for anabolic and energy-demanding pathways necessary for survival under nutrient-restricted conditions. Autophagy is upregulated during caloric restriction (CR) (reviewed in Bagherniya et al. 2018), which leads to lifespan extension across phyla (Taormina and Mirisola 2014) while also reducing the incidence of age-related disorders (Bordone and Guarente 2005; Escobar et al. 2019). Mice subjected to intermittent fasting, or complete food restriction between two daily meals, demonstrate activation of autophagy in liver, fat, brain, and muscle, leading to lower blood glucose and lipid levels (Martinez-Lopez et al. 2017). CR studies in non-human primates have shown lifespan improvements and delayed onset of age-related disorders (Mattison et al. 2017). The development of pharmacological mimetics of CR has been heralded as a strategy to extend lifespan in humans (Ingram et al. 2004; Roth et al. 2005). Treatment of mice or rats with the CR mimetics fisetin or metformin, a popular drug used to control diabetes and other metabolic ailments, offers neuroprotection against aging-related oxidative stress with a concurrent increase in autophagy gene expression via the AMPK pathway (Garg et al. 2016; Singh et al. 2018). Similarly, aging mice subjected to a cyclic ketogenic diet offers extended lifespan and healthspan (Roberts et al. 2017; Newman et al. 2017), which may be related to the support of the organism by increasing fatty acid oxidation and PPARα pathways and decreasing fatty acid and protein synthesis and TOR and insulin signalling. Organisms can survive periods of extended starvation by differentially inducing autophagy in an organ-specific manner in the liver, pancreas, kidney, and muscle while sparing the brain (Mizushima et al. 2004). Indeed, the breakdown of adipose tissue and muscle sustain the liver during its consumption by autophagy to export glucose and ketone bodies necessary to support the brain until the next feeding (Rabinowitz and White 2010). Blood glucose levels are maintained during starvation by AMPK activation of muscle autophagy, and aging-induced myopathy and mitochondrial dysfunction are accelerated if AMPK-mediated modulation is lost (Bujak Adam et al. 2015). Notably, exercise-induced autophagy (reviewed in Escobar et al. 2019) mediates beneficial metabolic effects in neurodegeneration, adult neurogenesis and cognitive function (He et al. 2012a, b), suggesting more broad and systemic effects of muscle-driven autophagy on physiology. Moderate running, for instance, has been shown to extend lifespan, purportedly through the induction of autophagy, among other metabolic, cardiovascular, musculoskeletal, and neuropsychiatric mechanisms (Lee et al. 2017). Thus, understanding how nutrient restriction- and exercise-induced autophagy can result in long-lasting impact on human lifespan remains a fertile field of study in aging research.
Metabolic integration of autophagy protein function
The activation of autophagy initiates an energetically-demanding process (Kaur and Debnath 2015) that is tightly regulated by the localization, accessibility, and proper modification of ATGs and associated proteins. In addition, many proteins that regulate autophagy undergo post-translational modifications (PTMs), including phosphorylation, ubiquitination, acetylation, O-linked attachment of β-N-acetyl-glucosamine (O-GlcNAc), and thiol modifications (reviewed in Wani et al. 2015). LC3 is a well-studied and key autophagy protein that is subjected to PTMs which govern its ability to localize properly to the autophagosome membrane. Under nutrient-depleted conditions, nuclear LC3 needs to be deacetylated, translocated to the cytoplasm, and lipidated in order for autophagy to progress (Huang et al. 2015). Catalyzed by protein kinase A (Cherra et al. 2010) and by hippo kinases STK3/STK4 (Wilkinson Deepti et al. 2015), LC3 phosphorylation regulates its lipidation and incorporation into maturing autophagosomes. While the lipidated form of LC3 (LC3-II) is elevated in old mice possibly via AMPK activity (Fritzen et al. 2016), the underlying mechanism related to LC3 dynamics in aging is not fully understood but likely involves compromised lysosomal capacity as mature LC3-II-containing autophagosomes accumulate in older animals and remain undegraded.
Selective autophagy of organelles and metabolic proteins
Autophagy can directly affect metabolism by degrading organelles that mediate energy production such as the mitochondria, or organelles that consists of large stores of energy such as lipid droplets. Selective autophagy of cargo is mediated by receptors that specifically recognize organelles or macromolecules bound for degradation (Deng et al. 2017). Mitophagy consists of the autophagic turnover of old or dysfunctional mitochondria, a process that can be induced as an adaptive metabolic response to prevent the build-up of reactive oxygen species (ROS) under prolonged hypoxia (Zhang et al. 2008). Mitochondrial biogenesis is modulated by PGC1α (Puigserver et al. 1998), and quality control by mitochondria clearance is regulated by PARKIN-mediated ubiquitination and high NAD+/NADH ratios (Jang et al. 2012; Wani et al. 2015). The proper balance between mitophagy and mitochondrial biogenesis is important for longevity (Palikaras et al. 2015) as the accumulation of damaged mitochondria accelerates aging. In addition, mitochondrial network dynamics is crucial for lifespan extension mediated by CR and AMPK activation (Weir et al. 2017). Compromised mitophagy is a hallmark of Parkinson’s disease (PD) as accumulation of damaged mitochondria render neurons vulnerable (Gao et al. 2017; Ryan et al. 2015). Impaired mitophagy is also observed in Alzheimer’s disease (AD) where levels of the phosphorylated mitophagy-associated proteins TBK1 and ULK1 are decreased, but cognitive decline is rescued upon mitophagy restoration (Fang et al. 2019), suggesting the accumulation of dysfunctional mitochondria and associated impaired energy metabolism could lead to the pathogenesis of AD. Control of the initiation, amplitude, and duration of the total mitophagic response is also modulated by the function of mitochondrial complex I where a shift toward oxidative phosphorylation (OXPHOS) increases the amplitude of autophagy while preference for aerobic glycolysis dampens autophagy (Thomas et al. 2018). Notably, mTOR inhibitors and glucose starvation can induce mitophagy while preserving complex I activity under OXPHOS conditions (Thomas et al. 2018). Thus, pharmacological and nutritional interventions aiming to enhance mitophagy while maintaining proper mitochondrial biogenesis and dynamics may provide an exploitable entry point to delay the onset of age-related diseases.
Lipophagy is the specific acquisition and breakdown of lipid droplets by the autophagy machinery (Singh et al. 2009). While the receptor for lipophagy remains elusive, lysosomal lipid breakdown is required for lifespan extension in various longevity models (Folick et al. 2015; Lapierre et al. 2011). Lysosomal-related lipid signalling can affect nuclear hormone receptor function to mediate changes in Iipid metabolism and lifespan (Folick et al. 2015; Seah et al. 2016), but such signals can also modulate mitochondrial activity (Ramachandran et al. 2019). Furthermore, non-cell autonomous modulation of lipophagy from the central nervous system (CNS) to the periphery has been recently described. The induction of autophagy in mouse hypothalmic POMC neurons induces lipophagy in brown adipose tissue (BAT) and the liver (Martinez-Lopez et al. 2016). Moreover, knock out of the autophagy gene Atg7 in POMC neurons blocks BAT and liver lipophagy (Martinez-Lopez and Singh 2016). This intricate CNS/periphery autophagic connection deserves further study as it could provide a new targeting mechanism to mitigate the development of metabolic disorders, fatty liver disease, as well as other neuronal and peripheral age-related diseases (Ward et al. 2016).
Other forms of selective autophagy such as CMA link metabolism to longevity. CMA, the lysosomal form of autophagy modulated by the lysosomal protein LAMP2A, can selectively and timely degrade proteins involved in metabolic pathways, such as glycolysis, lipolysis, and lipogenesis. This can in turn modulate the energetic flux through these specific pathways (reviewed in Madrigal-Matute and Cuervo 2016). To that point, the loss of CMA leads to increased glycolysis and lipogenesis while decreasing lipolysis and ATP levels in mouse livers (Tasset and Cuervo 2016). While CMA is selective, this begs the question if non-selective macroautophagy can also modulate the levels of metabolic enzymes and thereby guide the activity of metabolic pathways. Altogether, these recent findings on links between autophagy and metabolism highlight the need for systematic proteomic and metabolomics analyses as well as sub-cellular and tissue-specific analyses to properly resolve the autophagic and metabolic declines associated with the process of aging (Fig. 1).

Interplay between autophagy, metabolism and aging. Environmental cues influence nutrient-sensing pathways such as mTOR, AMPK, and insulin signalling in cells which modulate the expression of genes involved in autophagy by transcriptional and epigenetic regulation as well as by direct post-translational modification (PTM) of autophagy related (ATG) proteins. Functional autophagy, including its bulk and selective forms, ensures efficient organelle and macromolecular degradation and recycling which further impacts cell metabolism and homeostasis. Metabolic pathways feed back to nutrient-sensing pathways and autophagy regulation. In aging and neurodegeneration, defects (red) in several steps of autophagy regulation and execution lead to accumulation of damaged organelles and protein aggregates that adversely affect cell metabolism and homeostasis. This further exacerbates defective autophagy resulting in a vicious cycle that ends in cell death and neuronal loss. Emerging research also highlights non-cell autonomous, inter-tissue communication of autophagic status. Autophagy regulation is central to metabolism and aging and is a strong target for pharmacological strategies against age-related and neurodegenerative diseases
Autophagy regulation: transcriptional, epigenetic, and post-transcriptional
Transcriptional and epigenetic regulation of autophagy (ATG) genes are central mechanisms with impact on physiology and lifespan (Denzel et al. 2019; Lapierre et al. 2015). Here we highlight recent studies that have highlighted transcriptional, epigenomic, and post-transcriptional regulation of autophagy gene expression as well as advances on how nutrient availability controls transcription factor localization, autophagy induction, and lifespan.
Transcriptional regulation
Several transcription factors (TFs) have been shown to regulate the expression of core ATG genes involved in all stages of the process, from phagophore initiation to cargo degradation in autolysosomes (Fullgrabe et al. 2016). ATG gene transcription, akin to autophagy protein phosphorylation, is coordinately under strong regulation by nutrient and energy sensing pathways (reviewed in Efeyan et al. 2015). Similarly, TFs are subjected to regulatory phosphorylation that modulate their nucleocytoplasmic partitioning and thereby directly affect their ability to reach genomic sites to activate gene transcription.
The transcription factor EB (TFEB), acts as the master transcriptional regulator of autophagy and lysosomal biogenesis (Settembre et al. 2011). Under nutrient abundance, TFEB phosphorylation by the kinases mTOR and ERK at lysosomal membranes sequesters it in the cytoplasm via 14-3-3 proteins. Conversely, nutrient depletion inhibits mTOR and activates the phosphatase calcineurin, resulting in TFEB dephosphorylation and nuclear translocation. Nuclear localization of TFEB is also inducible under ER and mitochondrial stress (reviewed in Raben and Puertollano 2016). While importin-8 can mediate TFEB nuclear translocation (Perera et al. 2015), only recently has the regulated mechanism of TFEB exit to the cytoplasm been clarified. Research on genetic modifiers of TFEB nuclear localization uncovered the protein exportin-1 (XPO1/CRM1) as a key nuclear exporter of TFEB (Silvestrini et al. 2018). Originally suggested as a potential XPO1 cargo (Kirli et al. 2015), TFEB nuclear localization was seen under XPO1 inhibition, with concurrent autophagy gene induction and lifespan extension (Silvestrini et al. 2018) as observed in long-lived animals (Lapierre et al. 2013). Amino acids, refed after depletion, promote TFEB nuclear export via mTORC1-dependent phosphorylation of specific serine residues close to the NES in a sequential manner (Napolitano et al. 2018). Another recent study reported that the suppression of GSK3β by glucose deprivation-sensitive mTORC2 is responsible for TFEB nuclear export via a previously uncharacterized nuclear export signal (NES) (Li et al. 2018). Taken together, this suggests that the nucleocytoplasmic partitioning of TFEB to amino acid and glucose availability is dependent on mTORC1 and mTORC2, respectively. Besides phosphorylation, TFEB acetylation was shown to be required for its transcriptional induction of autophagy and lysosome synthesis (Zhang et al. 2018b). Being a regulator of autophagic flux, TFEB is the focus for autophagy induction in genetic and chemical screens for aging and age-related conditions like neurodegenerative diseases, metabolic syndrome and diabetes, and osteoarthritis (reviewed in Cortes and La Spada 2019; Lim et al. 2018; Martini-Stoica et al. 2016; Zheng et al. 2018).
Notably, the FOXO family TFs were also uncovered as autophagy regulators (Zhao et al. 2007). These include autophagy inducers FOXO1 and FOXO3, which increase autophagic flux through core ATG gene expression (Fullgrabe et al. 2016; Lapierre et al. 2015). Conversely, other FOXO TFs such as FOXK1 can inhibit autophagy by competing with FOXO3 for promoter binding at FOXO3-induced ATG genes and repressing gene expression (Bowman et al. 2014). FOXO TFs such as FOXO3 are inhibited by the insulin/insulin-like growth factor signalling (IIS) pathway via AKT phosphorylation, causing them to be sequestered in the cytoplasm by 14-3-3 proteins and preventing their transcriptional activity in the nucleus (reviewed in Calnan and Brunet 2008; Tzivion et al. 2011). FOXO acts as a longevity factor by inducing autophagy thought to be important for lifespan extension observed in IIS pathway mutants (reviewed in Kenyon 2010). Aside from the IIS pathway, FOXO TFs may also be regulated by other TFs such as GATA-1, XBP1, JNK, and REST (Kang et al. 2012; Lu et al. 2014; Xu et al. 2011; Zhao et al. 2013). Hence, these upstream regulators of FOXO TFs may indirectly affect autophagy. A recent study demonstrated that FOXO can, with TFEB, co-regulate many target genes to promote longevity and resistance to oxidative stress (Lin et al. 2018). Such combinatorial regulation highlights crosstalk between different TFs that modulate autophagy and further integrates signals from various nutrient-sensing pathways.
Besides FOXO TFs and TFEB, ATG gene expression can also be directly regulated by other TFs such as TP53, CREB, E2F1, NFκB, ZKSCAN, NRF2, PPARα, CEBP, XBP1, HIF1, JUN, and FXR. ATG gene expression appears to be induced by TFs such as E2F1, HIF1, and JUN, and repressed by FXR, whilst NFκB has a dual action. Details on these TFs and the genes they regulate have been reviewed recently (Fullgrabe et al. 2016). It is likely that a combination of these TF functions may transactivate subsets of autophagy genes to enhance the process, and understanding such interactions in conjunction with epigenetic changes compels further research.
Epigenetic regulation
Histone modifying enzymes regulate PTMs on histone proteins, dictating the extent of chromatin condensation and subsequent transcriptional activity of the underlying DNA. Autophagy genes have been shown to be regulated by epigenetic modifications such as histone acetylation and methylation, the most commonly studied histone modifications.
One such PTM is acetylation (ac), which is mediated by acetyltransferases. ATG gene expression can be regulated by the acetyltransferase KAT8/hMOF and its modified histone H4K16ac, as autophagy induction is associated with the downregulation of both components (Fullgrabe et al. 2013). Another transcription-activating mark H3 K56ac was demonstrated to regulate cell growth mediated by the yeast TORC1 ortholog of mTORC1, while H3K56ac mutants were hypersensitive to the mTOR inhibitor rapamycin (Chen et al. 2012). Given the inhibitory role of mTORC1 on autophagy in conditions of nutrient availability, this suggests an autophagy-regulating role of H3K56ac marks. In support of this, ATG genes were found to be enriched at genomic regions associated with increased H3K56ac and H3K27ac marks in nutrient-deprived HAP1 cells (Peeters et al. 2019).
Histone methylation (me) on lysine and arginine residues can regulate autophagy in opposing ways depending on which residue is methylated. For instance, autophagy induction is associated with the global reduction of the transcriptional-activating H3K4me3 mark (Fullgrabe et al. 2013) and upregulation of the repressive heterochromatic H4K20me3 modification (Kourmouli et al. 2004). Under glucose deprivation, autophagy can similarly be stimulated through arginine dimethylation at histone H3R17 (H3R17me2) as a genome-wide increase in H3R17me2 was found (Shin et al. 2016). This modification appears to be regulated by the sole H3R17 methyltransferase CARM1, which also acts as a coactivator for TFEB, as persistent starvation prevents its degradation by the SKP2-SCF E3 ubiquitin ligase and stabilizes it in the nucleus to co-activate TFEB-driven autophagy. Conversely, histone methylation can also repress autophagy, such as the H3K9me2 modification associated with the transcriptional repression of LC3B and WIPI1 through interactions at their gene promoters (Artal-Martinez de Narvajas et al. 2013). Similarly, a TFEB-independent transcriptional autophagic program can be repressed by the histone methyltransferase G9A and epigenetic reader BRD4 (Sakamaki et al. 2017). These epigenetic activities can be regulated by nutrient availability, as evidenced by starvation-induced dissociation of BRD4 from chromatin in an AMPK- and SIRT1-dependent manner. Unlike lysine and arginine methylation on histones, cytosine methylation by DNA methyltransferases (DNMTs) is an epigenetic mark that only represses gene expression. Indeed, the repressed ATG gene expression in macrophages from old mice via increased DNMT2 and hypermethylation of ATG5 and LC3B gene promoters was restored by chemical and genetic inhibition of DNMT2 (Khalil et al. 2016). Apart from acetylation and methylation, monoubiquitination on histone H2B (H2Bub1), a mark associated with highly expressed genes, is decreased under starvation via the deubiquitinase USP44 (Chen et al. 2017). This results in the induction of autophagy genes highlighting the role of other, less-studied epigenetic marks in regulating autophagy. Altogether, these studies indicate that epigenomic changes accompany autophagy induction, but the significance of specific modifications on cellular homeostasis and organismal aging remains to be clarified.
Post-transcriptional regulation
Various RNAs and RNA binding proteins (RBPs) are known to post-transcriptionally regulate the mRNAs of ATG components, either directly or indirectly by TF modulation. For instance, microRNA (miRNA/miR)-mediated repression of TFEB via miR128 impairs TFEB-activated ATG gene expression (Decressac et al. 2013). miRNAs can also target different stages of autophagy. For instance, early autophagy components such as ULK1 and Beclin1 are targeted by miR30a, miR30b, and miR471-5p, whilst ATG12, ATG5, ATG7 and LC3 in the LC3 recruitment stage are controlled by miR30b, miR30c, miR130a, miR471-5p, miR101. The selective autophagy receptor P62/SQSTM1 is targeted by multiple miRNAs such miR17, miR20, miR93, miR106, and miR372, whereas components involved in lysosomal function such as LAMP2 and SUMF1 are targeted by miR207 and miR95 (reviewed in Frankel et al. 2017; Hargarten and Williamson 2018). Understanding the regulation and physiological impact of these miRNAs is imperative to determine the extent of their function and druggability for autophagy modulation.
Long non-coding RNAs (lncRNAs), which comprise non-coding transcripts of more than 200 bases, were recently found to regulate autophagy. H19, a lncRNA that is inhibited under conditions of high glucose, suppresses autophagy by epigenetically repressing a GTPase DIRAS3 which regulates the PI3K/AKT/mTOR pathway as well as BECLIN1 and ATG7 gene expression in diabetic cardiomyopathy (Zhuo et al. 2016). Paradoxically, H19 induced autophagic cell death in cerebral ischemia–reperfusion injury by inhibiting DUSP5, a mitogen-activated protein kinase phosphatase (Wang et al. 2017b). Likewise, several lncRNAs such as NBR2, MEG3, HOTAIR, PTENP1, MALAT1, and H19 have been implicated in controlling genes involved in all autophagic stages (reviewed in Yang et al. 2017). Recently, the lncRNA NEAT1 was found to be upregulated in MPTP-induced PD in mice and neuroblastoma cells. NEAT1 induces autophagy by stabilizing PINK1, thus contributing to PINK1-mediated mitophagy in PD pathogenesis (Yan et al. 2018). Similarly, lncRNAs GAS5, CAIF, and DICER1-AS1 have been implicated in modulating autophagy in breast cancer, myocardial infarction, and osteosarcoma, respectively (Gu et al. 2018a, b; Liu et al. 2018). Taken together, these findings implicate changes in lncRNA-regulated autophagy in pathological conditions.
RBPs bind RNA and are required for the formation of ribonucleoproteins commonly involved in RNA splicing, polyadenylation, transport, and stability. The RBP HuD targets and induces ATG5 mRNA in pancreatic β cells and increases autophagosome formation (Kim et al. 2014), while tristetraprolin downregulates Beclin1 and LC3-II resulting in reduced autophagy and increased cell death in lung adenocarcinoma cells (Dong et al. 2018). TDP-43, the major defective protein implicated in ALS and FTD, has a low-complexity domain and is aggregation-prone. Depletion of this RBP has been shown to destabilize the mTORC1 adaptor protein RAPTOR, resulting in reduced mTOR activity and consequential TFEB nuclear localization and autophagy stimulation. Paradoxically, TDP-43 depletion can also hamper autophagic flux by impairing autophagosome-lysosome fusion in a dynactin 1-dependent manner (Xia et al. 2016). Thus, the role of RBPs in autophagy regulation is emerging and further investigation will provide mechanistic insights about how they control autophagy in pathologies such as neurodegeneration.
Transgenerational inheritance of starvation resistance
Another area that warrants attention is the transgenerational inheritance of stress resistance and the mechanisms involved therein. Heritable small RNAs in C. elegans (reviewed in Rechavi and Lev 2017) have been shown to be passed on for three consecutive generations upon starvation (Rechavi et al. 2014). Similarly, epigenetic modifications in the H3K4me3 regulatory complex leads to lifespan extension in three generations (Greer et al. 2010) and inheritance of stress-induced survival advantages in subsequent generations (Kishimoto et al. 2017). However, during acute starvation, AMPK blocks histone methyltransferases from methylating H3K4 thereby ensuring that chromatin marks are not established until nutrient conditions are replete (Demoinet et al. 2017). DNA methylation at 5-methylcytosine has been associated with transgenerational responses to high-fat diet in mammals (Ng et al. 2010) and at 6-methyladenine with mitochondrial stress response in nematodes (Ma et al. 2018). These studies underscore the importance of germline-soma communication of stress signals, but it remains to be elucidated whether inheritance of stress responses modulates autophagy in subsequent generations. Taken together, epigenetic and transcriptional modulation of autophagic genes is emerging as an essential level of robust autophagy regulation and understanding how aging compromises it (Fig. 1) will shed light onto how autophagy goes awry in metabolic syndromes and neurodegeneration.
Autophagy in aging and neurodegenerative diseases
Evidence of autophagy impairment in age and neurodegeneration
Numerous studies have demonstrated links between autophagy dysfunction and aging in both invertebrate and vertebrate model systems. Indeed, autophagic function deteriorates with age, and genes of the autophagy-lysosomal pathway are necessary for mediating lifespan extension in various mechanistically-different pro-longevity pathways (reviewed in Hansen et al. 2018). Notably, age is a major risk factor for many neurodegenerative diseases (NDs) such as AD and PD, polyglutamine (polyQ) expansion diseases such as Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration (FTLD), and tauopathies (Niccoli and Partridge 2012). The age-dependent functional decline of autophagy and the consequential attenuation of proteostasis and accrual of proteotoxicity over time is generally thought to have a significant contribution towards disease development and/or progression. Since they cannot segregate the proteotoxic damage from daughter cells upon mitosis, post-mitotic neurons are more susceptible to age-associated proteotoxicity. Multiple lines of evidence support the view that impaired autophagy is pathophysiological, although the exact mechanisms and roles of dysfunctional autophagy in each disease remain to be elucidated (reviewed in Menzies et al. 2017). On the genetics level, many disease causative and associated mutations uncovered in genes involved in the autophagic process provide insights into the onset of pathogenesis leading to neurodegeneration. For instance, such genes include those coding for autophagy effectors and receptors which trigger, recognize, and direct autophagic cargo to autophagosomes, suggesting impairments occurring upstream of autophagosome formation (reviewed in Deng et al. 2017). One such example is the SQSTM1 gene wherein ALS and FTLD mutations have been found (Fecto et al. 2011; Rubino et al. 2012). SQSTM1 encodes the aggrephagy receptor SQSTM1/P62, hence mutations in this gene could play an important role in disease pathogenesis by interfering with the selective clearance of protein aggregates. In the same vein, mitophagy defects arising from familial PD causative mutations in the phosphatase and tensin homolog-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin mitophagy effector genes supports the long-held view that the derailment of mitochondrial homeostasis is central to PD pathophysiology (Pickrell and Youle 2015). As autophagic cargoes are degraded upon fusion of autophagosomes with lysosomes, impairment at the lysosomal level also interferes with autophagy indirectly. This is exemplified by dominant familial early-onset AD mutations in the presenilin gene PSEN1, which disrupt lysosomal acidification and consequentially compromises lysosomal degradation of autophagic cargo (Lee et al. 2015). Autophagy impairments may also occur due to alterations at the epigenetics level. In AD brains for instance, reduced H4K16ac marks and enhanced ATG gene expression were found as compared to healthy aged brains (Nativio et al. 2018). However, how this corroborates separate findings of downregulated ATG gene expression that enhances Aβ-mediated neurotoxicity in AD is unclear (Omata et al. 2014). Aside from insights obtained from ND genetics, observations made at the pathological level provides complementary information about the pathophysiological mechanisms of autophagy. For instance, autophagosome accumulation observed in affected neurons from the brains of AD patients and mouse models suggests obstruction of autophagic flux in AD and potentially other NDs, although it remains a challenge to conclusively demonstrate this due to unavailable tools to monitor autophagic flux clinically (Nixon et al. 2005; Yu et al. 2005). Thus, the role of autophagic dysfunction as an initiator of neurodegenerative proteotoxicity remains to be clarified.
Further evidence has demonstrated that autophagy can be directly impaired by various ND-associated defective proteins, which form the major constituents of the characteristic proteinaceous aggregates in these diseases. For instance, PD Lewy body-associated mutant α-synuclein may repress autophagy by sequestering TFEB in the cytoplasm and preventing its translocation to the nucleus where it induces the expression of autophagy and lysosomal biogenesis genes (Decressac et al. 2013). In HD, mutant Huntingtin impairs autophagy in neurons and leads to AGO2 accumulation and global miRNA increase. However, AGO2 shuttles to and gets sequestered in stress granules, rendering miRNA inactive for gene silencing (Pircs et al. 2018). In polyQ expansion diseases such as HD and spinocerebellar ataxia type 3, respective polyQ-expanded mutant proteins Huntingtin and ataxin-3 may outcompete their wildtype counterparts for interaction with Beclin 1, impairing autophagy induction via their reduced abilities to protect Beclin 1 against proteasomal degradation as compared to the wildtype proteins (Ashkenazi et al. 2017). Aside from macroautophagy, CMA and microautophagy were additionally shown to be compromised by ND-associated proteins. Recent findings demonstrated that mutated forms of the tauopathy-associated microtubule-binding protein tau can, depending on the type of disease-associated mutation and biochemical properties, differentially alter their uptake and degradation through macroautophagy, CMA, and endosomal microautophagy (eMI), a selective type of microautophagy whereby cytosolic proteins are degraded in late endosomes (Caballero et al. 2018; Tekirdag and Cuervo 2018). This highlights the importance of understanding how other ND-associated proteins may also differentially affect the various forms of autophagy, which is important for improving the specificity of targeting autophagic processes for therapy. Overall, these findings highlight the importance of functional autophagy in ND proteinopathies as the interaction of defective proteins with autophagy can propagate a vicious cycle of proteotoxicity to aggravate disease.
An additional but important aspect to consider is how aberrant autophagy may play a role in selective neuronal vulnerability in NDs (reviewed in Fu et al. 2018). Levels of pathologically-affected proteins may be differentially expressed by neuronal populations. Moreover, levels of autophagic activity and its susceptibility to failure could vary between neurons. To complicate this further, autophagy appears to be differentially regulated in neurons and may have specialized roles in neuronal compartments (reviewed in Kulkarni and Maday 2018). Furthermore, autophagy impairment in glial cells, which have important homeostasis roles in the central nervous system, may affect autophagic activities in neurons (reviewed in Plaza-Zabala et al. 2017). Altogether, these factors can interact to give rise to the degeneration of specific neurons in separate NDs, suggesting that neuronal population-specific therapeutic approaches may be needed.
Impaired nucleocytoplasmic transport and loss of nuclear integrity may derail autophagy
The proper nucleocytoplasmic transport of autophagy-inducing TFs such as TFEB by RanGTP-dependent importins and exportins and the retention of such factors in the nucleus are important processes in proper autophagic regulation. In fact, nuclear pore complexes (NPCs), which form nucleocytoplasmic transport channels through the nuclear envelope, deteriorate with age and cause age-dependent nuclear pore leakiness in post-mitotic cells such as neurons (D’Angelo et al. 2009). The efficiency of TFEB nuclear retention may thus decrease with age, consequentially playing an important role in the age-dependent decline of autophagic activity. In the same vein, findings have highlighted the importance of proper nucleocytoplasmic transport in regulating and maintaining the correct subcellular localization of proteins and RNA, which are likely to include factors such as TFEB and autophagy components. For instance, various forms of stresses such as heat shock, oxidative stress, and UV irradiation have been shown to inhibit nuclear import of nuclear localization signal-containing substrates by preventing the nuclear export of the importin α subunit to the cytoplasm where it can form an active heterodimer with the β subunit to mediate cargo transport to the nucleus (Furuta et al. 2004; Miyamoto et al. 2004). Recent evidence has additionally provided further support of the importance of nucleocytoplasmic transport in health and disease by demonstrating that pathologically-affected proteins in NDs can disrupt this process by subcellularly mislocalizing proteins and RNA (reviewed in Fahrenkrog and Harel 2018). Mislocalized proteins included NPC components and nucleocytoplasmic transporters themselves which were aberrantly partitioned to the cytoplasm, and thus inhibited from performing their functions at the nucleus by phase separated stress granules in C9ORF72-mediated ALS and frontotemporal dementia (FTD) (Zhang et al. 2018c). Most recently, a novel mechanism of nucleocytoplasmic transport impairment by an FTD-associated form of mutant tau was attributed to disrupted microtubule dynamics and consequential deformation of the nuclear membrane (Paonessa et al. 2019). Nuclear pore dynamics was also recently found to be sensitive to tau levels as tau interaction with the nuclear pore protein Nup98 potentiated tau fibrilization (Eftekharzadeh et al. 2018). Interestingly, importin proteins have been recently shown to harbour disaggregase activity, which mitigate cytosolic FUS aggregation (Guo et al. 2018). In light of these findings, restoring proper nucleocytoplasmic transport or regulating specific partitioning with nuclear export modulators (Silvestrini et al. 2018) may resolve the proteotoxic stress associated with disordered proteins in NDs. Taken together, these findings additionally highlight that both nuclear integrity and proper nucleocytoplasmic partitioning of proteins and RNA are important to maintain autophagic dynamics and proteome health.
Emerging links between phase separation and autophagy
Cytoplasmic sequestration of nucleocytoplasmic transporters by phase separated stress granules may impair autophagy by attenuating the nuclear transport of autophagy-stimulating TFs such as TFEB. This not only provides further evidence for the pathophysiological mechanisms of aberrant phase separation in NDs but also suggests connections between this cellular process and autophagy defects. Autophagy regulation by the ALS- and FTD-associated and phase condensate competent TDP-43 RBP highlights such connection. Suggestive of further crosstalk between autophagy and phase separation, TDP-43 and other RBPs such as FUS, HNRNPA2B1, HNRNPA1, ATXN1 (ataxin 1) and ATXN2, and many components of autophagy and autophagy-regulating cellular stressors can also mediate phase separation (reviewed in Polymenidou 2018). For instance, heat stress-induced phase separation of PGL granules in C. elegans embryos via mTORC1 rendered the granules resistance to autophagy degradation (Zhang et al. 2018a). In the same vein, P62 promotes the phase separation of ubiquitin-positive proteins for aggrephagy to compensate for UPS malfunction (reviewed in Danieli and Martens 2018). As the highly dynamic nature of phase separated condensates can lead to transition to more rigid states, including aggregates, future work is needed to establish the spatiotemporal and mechanistic details of these transitions as they could be important pathological processes progressively affecting autophagy and proteostasis in aging and NDs (Fig. 1).
Concluding remarks
Autophagy is a crucial and versatile degradation mechanism for damaged macromolecules and dysfunctional organelles that accumulate during the process of aging. Cell-autonomous regulation of autophagic flux, therefore, results in direct improvement in proteostasis and somatic maintenance. Recent studies have demonstrated that tissue-specific induction of autophagy can also result in non-cell autonomous extension of organismal lifespan (Hansen et al. 2018). Although the mechanisms for how such non-cell autonomous autophagic regulation occurs is currently unclear, inter-tissue communication of stress responses such as the heat shock response and unfolded protein responses in the endoplasmic reticulum (UPRER) and mitochondrial (UPRmt) have been demonstrated (Taylor and Dillin 2013; van Oosten-Hawle et al. 2013; Zhang et al. 2018d). These organelle-specific and intracellular proteostatic mechanisms may potentially be involved in conveying autophagic status between tissues to regulate aging on a systemic level. Notably, autophagy proteins can also possess autophagy-independent functions (Cadwell and Debnath 2018), such as extracellular protein secretion (Cotzomi-Ortega et al. 2018), that may play a role in mediating inter-tissue integration of nutrient signalling, metabolism, and gene regulation to modulate proteostasis. As temporal and circadian considerations are becoming integrated in aging studies (reviewed in Fonseca Costa and Ripperger 2015; Hood and Amir 2017), the underlying mechanisms that jeopardize autophagy and proteostasis and lead to neurodegeneration are bound to be clarified and new pharmacologically-exploitable mechanisms are likely to be uncovered.
References
Artal-Martinez de Narvajas A, Gomez TS, Zhang JS, Mann AO, Taoda Y, Gorman JA, Herreros-Villanueva M, Gress TM, Ellenrieder V, Bujanda L, Kim DH, Kozikowski AP, Koenig A, Billadeau DD (2013) Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol 33:3983–3993
Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, Squitieri F, Hardenberg MC, Imarisio S, Menzies FM, Rubinsztein DC (2017) Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 545:108–111
Bagherniya M, Butler AE, Barreto GE, Sahebkar A (2018) The effect of fasting or calorie restriction on autophagy induction: a review of the literature. Ageing Res Rev 47:183–197
Bordone L, Guarente L (2005) Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol 6:298–305
Bowman CJ, Ayer DE, Dynlacht BD (2014) Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nat Cell Biol 16:1202–1214
Bujak Adam L, Crane Justin D, Lally James S, Ford Rebecca J, Kang Sally J, Rebalka Irena A, Green Alex E, Kemp Bruce E, Hawke Thomas J, Schertzer Jonathan D, Steinberg Gregory R (2015) AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab 21:883–890
Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, Mandelkow EM, Mandelkow E, Cuervo AM (2018) Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 17:e12692
Cadwell K, Debnath J (2018) Beyond self-eating: the control of nonautophagic functions and signaling pathways by autophagy-related proteins. J Cell Biol 217:813–822
Calnan DR, Brunet A (2008) The FoxO code. Oncogene 27:2276–2288
Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle RE, Mydock-McGrane L, Jiang X, van Eijkeren RJ, Davis OB, Louie SM, Perera RM, Covey DF, Nomura DK, Ory DS, Zoncu R (2017) Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 355:1306–1311
Chen H, Fan M, Pfeffer LM, Laribee RN (2012) The histone H3 lysine 56 acetylation pathway is regulated by target of rapamycin (TOR) signaling and functions directly in ribosomal RNA biogenesis. Nucleic Acids Res 40:6534–6546
Chen BJ, Ueberham U, Mills JD, Kirazov L, Kirazov E, Knobloch M, Bochmann J, Jendrek R, Takenaka K, Bliim N, Arendt T, Janitz M (2017) RNA sequencing reveals pronounced changes in the noncoding transcriptome of aging synaptosomes. Neurobiol Aging 56:67–77
Cherra SJ 3rd, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, Chu CT (2010) Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol 190:533–539
Cortes CJ, La Spada AR (2019) TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol Dis 122:83–93
Cotzomi-Ortega I, Aguilar-Alonso P, Reyes-Leyva J, Maycotte P (2018) Autophagy and its role in protein secretion: implications for cancer therapy. Mediators Inflamm 2018:4231591
D’Angelo MA, Raices M, Panowski SH, Hetzer MW (2009) Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in post-mitotic cells. Cell 136:284–295
Danieli A, Martens S (2018) p62-mediated phase separation at the intersection of the ubiquitin-proteasome system and autophagy. J Cell Sci 131:jcs214304
Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A (2013) TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci USA 110:E1817–E1826
Demoinet E, Li S, Roy R (2017) AMPK blocks starvation-inducible transgenerational defects in Caenorhabditis elegans. Proc Natl Acad Sci USA 114:E2689–E2698
Deng Z, Purtell K, Lachance V, Wold MS, Chen S, Yue Z (2017) Autophagy receptors and neurodegenerative diseases. Trends Cell Biol 27:491–504
Denzel MS, Lapierre LR, Mack HID (2019) Emerging topics in C. elegans aging research: transcriptional regulation, stress response and epigenetics. Mech Ageing Dev 177:4–21
Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL (2008) The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 4:851–865
Dong F, Li C, Wang P, Deng X, Luo Q, Tang X, Xu L (2018) The RNA binding protein tristetraprolin down-regulates autophagy in lung adenocarcinoma cells. Exp Cell Res 367:89–96
Efeyan A, Comb WC, Sabatini DM (2015) Nutrient-sensing mechanisms and pathways. Nature 517:302–310
Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ, Dujardin S, Amaral AS, Grima JC, Bennett RE, Tepper K, DeTure M, Vanderburg CR, Corjuc BT, DeVos SL, Gonzalez JA, Chew J, Vidensky S, Gage FH, Mertens J, Troncoso J, Mandelkow E, Salvatella X, Lim RYH, Petrucelli L, Wegmann S, Rothstein JD, Hyman BT (2018) Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99:925–940
Egan D, Kim J, Shaw RJ, Guan K-L (2011) The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7:643–644
Escobar KA, Cole NH, Mermier CM, VanDusseldorp TA (2019) Autophagy and aging: maintaining the proteome through exercise and caloric restriction. Aging Cell 18:e12876
Fahrenkrog B, Harel A (2018) Perturbations in traffic: aberrant nucleocytoplasmic transport at the heart of neurodegeneration. Cells 7:232
Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktaschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA (2019) Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22:401–412
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, Donkervoort S, Ajroud-Driss S, Sufit RL, Heller SL, Deng HX, Siddique T (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 68:1440–1446
Folick A, Oakley HD, Yu Y, Armstrong EH, Kumari M, Sanor L, Moore DD, Ortlund EA, Zechner R, Wang MC (2015) Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science 347:83
Fonseca Costa SS, Ripperger JA (2015) Impact of the circadian clock on the aging process. Front Neurol 6:43
Frankel LB, Lubas M, Lund AH (2017) Emerging connections between RNA and autophagy. Autophagy 13:3–23
Fritzen AM, Frøsig C, Jeppesen J, Jensen TE, Lundsgaard A-M, Serup AK, Schjerling P, Proud CG, Richter EA, Kiens B (2016) Role of AMPK in regulation of LC3 lipidation as a marker of autophagy in skeletal muscle. Cell Signal 28:663–674
Fu H, Hardy J, Duff KE (2018) Selective vulnerability in neurodegenerative diseases. Nat Neurosci 21:1350–1358
Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ, Joseph B (2013) The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 500:468–471
Fullgrabe J, Ghislat G, Cho DH, Rubinsztein DC (2016) Transcriptional regulation of mammalian autophagy at a glance. J Cell Sci 129:3059–3066
Furuta M, Kose S, Koike M, Shimi T, Hiraoka Y, Yoneda Y, Haraguchi T, Imamoto N (2004) Heat-shock induced nuclear retention and recycling inhibition of importin alpha. Genes Cells 9:429–441
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, Cuervo AM, Debnath J, Deretic V, Dikic I, Eskelinen EL, Fimia GM, Fulda S, Gewirtz DA, Green DR, Hansen M, Harper JW, Jaattela M, Johansen T, Juhasz G, Kimmelman AC, Kraft C, Ktistakis NT, Kumar S, Levine B, Lopez-Otin C, Madeo F, Martens S, Martinez J, Melendez A, Mizushima N, Munz C, Murphy LO, Penninger JM, Piacentini M, Reggiori F, Rubinsztein DC, Ryan KM, Santambrogio L, Scorrano L, Simon AK, Simon HU, Simonsen A, Tavernarakis N, Tooze SA, Yoshimori T, Yuan J, Yue Z, Zhong Q, Kroemer G (2017a) Molecular definitions of autophagy and related processes. EMBO J 36:1811–1836
Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G (2017b) Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov 16:487–511
Gao F, Yang J, Wang D, Li C, Fu Y, Wang H, He W, Zhang J (2017) Mitophagy in Parkinson’s disease: pathogenic and therapeutic implications. Front Neurol 8:527
Garg G, Singh S, Singh AK, Rizvi SI (2016) Antiaging Effect of metformin on brain in naturally aged and accelerated senescence model of rat. Rejuvenation Res 20:173–182
Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, Han S, Banko MR, Gozani O, Brunet A (2010) Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466:383–387
Gu J, Wang Y, Wang X, Zhou D, Wang X, Zhou M, He Z (2018a) Effect of the LncRNA GAS5-MiR-23a-ATG3 axis in regulating autophagy in patients with breast cancer. Cell Physiol Biochem 48:194–207
Gu Z, Hou Z, Zheng L, Wang X, Wu L, Zhang C (2018b) LncRNA DICER1-AS1 promotes the proliferation, invasion and autophagy of osteosarcoma cells via miR-30b/ATG5. Biomed Pharmacother 104:110–118
Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N, Zhang ZC, Coughlin M, Sweeny EA, DeSantis ME, Jackrel ME, Rodell CB, Burdick JA, King OD, Gitler AD, Lagier-Tourenne C, Pandey UB, Chook YM, Taylor JP, Shorter J (2018) Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173:677–692
Hansen M, Rubinsztein DC, Walker DW (2018) Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol 19:579
Hargarten JC, Williamson PR (2018) Epigenetic regulation of autophagy: a path to the control of autoimmunity. Front Immunol 9:1864
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460:392
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93
He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, Levine B (2012a) Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481:511–515
He C, Sumpter R Jr, Levine B (2012b) Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 8:1548–1551
Hood S, Amir S (2017) The aging clock: circadian rhythms and later life. J Clin Investig 127:437–446
Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, Liu B, Chang C, Zhou T, Lippincott-Schwartz J, Liu W (2015) Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell 57:456–466
Ingram DK, Anson RM, De Cabo R, Mamczarz J, Zhu MIN, Mattison J, Lane MA, Roth GS (2004) Development of calorie restriction mimetics as a prolongevity strategy. Ann N Y Acad Sci 1019:412–423
Jang SY, Kang HT, Hwang ES (2012) Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J Biol Chem 287:19304–19314
Kang YA, Sanalkumar R, O’Geen H, Linnemann AK, Chang CJ, Bouhassira EE, Farnham PJ, Keles S, Bresnick EH (2012) Autophagy driven by a master regulator of hematopoiesis. Mol Cell Biol 32:226–239
Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW-L, Thomas EL, Kockel L (2010) With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab 11:453–465
Kaur J, Debnath J (2015) Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16:461
Kaushik S, Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19:365–381
Kenyon C (2010) The genetics of aging. Nature 464:504–512
Khalil H, Tazi M, Caution K, Ahmed A, Kanneganti A, Assani K, Kopp B, Marsh C, Dakhlallah D, Amer AO (2016) Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics 11:381–388
Kim C, Kim W, Lee H, Ji E, Choe YJ, Martindale JL, Akamatsu W, Okano H, Kim HS, Nam SW, Gorospe M, Lee EK (2014) The RNA-binding protein HuD regulates autophagosome formation in pancreatic beta cells by promoting autophagy-related gene 5 expression. J Biol Chem 289:112–121
Kirli K, Karaca S, Dehne HJ, Samwer M, Pan KT, Lenz C, Urlaub H, Gorlich D (2015) A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. Elife 4:e11466
Kishimoto S, Uno M, Okabe E, Nono M, Nishida E (2017) Environmental stresses induce transgenerationally inheritable survival advantages via germline-to-soma communication in Caenorhabditis elegans. Nat Commun 8:14031
Kourmouli N, Jeppesen P, Mahadevhaiah S, Burgoyne P, Wu R, Gilbert DM, Bongiorni S, Prantera G, Fanti L, Pimpinelli S, Shi W, Fundele R, Singh PB (2004) Heterochromatin and tri-methylated lysine 20 of histone H4 in animals. J Cell Sci 117:2491–2501
Kulkarni VV, Maday S (2018) Compartment-specific dynamics and functions of autophagy in neurons. Dev Neurobiol 78:298–310
Lahiri V, Hawkins WD, Klionsky DJ (2019) Watch what you (self-) eat: autophagic mechanisms that modulate metabolism. Cell Metab 29:803–826
Lapierre LR, Gelino S, Melendez A, Hansen M (2011) Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr Biol 21:1507–1514
Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu CC, Visvikis O, Chang JT, Gelino S, Ong B, Davis AE, Irazoqui JE, Dillin A, Hansen M (2013) The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun 4:2267
Lapierre LR, Kumsta C, Sandri M, Ballabio A, Hansen M (2015) Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 11:867–880
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293
Lawrence RE, Zoncu R (2019) The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 21:133–142
Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U, Mitchell CH, Lloyd-Evans E, Nixon RA (2015) Presenilin 1 maintains lysosomal Ca2+ homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep 12:1430–1444
Lee DC, Brellenthin AG, Thompson PD, Sui X, Lee IM, Lavie CJ (2017) Running as a key lifestyle medicine for longevity. Prog Cardiovasc Dis 60:45–55
Li L, Friedrichsen HJ, Andrews S, Picaud S, Volpon L, Ngeow K, Berridge G, Fischer R, Borden KLB, Filippakopoulos P, Goding CR (2018) A TFEB nuclear export signal integrates amino acid supply and glucose availability. Nat Commun 9:2685
Lim H, Lim YM, Kim KH, Jeon YE, Park K, Kim J, Hwang HY, Lee DJ, Pagire H, Kwon HJ, Ahn JH, Lee MS (2018) A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat Commun 9:1438
Lin XX, Sen I, Janssens GE, Zhou X, Fonslow BR, Edgar D, Stroustrup N, Swoboda P, Yates JR 3rd, Ruvkun G, Riedel CG (2018) DAF-16/FOXO and HLH-30/TFEB function as combinatorial transcription factors to promote stress resistance and longevity. Nat Commun 9:4400
Liu CY, Zhang YH, Li RB, Zhou LY, An T, Zhang RC, Zhai M, Huang Y, Yan KW, Dong YH, Ponnusamy M, Shan C, Xu S, Wang Q, Zhang YH, Zhang J, Wang K (2018) LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat Commun 9:29
Lloyd-Evans E, Haslett LJ (2016) The lysosomal storage disease continuum with ageing-related neurodegenerative disease. Ageing Res Rev 32:104–121
Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang TH, Kim HM, Drake D, Liu XS, Bennett DA, Colaiacovo MP, Yankner BA (2014) REST and stress resistance in ageing and Alzheimer’s disease. Nature 507:448–454
Ma C, Niu R, Huang T, Shao LW, Peng Y, Ding W, Wang Y, Jia G, He C, Li CY, He A, Liu Y (2018) N6-methyldeoxyadenine is a transgenerational epigenetic signal for mitochondrial stress adaptation. Nat Cell Biol 21:319–327
Madrigal-Matute J, Cuervo AM (2016) Regulation of liver metabolism by autophagy. Gastroenterology 150:328–339
Martinez-Lopez N, Singh R (2016) Telemetric control of peripheral lipophagy by hypothalamic autophagy. Autophagy 12:1404–1405
Martinez-Lopez N, Garcia-Macia M, Sahu S, Athonvarangkul D, Liebling E, Merlo P, Cecconi F, Schwartz Gary J, Singh R (2016) Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab 23:113–127
Martinez-Lopez N, Tarabra E, Toledo M, Garcia-Macia M, Sahu S, Coletto L, Batista-Gonzalez A, Barzilai N, Pessin JE, Schwartz GJ, Kersten S, Singh R (2017) System-wide benefits of intermeal fasting by autophagy. Cell Metab 26:856–871
Martini-Stoica H, Xu Y, Ballabio A, Zheng H (2016) The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci 39:221–234
Mattison JA, Colman RJ, Beasley TM, Allison DB, Kemnitz JW, Roth GS, Ingram DK, Weindruch R, de Cabo R, Anderson RM (2017) Caloric restriction improves health and survival of rhesus monkeys. Nat Commun 8:14063
Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, Licitra F, Lopez Ramirez A, Pavel M, Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y, Skidmore J, Rubinsztein DC (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93:1015–1034
Miyamoto Y, Saiwaki T, Yamashita J, Yasuda Y, Kotera I, Shibata S, Shigeta M, Hiraoka Y, Haraguchi T, Yoneda Y (2004) Cellular stresses induce the nuclear accumulation of importin alpha and cause a conventional nuclear import block. J Cell Biol 165:617–623
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15:1101–1111
Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y (2009) Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 10:458
Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C, Monfregola J, Medina DL, Lippincott-Schwartz J, Ballabio A (2018) mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun 9:3312
Nativio R, Donahue G, Berson A, Lan Y, Amlie-Wolf A, Tuzer F, Toledo JB, Gosai SJ, Gregory BD, Torres C, Trojanowski JQ, Wang LS, Johnson FB, Bonini NM, Berger SL (2018) Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat Neurosci 21:497–505
Newman JC, Covarrubias AJ, Zhao M, Yu X, Gut P, Ng C-P, Huang Y, Haldar S, Verdin E (2017) Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab 26:547–557
Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ (2010) Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature 467:963–966
Niccoli T, Partridge L (2012) Ageing as a risk factor for disease. Curr Biol 22:R741–R752
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64:113–122
Omata Y, Lim Y-M, Akao Y, Tsuda L (2014) Age-induced reduction of autophagy-related gene expression is associated with onset of Alzheimer’s disease. Am J Neurodegener Dis 3:134–142
Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521:525
Paonessa F, Evans LD, Solanki R, Larrieu D, Wray S, Hardy J, Jackson SP, Livesey FJ (2019) Microtubules deform the nuclear membrane and disrupt nucleocytoplasmic transport in tau-mediated frontotemporal dementia. Cell Rep 26:582–593
Peeters JGC, Picavet LW, Coenen S, Mauthe M, Vervoort SJ, Mocholi E, de Heus C, Klumperman J, Vastert SJ, Reggiori F, Coffer PJ, Mokry M, van Loosdregt J (2019) Transcriptional and epigenetic profiling of nutrient-deprived cells to identify novel regulators of autophagy. Autophagy 15:98–112
Perera RM, Zoncu R (2016) The lysosome as a regulatory hub. Annu Rev Cell Dev Biol 32:223–253
Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, Settleman J, Stephanopoulos G, Dyson NJ, Zoncu R, Ramaswamy S, Haas W, Bardeesy N (2015) Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524:361–365
Pickrell AM, Youle RJ (2015) The roles of PINK1, Parkin and mitochondrial fidelity in Parkinson’s disease. Neuron 85:257–273
Pircs K, Petri R, Madsen S, Brattas PL, Vuono R, Ottosson DR, St-Amour I, Hersbach BA, Matusiak-Bruckner M, Lundh SH, Petersen A, Deglon N, Hebert SS, Parmar M, Barker RA, Jakobsson J (2018) Huntingtin aggregation impairs autophagy, leading to argonaute-2 accumulation and global MicroRNA dysregulation. Cell Rep 24:1397–1406
Platt FM, Boland B, van der Spoel AC (2012) Lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 199:723
Plaza-Zabala A, Sierra-Torre V, Sierra A (2017) Autophagy and microglia: novel partners in neurodegeneration and aging. Int J Mol Sci 18:598
Polymenidou M (2018) The RNA face of phase separation. Science 360:859–860
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92:829–839
Raben N, Puertollano R (2016) TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol 32:255–278
Rabinowitz JD, White E (2010) Autophagy and metabolism. Science 330:1344–1348
Ramachandran PV, Savini M, Folick AK, Hu K, Masand R, Graham BH, Wang MC (2019) Lysosomal signaling promotes longevity by adjusting mitochondrial activity. Dev Cell 48:685–696
Rechavi O, Lev I (2017) Principles of transgenerational small RNA inheritance in Caenorhabditis elegans. Curr Biol 27:R720–R730
Rechavi O, Houri-Ze’evi L, Anava S, Goh WSS, Kerk SY, Hannon GJ, Hobert O (2014) Starvation-induced transgenerational inheritance of small RNAs in C. elegans. Cell 158:277–287
Roberts MN, Wallace MA, Tomilov AA, Zhou Z, Marcotte GR, Tran D, Perez G, Gutierrez-Casado E, Koike S, Knotts TA, Imai DM, Griffey SM, Kim K, Hagopian K, McMackin MZ, Haj FG, Baar K, Cortopassi GA, Ramsey JJ, Lopez-Dominguez JA (2017) A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab 26:539–546
Roth GS, Lane MA, Ingram DK (2005) Caloric restriction mimetics: the next phase. Ann N Y Acad Sci 1057:365–371
Rubino E, Rainero I, Chiò A, Rogaeva E, Galimberti D, Fenoglio P, Grinberg Y, Isaia G, Calvo A, Gentile S, Bruni AC, St. George-Hyslop PH, Scarpini E, Gallone S, Pinessi L (2012) SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 79:1556–1562
Ryan BJ, Hoek S, Fon EA, Wade-Martins R (2015) Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci 40:200–210
Sakamaki JI, Wilkinson S, Hahn M, Tasdemir N, O’Prey J, Clark W, Hedley A, Nixon C, Long JS, New M, Van Acker T, Tooze SA, Lowe SW, Dikic I, Ryan KM (2017) Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol Cell 66(517–32):e9
Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 169:361–371
Seah NE, de Magalhaes Filho CD, Petrashen AP, Henderson HR, Laguer J, Gonzalez J, Dillin A, Hansen M, Lapierre LR (2016) Autophagy-mediated longevity is modulated by lipoprotein biogenesis. Autophagy 12:261–272
Settembre C, Di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433
Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, Kweon MN, Won KJ, Baek SH (2016) AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534:553–557
Silvestrini MJ, Johnson JR, Kumar AV, Thakurta TG, Blais K, Neill ZA, Marion SW, St Amand V, Reenan RA, Lapierre LR (2018) Nuclear export inhibition enhances HLH-30/TFEB activity, autophagy, and lifespan. Cell Rep 23:1915–1921
Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4:176–184
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458:1131
Singh S, Singh AK, Garg G, Rizvi SI (2018) Fisetin as a caloric restriction mimetic protects rat brain against aging induced oxidative stress, apoptosis and neurodegeneration. Life Sci 193:171–179
Taormina G, Mirisola MG (2014) Calorie restriction in mammals and simple model organisms. Biomed Res Int 2014:308690
Tasset I, Cuervo AM (2016) Role of chaperone-mediated autophagy in metabolism. FEBS J 283:2403–2413
Taylor RC, Dillin A (2013) XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153:1435–1447
Tekirdag K, Cuervo AM (2018) Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem 293:5414–5424
Thomas HE, Zhang Y, Stefely JA, Veiga SR, Thomas G, Kozma SC, Mercer CA (2018) Mitochondrial complex I activity is required for maximal autophagy. Cell Rep 24(2404–17):e8
Tzivion G, Dobson M, Ramakrishnan G (2011) FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta 1813:1938–1945
Valvezan AJ, Manning BD (2019) Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat Metab 1:321–333
van Oosten-Hawle P, Porter RS, Morimoto RI (2013) Regulation of organismal proteostasis by trans-cellular chaperone signaling. Cell 153:1366–1378
Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Müller F (2003) Influence of TOR kinase on lifespan in C. elegans. Nature 426:620
Wang F, Jia J, Rodrigues B (2017a) Autophagy, metabolic disease, and pathogenesis of heart dysfunction. Can J Cardiol 33:850–859
Wang J, Cao B, Han D, Sun M, Feng J (2017b) Long non-coding RNA H19 induces cerebral ischemia reperfusion injury via activation of autophagy. Aging Dis 8:71–84
Wani WY, Boyer-Guittaut M, Dodson M, Chatham J, Darley-Usmar V, Zhang J (2015) Regulation of autophagy by protein post-translational modification. Lab Invest 95:14–25
Ward C, Martinez-Lopez N, Otten EG, Carroll B, Maetzel D, Singh R, Sarkar S, Korolchuk VI (2016) Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim et Biophys Acta (BBA) 1861:269–284
Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, Laboy R, Hirschey MD, Mair WB (2017) Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab 26:884–896
Wilkinson Deepti S, Jariwala Jinel S, Anderson E, Mitra K, Meisenhelder J, Chang Jessica T, Ideker T, Hunter T, Nizet V, Dillin A, Hansen M (2015) Phosphorylation of LC3 by the hippo kinases STK3/STK4 is essential for autophagy. Mol Cell 57:55–68
Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, Condon KJ, Petri S, Kedir J, Scaria SM, Abu-Remaileh M, Frankel WN, Sabatini DM (2017) KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 543:438
Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, Ren H, Chen D, Han J, Ying Z, Wang G (2016) TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J 35:121–142
Xu P, Das M, Reilly J, Davis RJ (2011) JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 25:310–322
Yan W, Chen ZY, Chen JQ, Chen HM (2018) LncRNA NEAT1 promotes autophagy in MPTP-induced Parkinson’s disease through stabilizing PINK1 protein. Biochem Biophys Res Commun 496:1019–1024
Yang L, Wang H, Shen Q, Feng L, Jin H (2017) Long non-coding RNAs involved in autophagy regulation. Cell Death Dis 8:e3073
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA (2005) Macroautophagy—a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 171:87–98
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283:10892–10903
Zhang G, Wang Z, Du Z, Zhang H (2018a) mTOR regulates phase separation of PGL granules to modulate their autophagic degradation. Cell 174(1492–506):e22
Zhang J, Wang J, Zhou Z, Park JE, Wang L, Wu S, Sun X, Lu L, Wang T, Lin Q, Sze SK, Huang D, Shen HM (2018b) Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy 14:1043–1059
Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, Bowen KE, Wadhwa H, Yang P, Rigo F, Taylor JP, Gitler AD, Rothstein JD, Lloyd TE (2018c) Stress granule assembly disrupts nucleocytoplasmic transport. Cell 173:95871.e17
Zhang Q, Wu X, Chen P, Liu L, Xin N, Tian Y, Dillin A (2018d) The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent Wnt signaling. Cell 174:870–883
Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL (2007) FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6:472–483
Zhao Y, Li X, Cai M-Y, Ma K, Yang J, Zhou J, Fu W, Wei F-Z, Wang L, Xie D, Zhu W-G (2013) XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell Res 23:491
Zheng G, Zhan Y, Li X, Pan Z, Zheng F, Zhang Z, Zhou Y, Wu Y, Wang X, Gao W, Xu H, Tian N, Zhang X (2018) TFEB, a potential therapeutic target for osteoarthritis via autophagy regulation. Cell Death Dis 9:858
Zhou J, Chong SY, Lim A, Singh BK, Sinha RA, Salmon AB, Yen PM (2017) Changes in macroautophagy, chaperone-mediated autophagy, and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging 9:583–599
Zhuo C, Jiang R, Lin X, Shao M (2016) LncRNA H19 inhibits autophagy by epigenetically silencing of DIRAS3 in diabetic cardiomyopathy. Oncotarget 8:1429–1437
Acknowledgements
L.R.L is funded by a grant from the NIH/NIA (R01 AG051810) and a Glenn Award for Research in Biological Mechanisms of Aging from the Glenn Foundation for Medical Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wong, S.Q., Kumar, A.V., Mills, J. et al. Autophagy in aging and longevity. Hum Genet 139, 277–290 (2020). https://doi.org/10.1007/s00439-019-02031-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-019-02031-7