
Abstract
The major distinguishing feature of fibrosis is significant deposition of collagen and other extracellular matrix (ECM) proteins, which can result in scarring if sufficiently excessive. Fibrosis affects many tissue types, and thus contributes to a broad group of diseases which, with few exceptions, continues to lack specific therapy. It has been estimated that nearly 45% of deaths in the developed world are caused by fibroproliferative diseases, which contribute to cardiovascular disease, pulmonary, renal, gut and liver fibrosis, and scleroderma (Bitterman and Henke in Chest 99:81S–84S, [1]). Fibroblasts are the most common stromal cell type of the connective tissues found in the body, and are the primary source of ECM in physiological conditions, i.e. in the absence of disease. The conversion of fibroblasts or similar stromal cells to myofibroblasts is a principal mediator of pathological fibrosis in many tissue types, and frequently occurs in response to ongoing tissue injury and chronic inflammation. While the fibrotic response can occur in response to existing disease, the phenotype conversion of fibroblasts to myofibroblasts due to transient stress or damage may lead to the initiation of long-term fibrotic disease (Bagchi et al. in BMC Biol 14:21, [2]). Inflammation has been found to be a critical inducer of fibrosis, with immune cells generating a variety of growth factors and cytokines that play critical roles in fibroblast activation and subsequent tissue remodelling and fibrosis. A common cellular response to stress stimuli such as inflammation is autophagy, and recent studies have tightly linked the activation or inhibition of autophagy with fibrotic diseases in myriad tissues. Here, we discuss the inter-relationship of these pathways to provide insight into their potential as therapeutic targets in fibrotic disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
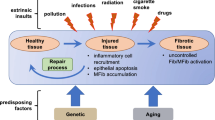
Fibrosis occurs due to the abnormal regulation of the synthesis and/or degradation of ECM, resulting in excessive extracellular deposition of fibrillar collagens (particularly type I and III) and other proteins and proteoglycans, and can alter the function of virtually every organ system in the body. While the precise impact of altered ECM production varies according to tissue type, fibrosis typically results in significant organ dysfunction, and frequently organ failure, contributing to patient morbidity and mortality. For example, idiopathic pulmonary fibrosis (IPF) progressively and severely compromises lung function, and shortens lifespan in affected individuals by many years [3]. Fibrosis frequently occurs secondary to other comorbidities—in the heart, hypertension and diabetes can both be significant drivers of fibrosis, as can smoking in the lung or acute or chronic toxic agent exposure in the liver [4,5,6,7,8]. Cardiac fibrosis can present in multiple patterns, but mid-wall fibrosis appears to be particularly dangerous, increasing the risk of death or hospitalization by up to 18-fold in dilated cardiomyopathy patients exhibiting this pattern compared to those without [9]. Thus, fibrosis is not only an outcome of pre-existing disease, it can also be a risk factor for further organ dysfunction, exacerbating adverse outcomes for patients. Clinical treatments for fibrosis in any organ remain scant, thus a better understanding of fibrosis pathogenesis is critically needed to enable the identification and development of novel therapeutics.
Fibroblasts are a polymorphic cell type which may arise from a variety of precursors such as endothelial or epithelial cells, depending on tissue type. Stromal fibroblasts play a variety of roles, including facilitating organ development, generating the supportive ECM of a tissue, and communicating with nearby parenchymal cells to maintain tissue homeostasis [10]. The ECM generated by fibroblasts consists of fibrous proteins such as collagen and elastin, gelatinous ground substance rich in glycosaminoglycans, and adhesive proteins such as laminin. This complex ECM provides overall tissue, organ and body integrity, and fibroblasts not only synthesize this material, they also play central roles in ECM maintenance and reabsorption. The specific composition of ECM can vary widely across tissues, depending on the specific mechanical stresses to which the tissue is subjected [11]. The critical importance of ECM, and thus of the fibroblasts that produce it, is reflected in the population of virtually all tissue types by fibroblasts or cells that fulfil a fibroblast function, such as hepatic stellate cells. Fibroblasts themselves are heterogeneous, even within tissues, and this is likely to help tune tissue integrity to local mechanical stresses [11, 12].
A general hypothesis that has gained significant consensus is that fibrosis represents a wound healing process that has somehow gone awry. In brief, the normal reparative process following tissue injury involves an initial inflammatory response, increased stromal tissue coupled with elevated ECM synthesis to support the healing injury, replacement of the dead or injured tissue with new parenchyma and a resolution phase in which the temporary ECM and stromal cells such as myofibroblasts are removed [13]. However, if the transition from the inflammatory phase to subsequent proliferation (of stroma and parenchyma) and wound remodelling (including ECM synthesis) fails to execute properly, fibrosis may occur.
A critical event during wound healing and the development of fibrosis is the activation of fibroblasts to a proliferative, migratory state, followed by a further phenotype transition to that of the myofibroblast [11, 14]. While fibroblasts synthesize and maintain ECM levels in homeostatic balance, myofibroblasts are the arbiters of excessive ECM production that occurs during tissue fibrosis. Not all myofibroblasts arise from fibroblasts, however they are largely responsible for disease progression and dysfunction in organs as varied as the heart, lungs, skin, kidneys, liver and gastrointestinal tract [12]. Myofibroblasts secrete high levels of ECM, and both secrete and are hypersensitive to a variety of cytokines, growth factors, and chemokines that promote and maintain the pro-fibrotic myofibroblast phenotype [15]. Their hallmark functional change is the acquisition of a contractile apparatus due to the induction of expression of α-smooth muscle actin which is incorporated into stress fibers, which may permit these cells to exert physical traction to help close or reduce the margins of wounds [12, 16, 17]. Cardiac myofibroblasts have been shown to also express the matricellular protein periostin, which itself has been implicated as a driver of fibrosis, and which is not expressed in non-activated fibroblasts [18, 19]. Fibroblast activation is unquestionably important in the wound healing process, but long-term maintenance of a myofibroblast-like phenotype contributes to pathological fibrosis [20, 21].
A number of cytokines and growth factors have been demonstrated to induce or facilitate the conversion of fibroblasts to myofibroblasts. While the most potent of these is TGFβ, a variety of other factors have also been implicated in fibroblast activation and fibrosis in multiple tissue types, including endothelin-1, Platelet-Derived Growth Factor (PDGF), angiotensin II, and Connective Tissue Growth Factor (CTGF/CCN2) [22,23,24,25,26,27]. Elevated TGFβ expression has long been associated with a host of fibrotic diseases including cystic fibrosis, scleroderma, and fibrosis of the lungs, heart, kidneys and liver [28,29,30,31,32,33,34]. TGFβ is produced and secreted to the extracellular matrix in a protein-bound, latent form by various cell types, including fibroblasts themselves as well as inflammatory cells [35, 36]. Various processes, including protease action or physical disruption, results in the release of active TGFβ, which acts via cell surface receptors to activate intracellular signalling cascades that can be Smad-dependent (canonical) or Smad-independent (non-canonical) [35, 37,38,39]. TGFβ-mediated expression of CTGF/CCN2, a matricellular protein, can further amplify pro-fibrotic processes, while upstream agents such as angiotensin II can up-regulate expression of TGFβ itself [40,41,42,43]. Antagonism of TGFβ signaling is effective in reducing evidence of fibrosis, and drugs that interfere with the renin–angiotensin–aldosterone system have been shown to exert anti-fibrotic effects [44,45,46]. It is unclear if these various factors act solely as a trigger to activate fibroblasts, or if their presence is required for ongoing maintenance of the myofibroblast phenotype. In either scenario, the transition of fibroblasts to myofibroblasts appears to be a critical step in the development and progression of fibrosis across tissue and organ types.
Immune Cells as Mediators of Fibrosis
Inflammation is a potent inducer of fibrosis across tissue types. While inflammation is an important initial step of the normal wound healing process, it is critical that inflammation resolves in a timely manner as a chronic inflammatory state leads to tissue remodelling and fibrosis [47]. Inflammation is mediated by a variety of immune cells including macrophages, mast cells, eosinophils, neutrophils, and CD4+ and CD8+ lymphocytes, and fibroblasts themselves can generate pro-inflammatory products such as growth factors and cytokines. Two primary types of immune responses can contribute to fibrosis. In type 1 immunity, Type 1 T helper (Th1) cells release factors such as interferon gamma (IFNγ) and interleukin-2 (IL-2). In turn, these factors stimulate phagocytic cells including mast cells and macrophages. In type 2 immunity, characterized by high antibody titers, Th2 cells secrete a variety of cytokines including IL-4, IL-5, IL-9, IL-10, and IL-13, resulting in eosinophil activation [48]. Fibrosis is initially characterized by the production of Th1 cytokines followed by the Th2 response, producing TGFβ and IL-13 and leading to activation of fibroblasts and their conversion to myofibroblasts to promote fibrosis [14, 49, 50]. Th17 cells have also been implicated in fibrosis of the skin and lung [51].
Mast Cells
Mast cells are involved in both innate and adaptive immunity, and play a pivotal role in inflammation as well as in tissue remodelling leading to fibrosis [52]. Mast cells produce, store and release various growth factors, inflammatory factors and cytokines which contribute to fibrosis, including TGFβ [53]. In addition, mast cells can produce proteoglycans such as hyaluronic acid which contribute to matrix composition directly, but which can also stimulate fibroblast activation [54]. Mast cells release a variety of proteases, including chymase, leukocyte elastase, and plasmin, that in turn release latent TGFβ from ECM niches to induce fibroblast activation [39]. Mice that were mast cell deficient were protected from bleomycin-induced pulmonary fibrosis, and this same study found that mast cell release of histamine and renin could activate fibroblasts either directly, or via the eventual generation of angiotensin II, respectively [55]. Thus, mast cells can trigger the initial activation of fibroblasts leading to their proliferation and ECM production via several complementary mechanisms, demonstrating their key role in the development of fibrosis.
Macrophages
Macrophages can be broadly classified into two phenotypes: the classical M1 phenotype that is pro-inflammatory and activated by cytokines such as IFNγ, and the alternative M2 phenotype that is involved in the resolution of inflammation and is activated by interleukins such as IL-4 and IL-13 [56, 57]. While M2 macrophages have been shown to specifically promote tissue remodelling and fibrosis, M1 macrophages can also drive fibrosis via the sustained maintenance of tissue inflammation, although the specific interplay of factors that determine whether M1 macrophages act in a pro- or anti-fibrosis fashion remain to be determined.
The pro-fibrotic role of M2 macrophages has been demonstrated in multiple tissue types [58]. When human monocyte THP-1 cells were polarized to M1 or M2 macrophages and exposed to human dermal fibroblasts, M2 macrophages, specifically, induced fibrotic responses including up-regulation of αSMA and expression of collagen [59]. However, the source of these macrophages appears to be important for their role in promoting fibrosis. Alveolar macrophages derived from monocytes were required for lung fibrosis in mice, whereas tissue-resident alveolar macrophages were not [60]. While M2 macrophages can serve as a source of TGFβ that subsequently acts directly on fibroblasts to induce their activation and promote fibrosis, TGFβ can instead act on bone marrow-derived macrophages to induce their conversion to myofibroblasts [61]. Macrophages thus exert a variety of effects on the induction of fibrosis across tissue types.
Interleukins
Interleukins are cytokines produced by white blood cells. They play a central role in the body’s immune and inflammatory responses, and their action is modulated by the inflammasome—a cytosolic multiprotein complex produced by myeloid cells which can contribute to tissue fibrosis [62]. In response to stress or injury, inflammasome assembly results in the activation of inflammatory caspases that in turn activate inflammatory cytokines via cleavage of inactive precursors [63]. The inflammasome acts as a binding site for a variety of caspases responsible for activating different interleukins, including IL-1 family members and related cytokines that stimulate fibrosis via the inflammatory response [63, 64].
Inflammatory cytokines including IL-1, Tissue Necrosis Factor and IL-33, can promote fibroblast activation, proliferation and collagen synthesis, potentially by increasing TGFβ expression [65,66,67]. Activated cytokines IL‐18 and IL‐33, along with IL-1, enhance inflammation and through the involvement of TGFβ can lead to the production of other cytokines such as IL-4 and IL-13, which in the setting of the lung can activate fibroblasts to behave as inflammatory cells, releasing pro-inflammatory cytokines and chemokines [68]. IL-5 is secreted by several inflammatory cell types, including mast cells and eosinophils, and eosinophils in turn are activated by IL-5 and Granulocyte–Macrophage Colony Stimulating Factor to contribute to activation of fibroblasts or other stromal cells to promote lung and intestinal fibrosis [69, 70].
In contrast to these pro-fibrotic cytokines, some demonstrate anti-fibrotic activity. IL-37 is an IL-1 family member, but has been found to attenuate the production of pro-inflammatory cytokines and chemokines by mast cells, macrophages or other immune cells that contribute to inflammation, and has shown promise as an anti-fibrotic in the lung and liver [71, 72]. Kim et al. reported that IL-37 reduced extracellular matrix protein expression in primary human lung fibroblasts, attenuated their proliferation in response to TGFβ, and was more highly expressed in alveolar epithelial cells and macrophages compared to cells from IPF patients [73]. In this study, the beneficial effect of IL-37 was dependent upon its ability to induce autophagy by inhibition of mTOR. IL-37 may also induce the expression of IL-10, and over-expression of IL-10 in the lung was able to reduce pulmonary fibrosis induced by bleomycin [74]. IL-10 has been reported to promote wound healing via activation of fibroblast-specific STAT3 and down-stream hyaluronan synthesis without driving fibrosis [75]. IL-22 is an IL-10 family member that has been found to act as an anti-inflammatory and anti-fibrotic cytokine in liver injury [76].
Autophagy
Autophagy is a critical process for the removal of excessive or defective cellular components including proteins and organelles, which can be recycled both to remove their detrimental effects on cell function and to provide energy and resources back to the cell. While autophagy can help the body to cope with stress, damage, injury, or pathogen infection, excessive levels of autophagy can be detrimental, potentially leading to cell apoptosis and tissue dysfunction.
Recent studies have linked autophagy, apoptosis and fibrosis in a variety of tissue types (Fig. 7.1). Stimulation of human atrial myofibroblasts with TGFβ induced both collagen synthesis and autophagy, while blockade of autophagy attenuated the fibrotic effect of TGFβ and in a separate study, prevented the conversion of rat cardiac fibroblasts to myofibroblasts [77, 78]. Over-expression of the TGFβ/Smad repressor Ski induced apoptosis in rat cardiac myofibroblasts, and this effect was increased if autophagy was simultaneously inhibited, suggesting that autophagy provides energy required for cell survival [79]. In normal human lung parenchymal and airway fibroblasts, TGFβ induced collagen synthesis and autophagy in parallel, however while the effect of TGFβ on collagen synthesis was increased in cells derived from IPF patients, autophagy induction was reduced demonstrating that the relationship between autophagy and fibrosis is variable [80]. Instead, the unfolded protein response was induced in IPF cells. These pathways may thus represent mechanisms to meet the high energy demands of ECM synthesis when fibroblasts become myofibroblasts, including in the setting of fibrosis. Conversely, apoptosis may be a means by which myofibroblasts are removed when energy levels are insufficient to support their ECM synthesizing function. These mechanisms may be highly context- and signalling pathway-dependent, however: in kidney mesangial cells, TGFβ induced both collagen synthesis and autophagy, while knockout or knockdown of the autophagy protein Beclin 1 also increased collagen synthesis [81].

The association of autophagy with TGFβ treatment is context-dependent. In response to tissue injury, chronic inflammation or impaired healing, fibroblasts undergo a phenotype conversion to myofibroblasts. The cytokine TGFβ can also independently induce this cellular change, however the induction of autophagy concomitantly with fibrosis is context-dependent. In a variety of cells including healthy lung fibroblasts, and atrial and ventricular cardiac fibroblasts, TGFβ induces both fibrotic gene expression and autophagy during conversion to myofibroblasts, and inhibition of autophagy typically attenuates fibrosis (lower panel). Conversely, in lung fibroblasts isolated from idiopathic pulmonary fibrosis (IPF) patients, autophagy decreases in response to TGFβ despite an induction in fibrotic gene expression during myofibroblast conversion. In kidney mesangial cells, TGFβ induced autophagy and collagen synthesis together, however reducing autophagy by decreasing Beclin-1 expression also stimulated collagen synthesis, further demonstrating that these pathways can be separated. Autophagy is positively correlated with fibrosis in other collagen-producing non-fibroblast cell types such as hepatocellular carcinoma cells, while in gut epithelium, attenuation or stimulation of autophagy increased or decreased fibrosis, respectively. The link between autophagy and fibrosis thus likely depends not only on cell type, but also on the specific environment and/or health of the cells
While autophagy may be required for fibroblast activation to myofibroblasts, the response of fibroblasts to the induction of autophagy may be distinct depending on the relative health status of the tissue, further supporting the idea that the specific cellular context is important in the relationship between autophagy and fibrosis. In normal lung fibroblasts, culturing on polymerized collagen can induce cell stress that results in apoptosis, but in contrast, IPF fibroblasts resist this stress and instead become proliferative and pro-fibrotic [82]. This was found to be due to alterations in PTEN/Akt/mTOR signalling in IPF fibroblasts compared to healthy lung fibroblasts, such that healthy cells undergo autophagy and subsequent apoptosis. Conversely, autophagy is down-regulated in IPF fibroblasts following culture on collagen resulting in increased cell survival [82]. A recent study suggests that IPF fibroblasts exhibit a senescent phenotype with reduced apoptosis and proliferation, rather than an activated fibroblast phenotype, with TGFβ inducing endoplasmic reticulum stress [83]. The disparity in these studies may represent differences in cell collection, nature or length of culture, biological variability across patients from which the cells are derived, or may simply be reflective of high levels of fibroblast heterogeneity. It is noteworthy, however, that both studies identified reduced apoptosis as a feature of IPF fibroblasts. It remains to be seen whether similar mechanisms linking fibrosis, autophagy and apoptosis occur in fibroblasts derived from other healthy or diseased tissues.
Regulation of Autophagy by TGFβ
TGFβ has been shown to regulate autophagy via both Smad-dependent and Smad-independent pathways, in a variety of disease contexts including cancer and fibrosis. In human hepatocellular carcinoma cells, for example, knockdown of Smad2/3 or Smad4 resulted in the inhibition of TGFβ-induced autophagy, although knockdown of c-Jun NH2-terminal kinase had a similar effect, implicating both canonical and non-canonical TGFβ pathways [84]. In several cancer cell lines, TGFβ induced pRb/E2F1-mediated up-regulation of autophagy genes and induction of autophagosome formation [85]. Conversely, berberine administration in a rat bleomycin model of IPF attenuated Smad and PI3K/Akt signalling, but also inhibited mTOR to increase autophagy [86]. As noted above, TGFβ both induced or inhibited autophagy in lung fibroblasts depending on whether they were derived from healthy or IPF donors, respectively, despite inducing collagen expression in both cell types [80]. Thus, the variable induction of canonical and non-canonical signalling pathways downstream of TGFβ may account, in part, for the differential effects noted on fibrotic gene expression and the induction of autophagy. In turn, alterations in the level of autophagy may be beneficial or detrimental with respect to fibrosis in different contexts. Clearly, additional investigation in this area is required.
Potential Therapeutic Targets for Fibrosis
Given the central roles of inflammation and autophagy in the induction and/or progression of fibrosis in various tissue types, it is tempting to consider these areas for exploitation in the quest for novel anti-fibrotic treatments, particularly given the current and conspicuous lack of such medications. Given its central role as a product of inflammatory cells and inducer of fibrosis and autophagy, TGFβ presents a tempting target. However, with its myriad roles within individual cells and across cell and tissue types, some of which may oppose one another depending on context or disease state, direct therapeutic interference with TGFβ itself is complicated at best [87].
Other targets may be more tractable for development [11]. As noted, IL-37 is an anti-inflammatory cytokine which can act directly by downregulating pro-inflammatory mediators and interfering with TGFβ signalling, and indirectly by promoting the production of other anti-inflammatory cytokines such as IL-10 [73, 74]. IL-37 may be of particular use in the setting of IPF. IL-10 and its family member IL-22 may similarly be useful therapeutically via their anti-inflammatory and anti-fibrotic properties [74, 76].
The regulation of autophagy provides another opportunity to reduce fibrosis. Autophagy may be required for the activation of fibroblasts to myofibroblasts, and autophagy inhibition leads to cardiac myofibroblast apoptosis [79]. However, the link between autophagy and fibrosis appears to be variable across tissues, and may further be influenced by not only the presence of disease, but also by the specific nature of the disease. For example, autophagy inhibition may actually increase activation of lung fibroblasts, with consequences for IPF [88]. Stimulation of autophagy by rapamycin in a gut fibrosis model reduced fibrosis, while autophagy inhibition with 3-methyladenine exacerbated fibrosis [89]. Thus, the positive or negative manipulation of autophagy to treat fibrosis will be critically dependent on the specific pathology involved.
Conclusion
Fibrosis is an aberrant wound healing process in which fibroblasts, in response to various stimuli including growth factors like TGFβ, which in turn is secreted and activated through inflammation-activated immune cells, increase collagen and extracellular matrix protein deposition. The end result is tissue remodeling leading to organ failure, with increased risk of patient morbidity and mortality. The interplay of fibrosis development and autophagy is complex, variable across cell and tissue types, and dependent on a variety of intracellular signaling pathways including mTOR/AKT/PTEN, Smads and others, despite the superficial similarity of fibrosis in different tissues.
The identification of novel anti-fibrotic medications is arguably one of the most urgent clinical challenges at present, given the widespread occurrence of fibrosis and dearth of treatments. Targeting the relationships between inflammation, autophagy and fibrosis provides an exciting new frontier for therapeutic development. However, caution is required given the heterogeneity of fibrotic disease mechanisms, and further mechanism-focused research in this area is critically needed.
References
Bitterman PB, Henke CA (1991) Fibroproliferative disorders. Chest 99:81S-84S
Bagchi RA, Roche P, Aroutiounova N, Espira L, Abrenica B, Schweitzer R, Czubryt MP (2016) The transcription factor scleraxis is a critical regulator of cardiac fibroblast phenotype. BMC Biol 14:21
Guenther A, Krauss E, Tello S, Wagner J, Paul B, Kuhn S, Maurer O, Heinemann S, Costabel U, Barbero MAN et al (2018) The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir Res 19:141
Silvestre JS, Heymes C, Oubenaissa A, Robert V, Aupetit-Faisant B, Carayon A, Swynghedauw B, Delcayre C (1999) Activation of cardiac aldosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation 99:2694–2701
Lee JY, Ihm HS, Kim JS, Hwang HS, Jeong KH, Ihm CG (2019) Baseline high blood pressure is associated with clinico-pathologic findings and later renal progression in chronic glomerulonephritis. Electrolyte Blood Press 17:54–61
Bian H, Zhu X, Xia M, Yan H, Chang X, Hu X, Pan B, Guo W, Li X, Gao X (2020) Impact of type 2 diabetes on nonalcoholic steatohepatitis and advanced fibrosis in patients with nonalcoholic fatty liver disease. Endocr Pract
Katzenstein AL, Mukhopadhyay S, Zanardi C, Dexter E (2010) Clinically occult interstitial fibrosis in smokers: classification and significance of a surprisingly common finding in lobectomy specimens. Hum Pathol 41:316–325
Tabet E, Genet V, Tiaho F, Lucas-Clerc C, Gelu-Simeon M, Piquet-Pellorce C, Samson M (2016) Chlordecone potentiates hepatic fibrosis in chronic liver injury induced by carbon tetrachloride in mice. Toxicol Lett 255:1–10
Leyva F, Taylor RJ, Foley PW, Umar F, Mulligan LJ, Patel K, Stegemann B, Haddad T, Smith RE, Prasad SK (2012) Left ventricular midwall fibrosis as a predictor of mortality and morbidity after cardiac resynchronization therapy in patients with nonischemic cardiomyopathy. J Am Coll Cardiol 60:1659–1667
Espira L, Czubryt MP (2009) Emerging concepts in cardiac matrix biology. Can J Physiol Pharmacol 87:996–1008
Czubryt MP (2019) Cardiac fibroblast to myofibroblast phenotype conversion-an unexploited therapeutic target. J Cardiovasc Dev Dis 6
Nagalingam RS, Al-Hattab DS, Czubryt MP (2019) What’s in a name? On fibroblast phenotype and nomenclature. Can J Physiol Pharmacol 97:493–497
Czubryt MP (2012) Common threads in cardiac fibrosis, infarct scar formation, and wound healing. Fibrogenesis Tissue Repair 5:19
Ma Y, Iyer RP, Jung M, Czubryt MP, Lindsey ML (2017) Cardiac fibroblast activation post-myocardial infarction: current knowledge gaps. Trends Pharmacol Sci 38:448–458
Blaauboer ME, Boeijen FR, Emson CL, Turner SM, Zandieh-Doulabi B, Hanemaaijer R, Smit TH, Stoop R, Everts V (2014) Extracellular matrix proteins: a positive feedback loop in lung fibrosis? Matrix Biol 34:170–178
Hinz B (2010) The myofibroblast: paradigm for a mechanically active cell. J Biomech 43:146–155
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA (2002) Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3:349–363
Zhao S, Wu H, Xia W, Chen X, Zhu S, Zhang S, Shao Y, Ma W, Yang D, Zhang J (2014) Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J Cardiol 63:373–378
Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ (2009) Origin of cardiac fibroblasts and the role of periostin. Circ Res 105:934–947
Kendall RT, Feghali-Bostwick CA (2014) Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol 5:123
Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC, JL, Aronow BJ, Tallquist MD et al (2016) Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 7:12260
Shi-Wen X, Chen Y, Denton CP, Eastwood M, Renzoni EA, Bou-Gharios G, Pearson JD, Dashwood M, du Bois RM, Black CM et al (2004) Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol Biol Cell 15:2707–2719
Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, Takigawa M, Nakanishi T, Takehara K (1999) Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: a mouse fibrosis model. J Cell Physiol 181:153–159
Shi-Wen X, Rodriguez-Pascual F, Lamas S, Holmes A, Howat S, Pearson JD, Dashwood MR, du Bois RM, Denton CP, Black CM et al (2006) Constitutive ALK5-independent c-Jun N-terminal kinase activation contributes to endothelin-1 overexpression in pulmonary fibrosis: evidence of an autocrine endothelin loop operating through the endothelin A and B receptors. Mol Cell Biol 26:5518–5527
Yu X, Xia Y, Zeng L, Zhang X, Chen L, Yan S, Zhang R, Zhao C, Zeng Z, Shu Y et al (2018) A blockade of PI3Kgamma signaling effectively mitigates angiotensin II-induced renal injury and fibrosis in a mouse model. Sci Rep 8:10988
Tanikawa AA, Grotto RM, Silva GF, Ferrasi AC, Sarnighausen VC, Pardini MI (2017) Platelet-derived growth factor A mRNA in platelets is associated with the degree of hepatic fibrosis in chronic hepatitis C. Rev Soc Bras Med Trop 50:113–116
Xu SW, Liu S, Eastwood M, Sonnylal S, Denton CP, Abraham DJ, Leask A (2009) Rac inhibition reverses the phenotype of fibrotic fibroblasts. PloS one 4:e7438
Wojnarowski C, Frischer T, Hofbauer E, Grabner C, Mosgoeller W, Eichler I, Ziesche R (1999) Cytokine expression in bronchial biopsies of cystic fibrosis patients with and without acute exacerbation. Eur Respir J: Off J Eur Soc Clin Respir Physiol 14:1136–1144
Ludwicka A, Ohba T, Trojanowska M, Yamakage A, Strange C, Smith EA, Leroy EC, Sutherland S, Silver RM (1995) Elevated levels of platelet derived growth factor and transforming growth factor-beta 1 in bronchoalveolar lavage fluid from patients with scleroderma. J Rheumatol 22:1876–1883
Rudnicka L, Varga J, Christiano AM, Iozzo RV, Jimenez SA, Uitto J (1994) Elevated expression of type VII collagen in the skin of patients with systemic sclerosis. Regulation by transforming growth factor-beta. J Clin Invest 93:1709–1715
Broekelmann TJ, Limper AH, Colby TV, McDonald JA (1991) Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc Natl Acad Sci U S A 88:6642–6646
Espira L, Lamoureux L, Jones SC, Gerard RD, Dixon IM, Czubryt MP (2009) The basic helix-loop-helix transcription factor scleraxis regulates fibroblast collagen synthesis. J Mol Cell Cardiol 47:188–195
Runyan CE, Schnaper HW, Poncelet AC (2003) Smad3 and PKCdelta mediate TGF-beta1-induced collagen I expression in human mesangial cells. Am J Physiol Renal Physiol 285:F413-422
Gressner AM, Weiskirchen R, Breitkopf K, Dooley S (2002) Roles of TGF-beta in hepatic fibrosis. Front Biosci 7:d793-807
Buscemi L, Ramonet D, Klingberg F, Formey A, Smith-Clerc J, Meister JJ, Hinz B (2011) The single-molecule mechanics of the latent TGF-beta1 complex. Curr Biol 21:2046–2054
Hinz B (2009) Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep 11:120–126
Zeglinski MR, Roche P, Hnatowich M, Jassal DS, Wigle JT, Czubryt MP, Dixon IM (2016) TGFbeta1 regulates Scleraxis expression in primary cardiac myofibroblasts by a Smad-independent mechanism. Am J Physiol Heart Circ Physiol 310:H239-249
Klingberg F, Chow ML, Koehler A, Boo S, Buscemi L, Quinn TM, Costell M, Alman BA, Genot E, Hinz B (2014) Prestress in the extracellular matrix sensitizes latent TGF-beta1 for activation. J Cell Biol 207:283–297
Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J (1995) Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem 270:4689–4696
Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH (2000) CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol 32:1805–1819
Gu J, Liu X, Wang QX, Tan HW, Guo M, Jiang WF, Zhou L (2012) Angiotensin II increases CTGF expression via MAPKs/TGF-beta1/TRAF6 pathway in atrial fibroblasts. Exp Cell Res 318:2105–2115
Yang F, Chung AC, Huang XR, Lan HY (2009) Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension 54:877–884
Campbell SE, Katwa LC (1997) Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol 29:1947–1958
Leask A (2010) Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res 106:1675–1680
Ma L, Hua J, He L, Li Q, Zhou J, Yu J (2012) Anti-fibrotic effect of Aliskiren in rats with deoxycorticosterone induced myocardial fibrosis and its potential mechanism. Bosn J Basic Med Sci/Udruzenje basicnih mediciniskih znanosti = Assoc Basic Med Sci 12:69–73
Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG (2010) Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res 107:418–428
Borthwick LA, Wynn TA, Fisher AJ (2013) Cytokine mediated tissue fibrosis. Biochim Biophys Acta 1832:1049–1060
Spellberg B, Edwards JE Jr (2001) Type 1/type 2 immunity in infectious diseases. Clin Infect Dis 32:76–102
Sandler NG, Mentink-Kane MM, Cheever AW, Wynn TA (2003) Global gene expression profiles during acute pathogen-induced pulmonary inflammation reveal divergent roles for Th1 and Th2 responses in tissue repair. J Immunol 171:3655–3667
Hoffmann KF, McCarty TC, Segal DH, Chiaramonte M, Hesse M, Davis EM, Cheever AW, Meltzer PS, Morse HC 3rd, Wynn TA (2001) Disease fingerprinting with cDNA microarrays reveals distinct gene expression profiles in lethal type 1 and type 2 cytokine-mediated inflammatory reactions. FASEB J 15:2545–2547
Lei L, Zhao C, Qin F, He ZY, Wang X, Zhong XN (2016) Th17 cells and IL-17 promote the skin and lung inflammation and fibrosis process in a bleomycin-induced murine model of systemic sclerosis. Clin Exp Rheumatol 34(Suppl 100):14–22
Conti P, Caraffa A, Mastrangelo F, Tettamanti L, Ronconi G, Frydas I, Kritas SK, Theoharides TC (2018) Critical role of inflammatory mast cell in fibrosis: Potential therapeutic effect of IL-37. Cell Prolif 51:e12475
Hugle T (2014) Beyond allergy: the role of mast cells in fibrosis. Swiss Med Wkly 144:w13999
Eggli PS, Graber W (1993) Cytochemical localization of hyaluronan in rat and human skin mast cell granules. J Invest Dermatol 100:121–125
Veerappan A, O’Connor NJ, Brazin J, Reid AC, Jung A, McGee D, Summers B, Branch-Elliman D, Stiles B, Worgall S et al (2013) Mast cells: a pivotal role in pulmonary fibrosis. DNA Cell Biol 32:206–218
Dona A, Abera M, Alemu T, Hawaria D (2018) Timely initiation of postpartum contraceptive utilization and associated factors among women of child bearing age in Aroressa District, Southern Ethiopia: a community based cross-sectional study. BMC Public Health 18:1100
Pechkovsky DV, Prasse A, Kollert F, Engel KM, Dentler J, Luttmann W, Friedrich K, Muller-Quernheim J, Zissel G (2010) Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin Immunol 137:89–101
Gieseck RL 3rd, Wilson MS, Wynn TA (2018) Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol 18:62–76
Zhu Z, Ding J, Ma Z, Iwashina T, Tredget EE (2017) Alternatively activated macrophages derived from THP-1 cells promote the fibrogenic activities of human dermal fibroblasts. Wound Repair Regen 25:377–388
Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN et al (2017) Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 214:2387–2404
Wang S, Meng XM, Ng YY, Ma FY, Zhou S, Zhang Y, Yang C, Huang XR, Xiao J, Wang YY et al (2016) TGF-beta/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 7:8809–8822
Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, Brown JH (2018) Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca(2+)/Calmodulin-Dependent Protein Kinase II delta Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation 138:2530–2544
Groslambert M, Py BF (2018) Spotlight on the NLRP3 inflammasome pathway. J Inflamm Res 11:359–374
Artlett CM (2018) The IL-1 family of cytokines. Do they have a role in scleroderma fibrosis? Immunol Lett 195:30–37
Lonnemann G, Shapiro L, Engler-Blum G, Muller GA, Koch KM, Dinarello CA (1995) Cytokines in human renal interstitial fibrosis. I. Interleukin-1 is a paracrine growth factor for cultured fibrosis-derived kidney fibroblasts. Kidney Int 47:837–844
Tan Z, Liu Q, Jiang R, Lv L, Shoto SS, Maillet I, Quesniaux V, Tang J, Zhang W, Sun B et al (2018) Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell Mol Immunol 15:388–398
Sullivan DE, Ferris M, Nguyen H, Abboud E, Brody AR (2009) TNF-alpha induces TGF-beta1 expression in lung fibroblasts at the transcriptional level via AP-1 activation. J Cell Mol Med 13:1866–1876
Doucet C, Brouty-Boye D, Pottin-Clemenceau C, Canonica GW, Jasmin C, Azzarone B (1998) Interleukin (IL) 4 and IL-13 act on human lung fibroblasts. Implication in asthma. J Clin Invest 101:2129–2139
Gharaee-Kermani M, Phan SH (1997) Lung interleukin-5 expression in murine bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 16:438–447
Takemura N, Kurashima Y, Mori Y, Okada K, Ogino T, Osawa H, Matsuno H, Aayam L, Kaneto S, Park EJ et al (2018) Eosinophil depletion suppresses radiation-induced small intestinal fibrosis. Sci Transl Med 10
Li Y, Gao Q, Xu K, Peng X, Yuan X, Jiang W, Li M (2018) Interleukin-37 attenuates bleomycin-induced pulmonary inflammation and fibrosis in mice. Inflammation 41:1772–1779
Feng XX, Chi G, Wang H, Gao Y, Chen Q, Ru YX, Luo ZL, Yan W, Li PY, Liu M et al (2019) IL-37 suppresses the sustained hepatic IFN-gamma/TNF-alpha production and T cell-dependent liver injury. Int Immunopharmacol 69:184–193
Kim MS, Baek AR, Lee JH, Jang AS, Kim DJ, Chin SS, Park SW (2019) IL-37 Attenuates lung fibrosis by inducing autophagy and regulating TGF-beta1 production in mice. J Immunol 203:2265–2275
Kurosaki F, Uchibori R, Sehara Y, Saga Y, Urabe M, Mizukami H, Hagiwara K, Kume A (2018) AAV6-mediated IL-10 expression in the lung Ameliorates bleomycin-induced pulmonary fibrosis in mice. Hum Gene Ther 29:1242–1251
Balaji S, Wang X, King A, Le LD, Bhattacharya SS, Moles CM, Butte MJ, de Jesus Perez VA, Liechty KW, Wight TN et al (2017) Interleukin-10-mediated regenerative postnatal tissue repair is dependent on regulation of hyaluronan metabolism via fibroblast-specific STAT3 signaling. FASEB J 31:868–881
Khawar MB, Azam F, Sheikh N, Abdul Mujeeb K (2016) How does interleukin-22 mediate liver regeneration and prevent injury and fibrosis? J Immunol Res 2016:2148129
Ghavami S, Cunnington RH, Gupta S, Yeganeh B, Filomeno KL, Freed DH, Chen S, Klonisch T, Halayko AJ, Ambrose E et al (2015) Autophagy is a regulator of TGF-beta1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis 6:e1696
Gupta SS, Zeglinski MR, Rattan SG, Landry NM, Ghavami S, Wigle JT, Klonisch T, Halayko AJ, Dixon IM (2016) Inhibition of autophagy inhibits the conversion of cardiac fibroblasts to cardiac myofibroblasts. Oncotarget 7:78516–78531
Zeglinski MR, Davies JJ, Ghavami S, Rattan SG, Halayko AJ, Dixon IM (2016) Chronic expression of Ski induces apoptosis and represses autophagy in cardiac myofibroblasts. Biochim Biophys Acta 1863:1261–1268
Ghavami S, Yeganeh B, Zeki AA, Shojaei S, Kenyon NJ, Ott S, Samali A, Patterson J, Alizadeh J, Moghadam AR et al (2018) Autophagy and the unfolded protein response promote profibrotic effects of TGF-beta1 in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 314:L493–L504
Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME (2012) Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1. J Biol Chem 287:11677–11688
Nho RS, Hergert P (2014) IPF fibroblasts are desensitized to type I collagen matrix-induced cell death by suppressing low autophagy via aberrant Akt/mTOR kinases. PloS one 9:e94616
Alvarez D, Cardenes N, Sellares J, Bueno M, Corey C, Hanumanthu VS, Peng Y, D’Cunha H, Sembrat J, Nouraie M et al (2017) IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol 313:L1164–L1173
Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, Sugimoto K, Miyazono K (2009) Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res 69:8844–8852
Korah J, Canaff L, Lebrun JJ (2016) The retinoblastoma tumor suppressor protein (pRb)/E2 promoter binding factor 1 (E2F1) pathway as a novel mediator of TGFbeta-induced autophagy. J Biol Chem 291:2043–2054
Chitra P, Saiprasad G, Manikandan R, Sudhandiran G (2015) Berberine inhibits Smad and non-Smad signaling cascades and enhances autophagy against pulmonary fibrosis. J Mol Med (Berl) 93:1015–1031
Al Hattab D, Czubryt MP (2017) A primer on current progress in cardiac fibrosis. Can J Physiol Pharmacol 95:1091–1099
Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M et al (2013) Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 304:L56-69
Cosin-Roger J, Canet F, Macias-Ceja DC, Gisbert-Ferrandiz L, Ortiz-Masia D, Esplugues JV, Alos R, Navarro F, Barrachina MD, Calatayud S (2019) Autophagy stimulation as a potential strategy against intestinal fibrosis. Cells 8
Acknowledgements
This work was supported by a Project Grant from the Canadian Institutes of Health Research (PJT-162422 to MPC).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Chattopadhyaya, S., Czubryt, M.P. (2022). Fibroblasts, Fibrosis and Autophagy. In: Kirshenbaum, L.A. (eds) Biochemistry of Apoptosis and Autophagy. Advances in Biochemistry in Health and Disease, vol 18. Springer, Cham. https://doi.org/10.1007/978-3-030-78799-8_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-78799-8_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-78798-1
Online ISBN: 978-3-030-78799-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)