
Abstract
Acute pulmonary embolism (aPE) is one of the great masqueraders in medicine. Therefore, a high index of clinical suspicion coupled with a detailed history and physical examination is invaluable when evaluating patients. Acute venous thromboembolism (VTE) that usually originates from the lower extremity deep veins (DVT) is clearly recognized as a significant healthcare problem. In the United States an estimated 900,000 cases of DVT and PE occur per year, causing approximately 300,000 deaths. Clinically, aPE is defined as massive, sub-massive, or non-massive. In the assessment and management of patients presenting with suspected aPE, clinical information is critical not only to the initial assessment of prognosis, but also to guide therapeutic decision-making. Hemodynamic stability and right ventricular (RV) function are found to be critically important in determining morbidity and mortality. Thus, risk stratification algorithms have been proposed to help physicians identify high- versus low-risk aPE patients in order to expedite diagnosis and treatment. Computed tomographic pulmonary angiography (CTPA) has been the imaging of choice in aPE patients not only for its higher sensitivity and specificity, but also for providing alternate diagnosis in patients with nonspecific signs and symptoms of aPE. More recently, echocardiography has been able to provide anatomical, functional, as well as mechanical information regarding RV function and RV-pulmonary unit interaction. This chapter intents to not only summarize the most important pathophysiological processes involved from clot formation to distal pulmonary embolization, but also describe potential hemodynamic implications and associated clinical manifestations. Current diagnostic and therapeutic algorithms are reviewed; available imaging modalities with most typically diagnostic aPE features are described; and mechanical characterization of the anatomical and functional abnormalities with regard to RV function are examined in terms of the hemodynamic derangement caused by acute obstruction to pulmonary flow.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Acute pulmonary embolism
- Deep venous thrombosis
- Venous thromboembolism
- Right ventricle
- RV outflow tract
- Pulmonary artery
- Pulmonary vascular resistance
- Pulmonary hypertension
- Transthoracic echocardiogram
- Computed tomographic pulmonary angiography
- Speckle tracking echocardiography
- Velocity vector imaging
Introduction
Acute pulmonary embolism (aPE) has perennially been considered one of the great masqueraders in medicine. Even though PE might be considered both a common and ubiquitous disorder, presenting symptoms and signs are often nonspecific; therefore, a high index of clinical suspicion coupled with a detailed history and physical examination is invaluable when evaluating patients.
A wealth of clinical and laboratory data has linked the development of deep venous thrombosis (DVT) with thromboembolic potential that may result in aPE [1,2,3,4,5,6,7,8,9,10,11]. Representative duplex images of a normal popliteal vein (Fig. 14.1a, b) and acute DVT (Fig. 14.1c, d) as well as a chronic DVT (Fig. 14.2a, b) are shown for comparison. Even though DVT is the most common source of embolization resulting in aPE, additional sources for potential embolization are shown in Fig. 14.3. A more complete list of potential recognized sources of thromboembolism is given in Table 14.1.


(a) A representative duplex image showing a normal popliteal vein before and after manual compression. (b) Normal color duplex signal in a normal popliteal vein filling the whole-vein contour. (c) Case of an acute DVT showing a dilated popliteal vein that lacks compressibility. (d) Color is not found due to the proximal acute DVT that impedes flow

(a) In sharp contrast, a chronic case of DVT is showing a visible organized clot. Please note that in most instances of a fresh clot as seen in (a), in acute DVT, the clot was not well visualized. (b) Color flow duplex signal within a popliteal vein with a partially filling clot that has distorted the main lumen

(a) Representative subcostal echocardiographic image showing a homogeneous density filling the inferior vena cava (IVC) denoted by the white arrow. (b) Four-chamber apical echocardiographic image showing a rather large tubular thrombus cast within the right atrium (RA), crossing the tricuspid valve and into the right ventricle (RV). (c) Transesophageal view from the bi-caval view at 90° showing the right atrium (RA) and catheter (arrow) in the superior vena cava and a thrombus mass due to catheter-related trauma encircled by (*). (d) Improved visualization of the catheter seen in the SVC and the mural thrombus seen in the right atrium (RA). (e) Transesophageal view from 45° showing the right atrium (RA), RV outflow tract (RVOT), left atrium (LA), as well as a globular homogeneous mass (arrow) attached to the interatrial septum that was resected and found to be a myxoma. (f) Transthoracic view from the RV inflow showing the right atrium (RA), right ventricle (RV), and tricuspid valve (TV) as well as a homogeneous mass found to be a vegetation (*). (g) Additional view showing the same tricuspid valve vegetation as seen in (f)
Most patients with acute venous thromboembolism (VTE) present with thrombosis in the legs as well as pulmonary thrombus at the time of diagnosis [12]. A high index of suspicion is required to recognize which patients are at risk of this otherwise deadly clinical entity. VTE is clearly recognized as a significant healthcare problem in the United States. It is estimated that 900,000 cases of DVT and PE occur per year and approximately 300,000 deaths are attributed to VTE [13]. Therefore, better understanding of the mechanisms regulating venous thrombosis and clot resolution is critical.
Although thrombophlebitis or DVT of the lower limbs was first reported in ancient Hindu medicine writings around the year 800 BCE [14], subsequent descriptions of venous thrombogenesis were unclear and remained elusive. Our current understanding of thrombus formation revolves around the well-described interplay of factors such as venous stasis, changes in the vessel wall, and thrombogenic changes within the blood in order to result in VTE. Though Virchow coined the word embolism and made significant advances in our understanding of thrombosis, he never formally proposed this triad—a concept that first appeared in the medical literature almost 100 years after his death [15].
It is estimated that the incidence of VTE in industrialized countries is 1–3 individuals per 1000 per year [8, 16,17,18]. However, a dramatic increase in the risk of VTE then occurs in individuals older than 85 years to 8 per 1000 persons/years, and for those over the age of 50 reaching as high as 1 in every 100 individuals annually [8]. These alarming statistics have led the United States Senate in 2005 to designate March as “DVT Awareness Month” followed by the Surgeon General’s call to action in 2008 to prevent DVT and PE.
Anticoagulation is the mainstay of treatment of symptomatic VTE. Anticoagulation prevents further thrombus deposition, allows established thrombus to undergo stabilization and/or endogenous lysis, and reduces the risk of interval recurrent thrombosis [12]. This chapter focuses on not only the pathophysiological and hemodynamic alterations that occur with aPE, but also the mechanical abnormalities that these processes have on the right ventricle (RV). This review intends to highlight the importance of the pulmonary-circulation-ventricular circuit in mediating cardiac performance and how the latter is the most critical factor determining both morbidity and mortality.
Mechanisms Regulating Thrombosis
Even though the molecular and translational pathways that regulate the dynamic balance between clot formation and lysis are well beyond the scope of this chapter, it is important to have a basic understanding of the individual elements responsible for these processes.
A healthy vascular endothelium is critical in maintaining adequate hemostasis. Under normal conditions, intact endothelial cells promote vasodilatation and local fibrinolysis. Hence, blood coagulation, platelet adhesion and activation, as well as inflammation and leukocyte activation are suppressed resulting in normal blood fluidity. A list of specific elements that maintain the natural nonthrombogenic state of the endothelial surface can be found in Table 14.2 [19, 20].
In contrast, during periods of direct vascular trauma or as a result of activation of the coagulation cascade, a prothrombotic and proinflammatory state ensues [20]. The latter is mainly characterized by an enhanced production of von Willebrand factor, tissue factor, plasminogen activator inhibitor-1, and factor V that augment thrombosis [20]. In addition, release of substances such as platelet-activating factor and endothelin-1 promotes vasoconstriction [21]. Finally, an exposed endothelial surface increases the expression of cell adhesion molecule, promoting accumulation and activation of leukocytes that further amplifies inflammation and thrombosis as shown in Fig. 14.4 [19, 22].

Proposed mechanisms for venous thrombosis. It has been proposed that the formation of a venous thrombosis can be divided into distinct steps. First, the endothelium is activated by hypoxia and/or inflammatory mediators and expresses the adhesion proteins P-selectin, E-selectin, and vWF. Second, circulating leukocytes, platelets, and TF+ MVs bind to the activated endothelium. Third, the bound leukocytes become activated and express TF. The local activation of the coagulation cascade overwhelms the protective anticoagulant pathways and triggers thrombosis. The fibrin-rich clot also contains platelets and red blood cells. (Reproduced with permission from Mackman N. New insights into the mechanisms of venous thrombosis. J Clin Invest. 2012; 122: 2331–2336)
Thrombus contains a mixture of platelets, fibrin, and in some cases red blood cells [23, 24]. The composition of thrombus depends on flow and vessel characteristics. Arterial clots are formed under high shear stress, typically after rupture of an atherosclerotic plaque or other damage to the blood vessel wall, and are largely platelet rich (white clots) and thus generally treated with antiplatelet drugs [20, 25, 26]. Venous clots form under lower shear stress and are mostly rich in fibrin (red clots) and are hence treated with anticoagulant drugs [12, 20, 27,28,29].
The coagulation cascade is regulated at several levels. Interaction of tissue factor exposed by vascular injury with plasma factor VIIa results in the formation of small amounts of thrombin. This thrombin production is then amplified through the intrinsic pathway, resulting in the formation of the fibrin clot. These reactions take place on phospholipid surfaces, usually the activated platelet surface [30]. In case of venous thrombosis, changes in blood flow and in endothelial cell lining of blood vessels, as initially proposed by Virchow, increase the risk of VTE [31]. A series of well-recognized risk factors, as shown in Table 14.3, have been associated with an increased risk of DVT and the potential for VTE [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51].
One group of patients that require some additional discussion are those with significant end-stage liver disease (ESLD). The previously held concept that ESLD patients are auto-anticoagulated as a result of a reduced hemostatic reserve given their reduced ability to synthesize procoagulant proteins and anticoagulation factors, hence at high risk for bleeding complications and consequently at a reduced risk for VTE, is no longer valid [51,52,53]. Close inspection reveals that initial reports were simply based on data obtained from relatively small single-center studies [54, 55]. Subsequent emergences of more robust data from much larger national and international databases have shed more relevant clinical information [56,57,58,59]. Specifically, based on these results it is now recommended that hospitalized and immobilized cirrhotic patients, certainly at increased risk of thrombotic events, should receive VTE prophylaxis with either low-molecular-weight heparin or unfractionated heparin (Level III, Grade C) [51]. In contrast, cirrhotic patients with VTE should be treated with anticoagulation similar to other medical patients without a specific recommendation. Most importantly, therapy selection should be determined on a case-by-case analysis (Level III, Grade C) [51]. Certainly, additional prospective randomized trials are urgently needed to advance our understanding of this complex group of patients and better guide clinical practices.
Furthermore, in cases of compression of the left common iliac vein either by the presence of the fetus during pregnancy or in patients with May-Thurner syndrome [60, 61], the most likely site of thrombus formation in DVT is the pocket of the valve sinus [62,63,64,65,66]. These sites are particularly prone to thrombosis because of the disrupted and irregular blood flow patterns [62]. Initial formation of thrombus within the venous valve pocket further disrupts the architecture of these valve pockets, disrupting pulsatility of venous flow and favoring local stasis of cellular elements [67]. Formation of a semi-solidified mass causes further activation of circulating platelets and leukocytes inducing additional fibrinogenesis favoring the growth of the thrombus beyond the confines of the valve pocket [67]. Amplification of this process causes further alteration of luminal flow dynamics resulting in progressive occlusion of the vein lumen [67]. Intraluminal growth of the thrombus has also been shown to lower local oxygen tension, as oxygen is being consumed by trapped cells resulting in luminal hypoxemia favoring cell death and contributing to thrombus growth [67]. Notwithstanding the fact that the luminal linear velocity of the blood continues to decline and further oxygen is being locally consumed, passing blood stream tugs additional layers of cells to the growing thrombus [67]. A graphic representation of this process is illustrated in Fig. 14.5 [62, 67,68,69].

Diagrammatic representation of a deep vein during normal flow, during acute thrombus formation (DVT), and during potential embolization (VTE). During normal flow blood cells freely move across vein valves. In the event of DVT, a clot forms on the underside of the vein valves. In the case of VTE, potential dislodgement of part of the clot material can be released into the venous circulation. Thrombosis is thought to be enhanced by a significant decline in oxygen tension in the region of the valve pocket sinus resulting in clot formation and the potential for VTE
Two simple anatomical considerations are important for understanding DVT and the potential for VTE. First, as the number of vein valves increases within any given vein segment, the risk of DVT increases. Second, during the stage of thrombus formation, not all portions of the thrombus anchor to the necrotic endothelium. Some portions of the growing thrombus are less strongly attached and can embolize in the direction of blood flow. Development of local symptoms of DVT depends on the extent of thrombosis, adequacy of collateral vessels, and also severity of associated vascular occlusion and inflammation. Phlegmasia cerulea and alba dolens refer to massive venous thrombus that can cause significant painful swelling of the calf and resultant pressure necrosis and gangrene. When the thrombus embolizes to the pulmonary vasculature, symptoms and hemodynamic adaptation largely depend on the underlying cardiopulmonary reserve of the individual [63,64,65]. A large thrombus is relatively well tolerated in the setting of normal cardiovascular reserve while a relatively smaller burden can cause hemodynamic compromise in those with limited reserve.
The early post-venous thrombosis stage is characterized by recruitments of neutrophils that are essential for thrombus resolution by promoting fibrinolysis and collagenolysis [70, 71]. This process then transitions over the course of a few days, peaking at approximately day 8, to a monocyte-predominant venous thrombus milieu that is then associated with plasmin and matrix metalloproteinase-mediated thrombus breakdown [72, 73].
Epidemiology of Venous Thromboembolism
It is important to realize that the current estimates of incidence of DVT and VTE represent an underestimate for several reasons. Milder cases may not be investigated and remain undiagnosed, while fatal undiagnosed events are never identified. The thrombosis cases treated outside the hospital setting (in the clinic or emergency room) are rarely included in studies. Since many studies use stringent validation criteria, patients with typical clinical findings but nondiagnostic radiologic studies are not reported. Finally, patients treated in long-term care facilities and cancer patients in hospice settings are usually not enrolled in clinical studies. It is also important to keep in mind that studies that include a large number of VTE cases diagnosed by autopsy have generally reported a higher proportion of PE than DVT cases likely due to the presence of asymptomatic PE.
Current estimates regarding incidence rates for VTE are recognized to be higher among African American populations when compared to Asian, Asian American, and Native American populations.
VTE is mostly seen in older individuals and is rare prior to late adolescence [74]. Even when incidence rates increase markedly for both men (130 per 100,000) and women (110 per 100,000) there is a clear difference that is important to be recognized [74]. Incidence rates of VTE are higher in women during childbearing years (16–44 years) compared to men of similar age; however, in individuals older than 45 years of age, VTE incidence rates are higher among men [74].
Interestingly, it has been reported that incident cases of VTE can occur without a specific recognizable trigger, in up to 40% of cases in populations of European and African origins [74].
Even when there is paucity of data relating VTE incidence, incidence rates for VTE, DVT, and PE either remained constant or increased between 1981 and 2000 with a substantial increase rate for VTE noted between 2001 and 2009 based on PE cases, the latter likely reflecting the increased utilization of better imaging techniques.
Data largely obtained from a case-controlled study that included 726 women with incident VTE between 1988 and 2000 in Olmsted County, MN, USA, identified the following case particulars as significant risk factors for the development of VTE. These are presented in the decreasing order of clinical impact [75]:
-
Major surgery (odds ratio (OR) 18.95)
-
Active cancer with or without concurrent chemotherapy (OR 14.64)
-
Neurological disease with leg paresis (OR 6.10)
-
Hospitalization for acute illness (OR 5.07)
-
Nursing-home confinement (OR 4.63)
-
Trauma or fracture (OR 4.56)
-
Pregnancy or puerperium (OR 4.24)
-
Oral contraception (OR 4.03)
-
Noncontraceptive estrogen plus progestin use (OR 2.53)
-
Estrogen (OR 1.81)
-
Progestin (OR 1.20)
-
Body mass index (OR 1.08)
Interestingly previously held notions accounting for VTE risk factors such as age, varicose veins, and progestin were not significantly associated with incident VTE in this studied population when included in the multivariate analysis [75].
Hospitalization remains a well-recognized risk factor for VTE conferring a greater than 100-fold risk regardless of the fact that if this hospitalization is due to medical illness (VTE risk 22%) or surgically related (24%) [74].
Recognized VTE risk factors among medical illness patients include [74]:
-
Age
-
Obesity
-
Previous VTE
-
Thrombophilia
-
Cancer
-
Recent trauma or surgery
-
Tachycardia
-
Acute myocardial infarction or stroke
-
Leg paresis or prolonged immobilization (bed rest)
-
Congestive heart failure
-
Acute infection
-
Rheumatological disorders
-
Hormone therapy
-
Central venous catheter
-
Admission to an intensive or coronary care unit
-
White blood cell and platelet count
In the case of surgical patients, VTE risk factors include [74]:
-
Age, particularly in those 65 years of age or older.
-
Type of surgery such as neurosurgical, major orthopedic procedures of the leg, renal transplantation, cardiovascular surgery, and thoracic, abdominal, or pelvic surgical interventions for cancer. Furthermore, obesity and a poor physical status are well-recognized risk factors after total-hip arthroplasty.
-
Smoking status.
-
Presence or absence of active cancer.
Additional elements that should be considered known to increase VTE risk factors include [76,77,78,79]:
-
Central venous catheters, particularly via femoral vein access
-
Prior superficial vein thrombosis
-
Long travels greater than 4 h
-
Hypertriglyceridemia in postmenopausal women
The risk of DVT associated with varicose veins is uncertain and seems to vary with patient age [80].
However, we would like to point out that generalizations should be avoided as all these recommendations require further validation as the fluidity of medical and surgical care as well as the complexity of cases have evolved.
It is important to point out that nursing-home residents account for an ever-growing number of patients for VTE risk. In fact, hospitalization and being a resident of a nursing home account for 60% of incident VTE with nursing-home residence independently responsible for 1/10th of all VTE cases [74].
Traditionally, cancer increases VTE risk and currently accounts for 20% of all incident cases occurring in the community [74]. Specific cancers associated to the increased VTE risk include pancreas, ovaries, colon, stomach, lung, kidney, bone, and brain [81, 82]. Furthermore, when cancer patients receive immunosuppressive or cytotoxic chemotherapy, particularly l-asparaginase, thalidomide, lenalidomide, or tamoxifen, these medications place these patients at even higher risk [83, 84].
With regard to cancer patients, VTE risk is particularly increased [85, 86]:
-
Gastric and pancreatic cancers
-
Platelet counts ≥350 × 109/l
-
Hemoglobin levels <100 g/l
-
Use of red cell growth factors
-
Leukocyte counts ≥11 × 109/l
-
BMI 35 kg/m2 or greater ≥35
-
Elevated plasma-soluble P-selectin and D-dimers
Aside from all these recognized risk factors and clinical situations associated with VTE, true estimates of the total number of VTE events that are either incident or recurrent occurring each year vary widely. Despite some limitations, attack rates have been estimated between 91 and 255 for DVT and between 51 and 75 for PE per 100,000 person years [87, 88].
Recurrent VTE events occur in up to 30% of cases within 10 years of the initial event with a reported rate of 19–39 per 100,000 person years [87, 89]. Based on the data originally published by Heit et al. the estimated cumulative incidence of the first overall VTE recurrence was 1.6% at 7 days, 5.2% at 30 days, 8.3% at 90 days, 10.1% at 180 days, 12.9% at 1 year, 16.6% at 2 years, 22.8% at 5 years, and 30.4% at 10 years with the risk of first recurrence being the highest close to the incident event and then decreasing as time goes on [90]. Although prophylaxis is crucial, duration of this initial treatment does not appear to affect the rate of recurrence beyond the initial 3 months of recommended prophylactic anticoagulation suggesting that VTE is in fact a chronic disease with episodic recurrence [91,92,93,94].
Recognized independent predictors of VTE recurrence include [32, 90, 95,96,97,98,99,100,101]:
-
Increasing age
-
Increasing BMI
-
Male sex
-
Active cancer
-
Neurological disease with leg paresis
-
Idiopathic VTE
-
Active lupus anticoagulant or antiphospholipid antibody
-
Deficiency of antithrombin, protein C, or protein S
-
Hyperhomocysteinemia
-
Elevated plasma D-dimers
-
In some cases, residual vein thrombosis
-
Factor V Leiden mutations combined with deficiencies of antithrombin, protein S, or protein C
-
Pancreatic, brain, lung, and ovarian cancer; myeloproliferative or myelodysplastic disorders; and stage IV cancers
Accurate assessments of case fatality rates after initial VTE are hindered by the nature of the analysis. First, retrospective data analysis is difficult to interpret when autopsy data is used because autopsies are not uniformly performed to confirm aPE diagnosis in patients dying unexpectedly. Second, prospective data collection is also difficult due to the declining rates of autopsies. Despite these limitations, it is believed that up to two-thirds of patients simply manifest DVT alone and death occurs in approximately 6% of these patients within a month of diagnosis, whereas a third of patients with symptomatic VTE develop PE and may experience up to a 12% mortality during the same time period [54]. Early mortality after VTE is strongly associated with presentation as PE, advanced age, cancer, and underlying cardiovascular disease [54, 102].
Finally, it is clear that VTE occurrence is not only influenced by multiple clinical factors and disease processes but also modified by several genetic-environmental interactions placing some individuals at greater risk than others and additional follow-up studies are certainly required.
Pulmonary Vasculature
The pulmonary vasculature consists of the arterial, venous, and bronchial arteries and to some extent albeit not a robust system the microvascular collateral circulation [103,104,105,106]. The pulmonary arterial (PA) system is the most relevant to a discussion of aPE and closely follows the bronchial pathways. From an anatomical perspective the main PA arises from the RV outflow tract (RVOT) and divides into the left and right pulmonary arteries. The left PA appears to be a continuation of the main PA as it arches over the left main stem bronchus and begins branching to supply the left upper and lower lobes. A representative view of the main PA with its main bifurcations is seen in Fig. 14.6. Typically, the right PA is longer and gives rise to the right upper lobe artery as it arches over the right main stem bronchus. It normally courses more caudal than the left, and its length is better visualized in chest radiography. The normal main PA caliber is less than 3 cm while both left and right PAs are usually 1.5 cm [107].

Volume-rendered reconstructions of a multi-detector-row computed tomographic scan elegantly showing the main pulmonary artery (MPA), left pulmonary artery (LPA), and right pulmonary artery (RPA) with proximal bifurcations. (Image reconstruction performed by Amy L. Smith, RT (R) (CT) and Dr. Robert O’Donnell, MD, University of Cincinnati Medical Center)
The right PA gives rise to an upper lobe artery, which then divides into an apical, a posterior, and an anterior segmental artery. The next right lobar artery branch is the middle lobe artery, which divides into a lateral and a medial lobe segmental artery. The right lower lobe artery divides into an apical, an anterior, a posterior, a medial, and a lateral segmental artery. Although this major branching pattern is typical and related to lobar and segmental lung development, other arterial branches may arise directly from the right and left PA [107]. In contrast, a typical left PA gives rise to an upper and a lower lobe artery. The left upper lobe artery gives off the apicoposterior, anterior, and lingular arteries that further divide into a superior and an inferior segmental artery. The left lower lobe artery divides into a superior, an anteromedial, a lateral, and a posterior basal segmental artery [107].
Final diagnostic documentation of the involved segment is based on the highest branching order resolved by the imaging technique. Therefore, main PA arising from the RVOT is recognized as the first order. The right and left PA are considered as second order. Visualization of lobar artery segments is considered third order. Finally, segmental arteries are recognized as fourth order, subsegmental arteries are fifth order, and the first branches of subsegmental arteries are described as sixth order [107]. Figure 14.7 demonstrates the proposed diameter-defined Strahler ordering system with regard to the PA system. Representative tomographic images of aPE cases are shown with a typical proximal saddle embolus shown in Fig. 14.8 and a subsegmental aPE shown in Fig. 14.9.

Illustration of diameter-defined Strahler ordering system. Vessel order numbers are determined by their connection and diameters. Arteries with smallest diameters are of order 1. A segment is a vessel between two successive points of bifurcation. When two segments meet, order number of confluent vessel is increased by 1. Horsfield and diameter-defined systems apply to arterial and venous trees only. They are not applicable to capillary network, the topology of which is not treelike. Each group of segments of the same order connected in series is lumped together and called an element. (Reproduced with permission from Huang W et al. J Appl Physiol 1996; 81: 2123–2133)

Chest tomographic image showing a large filling defect suggestive of a large saddle embolus seen (broken arrow). (Image provided by Amy L. Smith, RT (R) (CT) and Dr. Robert O’Donnell, MD, University of Cincinnati Medical Center)

Chest tomographic image showing two filling defects (shown by the arrows) on the left side and a defect on the right side representative of subsegmental regions of the PA vasculature. (Image provided by Amy L. Smith, RT (R) (CT) and Dr. Robert O’Donnell, MD, University of Cincinnati Medical Center)
Pulmonary Arterial System Network and the Right Ventricle
In the normal state, the pulmonary artery vasculature acts as an elastic reservoir for the stroke volume ejected from the RV, with minimal resistance provided from the main PA all the way down to subsegmental arteries (approximately 0.5 mm diameter). Exquisite flow resistance regulation takes place at the level of the muscular arteriole capillaries, which exert the greatest arterial flow and pressure control [108,109,110]. The diameter of these artery segments closely parallels that of the accompanying bronchi [110,111,112,113]. In the normal state, this dynamic process is instrumental in matching perfusion to ventilation in real time and in regulating RV systolic function.
RV cardiac output depends on proper calibration between RV myocardial contractility and impedance to blood flow through the lungs. The RV can accommodate large volumes without significant compromise in systolic performance as long as there is no impedance to ejection or if the impedance occurs slowly over a period of time, such as in chronic pulmonary hypertension. However, it has limited contractile reserve to match even minor increases in impedance to ejection (acute pulmonary hypertension). Clinically, the degree of hemodynamic compromise depends on the magnitude and timing of pulmonary vascular obstruction [114,115,116,117,118,119,120,121]. A small embolism (even if recurrent and multiple) can be asymptomatic or present with mild dyspnea. Due to these differences in clinical presentation, case fatality rate for aPE ranges from <1% to about 60% [122]. A more recent data analysis using figures from the Nationwide Inpatient Sample from 1999 to 2008 showed that all-cause case fatality rate decrease was primarily driven by a decrease in case fatality rates for stable patients [123]. There was no reduction of case fatality rate in unstable patients receiving thrombolytic therapy [123]. Though it is important to highlight the fact that the general case fatality rate for patients receiving thrombolytics and a vena cava filter was low, most of these unstable patients did not receive this combination therapy [123].
Even though the proposed pathways leading to RV injury are intrinsically different between chronic and acute pulmonary hypertension (PH) [124], RV dilatation and systolic dysfunction are common abnormalities shared by both clinical entities. These abnormalities in RV size and systolic performance are mainly explained by the Laplace relationship [125]. Specifically, an increased ratio between cavity radius and thickness will increase wall stress while a decrease in this ratio will decrease wall stress [126]. Acute elevations in PH (acute increase in impedance to flow in pulmonary artery, such as aPE) cause a sudden increase in cavity radius and since there is little time to compensate with increased wall thickness, wall stress increases. This principle is illustrated in Fig. 14.10 showing the relationship between RV wall dilatation and increased thickness and its effect on the left ventricle (LV).

(a) Echocardiographic short-axis view of the left ventricle at the papillary muscle level showing a normal curvature of the left ventricle in relationship to a normal-sized RV with normal wall thickness. (b) In contrast, a dilated and hypertrophied RV requires not only more energy to pump the same amount of blood as compared to the normal-sized RV but also the resultant higher wall tension and thus RV peak systolic pressure results in bowing of the interventricular septum causing systolic flattening during ventricular contraction
The right and left ventricles differ in their ability to compensate for the increase in chamber size. In the case of the LV, wall thickness is greater than the RV. The LV can compensate both to physiologically acute increases in afterload (such as increased BP during exercise) or pathological, chronic causes such as systemic hypertension or valvular lesions (albeit by undergoing compensatory hypertrophy) [127]. Increase in thickness allows the LV to provide adequate stroke volume without an unacceptable reduction in myocardial performance [127]. In the more compliant RV, the thin walls allow it to accommodate larger volumes without hemodynamic stress (such as the increased preload during exercise), while at the same time allowing it to eject the same stroke volume as the LV against approximately 25% of the afterload the LV faces [128]. This increased compliance does not allow the RV to tolerate acute increases in afterload, such as in aPE [125]. Direct mechanical obstruction of the PA by thrombus or any other embolic material accompanied by potent release of local vasoconstrictors will result in hypoxemia causing a rapid increase in pulmonary vascular resistance (PVR) and the resultant increase in PVR [129,130,131,132,133,134,135,136,137,138,139]. The latter will ultimately determine the compromise in RV performance and the degree of hemodynamic collapse in accordance with the location and extent of the acute thromboembolic occlusion as well as the generation of local vasoconstrictive mediators along the PA system network in relation to underlying functional state of the myocardium. In cases of gradual increase in RV afterload, such as chronic PH, the RV should be able to handle these remodeling changes more efficiently, as there is time to assemble new sarcomeres in parallel in order to increase wall thickness and compensate for the hemodynamic stress [125].
Clinically, PVR can be assessed either invasively by catheter measurement or noninvasively by using transthoracic echocardiography (TTE). TTE estimation relies on measuring the maximum tricuspid regurgitation (TR) jet velocity using continuous-wave Doppler and the velocity time integral (VTI) of the pulsed Doppler signal across the RVOT. Thus, TTE calculation uses the following formula: PVR (Woods unit [WU]) = (TR velocity max/RVOT VTI) × 10 + 0.16 to approximate values that are obtained during invasive right-heart catheterization. Representative TTE Doppler signals are shown in Fig. 14.11 [140].

Representative Doppler tracings used for PVR calculation using echocardiography. (a) Maximal TR velocity jet signal obtained from the apical four-chamber view measuring 3.3 m/s. (b) Pulsed Doppler signal across the RVOT showing a velocity time integral (VTI) of 18 cm resulting in a PVR of 2.0 WU. Echocardiographic estimation of PVR uses the following formula: PVR (Woods unit [WU]) = (TR velocity max/RVOT VTI) × 10 + 0.16
As mentioned earlier, this rise in PVR would depend on the location and extent of the acute thromboembolic occlusion and also generation of local vasoconstrictive mediators along the PA system network. In conjunction with the functional state of RV myocardium, the extent of increase of PVR is an important determinant of prognosis, particularly when aPE interferes with both the circulation and gas exchange.
While the presence of thrombus in the deep veins and pulmonary arteries has been discussed, it is not unusual to find intracardiac thrombi in those with suspected aPE. This finding signifies either a thrombus in transit from the leg veins to the pulmonary artery or a native intracardiac thrombus that can rarely cause aPE. Either way, presence of intracardiac thrombus is a poor prognostic sign. In the International Cooperative Pulmonary Embolism Registry (ICOPER), intracardiac thrombus was present in 42 of 1113 patients with a TTE at admission [141]. Patients with a demonstrable intracardiac thrombus at the time of the TTE were more hemodynamically compromised as suggested by lower systemic arterial pressure, higher prevalence of hypotension, faster heart rates, and frequent hypokinesis of the RV as determined by TTE than patients without a demonstrable intracardiac thrombus at the time of diagnosis [141].
From a pathophysiological point of view, the cascade of events resulting in aPE can be simplistically explained not only by interference of both the circulation and gas exchange at the pulmonary level caused by the embolic material but also by the disturbance created by that material along the pulmonary-ventricular unit that ultimately results in some variable degree of RV mechanic disruption. Primary cause of death in severe aPE has been singled out as RV failure [142].
When occlusion of more than 30–50% of the total cross-sectional pulmonary arterial bed by thromboembolic material and local release of vasoconstriction substances occur, we see elevation in pulmonary artery pressures as a result of an increase in PVR [133, 143,144,145]. A marked and sudden increase in PVR will dilate an otherwise normal thin-wall RV that by the mechanisms previously described would not be able to generate a mean pulmonary pressure above 40 mmHg and consequently fail [125, 126, 143].
From a clinical point of view the estimated in-hospital or 30-day mortality risk determines the severity of an aPE event mainly determined by the patient’s presentation. The latter will ultimately stratify patients into either high- (shock or persistent hypotension in the absence of arrhythmia, hypovolemia, or sepsis) or no high-risk PE with important diagnostic and therapeutic implications [143]. Differentiating patients based on this approach facilitates diagnostic and therapeutic strategies as reconciled on a recent guideline document that integrates previous knowledge into a more optimal and objective diagnostic and management strategies for those patients presenting with either a suspected or a confirmed aPE [143].
Clinical parameters based on severity index have shown to be quite useful in assessing prognosis and determining outcomes. The most validated score to date is the PESI in identifying patients’ worse 30-day outcomes [146,147,148,149]. A simplified sPESI version is also available and has been validated [150, 151]. These clinical parameters become clearly important in assessing the overall risk of adverse events and need for additional testing.
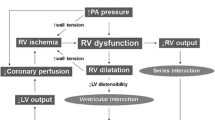
Traditionally, determination of the clinical magnitude of the thrombotic occlusion and its hemodynamic helps expedite therapeutic strategies [152]. A massive aPE is defined as a patient with hemodynamic compromise with significant RV dysfunction. Based on the location of thrombus, it is further categorized as saddle, main branch, or ≥2 lobar pulmonary emboli. It carries an estimated 60% mortality, two-thirds of which occur within the first hour after onset [122, 153, 154]. In contrast, sub-massive aPE occurs in hemodynamically stable patients who demonstrate evidence of right-heart strain, which is most commonly identified by TTE or elevation of cardiac enzymes [122, 154]. It carries an estimated 15–20% 30-day mortality rate and has been associated with chronic thromboembolic pulmonary hypertension and subsequent development of cor pulmonale [155]. Finally, a non-massive aPE is characterized by a normotensive patient with no evidence of RV dysfunction. A diagrammatic representation of this interrelationship is seen in Fig. 14.12. In summary, a high index of clinical suspicion is required to identify patients at risk as subtle findings can make the difference between life and death as shown in Fig. 14.13.

Diagrammatic representation of the sequence of events during aPE and the potential clinical implications of the thrombotic occlusion and resultant hemodynamic implications with associated mortality rates

Schema of relationship between suspected and actual cases of pulmonary embolism (PE). (From Ryu JH, Olson EJ, Pellikka PA. Clinical recognition of pulmonary embolism: problem of unrecognized and asymptomatic cases. Mayo Clin Proc 1998;73:877; with permission)
A recent publication by Jimenez and associates sheds light into most recent trends regarding hospital stay and 30-day mortality for aPE. Data from 23,858 patients from 2001 to 2013 taken from a large international aPE (RIETE) registry was used. For comparison purposes, results were divided into two time periods, first period between 2001 and 2005 and second period between 2010 and 2013. Using a multivariate regression model, these investigators found improvements in the overall length of stay (reduction from 13.6 to 9.3 days: 32% relative reduction, p < 0.001) and reductions in mortality (risk-adjusted rates for all-cause mortality from 6.6% to 4.9% and rates for aPE-related mortality from 3.3% to 1.8%, p < 0.01) [156].
Acute Pulmonary Embolism, Right Ventricular Architecture, and Myocardial Mechanics
In addition to causing an obvious sudden increase in PVR, an acute occlusive pulmonary thrombus also disrupts normal RV ejection due to RV myocyte lysis and induction of a proinflammatory phenotype [157,158,159]. Experimental data of aPE in the rat model suggests that neutrophils are present in the RVOT region between 6 h and 18 h of the thrombotic insult. Current data suggests that this inflammation is independent of the damage caused by the obstructive thrombus and also amplifies the initial thrombotic injury [160, 161]. In humans, an elevated concentration of circulating myeloperoxidase as well as other inflammatory biomarkers has been demonstrated in the aPE setting [162,163,164]. Hence, it has been suggested that an inflammatory response might be a primary part of the development of acute RV dysfunction in the setting of aPE in both clinical and experimental models [134].
Advances in the understanding of myocardial fiber orientation have been instrumental in advancing our knowledge of how the RV responds to an increase in afterload in response to aPE. Specifically, the RV free wall is mainly comprised of transverse fibers while the LV is encircled by oblique fibers [165]. Oblique fibers from the interventricular septum (IVS) also extend into the RVOT [166]. A schematic representation of this fiber orientation is shown in Fig. 14.14. It is now well established that the comparison of varying fiber orientations shows marked differences in contractile performance. Helical or oblique fibers are responsible for twisting and untwisting, while transverse fibers exert a compressive bellows-like activity [166]. From a mechanical perspective, helical or oblique fibers are at a considerable mechanical advantage compared with transverse fibers [167].

Schematic representation of the myocardial fiber orientation relationship of the IVS, composed of oblique fibers that arise from the descending and ascending segments of the apical loop, surrounded by the transverse muscle orientation of the basal loop that comprises the free RV wall. (Reproduced with permission from Buckberg G D, and the RESTORE Group Eur J Cardiothorac Surg 2006; 29: S272–S278)
In the healthy state, transverse fibers are the principal fiber group maintaining RV performance, with some assistance from the helical or oblique fibers from the LV (through the IVS) [168, 169]. In fact, left ventricular activity contributes to about 80% of the flow and up to two-thirds of the pressure generated by the RV during systole [168]. The influence of LV contractile function on RV contractile function via the shared IVS is part of the concept of ventricular interdependence.
However, in situations resulting in elevation in PVR, as it occurs in aPE, contribution from the IVS helical or oblique fibers becomes increasingly significant [170]. This contribution depends on the overall contractile state of the LV.
An acute increase in PVR results in systolic IVS flattening causing loss of the oblique orientation of the septal fibers as seen in Fig. 14.15 [24]. This contributes to the RV systolic dysfunction with aPE. In addition, the RV dilatation due to significant increases in PVR results in a shift of the IVS towards the LV causing LV underfilling [24,25,26,27,28]. This impaired LV performance and output add to the hemodynamic effects of aPE with catastrophic consequences as shown in Fig. 14.16.

Short-axis TTE view of the LV at the level of the papillary muscle showing a dilated RV that is causing significant flattening of the IVS as a result of an acute rise in PVR

Short-axis TTE view of the LV at the level of the papillary muscle showing a markedly dilated RV causing significant flattening of the IVS as a result of an acute rise in PVR resulting in both LV underfilling and impairing LV performance associated with hemodynamic compromise in cases of massive aPE. A pericardial effusion (PE) is also noted
Careful analysis of IVS in patients with chronic PH has led to the identification of two types of septal motions. Type A has been described as marked anterior motion in early systole while type B shows marked posterior motion in early diastole [171]. Type A IVS motion is mainly seen in patients with worse hemodynamic profiles and worse clinical outcomes when compared to those with type B IVS motion [171]. Deviation of the IVS towards the LV can thus compromise LV systolic as well as diastolic function [172,173,174].
The study of RV mechanics and IVS motion highlights not only the intricate hemodynamic and mechanical interdependence of the RV-pulmonary circulation unit, but also the importance of identifying RV dysfunction, particularly since aPE mortality is indisputably dependent on the presence and magnitude of RV strain [116, 119, 122, 142, 172, 175,176,177,178,179,180,181,182,183].
Diagnosis and Assessment of Right Ventricular Function in Acute Pulmonary Embolism
Unfortunately, the limited sensitivity and specificity of some signs and symptoms in certain clinical situations hinder aPE recognition [184,185,186,187]. Henceforth, a keen clinical judgment is critical at all times. The latter has certainly proven useful on several large series but lacks standardization for which prediction models have been developed [188,189,190,191,192,193,194,195]. Most importantly, whenever clinical presentation raises the suspicion for aPE, prompt objective testing is urgently required.
Introduction of multi-detector computed tomographic (MDCT) angiography with high spatial and temporal resolution has certainly revolutionized the assessment of suspected aPE patients and cemented its status as the preferred imaging method of choice for these patients [196,197,198]. With a reported sensitivity of 83% and a specificity of 96%, data from PIOPED II also highlighted the importance of combining clinical probability based on the Wells rule with CT findings [199]. For example, patients presenting with a low or intermediate clinical probability of aPE and a negative CT had a high negative predictive value for PE (96% and 89%, respectively) with only 60% in those with a high pretest probability [199]. On the other hand, the positive predictive value of a positive CT was high (92–96%) in patients with an intermediate or high clinical probability while it was only 58% in patients with a low pretest likelihood of aPE [199].
Additional CTPA findings in aPE are shown in Figs. 14.17, 14.18, and 14.19. In contrast, Figs. 14.20 and 14.21 show examples in which CTPA was useful to distinguish acute from chronic PE cases. Finally, Fig. 14.22 lists potential patient- and technique-related artifacts as well as anatomic mimickers that are important to be recognized when using multi-detector CT when evaluating patients with a presumptive diagnosis of aPE.

(a) Sagittal and (b) coronal reformatted images in a 56-year-old man with chest pain and dyspnea reveal thrombosed lower lobe posterior basal segmental artery branches. Notice the relative associated abnormal vascular enlargement (arrows) as compared with the adjacent patent vessels. (Reproduced with permission from Chhabra A, et al. Applied Radiology 2007)

(a) The donut sign in the left lower lobe pulmonary artery (arrow) and (b) tram track sign (arrows) in bilateral upper lobe segmental pulmonary arteries in cases of acute pulmonary emboli. (Reproduced with permission from Chhabra A, et al. Applied Radiology 2007)

(a, b) Acute right lower lobe subsegmental pulmonary embolus (arrows) in a 60-year-old man with peripheral pulmonary infarct. (Reproduced with permission from Chhabra A, et al. Applied Radiology 2007)

A 56-year-old man with a history of treated pulmonary embolism in the left lower lobe segmental arteries presented with acute chest pain. No evidence of acute embolism was seen. This volume-rendered coronal image reveals an abrupt cutoff in the left lower lobe segmental arteries (yellow arrow) with visualization of dilated bronchial collaterals. (Reproduced with permission from Chhabra A, et al. Applied Radiology 2007)

(a) Coronal and (b) sagittal reconstructions in a 70-year-old man with a history of bilateral acute pulmonary embolism and a 6-month history of warfarin treatment. These scans show chronic pulmonary emboli forming obtuse angles with the vessel wall in the left lower lobe pulmonary artery and its posterior segmental branch. (Images courtesy of Dr. Drew A. Torigian, MD, Hospital of the University of Pennsylvania, Philadelphia, PA; Reproduced with permission from Chhabra A, et al. Applied Radiology 2007)

List of potential patient- and technique-related artifacts as well as anatomic mimickers on multi-detector CT for aPE. (Reproduced with permission from Chhabra A, et al. Applied Radiology 2007)
In other words, MDCT documentation at segmental or more proximal levels is sufficient proof for aPE diagnosis when the clinical suspicion is high and a negative MDCT should with confidence exclude aPE in cases with low clinical suspicion; however, what to do in cases of high clinical suspicion and a negative MDCT is less clear and there is paucity of data if additional testing is needed.
Lastly, three instances involving CT require our attention. First, isolated identification of subsegmental aPE on CT: this has been reported in 4.7% of patients when imaged by single-detector CT angiography and in 9.4% of those undergoing MDCT [200]. Based on the lower positive predictive value and poor inter-observer agreement when distal pulmonary segments are being evaluated, the use of compression venous ultrasonography might be considered to exclude the presence of DVT that might require treatment [201]. Second, combination of computed tomographic venography with CT angiography as a single procedure using one intravenous injection of contrast is an alternative being proposed based on the PIOPED II data [199, 202]. Though this combination increased sensitivity for aPE diagnosis from 83% to 90% with similar specificity (95%), there was no significant increase in its negative predictive value [199, 202].
Furthermore, since CT venography yields similar results to the compression venous ultrasonography [202], the latter should be used instead as it would further reduce the total radiation dose. Finally, when an incidental pulmonary embolism is seen on CTs done among cancer, atrial fibrillation, and heart failure patients this might create some decision-making problem. Although accounting for only 1–2% of all CT cases [203,204,205,206], no robust treatment recommendations have been provided. However, it should be noted that most experts suggest that anticoagulation should be administered for incidental clots if found on lobar or more proximal level pulmonary circulation segments [207].
However, it is important to point out that the effectiveness of this imaging modality relies on identifying vascular obstruction and RV dilatation as a response of the former; however, it does not provide any information regarding RV function.
Ventilation–perfusion scintigraphy (V/Q scan) has been for some time a useful imaging modality used in the assessment of both acute and chronic PE considerations [208,209,210]. With lack of radiation and use of contrast material, V/Q scans have been preferentially used in outpatients with low clinical probability and for normal chest X-rays particularly among females, and in patients with a history of contrast-induced allergies, renal compromise, multiple myeloma, and paraproteinemic syndromes [211].
Though initial diagnostic criteria used for V/Q scan interpretation prompted revision into more useful management of high-probability scans and of normal perfusion V/Q scans, a high number of nondiagnostic intermediate probability scans continue to be problematic as additional diagnostic testing is required [212,213,214,215,216,217]. This problem might have been solved by using single-photon emission computed tomography (SPECT) data acquisition imaging with or without low-dose CT [218,219,220,221]; however, large-scale prospective studies are required to validate this approach. Once again, V/Q scan imaging also falls short in providing any information regarding RV function.
For decades, pulmonary angiography was seen as the “gold standard” imaging modality tool when assessing aPE patients [222]. Though currently replaced by the less invasive and equally effective method in providing diagnostic accuracy as CT angiography technique, pulmonary angiography is still used when guiding percutaneous catheter-directed treatment for aPE.
Associated procedure-related mortality for pulmonary angiography has been reported at 0.5% while minor and major complications have been quoted at 5% and 1%, respectively [223]. As expected, deaths are most likely related to either hemodynamic compromise or respiratory failure and bleeding-related complications if thrombolysis follows a diagnostic pulmonary angiogram [224].
Digital subtraction angiography is a fluoroscopic technique that improves visualization for a better accurate depiction of blood vessels by eliminating surrounding structures. Though this technique improves peripheral visualization of pulmonary arterial branches with identification of thrombi as small as 1–2 mm, main trunk imaging is compromised by cardiac motion [225, 226]. Direct visualization of a filling defect or amputation of a pulmonary arterial branch constitutes the only validated diagnostic sign of aPE as none of the proposed indirect signs have been validated [187].
Obviously, a heightened clinical suspicion when we encounter a patient with the right risk profile sets in motion a series of diagnostic testing modalities that according to the situation allow to exclude or confirm aPE in order to institute appropriate treatment. At times, due to the critical nature of patients, a simple bedside ultrasound detection of a venous thrombus in the lower extremity is often enough to initiate the treatment for presumed aPE. However, absence of DVT by ultrasound does not preclude aPE diagnosis while the presence of DVT by ultrasound is not confirmatory of aPE [227].
Once a diagnosis of aPE is made, determining the RV response to this challenge by clinical, laboratory, and imaging methods influences clinical management and prognosis. Prompt hemodynamic stability and RV function assessment are critical.
Once a diagnosis of aPE is made, determining the RV response to this challenge by clinical, laboratory, and imaging methods influences clinical management and prognosis. Prompt hemodynamic stability and RV function assessment are critical. Risk stratification algorithms have been proposed to help physicians identify high- versus low-risk aPE patients in order to expedite diagnosis and treatment, as shown in Fig. 14.23.

Pulmonary embolism severity index (PESI). (Modified with permission from Aujesky D, Perrier A, Roy PM, et al. Validation of a clinical prognostic model to identify low-risk patients with pulmonary embolism. J Intern Med. 2007; 261: 597–604 and Masotti L, et al. Prognostic stratification of acute pulmonary embolism: focus on clinical aspects, imaging, and biomarkers. Vasc Health Risk Manag. 2009; 5: 567–575)
While hemodynamic stability is often discernible by clinical examination, identification of high-risk features associated with significant RV strain and impending circulatory collapse with imaging can be challenging. Commonly used imaging modalities such as computed tomography and TTE are less than ideal in studying the true anatomic and functional derangements of the RV (Fig. 14.24) but are readily available and can detect simple markers that identify patients at risk of hemodynamic instability in aPE. Table 14.4 summarizes the most commonly associated variables currently used in the identification of abnormal RV strain. Figure 14.25 shows a pictorial representation of normal TTE RV findings that can be contrasted to abnormal RV strain findings as seen in Fig. 14.26.

(a) Open-cut macroscopic view of heart in a four-chamber view depicting the thin-walled RV in relation to the LV. (b) Computed tomographic view of a representative four-chamber view showing the same anatomical relationship of the macroscopic specimen. Image shows corresponding reformatted four-chamber contrast-enhanced ECG-gated MDCT scan. (Both (a) and (b) were reproduced with permission from Dupont MV, et al. Right ventricular function assessment by MKDCT. AJR 2011; 196: 77–86). (c) TTE representation of the RV and LV obtained from the same four-chamber apical view orientation as represented in (a, b)

Pictorial representation of normal TTE markers of RV size and systolic function. (a) Representative end diastolic four-chamber apical frame showing normal relation of RV size when compared to the LV; the right (RA) and left (LA) atria are also seen. (b) End-systolic four-chamber apical frame showing a normal-size relationship between the RV and LV denoted by straight white lines. (c) Representative end-diastolic four-chamber apical frame showing normal-size relationship between RV and LV (dashed lines) as well as a normal tricuspid annular size, denoted by the solid line. (d) Representative short-axis view at the level of the papillary muscles showing a normal relationship between RV and LV. Please note the crescent shape of the RV cavity at this level as well as the normal curvature of the IVS. (e) Representative tricuspid regurgitation signal that peaks early in systole with a peak velocity of 2.54 m/s. (f) Representative tricuspid annular plane systolic excursion from a normal patient with a maximum amplitude of 3 cm. (g) Representative tricuspid annular tissue Doppler imaging signal from a normal patient showing a normal systolic velocity of 0.14 m/s or 14 cm/s. (h) Representative pulsed Doppler signal across the pulmonic valve showing a normal RVOT envelope


Pictorial representation of abnormal TTE markers of RV strain. (a) Representative end-diastolic four-chamber apical frame from a patient with a confirmed aPE showing a dilated RV in comparison to the LV; a pericardial effusion (PE) is also appreciated. (b) End-systolic four-chamber apical frame showing a markedly dilated RV when compared to the LV denoted by the dashed white lines. (c) Representative end-diastolic four-chamber apical frame showing a dilated RV when compared to the LV. In addition, a dilated tricuspid annulus is also seen (solid line). Furthermore, stretched RV apex is also noted by the arrow and can be easily contrasted to the normal RV apex seen in Fig. 14.18b. (d) Representative short-axis view at the level of the papillary muscles showing a markedly dilated RV and small compressed LV. In addition, please note the flattened IVS that bows against the LV. (e) Representative tricuspid regurgitation signal that peaks late in systole with a velocity of 2.98 m/s in a patient with aPE. (f) Representative tricuspid annular plane systolic excursion from a patient with hemodynamically significant aPE that is hypotensive. Please note the significant reduction in the maximum amplitude of less than 1 cm. (g) Representative tricuspid annular tissue Doppler imaging signal from a patient with a hemodynamically significant aPE showing significant reduction in the systolic velocity of 5 cm/s. (h) Representative pulsed Doppler signal across the pulmonic valve showing a markedly abnormal RVOT envelope from a hypotensive patient with a confirmed aPE; please note the narrow width (lines) of the signal and the mid-systolic indentation in the signal (arrows)
Risk stratification algorithms have been proposed to help physicians identify high- versus low-risk aPE patients in order to expedite diagnosis and treatment [228,229,230,231,232]. While hemodynamic stability is often discernible by clinical examination, identification of high-risk features associated with significant RV strain and impending circulatory collapse with imaging can be challenging.
Although the electrocardiogram (ECG) has traditionally been one of the first diagnostic tests obtained from patients presenting with symptoms suggestive of aPE, it has not been incorporated into any risk stratification models. However, there are certain ECG findings in aPE that have been correlated with the presence of RHS [233,234,235,236]. Consequently, the American Heart Association has recommended risk stratification for aPE patients based on RHS [237]. Furthermore, in a study by Hariharan and associates, these investigators described 3-ECG findings that were independently associated with RHS [238]:
-
TWI in leads V1 through V3 (5 points)
-
S wave in lead I (2 points)
-
Sinus tachycardia (3 points)
Using this score, these investigators were able to risk-stratify 85% of the aPE they studied [238]. Specifically, when the score was 5 or greater, it was 93% specific for RHS while a score of 2 or less excluded the presence of RHS with 85% sensitivity [238]. These results were similar to findings reported by other investigators [239, 240]. Although these results might be encouraging further research should be undertaken not only to define the appropriate role of ECG in the assessment of RV involvement in aPE but also to clarify the potential role of ECG in aPE risk stratification models.
Biomarkers such as cardiac troponin, a well-known, reliable marker of injury, have been used in patients presenting with chest pain and dyspnea mainly with the aim of identifying an acute coronary syndrome; however, elevated cardiac troponins have been shown to occur in aPE as a result of acute RV dilatation and dysfunction [241]. Specifically, elevation in cardiac troponin levels can identify those aPE patients at higher risk of hemodynamic collapse [242]. Furthermore, the utility of cardiac troponin assessment among normotensive aPE patients has also been found useful to exclude RHS [243]. It is always important to keep in mind which clinical conditions affect cardiac troponin levels independently of cardiac injury [244].
In terms of the brain natriuretic peptide (BNP), a well-characterized peptide with numerous mechanistic actions known to primarily be secreted by the ventricles in response to stretching and wall tension, it is another biomarker with potential use in aPE patients [245,246,247,248,249]. Consequently, the value of measuring BNP levels in aPE patients with an initial stable hemodynamic status was considered. However, a meta-analysis of BNP levels found that while elevated BNP levels might be useful in identifying aPE at high risk of death and adverse outcome events, the high negative predictive value of a normal BNP level was certainly more useful in selecting those aPE patients with a likely uneventful follow-up [250].
In contrast to all the abovementioned imaging tools and blood markers, TTE provides a unique rapid, accurate, and reproducible opportunity to evaluate aPE. It could also potentially provide supportive evidence (such as presence of a thrombus in transit or presence of new RHS). As seen in Fig. 14.27 panels a and b depict the presence of a RV apical thrombus in a patient with biventricular dysfunction due to nonischemic cardiomyopathy. Two days later, the patient developed pleuritic chest pain and CTA of pulmonary arteries showed pulmonary embolism (Fig. 14.27c). Venous duplex of lower extremities was negative for thrombus.

(a, b) Depict the presence of a RV apical thrombus (red arrows) in a patient with biventricular dysfunction due to nonischemic cardiomyopathy. Two days later, the patient developed pleuritic chest pain and CTA of pulmonary arteries showed pulmonary embolism (c) as denoted by the red arrow. Venous duplex of lower extremities was negative for thrombus
In addition, it also aids in the hemodynamic evaluation of the extent and magnitude of the thrombotic burden that can be used to assess prognosis. Even though detection of clot burden in transit or identification of a proximal pulmonary artery thrombus is challenging by TTE on a regular basis, most routinely TTE’s role basically resides in hemodynamic assessment and evaluation of RV function.
Since clinical hemodynamic stability appears to be dependent on RV function rather than the magnitude of clot burden in aPE, particularly within the first hour, confirmation and assessment of mechanical stability of RV function are promptly required once this diagnosis is entertained (Fig. 14.28).

Diagrammatic representation of the relationship between clot burden and approximate degree of pulmonary bed obstruction and corresponding clinical effect in the absence of previous cardiopulmonary history and normal RV reserve
While echocardiography is clinically used to suggest the presence of aPE (via the presence of RV dysfunction in a patient with suspected aPE but unable to obtain a diagnostic test), several pitfalls in this setting need to be borne in mind. First, presence of RV strain may represent a chronic finding (such as COPD or chronic pulmonary HTN). Thus, without a prior echocardiogram showing normal RV function, assuming that the RV dysfunction is due to aPE may result in unnecessary and potentially dangerous treatment for aPE. Second, new-onset RV dysfunction can occur with ARDS or other pulmonary injuries. Finally, in the supine, ventilated, critically ill patients, echocardiographic windows are often poor and a partially visualized RV can be considered normal in size and aPE be falsely ruled out. Despite past skepticism regarding the predictive value of TTE in assessing RV dysfunction among hemodynamically stable aPE patients, TTE remains a useful imaging modality in the initial evaluation and follow-up of aPE patients [239, 240]. In view of the above, the 2011 American Society of Echocardiography appropriate use criteria (AUC) assign TTE a score of 2 (inappropriate) for diagnosis of aPE while assigning a score of 8 (appropriate) to guide therapy [251].
To become a serious imaging contender, TTE has to offer critical data that surpasses rudimentary information based on subjective interpretation of RV size and wall motions, abnormal IVS motion, presence of tricuspid regurgitation, and lack of collapse of the inferior vena cava during inspiration [252]. In general, RV systolic dysfunction can be easily quantified by measuring either tricuspid annular plane systolic excursion (TAPSE, <1.6 cm) or fractional area change (FAC <35%). In addition to these, other simple echo markers of RV strain have been described.
McConnell’s sign has been traditionally used to describe regional RV dysfunction that spares the apex and was initially felt to aid both in the diagnosis of aPE and in the detection of RV strain. Initial studies reported sensitivity of 77%, specificity of 94%, positive predictive value of 71%, and negative predictive value of 96% to diagnose aPE [253]. However when applied in routine clinical practice for the diagnosis of aPE, McConnell’s sign is not as reliable since similar pattern can be found in other right-heart pathologies such as acute RV infarct [254].
Doppler echocardiography is also useful in the evaluation of aPE patients. The acute increase in PVR often results in a rapid acceleration time and mid-systolic notching. An altered RVOT Doppler signal has been shown to be useful not only in characterizing PH severity, but also in overall RV systolic performance [255,256,257]. Representative RVOT images from a normal patient (Fig. 14.29a, b) and a patient with a confirmed aPE are shown (Fig. 14.30a, b).

Representative short-axis views at the level of the aortic valve demonstrating (a) RVOT end-diastolic length and (b) end-systolic length by the white arrows from a normal patient demonstrating excellent RVOT systolic function (RVOT end-diastolic length – RVOT end-systolic length)/RVOT end-diastolic length × 100

Representative short-axis views at the level of the aortic valve demonstrating (a) RVOT end-diastolic length and (b) end-systolic length by the dashed white arrows from a patient with confirmed aPE. No significant difference between RVOT end-diastolic and end-systolic lengths corresponding to a reduced RVOT systolic excursion based on the formula listed in the figure legend of Fig. 14.29
The 60/60 sign refers to the presence of a short RVOT signal acceleration time (<60 ms) along with an estimated RV systolic pressure <60 mm of Hg [258]. This sign reflects the acute increase in PVR compared to a more chronic cause such as chronic pulmonary arterial hypertension. Similar to McConnell’s, this sign is insensitive for the diagnosis of aPE.
As noted above, the clinical effects of aPE depend both on clot burden and the underlying cardiac reserve—some patients with a massive aPE but excellent cardiac reserve might behave similarly to patients with sub-massive aPE but compromised cardiac reserve [259, 260]. Untreated aPE can be fatal in up to 30% of patients largely depending on the degree of RV compromise [176, 261,262,263]. Since clinical hemodynamic stability appears to be dependent on the RV function rather than the magnitude of clot burden in aPE, particularly within the first hour, confirmation and assessment of mechanical stability of RV function are promptly required once this diagnosis is entertained [264,265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287].
New Frontiers in the Assessment of Right Ventricular Function in Acute Pulmonary Embolism
Integrity of RV function plays a pivotal role in determining the prognosis after aPE [124, 177, 179, 264,265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287]. Experimental data using healthy dogs has revealed that acute embolization using microbead injections induces dramatic stiffening of the PA leading to increased RV stroke work [288]. In this particular model, the use of both invasive and magnetic resonance imaging measures to assess PA stiffness and RV systolic function revealed that the RV was able to shift its working conditions, preserve function, and maintain hemodynamic coupling with the PA despite the acute insult [288]. This however occurs at the expense of a reduction in RV efficiency [288]. This interaction between RV and PA has clarified our understanding of RV function and it is now clear that RV systolic function cannot be studied in isolation, as both RV and PA work in series [289]. Evolution of RV pathology from normal to a decompensated state parallels the evolution of pulmonary vascular pathology; hence studying the RV and the PA as a unit will be critical to understanding the true performance of the RV [289].
RV-PA coupling is critically important, and this relationship is best demonstrated by changes in hydraulic load occurring in the setting of PA stiffening as it will occur in aPE. As the RV is met with increased hydraulic wave reflection, its workload increases to maintain forward flow. It is important to remember that up to one-half of the hydraulic power in the main PA is contained in the pulsatile components of flow. Thus, acute changes in PA impedance, a measure of opposition to these pulsatile components of flow, would be crucial to study, though unrealistic, unless they are measured in animal models. This is however unrealistic except when measured in animal models. The elastic properties of the pulmonary vasculature are vitally important as the heart would not be able to generate forward flow without them [290,291,292].
From a mechanistic perspective, RV ejection is dependent on PVR as well as on the oscillatory or pulsatile component of flow that is dissipated as wasted energy through the PA system with each RV ejection [292,293,294,295]. Hence, PA elasticity is crucial to maintain RV efficiency [292,293,294,295]. Since hemodynamic instability unmistakably predicts adverse outcomes and 30-day mortality in aPE patients when RV dysfunction is confirmed, a proposed scheme of high-risk features for the potential development of hemodynamic instability in aPE is listed in Fig. 14.31 [234, 265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287, 296,297,298,299,300].

Schematic diagram demonstrating high-risk features on admission that helps in the identification of poor prognosis in aPE
As previously outlined above, the McConnell’s sign suggests a regional rather than a global RV dysfunction in aPE [253, 254]. However, assessment of myocardial deformation with the use of velocity vector imaging often detects significant reduction in global myocardial strain generation of all main RV chamber segments, including the RV apex rather than regional abnormality [301]. Why only some patients present with regional compared to global RV dysfunction remains unknown and the role of the RV apex in aPE and its influence on overall prognosis have not been fully elucidated [301,302,303]. A representative velocity vector imaging sample from a normal RV showing normal longitudinal strain of all segments, including the RV apex, is shown in Fig. 14.32a–f. In sharp contrast, a representative velocity vector image is also shown from a patient with confirmed aPE (Fig. 14.33a–f) that shows significant reduction in overall systolic deformation from all segments, a very abnormal RV apical segment signature, and significant temporal dyssynchrony among all six RV segments when compared to the well-coordinated peaking of all RV segments in Fig. 14.32.



Representative LV speckle tracking imaging signals obtained from the short-axis view at the level of the papillary muscles from an individual with normal biventricular systolic function. Color-coded representation is similar for all images and the green corresponds to the anterior septum; light purple to anterior wall; light blue to the lateral wall; dark blue to the posterior wall; yellow to the inferior wall; and deep purple to the inferior septum. (a) Radial velocity is shown for all six segments with the systolic component represented by S, early diastole by E, and late diastole by A. (b) Representative rotation rate showing normal synchronous counterclockwise as well as clockwise rotation of all segments. (c) Normal radial displacement curves of all six LV segments. (d) Representative radial rotation displacement curves showing normal synchronous counterclockwise as well as clockwise rotation of all segments. (e) Normal radial strain curves for all six LV segments. (f) Normal circumferential strain curves for all six LV segments



Representative LV speckle tracking imaging signals obtained from the short-axis view at the level of the papillary muscles from a patient with confirmed aPE and RV dysfunction and abnormal strain. Color-coded representation is similar for all images as listed in Fig. 14.30. Green corresponds to the anterior septum; light purple to anterior wall; light blue to the lateral wall; dark blue to the posterior wall; yellow to the inferior wall; and deep purple to the inferior septum. (a) Radial velocity is shown for all six segments with the systolic component represented by S, early diastole by E, and late diastole by A. (b) Representative rotation rate showing dyssynchronous counterclockwise and clockwise rotation abnormalities of all segments. (c) Abnormal radial displacement curves of all six LV segments. (d) Representative radial rotation displacement curves showing dyssynchronous counterclockwise and clockwise rotation abnormalities of all segments. (e) Abnormal radial strain curves for all six LV segments. (f) Abnormal circumferential strain curves for all six LV segments
As previously illustrated, RVOT systolic excursion has also been a useful echocardiographic variable used in estimating global impairment in RV contractility as well as acute hemodynamic derangement [304]. However, the ratio between main chamber RV fractional area change and RVOT systolic excursion appears to be markedly abnormal in patients with proven bilateral proximal embolus by CTPA. Furthermore, this ratio was also found to be a very useful TTE finding when trying to distinguish aPE from chronic PH [305].
Furthermore, speckle tracking strain imaging has been shown to be not only extremely accurate and reproducible, but also extremely useful in identifying subtle wall motion as well as systolic function abnormalities [306,307,308,309]. Tissue Doppler and speckle tracking imaging have been invaluable for assessing myocardial mechanics in chronic pulmonary hypertension [172, 310,311,312,313,314], especially in quantifying global as well as segmental longitudinal RV peak systolic strain and defining the presence of RV dyssynchrony in aPE patients [315]. Moreover, these abnormalities in RV heterogeneity and dyssynchrony resolve after the acute aPE insult and return to normal values [315]. Thus, speckle tracking as a portable, accurate, and reproducible imaging modality holds particular promise in offering prompt and reliable information that can be used not only at the bedside for diagnostic purpose, but also for follow-up of aPE patients once appropriate therapy is instituted. Representative speckle tracking images from a normal individual (Fig. 14.34) and from a patient with confirmed aPE (Fig. 14.35) are shown.

Representative velocity vector imaging from a normal RV showing normal longitudinal strain curves from all six segments. Individual segment recognition is as follows: RV free wall annulus side is green; mid RV free wall is lilac; apical side of the RV wall is light blue; IVS base is dark blue; mid IVS is yellow; and apical side of the IVS is purple. Please note that all six segments have a normal negative deflection in systole with normal peak values of approximately −20%. Specifically, note the normal movement of the RV apical segment

Representative velocity vector imaging from an aPE patient showing abnormal longitudinal strain curves from all six segments. Individual segment recognition is similar as noted for Fig. 14.34. Please note that all six segments have significant reduction in the overall systolic deformation. Specifically, note the very abnormal RV apical segment signature. Finally, a significant amount of dyssynchrony is also noted as time to peak of all six segments is different when compared to the well-coordinated peaking of all segments in Fig. 14.34
In order to link the RV-PA unit with the LV, it will then be useful to review our current understanding of myocardial fiber direction. As proposed by the late Torrent-Guasp, if indeed there is a continuum of subepicardial RV muscle fibers forming a plane along the posterior LV to the region of the left fibrous trigone and these fibers do descend from the left fibrous trigone to the LV apex [316, 317], then whichever process affects the RV will certainly impact the LV. It has been shown that potential traction on this muscular band by RV dilatation adversely affects basal LV rotation in cases of chronic PH [172]. Whether this represents a functional continuity between the ventricles or is merely a static anatomical entity mediated by the IVS remains largely unknown. Therefore, it is reasonable to believe that similar interactions do occur with sudden increase in RV afterload in aPE. In addition, bowing of the IVS towards the LV has been shown to alter global LV kinetics in the rat model, even if intrinsic LV systolic function is normal [318]. In humans, aPE has shown to induce reversible global LV dysfunction; nonuniformity of LV contractility in the radial, longitudinal, and circumferential directions; and discoordination of radial LV wall motion [315]. Once again, all these abnormalities were found to be reversible and normalized with treatment after patient stabilization [315]. Recent evidence in experimental animal models suggests that aPE mainly impairs LV performance by primarily altering septal strain and apical rotation [319]. Similarly, even though mainly recognized in case of human chronic PH, abnormal LV diastolic function might be likely affected by the similar mechanisms during aPE [172, 173, 320, 321]. Unfortunately, to date none of these mechanisms that directly affect LV diastolic function in cases of aPE have been clinically investigated.
After Acute Pulmonary Embolism Diagnosis
In summary, it is clear that certain clinical conditions predispose to the development of DVT and also aPE; however, these conditions are also found in otherwise healthy people, and aggressive investigation for potential pulmonary embolism is likely to find an incidental clot without clinical consequence [322]. In fact, in up to 90% of autopsy reports an identifiable new or old PE might be found, particularly if microscopic examination is done on numerous blocks of lung tissue [323,324,325]. Therefore, it becomes sometimes difficult to determine how often any of these clots are clinically significant. Similarly, the advent of advanced diagnostic modalities has allowed detection of very small thrombi of otherwise unknown clinical significance [297], particularly when healthy outpatients present with severe pleuritic chest pain.
Several developments have taken place with regard to risk, diagnosis, and management of VTE since the last European Society of Cardiology (ESC) guidelines and update of the American College of Chest Physicians in 2016. In particular, it is important to take into consideration the very comprehensive 2019 ESC guidelines which were developed in collaboration with the European Respiratory Society [326]. Here are the most remarkable highlights:
Risk Assessment
-
1.
Pretest probability scores include the revised Geneva score and the Wells rule [188, 327]. Based on the current data, aPE is expected to be confirmed according to pretest probability in 10% of cases when pretest probability is low compared to 30% in moderate and 65% in high pretest probability.
-
2.
The Pulmonary Embolism Rule-out Criteria (PERC) score should be limited to patients seen in the emergency department with low pretest probability that any additional diagnostic testing is intended. This PERC score includes eight parameters [328]:
-
(a)
Age <50 years
-
(b)
Pulse <100 beats/min
-
(c)
Oxygen saturation >94%
-
(d)
No unilateral leg swelling
-
(e)
No hemoptysis
-
(f)
No recent trauma or surgery
-
(g)
No VTE history
-
(h)
No previous oral hormone use
-
(a)
-
3.
D-dimer [329]:
-
(a)
Point-of-care D-dimers only to be used in patients with a low pretest probability due to their reduced sensitivity (88%) compared with the standard laboratory-based assay (95%).
-
(b)
In patients with low-to-intermediate clinical probability of aPE, D-dimer should be the initial test. If negative, no treatment is indicated. If positive, CTPA should be performed for definitive diagnosis.
-
(c)
Age-adjusted D-dimer (age × 10 mcg/l) for patients older than 50 years should be considered to identify low-risk patients (Class IIa).
-
(d)
D-dimer cutoff values adjusted for age or clinical probability can be used as an alternative to the fixed cutoff value.
-
(a)
-
4.
Definition of hemodynamic instability:
-
(a)
Cardiac arrest
-
(b)
Obstructive shock (systolic blood pressure [BP] <90 mmHg or need for vasopressors to achieve BP ≥90 mmHg and end-organ hypoperfusion)
-
(c)
Persistent hypotension (systolic BP <90 mmHg or systolic BP drop ≥40 mmHg lasting longer than 15 min and not due to another identifiable cause)
-
(a)
-
5.
Initial test to be obtained in patients deemed to be at a high clinical probability of PE is CTPA.
Imaging
-
1.
Accepted diagnosis for VTE and aPE: Proximal DVT confirmed by compressive ultrasonography in a patient with clinical suspicion for aPE.
-
2.
TTE alone cannot be used to rule out aPE. However, it is quite useful in suspected high-risk aPE, in which the absence of echocardiographic signs of RV overload or dysfunction essentially rules out aPE as the reason for hemodynamic instability.
-
3.
Availability of imaging studies for aPE diagnosis:
-
(a)
CTPA is the most accessible not only with an excellent specificity (96%) but also helping to provide an alternative diagnosis. Main concerns are concern for breast radiation exposure (3–10 mSv) for young women, renal dysfunction, and iodinated contrast allergy.
-
(b)
Even though planar V/Q scan is inexpensive with few contraindications and relatively low radiation exposure (2 mSv), it is unable to provide an alternative diagnosis and it could be inconclusive in 50% of all cases.
-
(c)
V/Q single-photon emission CT provides the lowest rate of nondiagnostic tests (<3%), has few contraindications, and uses low radiation (2 mSv). Unfortunately, it does not provide an alternative diagnosis and to date there has been no validated outcome data.
-
(d)
Pulmonary angiography is the gold standard, but it is an invasive procedure with the highest radiation dose (10–20 mSv).
-
(a)
Prognosis
-
1.
An initial risk stratification is to be performed in all patients with suspected or confirmed aPE. Prognostic assessment should include clinical parameters (simplified Pulmonary Embolism Severity Index [PESI] score, RV function, hemodynamics, and elevated biomarkers) [147, 149, 330, 331].
-
2.
Markers associated with an unfavorable short-term prognosis in aPE have been associated with the presence of tachycardia, low systolic BP, respiratory insufficiency, and syncope.
-
3.
TTE findings associated with a poor prognosis are an RV/left ventricular diameter ratio ≥1.0 and TAPSE <1.6 cm.
-
4.
An RV/left ventricular diameter ratio ≥1.0 on CT is associated with a 2.5-fold increased risk of all-cause mortality and 5-fold increased aPE-related mortality.
-
5.
High-sensitivity troponin has a 98% negative predictive value when <14 pg/mL for excluding an adverse in-hospital clinical outcome.
-
6.
B-type natriuretic peptide, N-terminal pro-B-type natriuretic peptide, and troponin have low specificity and positive predictive value for early mortality for normotensive patients with PE.
-
7.
Elevated lactate, high-serum creatinine, and hyponatremia are some of the other laboratory markers of adverse prognosis.
-
8.
Prognostic assessment strategy is recommended for patients without hemodynamic instability:
-
(a)
Low risk (PESI Classes I–II, simplified PESI of 0)
-
(b)
Intermediate low risk (elevated PESI score with or without RV dysfunction or elevated troponin)
-
(c)
Intermediate high risk (elevated PESI score with both RV dysfunction and elevated troponin)
-
(d)
High risk (hemodynamic instability)
-
(a)
In addition, the 2019 ESC writing group also recommended the following regarding which patients are at increased risk of recurrence and how to institute follow-up of these patients [326].
Groups at Increased Risk of Recurrence (High at >8%/Year)
-
1.
A strong (major) attributable transient or reversible risk factor, such as major surgery or trauma
-
2.
Persistence of a weak (minor) transient or reversible risk factor or a nonmalignant risk factor for thrombosis
-
3.
Index episode that occurred in the absence of any identifiable risk factor
-
4.
One or more previous episodes of VTE
-
5.
Major persistent prothrombotic condition
-
6.
Active cancer
In terms of follow-up of these patients, the 2019 ESC authors recommend performing either the Medical Research Council Scale or the World Health Organization Functional Scale Assessment 3–6 months after aPE diagnosis with dyspnea (Class I) [332, 333]. Furthermore, a TTE should be ordered if the patient has persistent dyspnea at 3–6 months (Class IIa) post-aPE. Finally, a V/Q scan should be considered at 3–6 months if there are concerns for PH to be assessed for chronic thromboembolic PH (Class IIa), with referral to a PH or chronic thromboembolic PH specialist if abnormal (Class I).
Finally, treatment was also addressed by this 2019 document as follows [326]:
-
1.
Prompt institution of anticoagulation is required in all high- or intermediate-probability aPE while awaiting results of diagnostic testing (Class I):
-
(a)
Low-molecular-weight heparin (LMWH) and fondaparinux are preferred over unfractionated heparin (UFH) given lower risks of major bleeding or heparin-induced thrombocytopenia (Class I).
-
(b)
UFH should be reserved for hemodynamically unstable patients or patients awaiting reperfusion therapy.
-
(a)
-
2.
Oral anticoagulants:
-
(a)
Based on current data, direct-acting oral anticoagulants (DOACs) are noninferior to LMWH and vitamin K antagonist (VKA) with lower rates of major bleeding and are recommended over VKA in eligible patients (Class I).
-
(b)
Recommended dosing regimens:
-
Dabigatran parenteral anticoagulation for ≥5 days followed by dabigatran 150 mg BID
-
Rivaroxaban 15 mg BID for 3 weeks followed by 20 mg QD
-
Apixaban 10 mg BID for 7 days followed by 5 mg BID
-
Edoxaban UFH or LMWH for ≥5 days followed by 60 mg daily or 30 mg daily if creatinine clearance = 30–50 mL/min or body weight <60 kg
-
-
(c)
If VKA is given, UFH, LMWH, or fondaparinux should be continued for ≥5 days and until international normalized ratio is 2.5 (range 2.0–3.0) (Class I).
-
(d)
Recommended initial starting dose of warfarin may be 10 mg when <60 years in healthy patients while 5 mg could be used initially in all others.
-
(e)
Novel oral anticoagulants are contraindicated with severe renal impairment, during pregnancy and lactation, and in patients with antiphospholipid syndrome (Class III).
-
(a)
-
3.
In terms of systemic thrombolytic therapy this should be reserved for high-risk, hemodynamically unstable patients (Class I). When thrombolytic therapy fails or patients are not considered adequate candidates, then surgical embolectomy should be recommended (Class I). Catheter-directed thrombolysis should be reserved for high-risk patients in whom thrombolysis has failed or is contraindicated (Class IIa).
-
4.
Routine use of inferior vena cava filters is still not routinely recommended (Class III), though it could be considered if there are absolute contraindications to anticoagulation (Class IIa) or in cases of recurrent PE despite therapeutic anticoagulation (Class IIa).
-
5.
Early discharge from hospital with continuation of home anticoagulation therapy should be considered in the following cases:
-
(a)
Use of the Hestia exclusion criteria that utilizes a checklist of clinical parameters or questions integrating aspects of aPE severity, comorbidity, and feasibility of home treatment. The use of this bedside tool gives the healthcare professional the ability of identifying that when one or more of the questions are answered with a “yes,” then the patient cannot be discharged early. Consequently, when all Hestia criteria are negative, the risk of early aPE-related death or serious complication is low [326].
-
(b)
No serious comorbidity or aggravating conditions warranting hospitalization.
-
(c)
Logistically intact and identifiable proper outpatient care resources and follow-up ability.
-
(a)
Duration of Anticoagulation
-
1.
Based on current data, all patients should receive 3 months of therapy (Class I).
-
2.
Discontinuation of therapy is recommended after 3 months if index PE/DVT due to major transient or reversible risk factor (Class I).
-
3.
The risk of recurrence is similar when therapy is withdrawn at 3–6 months versus 12–24 months.
-
4.
Extended duration of anticoagulation increases bleeding risk but decreases recurrence risk by ≤90%.
-
5.
Indefinite anticoagulation for recurrent VTE not related to a major transient or reversible risk factor (Class I).
-
6.
Indefinite anticoagulation with VKA is recommended for patients with antiphospholipid syndrome (Class I).
Currently recommended algorithm for risk stratification, diagnostic, and treatment strategy to assess aPE is shown in Fig. 14.36 [179, 334,335,336,337]. As already reviewed it is critically important that objective imaging assessment of RV dysfunction (Fig. 14.37) or RV strain is done with the use of elevated cardiac biomarkers (Fig. 14.38) [334,335,336,337,338,339,340,341,342,343]. Pooled diagnostic sensitivities, specificities, and negative and positive predictive values for the use of TTE, CTPA, and cardiac biomarkers are shown in Fig. 14.39.

Risk stratification, diagnostic, and treatment algorithm for patients with aPE. BNP brain natriuretic peptide, Fx fondaparinux, ICU intensive care unit, LMWH low-molecular-weight heparin, PESI pulmonary embolism severity index, RV right ventricle, tropo troponin, UFH unfractionated heparin. (Reprinted with permission from Penaloza A, et al. Curr Opin Crit Care. 2012; 18: 318–325)

Prognostic value of right ventricular dysfunction for mortality in patients with pulmonary embolism without shock. The outcome was in-hospital mortality for all studies, except two: (*) 40-day mortality and (†) 90-day mortality. (Reprinted with permission from Sanchez, O, et al. Eur Heart J. 2008; 29: 1569–1577)

Prognostic value of cardiac biomarkers for mortality in patients with pulmonary embolism without shock. The outcome was in-hospital mortality for all studies, except two: (*) 40-day mortality and (†) 90-day mortality. (Reprinted with permission from Sanchez, O, et al. Eur Heart J. 2008; 29: 1569–1577)

Pooled diagnostic indexes for echocardiography, computed tomography, brain natriuretic peptide (BNP), pro-BNP, and cardiac troponin. (Reprinted with permission from Sanchez, O, et al. Eur Heart J. 2008; 29: 1569–1577)
Chronic Pulmonary Embolism
It is important to now shift gears and discuss a prevalent clinical entity found in survivors of aPE: a condition known as chronic thromboembolic pulmonary hypertension (CTEPH). The latter results from either a single or a recurrent pulmonary embolism event(s) that for the most part originates from a lower extremity vein DVT, although any venous thrombosis site can be responsible.
Even though the majority of aPE episodes typically resolve and restoration of normal pulmonary hemodynamics, gas exchange, and exercise tolerance occurs, in some patients there is incomplete embolic resolution with some residual degree of obstruction or significant narrowing of the central pulmonary vessels by organized scar tissue [344]. The latter would then result in CTEPH. When CTEPH is left untreated progressive RV failure and even death can occur [345].
Even though the full spectrum leading to CTEPH has not been fully defined, it appears that incomplete recovery of perfusion after the initial aPE insult is a common requisite. However, it is now known that changes in the distal pulmonary vascular bed, referred to as a secondary small-vessel vasculopathy, are also critically important for the evolution of this clinical entity in conjunction with a series of other characterized steps such as impaired fibrinolysis and fibrinogen mutations, endothelial dysfunction and defects in neoangiogenesis, differential gene expression, platelet dysfunction, and inflammation [346, 347]. However, it is important to point out that in approximately 25% of all CTEPH cases no identified preceding PE can be found [348].
As already established, for the majority of CTEPH cases to occur aPE patients must survive the initial event and complete adequate anticoagulation resulting in a whole spectrum of cases in which patients may have complete restoration of normal perfusion to significant residual chronic clot rising that raises the pulmonary pressures [349]. Recent data suggest that after 6 months of anticoagulation in patients treated for an initial aPE event, abnormal perfusion scans can be found between 30% and 50% of cases [350, 351]. So not only a persistent residual perfusion is a well-known requisite to develop CTEPH but also the larger the residual defect the more likely it is to progress to CTEPH [351]. Though the majority of patients with persistent perfusion defects after an aPE are for the most part either asymptomatic or mildly limited with no evidence of resting PH, in 10% of aPE patients with persistent perfusion defects, resting PH develops [350].
Even though there is paucity of data regarding true estimates of the incidence of aPE events, it has been suggested that approximately 300,000 aPE events occur yearly in the United States; of these aPE survivors some 3000 new cases of CTEPH should prospectively be diagnosed per year [352]. These estimates do not include the potential 25% of all CTEPH cases, not associated to a prior PE event [348], simply stating the fact that CTEPH is largely underdiagnosed.
To complicate matters even further, rates for identifying CTEPH patients might be complicated by the way studies have been conducted and what definition was employed in each clinical trial [353,354,355,356]. Specifically, estimates for the incidence of CTEPH using data from active surveillance studies following aPE patients range from 0.1% to 8.8% within 2 years of initial diagnosis [353,354,355,356].
Based on current data, CTEPH patients could be divided into those who survive aPE and those patients in whom a particular profile is seen more frequently with CTEPH rather than with idiopathic pulmonary arterial hypertension. Consequently, CTEPH has been associated with the following [357,358,359,360]:
-
Unprovoked aPE cases (odds ratio [OR]: 20.0; 95% CI: 2.7–100)
-
When there is a delay in diagnosis of greater than 2 weeks from symptom onset (OR: 7.9; 95% CI: 3.3–19.0) (28)
-
RV dysfunction at the time of aPE
-
Initial estimated RV systolic pressure by TTE >50 mmHg (3.3 times higher odds of persistent PH at 12 months)
-
Antiphospholipid antibodies or the lupus anticoagulant is the only hypercoagulable state more commonly seen in CTEPH
-
Infected ventriculoatrial shunts
-
Splenectomy
-
History of malignancy
-
Hypothyroidism
In terms of diagnosing CTEPH, identification of chronic thromboembolic material obstructing the pulmonary arteries is required with a precapillary PH mean pulmonary artery pressure >20 mmHg and a PVR >3 Wood units in the setting of a pulmonary artery occlusion pressure <15 mmHg [361].
However, in order to get diagnosed a high index of suspicion is required from the onset. Unfortunately, the current median time between symptom onset and final CTEPH diagnosis is 14.1 months [348, 360]. A delay that likely contributes is secondary vasculopathy [362]. Therefore, it is important that once signs and/or symptoms of pulmonary hypertension are encountered in patients (regardless of a prior history of PE or not), obtain a TTE. This imaging modality, as previously mentioned, will not only provide objective evidence of PH while giving an estimate of RV size and function but also give an overall assessment of left ventricular size and function, valvular structures, and presence of intracardiac shunts [362].
If PH is identified, a V/Q scan continues to be a very useful imaging modality in the continued evaluation of these patients with suspected CTEPH. Diagnosis requires identification of at least one but more commonly several segmental or larger mismatched perfusion defects. Unfortunately, V/Q scans might underestimate the extent of pulmonary artery obstruction if recanalization of previously occluded segments has occurred [363]. If no mismatched segmental segment is identified, the patient might be referred for further evaluation to a pulmonary hypertension center for additional management. If, on the other hand, with mismatched defects, a CTPA should be considered unless a nonthrombotic condition is identified as the cause of the PH. CTPA is an excellent diagnostic modality for detecting CTEPH when performed at specialized centers as interpretive scanning difficulties are known to occur otherwise leading to a falsely low sensitivity for CT-PA [364,365,366]. However, even when properly interpreted, a negative CT-PA scan cannot exclude the possibility of CTEPH and consequently a V/Q scan remains as the preferred initial imaging test for screening [362].
Final CTEPH confirmation requires a right-heart catheterization with selective digital subtraction pulmonary angiography (DS-PA) that not only provides a comprehensive radiographic and hemodynamic assessment but can also be done with or without exhaled gas analysis.
Once CTEPH has been diagnosed, surgical consideration is given to every patient with the intention of offering the potential for cure with a low perioperative mortality (<5%) at experienced surgical centers [367].
Surgical resection of the occluding thromboembolic material to re-establish flow requires experienced surgeons that can result in significant improvement for most patients immediately post-surgery; however, the presence of residual PH can result in significant postoperative morbidity for which a number of pharmacological treatment options remain available for those patients and for inoperable CTEPH [362].
Conclusion
Acute pulmonary embolism encompasses a very heterogeneous patient population whose management continues to evolve with not only several unresolved issues to be answered by future or ongoing research but also better risk stratification strategies. Considerable progress has been made regarding identification of intermediate-high-risk patients, but ongoing discussion still debates whether patients classified as low risk based on clinical parameters should undergo additional imaging, particularly testing for RV function. Surely many will argue one way or another; some opponents will state that routine testing has not been shown to have therapeutic implications while others will cite time- and cost-intensive data. The fact of the matter is that this is still a fluid field and as new data continues to be collected, we should be able to change our position, if needed.
For the time being it is thoroughly clear that aPE is as much a disease of the RV as it is of the pulmonary arteries. While the pulmonary burden may predict the severity of symptoms, it is the RV that determines hemodynamic response and survival. Prompt assessment of RV function can direct treatment and influence prognosis.
Abbreviations
- aPE:
-
Acute pulmonary embolism
- AUC:
-
Appropriate use criteria
- BP:
-
Blood pressure
- BNP:
-
Brain natriuretic peptide
- CDMT:
-
Catheter-directed mechanical thrombectomy
- CTEPH:
-
Chronic thromboembolic pulmonary hypertension
- CTPA:
-
Computed tomographic pulmonary angiography
- DVT:
-
Deep venous thrombosis
- DOACs:
-
Direct-acting oral anticoagulants
- ECG:
-
Electrocardiogram
- ESLD:
-
End-stage liver disease
- ESC:
-
European Society of Cardiology
- FAC:
-
Fractional area change
- IVS:
-
Interventricular septum
- LV:
-
Left ventricle
- LMWH:
-
Low-molecular-weight heparin
- OR:
-
Odds ratio
- PIOPED:
-
Prospective investigation of pulmonary embolism diagnosis
- PA:
-
Pulmonary artery
- PERC:
-
Pulmonary Embolism Rule-out Criteria
- PH:
-
Pulmonary hypertension
- PVR:
-
Pulmonary vascular resistance
- RHS:
-
Right-heart strain
- RV:
-
Right ventricle
- RVOT:
-
RV outflow tract
- TTE:
-
Transthoracic echocardiogram
- TAPSE:
-
Tricuspid annular plane systolic excursion
- TR:
-
Tricuspid regurgitation
- UFH:
-
Unfractionated heparin
- VTI:
-
Velocity time integral
- VTE:
-
Venous thromboembolism
- V/Q scan:
-
Ventilation-perfusion scintigraphy
- VKA:
-
Vitamin K antagonist
References
Geerts WH, Pineo GF, Heit JA, Bergqvist D, Lassen MR, Colwell CW, Ray JG. Prevention of venous thromboembolism: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest. 2004;126(Suppl. 3):338–400S.
Den Heijer M, Rosendaal FR, Blom HJ, Gerrits WB, Bos GM. Hyperhomocysteinemia and venous thrombosis: a meta-analysis. Thromb Haemost. 1998;80:566–9.
Martinelli I, Mannucci PM, De Stefano V, Taioli E, Rossi V, Crosti F, Paciaroni K, Leone G, Faioni EM. Different risks of thrombosis in four coagulation defects associated with inherited thrombophilia: a study of 150 families. Blood. 1998;92:2353–8.
Heit JA, Silverstein MD, Mohr DN, Petterson TM, Lohse CM, O’Fallon WM, Melton LJ 3rd. The epidemiology of venous thromboembolism in the community. Thromb Haemost. 2001;86:452–63.
Galli M, Luckiani D, Bertolini G, Barbui T. Lupus anticoagulants are stronger risk factors for thrombosis than anticardiolipin antibodies in the antiphospholipid syndrome: a systematic review of the literature. Blood. 2003;101:1827–32.
White RH, Zhou H, Romano PS. Incidence of symptomatic venous thromboembolism after different elective or urgent surgical procedures. Thromb Haemost. 2003;90:446–55.
Alikhan R, Cohen RT, Combe S, Samama MM, Desjardins L, Eldor A, Janbon C, Leizorovicz A, Olsson CG, Turpie AG. Risk factors for venous thromboembolism in hospitalized patients with acute medical illness: analysis of the MEDENOX Study. Arch Intern Med. 2004;164:963–8.
Hoffman M, Monroe DM. Coagulation 2006: a modern view of hemostasis. Hematol Oncol Clin North Am. 2007;21(1):1–11. https://doi.org/10.1016/j.hoc.2006.11.004.
Cina G, Marra R, Di Stasi C, Macis G. Epidemiology, pathophysiology and natural history of venous thromboembolism. Rays. 1996;21:315–27.
Wilkins RW, Stanton JR. Elastic stockings in the prevention of pulmonary embolism: a progress report. N Engl J Med. 1953;248:1087–90.
Ramzi DW, Leeper KV. DVT and pulmonary embolism: part II. Treatment and prevention. Am Fam Physician. 2004;69:2841–8.
Kearon C. Natural history of venous thromboembolism. Circulation. 2003;107(23 Suppl 1):I22–30.
Heit JA, Cohen AT, Anderson FJ. Estimated annual number of incident and recurrent, non-fatal venous thromboembolism (VTE) events in the US. Blood. 2005;106:11.
Boulay F, Berthier F, Schoukroun G, et al. Seasonal variations in hospital admission for deep vein thrombosis and pulmonary embolism: analysis of discharge data. BMJ. 2001;323:601–2.
Cervantes J, Rojas G. Virchows legacy: deep vein thrombosis and pulmonary embolism. World J Surg. 2005;29:S30–4.
Naess IA, Christiansen SC, Romundstad P, Cannegieter SC, Rosendaal FR, Hammerstrom J. Incidence and mortality of venous thrombosis: a population based study. J Thromb Haemost. 2007;5:692–9.
Cushman M, Tsai AW, White RH, Heckbert SR, Rosamond WD, Enright P, Folsom AR. Deep vein thrombosis and pulmonary embolism in two cohorts: the longitudinal investigation of thromboembolism etiology. Am J Med. 2004;117:19–25.
Rosendaal FR, Reitsma PH. Genetics of venous thrombosis. J Thromb Haemost. 2009;7(suppl 1):301–4.
Wakefield TW, Myers DD, Henke PK. Mechanisms of venous thrombosis and resolution. Arterioscler Thromb Vasc Biol. 2008;28:387–91.
Becker BF, Heindl B, Kupatt C, Zahler S. Endothelial function and hemostasis. Z Kardiol. 2000;89:160–7.
Gross PL, Aird WC. The endothelium and thrombosis. Semin Thromb Hemost. 2000;26:463–78.
Mackman N. New insights into the mechanisms of venous thrombosis. J Clin Invest. 2012;122:2331–6.
Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008;451:914–8.
Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–49.
Lippi G, Franchini M, Targher G. Arterial thrombus formation in cardiovascular disease. Nat Rev Cardiol. 2011;8:502–12.
Jackson SP. Arterial thrombosis-insidious, unpredictable and deadly. Nat Med. 2011;17:1423–36.
Undas A, Ariëns RA. Fibrin clot structure and function: a role in the pathophysiology of arterial and venous thromboembolic diseases. Arterioscler Thromb Vasc Biol. 2011;31:e88–99.
Wolberg AS. Plasma and cellular contributions to fibrin network formation, structure, and stability. Haemophilia. 2010;16(Suppl 3):7–12.
Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24:1015–22.
Hoffbrand AV, Pettit JE. Essential hematology. 3rd ed. Oxford: Blackwell Scientific Publications; 1993.
Virchow RLK. Gesammelte Abhandlungen zur wissenschaftlichen Medicin. Von Meidinger & Sohn: Frankfurt; 1856.
Iorio A, et al. Risk of recurrence after a first episode of symptomatic venous thromboembolism provoked by a transient risk factor: a systematic review. Arch Intern Med. 2010;170:1710–6.
Rosendaal FR, van Hylckama VA, Doggen CJ. Venous thrombosis in the elderly. J Thromb Haemost. 2007;5(suppl 1):310–7.
Lowe GD, et al. Epidemiology of coagulation factors, inhibitors and activation markers: the Third Glasgow MONICA Survey. I. Illustrative reference ranges by age, sex and hormone use. Br J Haematol. 1997;97:775–84.
Li C, Ford ES, McGuire LC, Mokdad AH. Increasing trends in waist circumference and abdominal obesity among US adults. Obesity (Silver Spring). 2007;15:216–24.
Smeeth L, Cook C, Thomas S, Hall AJ, Hubbard R, Vallance P. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet. 2006;367:1075–9.
Osterud B, Due J Jr. Blood coagulation in patients with benign and malignant tumours before and after surgery. Special reference to thromboplastin generation in monocytes. Scand J Haematol. 1984;32:258–64.
Johnson GJ, Leis LA, Bach RR. Tissue factor activity of blood mononuclear cells is increased after total knee arthroplasty. Thromb Haemost. 2009;102:728–34.
White RH, Romano PS, Zhou H, Rodrigo J, Bargar W. Incidence and time course of thromboembolic outcomes following total hip or knee arthroplasty. Arch Intern Med. 1998;158:1525–31.
James AH. Venous thromboembolism in pregnancy. Arterioscler Thromb Vasc Biol. 2009;29:326–31.
Bremme KA. Haemostatic changes in pregnancy. Best Pract Res Clin Haematol. 2003;16:153–68.
James AH, Jamison MG, Brancazio LR, Myers ER. Venous thromboembolism during pregnancy and the postpartum period: incidence, risk factors, and mortality. Am J Obstet Gynecol. 2006;194:1311–5.
Middeldorp S, et al. Effects on coagulation of levonorgestrel- and desogestrel-containing low dose oral contraceptives: a cross-over study. Thromb Haemost. 2000;84:4–8.
Vandenbroucke JP, et al. Oral contraceptives and the risk of venous thrombosis. N Engl J Med. 2001;344:1527–35.
Abdollahi M, Cushman M, Rosendaal FR. Obesity: risk of venous thrombosis and the interaction with coagulation factor levels and oral contraceptive use. Thromb Haemost. 2003;89:493–8.
Ayer JG, Song C, Steinbeck K, Celermajer DS, Ben Freedman S. Increased tissue factor activity in monocytes from obese young adults. Clin Exp Pharmacol Physiol. 2010;37:1049–54.
Khorana AA. Venous thromboembolism and prognosis in cancer. Thromb Res. 2010;125:490–3.
Caine GJ, Stonelake PS, Lip GY, Kehoe ST. The hypercoagulable state of malignancy: pathogenesis and current debate. Neoplasia. 2002;4:465–73.
Allman-Farinelli MA. Obesity and venous thrombosis: a review. Semin Thromb Hemost. 2011;37:903–7.
Noble S, Pasi J. Epidemiology and pathophysiology of cancer-associated thrombosis. Br J Cancer. 2010;102:S2–9.
Verbeek TA, Jonathan SG, Saner FH, et al. Hypercoagulability in end-stage liver disease: review of epidemiology, etiology, and management. Transplant Direct. 2018;4(11):e403. https://doi.org/10.1097/TXD.0000000000000843.
Silverstein MD, Heit JA, Mohr DN, et al. Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med. 1998;158:585–93.
Northup PG, McMahon MM, Ruhl AP, et al. Coagulopathy does not fully protect hospitalized cirrhosis patients from peripheral venous thromboembolism. Am J Gastroenterol. 2006;101:1524–8.
Heit JA, Silverstein MD, Mohr DN, et al. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med. 2000;160:809–15.
Huerta C, Johansson S, Wallander MA, et al. Risk factors and short-term mortality of venous thromboembolism diagnosed in the primary care setting in the United Kingdom. Arch Intern Med. 2007;167:935–43.
Gulley D, Teal E, Suvannasankha A, et al. Deep vein thrombosis and pulmonary embolism in cirrhosis patients. Dig Dis Sci. 2008;53:3012–7.
Wu H, Nguyen GC. Liver cirrhosis is associated with venous thromboembolism among hospitalized patients in a nationwide US study. Clin Gastroenterol Hepatol. 2010;8:800–5.
Northup PG, Caldwell SH. Coagulation in liver disease: a guide for the clinician. Clin Gastroenterol Hepatol. 2013;11:1064–74.
Dabbagh O, Oza A, Prakash S, et al. Coagulopathy does not protect against venous thromboembolism in hospitalized patients with chronic liver disease. Chest. 2010;137:1145–9.
Cockett FB, Thomas ML. The iliac compression syndrome. Br J Surg. 1965;52:816–21.
Moudgill N, Hager E, Gonsalves C, Larson R, Lombardi J, DiMuzio P. May-Thurner syndrome: case report and review of the literature involving modern endovascular therapy. Vascular. 2009;17:330–5.
Bovill EG, van der Vliet A. Venous valvular stasis-associated hypoxia and thrombosis: what is the link? Annu Rev Physiol. 2011;73:527–45.
Nicolaides AN, Kakkar VV, Field ES, et al. The origin of deep vein thrombosis: a venographic study. Br J Radiol. 1971;44:653–63.
Kakkar VV, Howe CT, Flanc C, et al. Natural history of postoperative deep-vein thrombosis. Lancet. 1969;2:230–2.
Cogo A, Lensing AWA, Prandoni P, et al. Distribution of thrombosis in patients with symptomatic deep-vein thrombosis: implications for simplifying the diagnostic process with compression ultrasound. Arch Intern Med. 1993;153:2777–80.
Moser KM, LeMoine JR. Is embolic risk conditioned by location of deep venous thrombosis? Ann Intern Med. 1981;94:439–44.
Malone PC, Agutter PS. The aetiology of deep venous thrombosis. QJM. 2006;99:581–93.
Hamer JD, Malone PC, Silver IA. The PO2 in venous valve pockets: its possible bearing on thrombogenesis. Br J Surg. 1981;68(3):166–70.
Liu GC, Ferris EJ, Reifsteck JR, Baker ME. Effect of anatomic variations on deep venous thrombosis of the lower extremity. Am J Roentgenol. 1986;146(4):845–8.
Varma MR, Varga AJ, Knipp BS, Sukheepod P, Upchurch GR, Kunkel SL, Wakefield TW, Henke PK. Neutropenia impairs venous thrombosis resolution in the rat. J Vasc Surg. 2003;38:1090–8.
Stewart GJ. Neutrophils and deep venous thrombosis. Haemostasis. 1993;23(Suppl 1):127–40.
Henke PK, Pearce CG, Moaveni DM, Moore AJ, Lynch EM, Longo C, Varma M, Dewyer NA, Deatrick KB, Upchurch GR Jr, Wakefield TW, Hogaboam C, Kunkel SL. Targeted deletion of CCR2 impairs deep vein thrombosis resolution in a mouse model. J Immunol. 2006;177:3388–97.
Henke PK, Varma MR, Moaveni DK, Dewyer NA, Moore AJ, Lynch EM, Longo C, Deatrick CB, Kunkel SL, Upchurch GR Jr, Wakefield TW. Fibrotic injury after experimental deep vein thrombosis is determined by the mechanism of thrombogenesis. Thromb Haemost. 2007;98:1045–55.
Heit JA. Epidemiology of venous thromboembolism. Nat Rev Cardiol. 2015;12(8):464–74. https://doi.org/10.1038/nrcardio.2015.83.
Barsoum MK, Heit JA, Ashrani AA, Leibson CL, Petterson TM, Bailey KR. Is progestin an independent risk factor for incident venous thromboembolism? A population-based case-control study. Thromb Res. 2010;126(5):373–8.
Merrer J, et al. Complications of femoral and subclavian venous catheterization in critically ill patients: a randomized controlled trial. JAMA. 2001;286:700–7.
Cogo A, et al. Acquired risk factors for deep-vein thrombosis in symptomatic outpatients. Arch Intern Med. 1994;154:164–8.
Kuipers S, et al. The absolute risk of venous thrombosis after air travel: a cohort study of 8,755 employees of international organisations. PLoS Med. 2007;4:e290.
Doggen CJ, et al. Serum lipid levels and the risk of venous thrombosis. Arterioscler Thromb Vasc Biol. 2004;24:1970–5.
Decousus H, et al. Superficial venous thrombosis and venous thromboembolism: a large, prospective epidemiologic study. Ann Intern Med. 2010;152:218–24.
Noboa S, Mottier D, Oger E, EPI-GETBO Study Group. Estimation of a potentially preventable fraction of venous thromboembolism: a community-based prospective study. J Thromb Haemost. 2006;4(12):2720–2.
Chew HK, Wun T, Harvey D, Zhou H, White RH. Incidence of venous thromboembolism and its effect on survival among patients with common cancers. Arch Intern Med. 2006;166:458–64.
Blom JW, et al. Incidence of venous thrombosis in a large cohort of 66,329 cancer patients: results of a record linkage study. J Thromb Haemost. 2006;4:529–35.
Heit JA, et al. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med. 2000;160:809–15.
Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood. 2008;111:4902–7.
Ay C, et al. Prediction of venous thromboembolism in cancer patients. Blood. 2010;116:5377–82.
Huang W, Goldberg RJ, Anderson FA, Kiefe CI, Spencer FA. Secular trends in occurrence of acute venous thromboembolism: the Worcester VTE study (1985–2009). Am J Med. 2014;127:829.e5–39.e5.
Heit JA. Estimating the incidence of symptomatic postoperative venous thromboembolism: the importance of perspective. JAMA. 2012;307:306–7.
Spencer FA, et al. Incidence rates, clinical profile, and outcomes of patients with venous thromboembolism. The Worcester VTE study. J Thromb Thrombolysis. 2009;28:401–9.
Heit JA, et al. Predictors of recurrence after deep vein thrombosis and pulmonary embolism: a population-based cohort study. Arch Intern Med. 2000;160:761–8.
van Dongen CJ, Vink R, Hutten BA, Büller HR, Prins MH. The incidence of recurrent venous thromboembolism after treatment with vitamin K antagonists in relation to time since first event: a meta-analysis. Arch Intern Med. 2003;163:1285–93.
Schulman S, et al. Post-thrombotic syndrome, recurrence, and death 10 years after the first episode of venous thromboembolism treated with warfarin for 6 weeks or 6 months. J Thromb Haemost. 2006;4:734–42.
Prandoni P, et al. Residual venous thrombosis as a predictive factor of recurrent venous thromboembolism. Ann Intern Med. 2002;137:955–60.
Kyrle PA, Eichinger S. The risk of recurrent venous thromboembolism: the Austrian Study on Recurrent Venous Thromboembolism. Wien Klin Wochenschr. 2003;115:471–4.
Baglin T, et al. Does the clinical presentation and extent of venous thrombosis predict likelihood and type of recurrence? A patient-level meta-analysis. J Thromb Haemost. 2010;8:2436–42.
Kovacs MJ, et al. Patients with a first symptomatic unprovoked deep vein thrombosis are at higher risk of recurrent venous thromboembolism than patients with a first unprovoked pulmonary embolism. J Thromb Haemost. 2010;8:1926–32.
Schulman S, Svenungsson E, Granqvist S. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Duration of Anticoagulation Study Group. Am J Med. 1998;104:332–8.
Garcia D, Akl EA, Carr R, Kearon C. Antiphospholipid antibodies and the risk of recurrence after a first episode of venous thromboembolism: a systematic review. Blood. 2013;122:817–24.
Jayakody Arachchillage D, Greaves M. The chequered history of the antiphospholipid syndrome. Br J Haematol. 2014;165:609–17.
Brouwer JL, et al. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost. 2009;101:93–9.
Chee CE, et al. Predictors of venous thromboembolism recurrence and bleeding among active cancer patients: a population-based cohort study. Blood. 2014;123:3972–8.
White RH, Gettner S, Newman JM, Trauner KB, Romano PS. Predictors of rehospitalization for symptomatic venous thromboembolism after total hip arthroplasty. N Engl J Med. 2000;343:1758–64.
Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–35.
Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–8.
Larsen WJ. Human embryology. New York: Churchill Livingstone; 1993. p. 111–204.
Moore KL, Persaud TV. The developing human: clinically oriented embryology. Philadelphia, PA: WB Saunders; 1998. p. 241–53, 329–80.
Murillo H, Cutalo MJ, Jones RP, Lane MJ, Fleischmann D, Restrepo CS. Pulmonary circulation imaging: embryology and normal anatomy. Semin Ultrasound CT MR. 2012;33:473–84.
Gao Y, Raj JU. Regulation of the pulmonary circulation in the fetus and newborn. Physiol Rev. 2010;90:1291–335.
Burri PH. Structural aspects of postnatal lung development—alveolar formation and growth. Biol Neonate. 2006;89:313–22.
Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012;92:367–520.
Berrocal T, Madrid C, Novo S, et al. Congenital anomalies of the tracheobronchial tree, lung, and mediastinum: embryology, radiology, and pathology. Radiographics. 2003;24:e17.
Castañer E, Gallardo X, Rimola J, et al. Congenital and acquired pulmonary artery anomalies in the adult: radiologic overview. Radiographics. 2006;26:349–71.
Grosse C, Grosse A. CT findings in diseases associated with pulmonary hypertension: a current review. Radiographics. 2010;30:1753–77.
Ghio S, Gavazzi A, Campana C, Inserra C, Klersy C, Sebastiani R, et al. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol. 2001;37:183–8.
Becattini C, Agnelli G. Predictors of mortality from pulmonary embolism and their influence on clinical management. Thromb Haemost. 2008;100:747–51.
Sanchez O, Trinquart L, Colombet I, et al. Prognostic value of right ventricular dysfunction in patients with haemodynamically stable pulmonary embolism: a systematic review. Eur Heart J. 2008;29:1569–77.
Stevinson BG, Hernandez-Nino J, Rose G, Kline JA. Echocardiographic and functional cardiopulmonary problems six months after first-time pulmonary embolism in previously healthy patients. Eur Heart J. 2007;28:2517–24.
Haddad F, Hunt SA, Rosenthal DN, Murphy DJ. Right ventricular function in cardiovascular disease, part I: anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation. 2008;117:1436–48.
Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, et al. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114:1883–91.
Hemnes AR, Champion HC. Right heart function and haemodynamics in pulmonary hypertension. Int J Clin Pract. 2008;62(Suppl 160):11–9.
McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119:2250–94.
Goldhaber SZ, Visani L, De Rosa M. Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER). Lancet. 1999;353:1386–9.
Stein PD, Matta F, Alrifai A, Rahman A. Trends in case fatality rate in pulmonary embolism according to stability and treatment. Thromb Res. 2012;130(6):841–6.
Watts JA, Marchick MR, Kline JA. Right ventricular heart failure from pulmonary embolism: key distinctions from chronic pulmonary hypertension. J Card Fail. 2010;16:250–9.
Bristow MR, Zisman LS, Lowes BD, Abraham WT, Badesch DB, Groves BM, Voelkel NF, Lynch DM, Quaife RA. The pressure-overloaded right ventricle in pulmonary hypertension. Chest. 1998;114(1 Suppl):101S–6S.
Carabello BA. The relationship of left ventricular geometry and hypertrophy to left ventricular function in valvular heart disease. J Heart Valve Dis. 1995;4(Suppl 2):S132–8.
Konstam MA, Cohen SR, Salem DN, et al. Comparison of left and right ventricular end-systolic pressure-volume relations in congestive heart failure. J Am Coll Cardiol. 1985;5:1326–34.
Nakamura H, Adachi H, Sudoh A, Yagyu H, Kishi K, Oh-ishi S, et al. Subacute cor pulmonale due to tumor embolism. Intern Med. 2004;43:420–2.
Archer S, Michelakis E. The mechanism(s) of hypoxic pulmonary vasoconstriction: potassium channels, redox O(2) sensors, and controversies. News Physiol Sci. 2002;17:131–7.
Memtsoudis SG, Rosenberger P, Walz JM. Critical care issues in the patient after major joint replacement. J Intens Care Med. 2007;22:92–104.
Toledo LS, Mauad R. Complications of body sculpture: prevention and treatment. Clin Plastic Surg. 2006;33:1–11.
Mirski MA, Lele AV, Fitzsimmons L, Toung TJ. Diagnosis and treatment of vascular air embolism. Anesthesiology. 2007;106:164–77.
Smulders YM. Pathophysiology and treatment of haemodynamic instability in acute pulmonary embolism: the pivotal role of pulmonary vasoconstriction. Cardiovasc Res. 2000;48:23–33.
Jones AE, Watts JA, Debelak JP, Thornton LR, Younger JG, Kline JA. Inhibition of prostaglandin synthesis during polystyrene microsphere-induced pulmonary embolism in the rat. Am J Physiol Lung Cell Mol Physiol. 2003;284:L1072–81.
Reeves WC, Demers LM, Wood MA, Skarlatos S, Copenhaver G, Whitesell L, et al. The release of thromboxane A2 and prostacyclin following experimental acute pulmonary embolism. Prostagland Leukotrienes Med. 1983;11:1–10.
Todd MH, Forrest JB, Cragg DB. The effects of aspirin and methysergide, singly and in combination, on systemic haemodynamic responses to pulmonary embolism. Can Anaesth Soc J. 1981;28:373–80.
Breuer J, Meschig R, Breuer HW, Arnold G. Effects of serotonin on the cardiopulmonary circulatory system with and without 5-HT2-receptor blockade by ketanserin. J Cardiovasc Pharmac. 1985;7:64–6.
Battistini B. Modulation and roles of the endothelins in the pathophysiology of pulmonary embolism. Can J Physiol Pharmacol. 2003;81:555–69.
Kapsch DN, Metzler M, Silver D. Contributions of prostaglandin F2 alpha and thromboxane A2 to the acute cardiopulmonary changes of pulmonary embolism. J Surg Res. 1981;30:522–9.
Kim SH, Yi MZ, Kim DH, Song JM, Kang DH, Lee SD, Song JK. Prognostic value of echocardiographic estimation of pulmonary vascular resistance in patients with acute pulmonary thromboembolism. J Am Soc Echocardiogr. 2011;24:693–8.
Torbicki A, Galié N, Covezzoli A, Rossi E, De Rosa M, Goldhaber SZ, ICOPER Study Group. Right heart thrombi in pulmonary embolism: results from the International Cooperative Pulmonary Embolism Registry. J Am Coll Cardiol. 2003;41:2245–51.
Konstantinides SV, Torbicki A, Agnelli G, et al. 2014 ESC guidelines on the diagnosis and management of acute pulmonary embolism: the task force for the diagnosis and management of acute pulmonary embolism of the European Society of Cardiology (ESC). Endorsed by the European Respiratory Society (ERS). Eur Heart J. 2014;35(43):3033–80.
McIntyre KM, Sasahara AA. The hemodynamic response to pulmonary embolism in patients without prior cardiopulmonary disease. Am J Cardiol. 1971;28(3):288–94.
Delcroix M, Mélot C, Lejeune P, Leeman M, Naeije R. Effects of vasodilators on gas exchange in acute canine embolic pulmonary hypertension. Anesthesiology. 1990;72(1):77–84.
Lankhaar JW, Westerhof N, Faes TJ, Marques KM, Marcus JT, Postmus PE, Vonk-Noordegraaf A. Quantification of right ventricular afterload in patients with and without pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2006;291(4):H1731–7.
Chan CM, Woods C, Shorr AF. The validation and reproducibility of the pulmonary embolism severity index. J Thromb Haemost. 2010;8(7):1509–14.
Donzé J, Le Gal G, Fine MJ, et al. Prospective validation of the Pulmonary Embolism Severity Index. A clinical prognostic model for pulmonary embolism. Thromb Haemost. 2008;100(5):943–8.
Vanni S, Nazerian P, Pepe G, et al. Comparison of two prognostic models for acute pulmonary embolism: clinical vs. right ventricular dysfunction-guided approach. J Thromb Haemost. 2011;9(10):1916–23.
Aujesky D, Obrosky DS, Stone RA, et al. Derivation and validation of a prognostic model for pulmonary embolism. Am J Respir Crit Care Med. 2005;172(8):1041–6.
Jimenez D, Aujesky D, Moores L, et al. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med. 2010;170(15):1383–9.
Righini M, Roy PM, Meyer G, et al. The Simplified Pulmonary Embolism Severity Index (PESI): validation of a clinical prognostic model for pulmonary embolism. J Thromb Haemost. 2011;9(10):2115–7.
Nassiri N, Jain A, McPhee D, Mina B, Rosen RJ, Giangola G, et al. Massive and submassive pulmonary embolism: experience with an algorithm for catheter-directed mechanical thrombectomy. Ann Vasc Surg. 2012;26(1):18–24.
Silverstein MD, Heit JA, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med. 1998;158:585–93.
Agnelli G, Becattini C. Acute pulmonary embolism. N Engl J Med. 2010;363:266–74.
Piazza G, Goldhaber SZ. Current concepts: chronic thromboembolic pulmonary hypertension. New Engl J Med. 2011;364:351–60.
Jiménez D, de Miguel-Díez J, Guijarro R, et al., RIETE Investigators. Trends in the management and outcomes of acute pulmonary embolism: analysis from the RIETE Registry. J Am Coll Cardiol. 2016;67(2):162–70.
Hsiao SH, Lee CY, Chang SM, Yang SH, Lin SK, Huang WC. Pulmonary embolism and right heart function: insights from myocardial Doppler tissue imaging. J Am Soc Echocard. 2006;19:822–8.
Iwadate K, Tanno K, Doi M, Takatori T, Ito Y. Two cases of right ventricular ischemic injury due to massive pulmonary embolism. Forensic Sci Int. 2001;116:189–95.
Begieneman MP, van de Goot FR, van der Bilt I, Noordegraaf AV, Spreeuwenberg MD, Paulus WJ, et al. Pulmonary embolism causes endomyocarditis in the human heart. Heart. 2008;94:450–6.
Zagorski J, Gellar MA, Obraztsova M, Kline JA, Watts JA. Inhibition of CINC-1 decreases right ventricular damage caused by experimental pulmonary embolism in rats. J Immunol. 2007;179:7820–6.
Watts JA, Zagorski J, Gellar MA, Stevinson BG, Kline JA. Cardiac inflammation contributes to right ventricular dysfunction following experimental pulmonary embolism in rats. J Molec Cell Cardiol. 2006;41:296–307.
Kline JA, Zeitouni R, Marchick MR, Hernandez-Nino J, Rose GA. Comparison of 8 biomarkers for prediction of right ventricular hypokinesis 6 months after submassive pulmonary embolism. Am Heart J. 2008;156:308–14.
Nordenholz KE, Mitchell AM, Kline JA. Direct comparison of the diagnostic accuracy of fifty protein biological markers of pulmonary embolism for use in the emergency department. Acad Emerg Med. 2008;15:795–9.
Mitchell AM, Nordenholz KE, Kline JA. Tandem measurement of D dimer and myeloperoxidase of C-reactive protein to effectively screen for pulmonary embolism in the emergency department. Acad Emerg Med. 2008;15:800–5.
Torrent-Guasp F, Whimster WF, Redmann K. A silicone rubber mould of the heart. Technol Health Care. 1997;5:13–20.
Buckberg GD, RESTORE Group. The ventricular septum: the lion of right ventricular function, and its impact on right ventricular restoration. Eur J Cardiothorac Surg. 2006;29(Suppl 1):S272–8.
Sallin EA. Fiber orientation and ejection fraction in the human left ventricle. Biophys J. 1969;9:954–64.
Damiano RJ Jr, La Follette P Jr, Cox JL, Lowe JE, Santamore WP. Significant left ventricular contribution to right ventricular systolic function. Am J Physiol. 1991;261(5 Pt 2):H1514–24.
Osculati G, Malfatto G, Chianca R, Perego GB. Left-to-right systolic ventricular interaction in patients undergoing biventricular stimulation for dilated cardiomyopathy. J Appl Physiol. 2010;109:418–23.
Schwarz K, Singh S, Dawson D, Frenneaux MP. Right ventricular function in left ventricular disease: pathophysiology and implications. Heart Lung Circ. 2013;22:507–11.
Mori S, Nakatani S, Kanzaki H, Yamagata K, Take Y, Matsuura Y, et al. Patterns of the interventricular septal motion can predict conditions of patients with pulmonary hypertension. J Am Soc Echocardiogr. 2008;21:386–93.
Ramani GV, Bazaz R, Edelman K, López-Candales A. Pulmonary hypertension affects left ventricular basal twist: a novel use for speckle-tracking imaging. Echocardiography. 2009;26:44–51.
López-Candales A, Edelman K. Chronic pulmonary hypertension causes significant interventricular spatiotemporal dyssynchrony when onset of diastolic flow signals are assessed by color M-mode. Echocardiography. 2012;29:653–60.
Piazza G. Submassive pulmonary embolism. JAMA. 2013;309(2):171–80.
Konstantinides S. Should thrombolytic therapy be used in patients with pulmonary embolism? Am J Cardiovasc Drugs. 2004;4:69–74.
Kasper W, Konstantinides S, Geibel A, Olschewski M, Heinrich F, Grosser KD, et al. Management strategies and determinants of outcome in acute major pulmonary embolism: results of a multicenter registry. J Am Coll Cardiol. 1997;30:1165–71.
Wood KE. Major pulmonary embolism: review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. Chest. 2002;121:877–905.
Goldhaber SZ, Haire WD, Feldstein ML, Miller M, Toltzis R, Smith JL, et al. Alteplase versus heparin in acute pulmonary embolism: randomized trial assessing right-ventricular function and pulmonary perfusion. Lancet. 1993;314:507–11.
Grifoni S, Olivotto I, Cecchini P, Pieralli F, Camaiti A, Santoro G, et al. Short-term clinical outcome of patients with pulmonary embolism, normal blood pressure, and echocardiographic right ventricular dysfunction. Circulation. 2000;101:2817–22.
Frémont B, Pacouret G, Jacobi D, Puglisi R, Charbonnier B, de Labriolle A. Prognostic value of echocardiographic right/left ventricular end-diastolic diameter ratio in patients with acute pulmonary embolism: results from a monocenter registry of 1,416 patients. Chest. 2008;133:358–62.
Sanchez O, Trinquart L, Caille V, Couturaud F, Pacouret G, Meneveau N, et al. Prognostic factors for pulmonary embolism: the PREP Study, a prospective multicenter cohort study. Am J Respir Crit Care Med. 2010;181:168–73.
Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Prognostic role of echocardiography among patients with acute pulmonary embolism and a systolic arterial pressure of 90 mm Hg or higher. Arch Intern Med. 2005;165:1777–81.
Stein PD, Henry JW. Prevalence of acute pulmonary embolism among patients in a general hospital and at autopsy. Chest. 1995;108:978–81.
Miniati M, Pistolesi M, Marini C, et al. Value of perfusion lung scan in the diagnosis of pulmonary embolism: results of the Prospective Investigative Study of Acute Pulmonary Embolism Diagnosis (PISA-PED). Am J Respir Crit Care Med. 1996;154(5):1387–93.
Musset D, Parent F, Meyer G, et al. Diagnostic strategy for patients with suspected pulmonary embolism: a prospective multicentre outcome study. Lancet. 2002;360(9349):1914–20.
Le Gal G, Righini M, Roy PM, et al. Prediction of pulmonary embolism in the emergency department: the revised Geneva score. Ann Intern Med. 2006;144(3):165–71.
PIOPED Investigators. Value of the ventilation/perfusion scan in acute pulmonary embolism. Results of the prospective investigation of pulmonary embolism diagnosis (PIOPED). JAMA. 1990;263(20):2753–9.
Wells PS, Anderson DR, Rodger M, et al. Derivation of a simple clinical model to categorize patients probability of pulmonary embolism: in- creasing the models utility with the SimpliRED D-dimer. Thromb Haemost. 2000;83(3):416–20.
Anderson DR, Kovacs MJ, Dennie C, et al. Use of spiral computed tomography contrast angiography and ultrasonography to exclude the diagnosis of pulmonary embolism in the emergency department. J Emerg Med. 2005;29(4):399–404.
Kearon C, Ginsberg JS, Douketis J, et al. An evaluation of D-dimer in the diagnosis of pulmonary embolism: a randomized trial. Ann Intern Med. 2006;144(11):812–21.
Sohne M, Kamphuisen PW, van Mierlo PJ, et al. Diagnostic strategy using a modified clinical decision rule and D-dimer test to rule out pulmonary embolism in elderly in- and outpatients. Thromb Haemost. 2005;94(1):206–10.
van Belle A, Buller HR, Huisman MV, et al. Effectiveness of managing suspected pulmonary embolism using an algorithm combining clinical probability, D-dimer testing, and computed tomography. JAMA. 2006;295(2):172–9.
Wells PS, Anderson DR, Rodger M, et al. Excluding pulmonary embolism at the bedside without diagnostic imaging: management of patients with suspected pulmonary embolism presenting to the emergency department by using a simple clinical model and d-dimer. Ann Intern Med. 2001;135(2):98–107.
Rodger MA, Maser E, Stiell I, et al. The interobserver reliability of pretest probability assessment in patients with suspected pulmonary embolism. Thromb Res. 2005;116(2):101–7.
Runyon MS, Webb WB, Jones AE, et al. Comparison of the unstructured clinician estimate of pretest probability for pulmonary embolism to the Canadian score and the Charlotte rule: a prospective observational study. Acad Emerg Med. 2005;12(7):587–93.
Ghaye B, Szapiro D, Mastora I, et al. Peripheral pulmonary arteries: how far in the lung does multi-detector row spiral CT allow analysis? Radiology. 2001;219(3):629–36.
Patel S, Kazerooni EA, Cascade PN. Pulmonary embolism: optimization of small pulmonary artery visualization at multi-detector row CT. Radiology. 2003;227(2):455–60.
Remy-Jardin M, Remy J, Wattinne L, et al. Central pulmonary thromboembolism: diagnosis with spiral volumetric CT with the single-breath-hold technique: comparison with pulmonary angiography. Radiology. 1992;185(2):381–7.
Stein PD, Fowler SE, Goodman LR, et al. Multidetector computed tomography for acute pulmonary embolism. N Engl J Med. 2006;354(22):2317–27.
Carrier M, Righini M, Wells PS, et al. Subsegmental pulmonary embolism diagnosed by computed tomography: incidence and clinical implications. A systematic review and meta-analysis of the management outcome studies. J Thromb Haemost. 2010;8(8):1716–22.
Stein PD, Goodman LR, Hull RD, et al. Diagnosis and management of isolated subsegmental pulmonary embolism: review and assessment of the options. Clin Appl Thromb Hemost. 2012;18(1):20–6.
Goodman LR, Stein PD, Matta F, et al. CT venography and compression sonography are diagnostically equivalent: data from PIOPED II. AJR Am J Roentgenol. 2007;189(5):1071–6.
Farrell C, Jones M, Girvin F, et al. Unsuspected pulmonary embolism identified using multidetector computed tomography in hospital outpatients. Clin Radiol. 2010;65(1):1–5.
Jia CF, Li YX, Yang ZQ, et al. Prospective evaluation of unsuspected pulmonary embolism on coronary computed tomographic angiography. J Comput Assist Tomogr. 2012;36(2):187–90.
Palla A, Rossi G, Falaschi F, et al. Is incidentally detected pulmonary embolism in cancer patients less severe? A case-control study. Cancer Invest. 2012;30(2):131–4.
Sahut D’Izarn M, Caumont Prim A, Planquette B, et al. Risk factors and clinical outcome of unsuspected pulmonary embolism in cancer patients: a case-control study. J Thromb Haemost. 2012;10(10):2032–8.
Kearon C, Akl EA, Comerota AJ, et al. Antithrombotic therapy for VTE disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e419S–94S.
Alderson PO. Scintigraphic evaluation of pulmonary embolism. Eur J Nucl Med. 1987;13(Suppl):S6–10.
Bajc M, Neilly JB, Miniati M, Schuemichen C, Meignan M, Jonson B, EANM Committee. EANM guidelines for ventilation/perfusion scintigraphy: part 1. Pulmonary imaging with ventilation/perfusion single photon emission tomography. Eur J Nucl Med Mol Imaging. 2009;36(8):1356–70.
Bajc M, Neilly JB, Miniati M, Schuemichen C, Meignan M, Jonson B. EANM guidelines for ventilation/perfusion scintigraphy: part 2. Algorithms and clinical considerations for diagnosis of pulmonary emboli with V/P(SPECT) and MDCT. Eur J Nucl Med Mol Imaging. 2009;36(9):1528–38.
Reid JH, Coche EE, Inoue T, et al. Is the lung scan alive and well? Facts and controversies in defining the role of lung scintigraphy for the diagnosis of pulmonary embolism in the era of MDCT. Eur J Nucl Med Mol Imaging. 2009;36(3):505–21.
Gottschalk A, Sostman HD, Coleman RE, et al. Ventilation-perfusion scintigraphy in the PIOPED study. Part II. Evaluation of the scintigraphic criteria and interpretations. J Nucl Med. 1993;34(7):1119–26.
Sostman HD, Coleman RE, DeLong DM, et al. Evaluation of revised criteria for ventilation-perfusion scintigraphy in patients with suspected pulmonary embolism. Radiology. 1994;193(1):103–7.
Bajc M, Olsson B, Palmer J, et al. Ventilation/perfusion SPECT for diagnostics of pulmonary embolism in clinical practice. J Intern Med. 2008;264(4):379–87.
Glaser JE, Chamarthy M, Haramati LB, et al. Successful and safe implementation of a trinary interpretation and reporting strategy for V/Q lung scintigraphy. J Nucl Med. 2011;52(10):1508–12.
Sostman HD, Stein PD, Gottschalk A, et al. Acute pulmonary embolism: sensitivity and specificity of ventilation-perfusion scintigraphy in PIOPED II study. Radiology. 2008;246(3):941–6.
Stein PD, Terrin ML, Gottschalk A, et al. Value of ventilation/perfusion scans vs. perfusion scans alone in acute pulmonary embolism. Am J Cardiol. 1992;69(14):1239–41.
Collart JP, Roelants V, Vanpee D, et al. Is a lung perfusion scan obtained by using single photon emission computed tomography able to improve the radionuclide diagnosis of pulmonary embolism? Nucl Med Commun. 2002;23(11):1107–13.
Corbus HF, Seitz JP, Larson RK, et al. Diagnostic usefulness of lung SPET in pulmonary thromboembolism: an outcome study. Nucl Med Commun. 1997;18(10):897–906.
Reinartz P, Wildberger JE, Schaefer W, et al. Tomographic imaging in the diagnosis of pulmonary embolism: a comparison between V/Q lung scintigraphy in SPECT technique and multislice spiral CT. J Nucl Med. 2004;45(9):1501–8.
Gutte H, Mortensen J, Jensen CV, et al. Detection of pulmonary embolism with combined ventilation-perfusion SPECT and low-dose CT: head-to-head comparison with multidetector CT angiography. J Nucl Med. 2009;50(12):1987–92.
van Beek EJ, Reekers JA, Batchelor DA, et al. Feasibility, safety and clinical utility of angiography in patients with suspected pulmonary embolism. Eur Radiol. 1996;6(4):415–9.
Stein PD, Athanasoulis C, Alavi A, et al. Complications and validity of pulmonary angiography in acute pulmonary embolism. Circulation. 1992;85(2):462–8.
Wan S, Quinlan DJ, Agnelli G, et al. Thrombolysis compared with heparin for the initial treatment of pulmonary embolism: a meta-analysis of the randomized controlled trials. Circulation. 2004;110(6):744–9.
Diffin DC, Leyendecker JR, Johnson SP, et al. Effect of anatomic distribution of pulmonary emboli on interobserver agreement in the interpretation of pulmonary angiography. AJR Am J Roentgenol. 1998;171(4):1085–9.
Stein PD, Henry JW, Gottschalk A. Reassessment of pulmonary angiography for the diagnosis of pulmonary embolism: relation of interpreter agreement to the order of the involved pulmonary arterial branch. Radiology. 1999;210(3):689–91.
Zhang Y, Xia H, Wang Y, et al. The rate of missed diagnosis of lower-limb DVT by ultrasound amounts to 50% or so in patients without symptoms of DVT: a meta-analysis [published correction appears in Medicine (Baltimore)]. Medicine (Baltimore). 2019;98(37):e17103. https://doi.org/10.1097/MD.0000000000017103.
Aujesky D, Perrier A, Roy PM, Stone RA, Cornuz J, Meyer G, Obrosky DS, Fine MJ. Validation of a clinical prognostic model to identify low-risk patients with pulmonary embolism. J Intern Med. 2007;261:597–604.
Barra SN, Paiva L, Providência R, Fernandes A, Marques AL. A review on state-of-the-art data regarding safe early discharge following admission for pulmonary embolism: what do we know? Clin Cardiol. 2013;36:507–15.
Barra S, Paiva L, Providência R, Fernandes A, Nascimento J, Marques AL. LR-PED rule: low risk pulmonary embolism decision rule—a new decision score for low risk pulmonary embolism. Thromb Res. 2012;130:327–33.
Jiménez D, Aujesky D, Moores L, Gómez V, Lobo JL, Uresandi F, Otero R, Monreal M, Muriel A, Yusen RD, RIETE Investigators. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med. 2010;170:1383–9.
Rudski LG, Lai WW, Afilalo J, Hua L, Handschumacher MD, Chandrasekaran K, et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr. 2010;23:685–713.
Choi BY, Park DG. Normalization of negative T-wave on electrocardiography and right ventricular dysfunction in patients with an acute pulmonary embolism. Korean J Intern Med. 2012;27:53–9.
Golpe R, Castro-Anon O, Perez-de-Llano LA, et al. Electrocardiogram score predicts severity of pulmonary embolism in hemodynamically stable patients. J Hosp Med. 2011;6:285–9.
Kukla P, Dlstrokugopolski R, Krupa E, et al. Electrocardiography and prognosis of patients with acute pulmonary embolism. Cardiol J. 2011;18:648–53.
Daniel KR, Courtney DM, Kline JA. Assessment of cardiac stress from massive pulmonary embolism with 12-lead ECG. Chest. 2001;120:474–81.
Jaff MR, McMurtry MS, Archer SL, et al. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association [published corrections appear in Circulation. 2012;125:e495 and Circulation. 2012;126:e104]. Circulation. 2011;123:1788–830.
Hariharan P, Dudzinski DM, Okechukwu I, et al. Association between electrocardiographic findings, right heart strain, and short-term adverse clinical events in patients with acute pulmonary embolism. Clin Cardiol. 2015;38(4):236–42. https://doi.org/10.1002/clc.22383.
Kukla P, Długopolski R, Krupa E, et al. The value of ECG parameters in estimating myocardial injury and establishing prognosis in patients with acute pulmonary embolism. Kardiol Pol. 2011;69:933–8.
Punukollu G, Gowda RM, Vasavada BC, et al. Role of electrocardiography in identifying right ventricular dysfunction in acute pulmonary embolism. Am J Cardiol. 2005;96:450–2.
Douketis JD, Crowther MA, Stanton EB, Ginsberg JS. Elevated cardiac troponin levels in patients with submassive pulmonary embolism. Arch Intern Med. 2002;162(1):79–81. https://doi.org/10.1001/archinte.162.1.79.
Andrews J, MacNee W, Murchison J. Measurement of cardiac troponin identifies patients with moderate to large pulmonary emboli and right ventricular strain. Eur Respir J. 2014;44:P2407.
Keller K, Beule J, Schulz A, Coldewey M, Dippold W, Balzer JO. Cardiac troponin I for predicting right ventricular dysfunction and intermediate risk in patients with normotensive pulmonary embolism. Neth Heart J. 2015;23(1):55–61. https://doi.org/10.1007/s12471-014-0628-7.
Tanindi A, Cemri M. Troponin elevation in conditions other than acute coronary syndromes. Vasc Health Risk Manag. 2011;7:597–603. https://doi.org/10.2147/VHRM.S24509.
Sudoh T, Kangawa K, Minamino N, Matsuo H. A new natriuretic peptide in porcine brain. Nature. 1988;332:78–81.
Holmes SJ, Espiner EA, Richards AM, Yandle TG, Frampton C. Renal, endocrine, and hemodynamic effects of human brain natriuretic peptide in normal man. J Clin Endocrinol Metab. 1993;76:91–6.
Levin ER, Gardner DG, Samson WK. Natriuretic peptides. N Engl J Med. 1998;339:321–8.
Yoshimura M, Yasue H, Morita E, Sakaino N, Jougasaki M, Kurose M, et al. Hemodynamic, renal, and hormonal responses to brain natriuretic peptide infusion in patients with congestive heart failure. Circulation. 1991;84:1581–8.
Yasue H, Yoshimura M, Sumida H, Kikuta K, Kugiyama K, Jougasaki M, et al. Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation. 1994;90:195–203.
Coutance G, Le Page O, Lo T, Hamon M. Prognostic value of brain natriuretic peptide in acute pulmonary embolism. Crit Care. 2008;12(4):R109. https://doi.org/10.1186/cc6996.
Douglas PS, Garcia MJ, Haines DE, Lai WW, Manning WJ, Patel AR, et al. ACCF/ASE/AHA/ASNC/HFSA/HRS/SCAI/SCCM/SCCT/SCMR 2011 appropriate use criteria for echocardiography. J Am Soc Echocardiogr. 2011;24:229–67.
Ten Wolde M, Söhne M, Quak E, Mac Gillavry MR, Büller HR. Prognostic value of echocardiographically assessed right ventricular dysfunction in patients with pulmonary embolism. Arch Intern Med. 2004;164:1685–9.
McConnell MV, Solomon SD, Rayan ME, Come PC, Goldhaber SZ, Lee RT. Regional right ventricular dysfunction detected by echocardiography in acute pulmonary embolism. Am J Cardiol. 1996;78:469–73.
Casazza F, Bongarzoni A, Capozi A, Agostoni O. Regional right ventricular dysfunction in acute pulmonary embolism and right ventricular infarction. Eur J Echocardiogr. 2005;6:11–4.
Lopez-Candales A, Eleswarapu A, Shaver J, Edelman K, Gulyasy B, Candales MD. Right ventricular outflow tract spectral signal: a useful marker of right ventricular systolic performance and pulmonary hypertension severity. Eur J Echocardiogr. 2010;11:509–15.
Lopez-Candales A, Edelman K, Gulyasy B, Candales MD. Differences in the duration of total ejection between right and left ventricles in chronic pulmonary hypertension. Echocardiography. 2011;28:509–15.
López-Candales A, Edelman K. Shape of the right ventricular outflow Doppler envelope and severity of pulmonary hypertension. Eur Heart J Cardiovasc Imaging. 2012;13:309–16.
Kurzyna M, Torbicki A, Pruszczyk P, Burakowska B, Fijałkowska A, Kober J, Oniszh K, Kuca P, Tomkowski W, Burakowski J, Wawrzyńska L. Disturbed right ventricular ejection pattern as a new Doppler echocardiographic sign of acute pulmonary embolism. Am J Cardiol. 2002;90(5):507–11.
Gorham LW. A study of pulmonary embolism: two the mechanism of death based on a clinical pathological investigation of 100 cases of massive and 285 cases of minor embolism of the pulmonary artery. Arch Intern Med. 1961;108:76–90.
Del Guercio LRM, Cohn JDFNR. Pulmonary embolism shock: physiologic basis of a bedside screening test. JAMA. 1960;196:751–6.
Urokinase Pulmonary Embolism Trial. Phase 1 results: a cooperative study. JAMA. 1970;214:2163–72.
Alpert JS, Smith R, Carlson J, et al. Mortality in patients treated for pulmonary embolism. JAMA. 1976;236:1477–80.
Calder KK, Herbert M, Henderson SO. The mortality of untreated pulmonary embolism in emergency department patients. Ann Emerg Med. 2005;45:302–10.
Golpe R, Testa-Fernández A, Pérez-de-Llano LA, Castro-Añón O, González-Juanatey C, Pérez-Fernández R, Fariñas MC. Long-term clinical outcome of patients with persistent right ventricle dysfunction or pulmonary hypertension after acute pulmonary embolism. Eur J Echocardiogr. 2011;12:756–61.
Lo A, Stewart P, Younger JF, Atherton J, Prasad SB. Usefulness of right ventricular myocardial strain in assessment of response to thrombolytic therapy in acute pulmonary embolism. Eur J Echocardiogr. 2010;11:892–5.
McIntyre KM, Sasahara AA. Correlation of pulmonary photoscan and angiogram as measures of the severity of pulmonary embolic involvement. J Nucl Med. 1971;12:732–8.
McDonald IG, Hirsh J, Hale GS, et al. Major pulmonary embolism, a correlation of clinical findings, haemodynamics, pulmonary angiography, and pathological physiology. Br Heart J. 1972;34:356–64.
Dalen JE, Haynes FW, Hoppin FG, et al. Cardiovascular responses to experimental pulmonary embolism. Am J Cardiol. 1967;20:3–9.
McIntyre KM, Sasahara AA. Hemodynamic and ventricular responses to pulmonary embolism. Prog Cardiovasc Dis. 1974;17:175–90.
Parker BM, Smith JR. Pulmonary embolism and infarction: a review of the physiologic consequences of pulmonary artery obstruction. Am J Med. 1958;24:402–27.
Stein M, Levy SE. Reflex and humoral responses to pulmonary embolism. Prog Cardiovasc Dis. 1974;17:167–74.
Malik AB. Pulmonary microembolism. Physiol Rev. 1983;63:1114–207.
Alpert JS, Godtfredsen J, Ockene IS, et al. Pulmonary hypertension secondary to minor pulmonary embolism. Chest. 1978;73:795–7.
Calvin JE Jr, Baer RW, Glantz SA. Pulmonary artery constriction produces a greater right ventricular dynamic afterload than lung microvascular injury in the open chest dog. Circ Res. 1985;56:40–56.
Stein PD, Sabbah HN, Anbe DT, et al. Performance of the failing and nonfailing right ventricle of patients with pulmonary hypertension. Am J Cardiol. 1979;44:1050–5.
Calvin JE, Quinn B. Right ventricular pressure overload during acute lung injury: cardiac mechanisms and the pathophysiology of right ventricular systolic dysfunction. J Crit Care. 1989;4:251–65.
Calvin JE Jr. Acute right heart failure: pathophysiology, recognition, and pharmacological management. J Cardiothorac Vasc Anesth. 1991;5:507–13.
Taylor RR, Covell JW, Sonnenblick EH, et al. Dependence of ventricular distensibility on filling of the opposite ventricle. Am J Physiol. 1967;213:711–8.
Stein PD, Fowler SE, Goodman LR, Gottschalk A, Hales CA, Hull RD, Leeper KV Jr, Popovich J Jr, Quinn DA, Sos TA, Sostman HD, Tapson VF, Wakefield TW, Weg JG, Woodard PK, PIOPED II Investigators. Multidetector computed tomography for acute pulmonary embolism. N Engl J Med. 2006;354:2317–27.
Martins SR. Pulmonary CT angiography in pulmonary embolism: beyond diagnosis. Rev Port Cardiol. 2012;31:697–9.
Apfaltrer P, Bachmann V, Meyer M, Henzler T, Barraza JM, Gruettner J, Walter T, Schoepf UJ, Schoenberg SO, Fink C. Prognostic value of perfusion defect volume at dual energy CTA in patients with pulmonary embolism: correlation with CTA obstruction scores, CT parameters of right ventricular dysfunction and adverse clinical outcome. Eur J Radiol. 2012;81:3592–7.
Kang DK, Sun JS, Park KJ, Lim HS. Usefulness of combined assessment with computed tomographic signs of right ventricular dysfunction and cardiac troponin T for risk stratification of acute pulmonary embolism. Am J Cardiol. 2011;108:133–40.
Becattini C, Vedovati MC, Agnelli G. Prognostic value of troponins in acute pulmonary embolism: a meta-analysis. Circulation. 2007;116:427–33.
Jiménez D, Uresandi F, Otero R, Lobo JL, Monreal M, Martí D, Zamora J, Muriel A, Aujesky D, Yusen RD. Troponin-based risk stratification of patients with acute nonmassive pulmonary embolism: systematic review and metaanalysis. Chest. 2009;136:974–82.
Hunt JM, Bull TM. Clinical review of pulmonary embolism: diagnosis, prognosis, and treatment. Med Clin North Am. 2011;95:1203–22.
Stergiopoulos K, Bahrainy S, Strachan P, Kort S. Right ventricular strain rate predicts clinical outcomes in patients with acute pulmonary embolism. Acute Card Care. 2011;13:181–8.
Jiménez D, Aujesky D, Moores L, Gómez V, Martí D, Briongos S, Monreal M, Barrios V, Konstantinides S, Yusen RD. Combinations of prognostic tools for identification of high-risk normotensive patients with acute symptomatic pulmonary embolism. Thorax. 2011;66:75–81.
Bellofiore A, Roldán-Alzate A, Besse M, Kellihan HB, Consigny DW, Francois CJ, Chesler NC. Impact of acute pulmonary embolization on arterial stiffening and right ventricular function in dogs. Ann Biomed Eng. 2013;41:195–204.
Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation. 2009;120:992–1007.
Kussmaul WG, Noordergraaf A, Laskey WK. Right ventricular-pulmonary arterial interactions. Ann Biomed Eng. 1992;20:63–80.
Parmley WW, Tyberg JV, Glantz SA. Cardiac dynamics. Annu Rev Physiol. 1977;39:277–99.
Piene H. Pulmonary arterial impedance and right ventricular function. Physiol Rev. 1986;66:606–52.
Milnor WR, Bergel DH, Bargainer JD. Hydraulic power associated with pulmonary blood flow and its relation to heart rate. Circ Res. 1966;19:467–80.
Piene H, Sund T. Flow and power output of right ventricle facing load with variable input impedance. Am J Physiol. 1979;237:H125–30.
O’Rourke MF. Vascular impedance in studies of arterial and cardiac function. Physiol Rev. 1982;62:570–623.
Giannitsis E, Muller-Bardorff M, Kurowski V, et al. Independent prognostic value of cardiac troponin T in patients with confirmed pulmonary embolism. Circulation. 2000;102:211–7.
Konstantinides S, Geibel A, Olschewski M, et al. Importance of cardiac troponins I and T in risk stratification of patients with acute pulmonary embolism. Circulation. 2002;106:1263–8.
Kucher N, Goldhaber SZ. Cardiac biomarkers for risk stratification of patients with acute pulmonary embolism. Circulation. 2003;108:2191–4.
Kucher N, Goldhaber SZ. Risk stratification of acute pulmonary embolism. Semin Thromb Hemost. 2006;32:838–47.
Meyer T, Binder L, Hruska N, Luthe H, Buchwald AB. Cardiac troponin I elevation in acute pulmonary embolism is associated with right ventricular dysfunction. J Am Coll Cardiol. 2000;36:1632–6.
López-Candales A, Edelman K, Candales MD. Right ventricular apical contractility in acute pulmonary embolism: the McConnell sign revisited. Echocardiography. 2010;27:614–20.
Platz E, Hassanein AH, Shah A, Goldhaber SZ, Solomon SD. Regional right ventricular strain pattern in patients with acute pulmonary embolism. Echocardiography. 2012;29:464–70.
Descotes-Genon V, Chopard R, Morel M, Meneveau N, Schiele F, Bernard Y. Comparison of right ventricular systolic function in patients with low risk and intermediate-to-high risk pulmonary embolism: a two-dimensional strain imaging study. Echocardiography. 2013;30:301–8.
López-Candales A, Edelman K. Right ventricular outflow tract systolic excursion: a distinguishing echocardiographic finding in acute pulmonary embolism. Echocardiography. 2013;30(6):649–57.
Lopez-Candales A. Marked reduction in the ratio of main right ventricular chamber to outflow tract function in patients with proximal bilateral acute pulmonary embolism. Int J Cardiol. 2013;168(1):592–3.
Gorcsan J 3rd, Tanaka H. Echocardiographic assessment of myocardial strain. J Am Coll Cardiol. 2011;58:1401–13.
Huang SJ, Orde S. From speckle tracking echocardiography to torsion: research tool today, clinical practice tomorrow. Curr Opin Crit Care. 2013;19:250–7.
Amundsen BH, Helle-Valle T, Edvardsen T, Torp H, Crosby J, Lyseggen E, Støylen A, Ihlen H, Lima JA, Smiseth OA, Slørdahl SA. Noninvasive myocardial strain measurement by speckle tracking echocardiography: validation against sonomicrometry and tagged magnetic resonance imaging. J Am Coll Cardiol. 2006;47:789–93.
Helle-Valle T, Crosby J, Edvardsen T, Lyseggen E, Amundsen BH, Smith HJ, Rosen BD, Lima JA, Torp H, Ihlen H, Smiseth OA. New noninvasive method for assessment of left ventricular rotation: speckle tracking echocardiography. Circulation. 2005;112:3149–56.
López-Candales A, Rajagopalan N, Gulyasy B, Edelman K, Bazaz R. Differential strain and velocity generation along the right ventricular free wall in pulmonary hypertension. Can J Cardiol. 2009;25:73–7.
López-Candales A, Dohi K, Bazaz R, Edelman K. Relation of right ventricular free wall mechanical delay to right ventricular dysfunction as determined by tissue Doppler imaging. Am J Cardiol. 2005;96:602–6.
López-Candales A, Dohi K, Rajagopalan N, Suffoletto M, Murali S, Gorcsan J 3rd, Edelman K. Right ventricular dyssynchrony in patients with pulmonary hypertension is associated with disease severity and functional class. Cardiovasc Ultrasound. 2005;3(1):23.
Rajagopalan N, Dohi K, Simon MA, Suffoletto M, Edelman K, Murali S, López-Candales A. Right ventricular dyssynchrony in heart failure: a tissue Doppler imaging study. J Card Fail. 2006;12:263–7.
Dohi K, Onishi K, Gorcsan J 3rd, López-Candales A, Takamura T, Ota S, Yamada N, Ito M. Role of radial strain and displacement imaging to quantify wall motion dyssynchrony in patients with left ventricular mechanical dyssynchrony and chronic right ventricular pressure overload. Am J Cardiol. 2008;101:1206–12.
Sugiura E, Dohi K, Onishi K, Takamura T, Tsuji A, Ota S, Yamada N, Nakamura M, Nobori T, Ito M. Reversible right ventricular regional non-uniformity quantified by speckle-tracking strain imaging in patients with acute pulmonary thromboembolism. J Am Soc Echocardiogr. 2009;22:1353–9.
Taccardi B, Lux RL, Ershler PR, et al. Anatomical architecture and electrical activity of the heart. Acta Cardiol. 1997;52:91–105.
Torrent-Guasp F, Buckberg GD, Clemente C, et al. The structure and function of the helical heart and its buttress wrapping: I. The normal macroscopic structure of the heart. Semin Thorac Cardiovasc Surg. 2001;134:301–19.
Sullivan DM, Watts JA, Kline JA. Biventricular cardiac dysfunction after acute massive pulmonary embolism in the rat. J Appl Physiol. 2001;90:1648–56.
Chua JH, Zhou W, Ho JK, Patel NA, Mackensen GB, Mahajan A. Acute right ventricular pressure overload compromises left ventricular function by altering septal strain and rotation. J Appl Physiol. 2013;115:186–93.
Ichikawa K, Dohi K, Sugiura E, Sugimoto T, Takamura T, Ogihara Y, Nakajima H, Onishi K, Yamada N, Nakamura M, Nobori T, Ito M. Ventricular function and dyssynchrony quantified by speckle-tracking echocardiography in patients with acute and chronic right ventricular pressure overload. J Am Soc Echocardiogr. 2013;26:483–92.
López-Candales A, Bazaz R, Edelman K, Gulyasy B. Altered early left ventricular diastolic wall velocities in pulmonary hypertension: a tissue Doppler study. Echocardiography. 2009;26:1159–66.
Egermayer P, Town GI. The clinical significance of pulmonary embolism: uncertainties and implications for treatment: a debate. J Intern Med. 1997;241:5–10.
Freiman DG, Suyemoto J, Wessler S. Frequency of pulmonary thromboembolism in man. N Engl J Med. 1965;272:1278–80.
Havig O. Deep vein thrombosis and pulmonary embolism: an autopsy study with multiple regression analysis of possible risk factors. Acta Chir Scand. 1977;478(Suppl):1–108.
Wagenvoort CA. Pathology of pulmonary thromboembolism. Chest. 1995;107(Suppl):10S–7S.
Konstantinides SV, Meyer G, Becattini C, et al. 2019 ESC guidelines for the diagnosis and management of acute pulmonary embolism developed in collaboration with the European Respiratory Society (ERS): the task force for the diagnosis and management of acute pulmonary embolism of the European Society of Cardiology (ESC). Eur Respir J. 2019;54:1901647.
Klok FA, Mos IC, Nijkeuter M, et al. Simplification of the revised Geneva score for assessing clinical probability of pulmonary embolism. Arch Intern Med. 2008;168:2131–6.
Kline JA, Mitchell AM, Kabrhel C, et al. Clinical criteria to prevent unnecessary diagnostic testing in emergency department patients with suspected pulmonary embolism. J Thromb Haemost. 2004;2:1247–55.
Geersing GJ, Janssen KJ, Oudega R, et al. Excluding venous thromboembolism using point of care D-dimer tests in outpatients: a diagnostic meta-analysis. BMJ. 2009;339:b2990.
Elias A, Mallett S, Daoud-Elias M, et al. Prognostic models in acute pulmonary embolism: a systematic review and meta-analysis. BMJ Open. 2016;6:e010324.
Kohn CG, Mearns ES, Parker MW, et al. Prognostic accuracy of clinical prediction rules for early post-pulmonary embolism all-cause mortality: a bivariate meta-analysis. Chest. 2015;147:1043–62.
Guerin L, Couturaud F, Parent F, et al. Prevalence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism. Prevalence of CTEPH after pulmonary embolism. Thromb Haemost. 2014;112:598–605.
Galie N, Humbert M, Vachiery JL, Gibbs S, et al., ESC Scientific Document Group. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67–119.
Vonk Noordegraaf A, Marcus JT, Roseboom B, Postmus PE, Faes TJ, de Vries PM. The effect of right ventricular hypertrophy on left ventricular ejection fraction in pulmonary emphysema. Chest. 1997;112:640–5.
Pieralli F, Olivotto I, Vanni S, Conti A, Camaiti A, Targioni G, Grifoni S, Berni G. Usefulness of bedside testing for brain natriuretic peptide to identify right ventricular dysfunction and outcome in normotensive patients with acute pulmonary embolism. Am J Cardiol. 2006;97:1386–90.
Kostrubiec M, Pruszczyk P, Bochowicz A, Pacho R, Szulc M, Kaczynska A, Styczynski G, Kuch-Wocial A, Abramczyk P, Bartoszewicz Z, Berent H, Kuczynska K. Biomarker-based risk assessment model in acute pulmonary embolism. Eur Heart J. 2005;26:2166–72.
Ghuysen A, Ghaye B, Willems V, Lambermont B, Gerard P, Dondelinger RF, D’Orio V. Computed tomographic pulmonary angiography and prognostic significance in patients with acute pulmonary embolism. Thorax. 2005;60:956–61.
Tulevski II, ten Wolde M, van Veldhuisen DJ, Mulder JW, van der Wall EE, Büller HR, Mulder BJ. Combined utility of brain natriuretic peptide and cardiac troponin T may improve rapid triage and risk stratification in normotensive patients with pulmonary embolism. Int J Cardiol. 2007;116:161–6.
Tulevski II, Hirsch A, Sanson BJ, Romkes H, van der Wall EE, van Veldhuisen DJ, Büller HR, Mulder BJ. Increased brain natriuretic peptide as a marker for right ventricular dysfunction in acute pulmonary embolism. Thromb Haemost. 2001;86:1193–6.
Vieillard-Baron A, Page B, Augarde R, Prin S, Qanadli S, Beauchet A, Dubourg O, Jardin F. Acute cor pulmonale in massive pulmonary embolism: incidence, echocardiographic pattern, clinical implications and recovery rate. Intensive Care Med. 2001;27:1481–6.
Kucher N, Printzen G, Doernhoefer T, Windecker S, Meier B, Hess OM. Low pro-brain natriuretic peptide levels predict benign clinical outcome in acute pulmonary embolism. Circulation. 2003;107:1576–8.
ten Wolde M, Tulevski II, Mulder JW, Sohne M, Boomsma F, Mulder BJ, Buller HR. Brain natriuretic peptide as a predictor of adverse outcome in patients with pulmonary embolism. Circulation. 2003;107:2082–4.
Pruszczyk P, Kostrubiec M, Bochowicz A, Styczynski G, Szulc M, Kurzyna M, Fijalkowska A, Kuch-Wocial A, Chlewicka I, Torbicki A. N-terminal pro-brain natriuretic peptide in patients with acute pulmonary embolism. Eur Respir J. 2003;22:649–53.
Moser KM, Auger WR, Fedullo PF. Chronic major-vessel thromboembolic pulmonary hypertension. Circulation. 1990;81:1735–43.
Matthews DT, Hemnes AR. Current concepts in the pathogenesis of chronic thromboembolic pulmonary hypertension. Pulm Circ. 2016;6:145–54.
Moser KM, Bloor CM. Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension. Chest. 1993;103:685–92.
Galiè N, Kim NH. Pulmonary microvascular disease in chronic thromboembolic pulmonary hypertension. Proc Am Thorac Soc. 2006;3:571–6.
Pepke-Zaba J, Delcroix M, Lang I, et al. Chronic Thromboembolic Pulmonary Hypertension (CTEPH): results from an international prospective registry. Circulation. 2011;124:1973–81.
Ende-Verhaar YM, Cannegieter SC, Vonk Noordegraaf A, et al. Incidence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism: a contemporary view of the published literature. Eur Respir J. 2017;49:1601972.
Sanchez O, Helley D, Couchon S, et al. Perfusion defects after pulmonary embolism: risk factors and clinical significance. J Thromb Haemost. 2010;8:1248–55.
Pesavento R, Filippi L, Palla A, et al. Impact of residual pulmonary obstruction on the long-term outcome of patients with pulmonary embolism. Eur Respir J. 2017;49:1601980.
Heit JA, Spencer FA, White RH. The epidemiology of venous thromboembolism. J Thromb Thrombolysis. 2016;41:3–14.
Pengo V, Lensing AWA, Prins MH, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med. 2004;350:2257–64.
Becattini C, Agnelli G, Pesavento R, et al. Incidence of chronic thromboembolic pulmonary hypertension after a first episode of pulmonary embolism. Chest. 2006;130:172–5.
Klok FA, van Kralingen KW, van Dijk AP, et al. Prospective cardiopulmonary screening program to detect chronic thromboembolic pulmonary hypertension in patients after acute pulmonary embolism. Haematologica. 2010;95:970–5.
Dentali F, Donadini M, Gianni M, et al. Incidence of chronic pulmonary hypertension in patients with previous pulmonary embolism. Thromb Res. 2009;124:256–8.
Klok FA, Dzikowska-Diduch O, Kostrubiec M, et al. Derivation of a clinical prediction score for chronic thromboembolic pulmonary hypertension after acute pulmonary embolism. J Thromb Haemost. 2016;14:121–8.
Ribeiro A, Lindmarker P, Johnsson H, et al. Pulmonary embolism: one-year follow-up with echocardiography doppler and five-year survival analysis. Circulation. 1999;99:1325–30.
Bonderman D, Wilkens H, Wakounig S, et al. Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir J. 2009;33:325–31.
Klok FA, Barco S, Konstantinides SV, et al. Determinants of diagnostic delay in chronic thromboembolic pulmonary hypertension: results from the European CTEPH Registry. Eur Respir J. 2018;52:1801687.
Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2018;53:1801913.
Papamatheakis GD, Poch DS, Fernandes TM, et al. Chronic thromboembolic pulmonary hypertension. J Am Coll Cardiol. 2020;76:2155–69.
Ryan KL, Fedullo PF, Davis GB, et al. Perfusion scan findings understate the severity of angiographic and hemodynamic compromise in chronic thromboembolic pulmonary hypertension. Chest. 1988;93:1180–5.
He J, Fang W, Lv B, et al. Diagnosis of chronic thromboembolic pulmonary hypertension: comparison of ventilation/perfusion scanning and multidetector computed tomography pulmonary angiography with pulmonary angiography. Nucl Med Commun. 2012;33:459–63.
Sugiura T, Tanabe N, Matsuura Y, et al. Role of 320-slice CT imaging in the diagnostic workup of patients with chronic thromboembolic pulmonary hypertension. Chest. 2013;143:1070–7.
Rogberg AN, Gopalan D, Westerlund E, et al. Do radiologists detect chronic thromboembolic disease on computed tomography. Acta Radiol. 2019;60:1576–83.
Hoeper MM, Madani M, Nakanishi N, et al. Chronic thromboembolic pulmonary hypertension. Lancet Respir Med. 2014;2:573–82.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
López-Candales, A., Vallurupalli, S. (2021). Pulmonary Embolism. In: Gaine, S.P., Naeije, R., Peacock, A.J. (eds) The Right Heart. Springer, Cham. https://doi.org/10.1007/978-3-030-78255-9_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-78255-9_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-78254-2
Online ISBN: 978-3-030-78255-9
eBook Packages: MedicineMedicine (R0)