
Abstract
Liquid ophthalmic drug products are the most common presentation for pharmacotherapy used to treat a variety of anterior and posterior segment diseases of the eye. Their attributes largely mirror those of parenteral formulations, but specifically consider certain qualities for drug substance and product from a perspective of compatibility and delivery to a biologically and physiologically distinct environment in and around the eye. Features such as formulation pH and osmolarity, or properties of all inactive ingredients, play a critical role when considering the route of ocular administration. This chapter provides an overview of physical chemistry, formulation, and manufacturing considerations as they relate to the anatomical characteristics and physiology of the eye from a pragmatic, historical, case-study-driven, and biosystem-based perspective.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Preface
Liquid ophthalmic products are categorized as parenteral formulations; however, they are within a highly specialized subclass of their own. To provide a sensible and comprehensive analysis of physical chemistry, compounding pharmacy or formulation, and manufacturing science that’s entailed within the larger scope of all liquid ophthalmic drug products, the objectives of this chapter are twofold: first, to briefly visit key topics and critical attributes, which in turn (second) provide examples with references to benefit newcomers into the field for subsequent development of deeper expertise. Liquid ophthalmic drug products can be defined and classified by several differentiating attributes from other dosage forms that are administered into the body. From a global perspective, these characteristics stem out of three ocular biopharmaceutics blueprint attributes. Features include the qualities of drug substance or active pharmaceutical ingredient; the drug product or formulation from a perspective of aqueous solution pH, total concentration of osmolytes, and properties related to the actual vehicle composition taking into account all inactive ingredients (e.g., excipients); and finally, precise route of administration into the eye (e.g., topical eye drops vs. intraocular or periorbital injections) as it relates to the anatomical characteristics and physiology of this organ.
Considerations for Drug Substance
Relating to the active ingredient or drug substance, an inclusive examination of precedence in liquid ophthalmic products (see Table 1) suggests existence of two general categories. There are some liquid ophthalmic products that stem from pure leads. In other words, they contain an active ingredient that was discovered and developed solely for an ophthalmic indication. Moreover, most liquid ophthalmic products are carefully designed reformulations of existing active ingredients repurposed from other therapeutic indications. Molecular-drug profiling for ophthalmic repositioning, in this case, involves development of a preexisting compound into a new liquid ophthalmic drug product for use in an ocular disease. Such compound repurposing capitalizes on the fact that approved drugs and many compounds in the pipeline (note that clinical development candidates that are not yet approved could come from active or even abandoned programs in the pipeline) have achieved human testing and are accompanied with an understanding of pharmacology, defined systemic pharmacokinetics and safety data, and possibly a proof (or in vivo validation) of a mechanism of action. While there are close to 600 ophthalmic drug products captured in the current edition of the FDA’s Orange Book (https://www.hhs.gov/ 2019), about 80% of these are drug repositioning examples underpinned by the fact that common molecular pathways contribute to different disease phenotypes. Furthermore, approximately the same proportion of Orange Book listed ocular products (~80%) are variations on ophthalmic formulations of the same drug or active ingredient, with more than half of those (approximately 200 reference listed drugs) qualifying as liquid ophthalmic drug products (https://www.hhs.gov/ 2019).
Physical and Chemical Considerations
Conventional physicochemical characterization approaches also apply to all active pharmaceutical ingredients used in liquid ophthalmic products; however, other distinctive requirements exist. Physical and chemical properties include those of small organic molecules as well as large macromolecules derived from biotechnology (e.g., biophysical considerations). Understanding of crystal structure and disposition thereof, single crystal data (molecular orientation and long-range packing, or that of salts and hydrates/solvates from the same perspective as isolated from the final step in process chemistry), solid state polymorphisms and solid form as it impacts thermodynamic stability and solubility in aqueous liquids, drug substance morphology including particle size distributions, and other properties which are related to manufacturability of a downstream product—e.g., melting point or glass transition temperature, hygroscopic tendencies, absolute density of substance, and any latent process chemistry or recombinant/fermentation-related impurities (Hilfiker et al. 2006). Intuitively, the aforementioned properties relate to the quality of a downstream product, e.g., controls around stability and purity; however, in some cases they can also directly impact performance and hence potentially affect safety and efficacy. Furthermore, drug substance chemical and biophysical properties in a selected ophthalmic candidate must also be fully characterized as they can relate to and influence the nature of previously listed physical considerations. Chemistry and (bio)physics can also impact the biopharmaceutical aspects which typically address liquid formulations and absorption mechanisms for a given dose and route of ocular delivery: for example, the balance between equilibrium solubility values in an aqueous environment vs. in oil, e.g., the oil/water partition coefficient—Po/w (Schoenwald and Huang 1983b; Wang et al. 1991); the ionization constant if the molecule has one within the relevant ocular physiological pH range (discussed later)—pKa, pKb, or pI values for acids, bases, and zwitterions, resp. (Pawar et al. 2013; Schoenwald and Huang 1983b); and finally, the molecules’ absolute or thermodynamic aqueous solubility with a defined pH-dependent solubility profile, or an equilibrium solubility product rate constant (Ksp) if ionic drug substance is being considered (Breda et al. 2009; Diehl and Markuszewski 1985; Maren et al. 1990; Pawar et al. 2013; Scozzafava et al. 1999; Shirasaki 2008; Shoghi et al. 2013; Sieg and Robinson 1977; Zhang et al. 2013). For classes of liquid ophthalmic suspension products, e.g., PRED FORTE® (https://www.accessdata.fda.gov/ 1973) 10 mg/mL prednisolone acetate topical microfine suspension indicated for treatment of steroid-responsive inflammation in anterior ocular segment tissues or TRIESSENCE® (https://www.accessdata.fda.gov/ 2007) 40 mg/mL injectable triamcinolone acetonide suspension indicated for posterior ocular inflammatory conditions unresponsive to topical corticosteroids and visualization during vitrectomy, the final particle size distribution plays a key role in precorneal residence time (a combination of turnover due to tear fluid secretion and nasolacrimal drainage) and intensity plus durability of intravitreal exposure, respectively (Missel et al. 2010; Sieg and Robinson 1975). Particle size characterization studies in topical liquid ophthalmic suspensions support the belief that moderate dilution of a suspension of a poorly soluble drug (such as the steroidal anti-inflammatory examples given earlier) does not diminish aqueous humor drug levels or, conversely, that the use of a higher drug particle count within a suspension increases aqueous humor (typical ocular pharmacokinetic sampling compartment) drug concentration-time profiles (Sieg and Robinson 1975). An order-of-magnitude lower dose (vs. PRED FORTE® (https://www.accessdata.fda.gov/ 1973)), 0.1% fluorometholone suspension, compared to a saturated solution of the same drug did not produce sustaining pharmacokinetic effects, suggesting that the conjunctival cul-de-sac retains suspended particles within a topical liquid ophthalmic eye drop and contributes significantly to the overall extent of steroid penetrating across the cornea (Sieg and Robinson 1975). Furthermore, investigations of various particle sizes and concentrations (e.g., 77–428 μm and 40–160 mg/mL) and their effect on intraocular residence time suggested that performance of liquid intravitreal-injectable suspension depots is insensitive to these physical and pharmaceutical parameters (Missel et al. 2010).
Chemical Characteristics
For small molecules, information on the lipophilicity, ionization state, and aqueous solubility forms a trifecta of physicochemical properties relating to the oil/water partition coefficient (Po/w or more commonly reported as log P). A known relationship exists with permeability across various ocular epithelial tissue barriers (note here one must consider the actual physiological route of administration for rationale in the final selection of drug substance for a liquid ophthalmic product design) or in other words absorption into the eye and intraocular target tissues (Chien et al. 1990; Edward and Prausnitz 2001; Friedrich et al. 1997; Hamalainen et al. 1997; Kidron et al. 2010; Pitkanen et al. 2005; Prausnitz and Noonan 1998; Ramsay et al. 2018, 2017; Schoenwald and Huang 1983a, b; Tai et al. 2003; Wang et al. 1991; Yoshida and Topliss 1996; Ahmed et al. 1987; Shirasaki 2008; Gukasyan et al. 2019a, b). The hydrophobic or hydrophilic nature of the active pharmaceutical ingredient can also be carefully used in delivery vehicle design, choice, and respective amounts of inactive ingredients used, and (bio)chemical specifications such as final pH, buffer capacity, and ionic strength or osmolyte content (Breda et al. 2009; He et al. 2003; Leibowitz et al. 1978; Mitra 1993; Palkama et al. 1985; Pawar et al. 2013; Sieg and Robinson 1975, 1977, 1979; Zhang et al. 2013; Singh et al. 2009). Formulation design, at least partly related to choices of inactive ingredient selection, will be discussed in detail in the following sections; however, it is noteworthy to mention that physicochemical properties like log P and pKa (or log D which combines log P value with an acid or base dissociation constant at a particular pH) are important toward the selection of appropriate solubilizing excipients. Ionization constants (e.g., pKa or pKb) are similarly related to multiple biopharmaceutical dimensions as they influence molecules’ final dose and overall absorption efficiency into the eye (Gukasyan et al. 2019b; Shirasaki 2008). Chiefly, these include the required dose and its inherent (pH)-solubility ratio, and also dissolved active pharmaceutical ingredient fraction within a total dose that’s molecularly and thermodynamically eligible and available to present a chemical driving force (gradient) for flux across ocular tissue barriers (Mortimer and Eyring 1980). It is generally accepted that the neutral form of any drug substance is favored in terms of transcellular flux across biological membrane barriers; hence, within this context the physiological properties of tear fluid and intraocular compartments must be considered in conjunction with formulation attributes and how they would influence the degree of ionization of a molecule (if any) temporally from the time point of introduction into ocular space (Hogben et al. 1959; Kansy et al. 1998; Mortimer and Eyring 1980). This is an important theoretical concept with several practical examples in liquid ophthalmic dosage forms (e.g., those of brimonidine (https://www.accessdata.fda.gov/ 2001, 2006, 1996)) which will be discussed in the drug product pH considerations section.
Since the eye is exposed to direct light, as it relates to the circadian rhythm, diurnal and nocturnal changes in several physiological factors, esp. in topical ocular drug delivery, it is important to understand the chemical photosensitivity of liquid ophthalmic candidates. On a molecular level in solution, the absorbance of sunlight energy in the visible, UVA, and partially UVB radiation range is a common characteristic which can potentially lead to photoirritation and photoallergy. Hence, it is essential to characterize light absorbance profiles of liquid ophthalmic candidates and identify wavelengths within the relevant spectrum which achieve the maximum absorption (e.g., at λmax value the molar extinction coefficient > 1000 L × mol−1 × cm−1 (https://www.ich.org/products/guidelines/ 2019)), and if needed evaluate the prevalence and phototoxic activity of light-excitable drug substances. Several mechanisms for light-induced ocular drug toxicity have been proposed and are equally helpful in in vitro or in vivo ocular models designed toward simple, inexpensive testing of developmental stage compounds as a screen for their potential ocular phototoxicity (Fishman 1986; Roberts 2002). For example, fluoroquinolone class of antibiotics commonly used in topical ophthalmic formulations, and via intravitreal or intracameral injections, are known to cause various degrees of phototoxicity (with an established structure activity relationship for their potential to cause photoinstability and photocarcinogenic effect, as well as chemical mechanisms of action) when exposed to ultraviolet (UV) light (Pawar et al. 2013; Thompson 2007). The UV-fluoroquinolone phototoxicity is associated with the formation of reactive oxygen species (ROS ), where excitation by light energy produces both singlet oxygen and superoxide, followed by ocular-cellular damage (Thompson 2007). A related (e.g., via ROS mechanism) notable mechanism of action is the effect of such drugs (or even some inactive ingredients found in liquid ophthalmic compositions, to be discussed in the subsequent section) in liquid ophthalmic drug products on equilibrium concentrations of reduced glutathione (GSH) within physiological ocular fluids (e.g., tear fluid, aqueous humor, vitreous humor) or cells that comprise tissues which come into immediate contact with the product (Aguirre et al. 2012; Gurbay and Hincal 2004). For example, reduction of tear fluid or aqueous humor GSH concentration is known to trigger undesirable changes in corneal endothelial cell permeability (Green et al. 2001). Similarly, S-(1,2-dicarboxyethyl)glutathione (DCE-GS), which is biosynthesized in an enzyme-mediated reaction utilizing reduced glutathione and l-malate, is found at highest known concentrations in mammalian lens tissue and thought to play several key ocular physiological roles (Green et al. 2001; Tsuboi et al. 1990a, b). Within the context of liquid ophthalmic products, the extent of phototoxic damage would be a function of both the drug concentration (which is a known factor for the fluoroquinolone class) and total UV-light dose. Moreover, despite the availability of relatively more photostable fluoroquinolones such as 8-methoxy analogs of gatifloxacin and moxifloxacin vs. the photo-unstable ciprofloxacin, plus a paucity of data supporting human fluoroquinolone-induced photocarcinogenicity, in clinical use an advisory to avoid sunlight exposure for the duration of therapy with these agents is persistent (Thompson 2007; Gurbay and Hincal 2004).
Physical Characteristics
Drug substance solid form is an important consideration for liquid ophthalmic formulation development, and it warrants a brief discussion using a case study to exemplify challenges in drug repurposing for ophthalmic use as well as bridging and bioequivalence understanding form a pharmaco- and toxico-kinetic point of view. Studies with gatifloxacin (Table 1, fluoroquinolone broad-spectrum antibiotic) have provided the pharmaceutical industry with ample reasons and rationale to devote enough attention to identification and understanding of inter-relationships between all possible crystalline solid forms and how the polymorph landscape would impact the desired dosage form and development plans. Gatifloxacin was initially discovered as a hemihydrate crystallized from methanol (Masuzawa et al. 1991). Since this particular crystal form displayed poor characteristics for tableting, e.g., extremely hygroscopic with slow disintegration and dissolution for original therapeutic indication using enteral delivery route, this directed several subsequent polymorph screens and identification of 14 additional solid forms for gatifloxacin (Matsumoto et al. 1999; Raghaven et al. 2002). Briefly, all these studies added considerable challenges to the overall development pathway of the molecule to an oral product, called Tequin®, which was ironically withdrawn from major markets in 2006 for systemic safety reasons. As an appropriate segue to the next section, a highly soluble sesquihydrate (Raghaven et al. 2002) of gatifloxacin was ultimately chosen/repurposed and utilized for production of ophthalmic topical solutions called Zymar® followed by Zymaxid® (which differs at least based on label claim in active ingredient concentration, 0.3% (3 mg/mL) and 0.5% (5 mg/mL) gatifloxacin, resp., with benzalkonium chloride at 0.005%, EDTA, purified water, and sodium chloride in both), and as the compound went off-patent sometime in 2010, the generic maker Apotex Inc. started using the hemihydrate in their version of the topical drug product (Newman and Wenslow 2016). While several reports exist, the aqueous solubility relationship among known forms of gatifloxacin is understood to parallel its thermodynamic stability, with the pentahydrate having the lowest solubility at 25 °C (Raghaven et al. 2002). As a general best practice, an approach which evaluates (or identifies, if unknown) the risks and benefits associated with all solid forms of a given drug substance being considered for liquid ophthalmic product development should be adopted within the context of the proposed ocular dose and route of delivery. While it would be prudent to identify the form with lowest free energy and propose a process of isolating it from the last step in drug substance synthesis, for liquid ophthalmic products it is also important to address any risks of potentially forming less soluble hydrates or salts from common physiological or buffer ions. A full polymorphic landscape analysis will dictate also the complete interconversion mechanisms between known solid forms, ideally allowing for establishment of tight process controls and analytical methodology to produce crystalline material with high homogeneity (i.e., no detectable presence of other known polymorphs). If lower solubility forms exist than the one used in liquid ophthalmic product development, a potential supersaturated state is rendered and conversion during storage (or after introduction into intraocular compartments) toward lower-soluble forms can occur. While this is a temporally kinetic phenomenon, it is a risk which could impact the quality (e.g., formation of a precipitate) and performance (e.g., dissolution and absorption) of a liquid ophthalmic product. Unless there is a clear reason related to a medical benefit which suggests that a metastable or amorphous form for a drug substance is desired for product development, only the most stable solid form should be selected/developed. If the former exception is not applicable, and a less thermodynamically stable form is used for manufacturing ease (or other nonscience-related or regulatory strategic reasons), then it is incumbent upon the pharmaceutical developer to minimize and mitigate risk to patients from a performance and quality point of view (Singhal and Curatolo 2004).
Drug Product Considerations
The next layer of classification in liquid ophthalmic products relates to the design of delivery vehicle itself. While several strata of complexity exist in liquid ophthalmic formulation design from a physiologically based route of administration perspective, here the focus will be agnostic of site of ocular drug deposition. Progressive understanding of barriers presented by ocular anatomical features on drug delivery impart parallel protective mechanisms that help this organ to perform its primary function of ensuring proper vision. These protective mechanisms include clearance of exogenous chemicals (such as drug molecules) into the systemic circulation via fluid drainage and lacrimation. Liquid ophthalmic formulation design must consider these physiological attributes and find a logical balance between those and physicochemical ones that govern boundaries in product design. While a finite collection of different configurations exists, a deep understanding of all overlapping physiological and physicochemical characteristics is required to nominate possible formulation contenders for initial in vivo prototyping and testing.
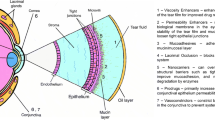
All liquid ophthalmic dosage forms face a primary challenge that’s related to the limited amount of space available for drug delivery to the eye. A typical eye drop volume is thought to be approximately 30 μL, although reports indicate a range between 25 and 56 μL with a key importance on dropper tip inner/outer diameter (as opposed to liquid formulation properties like viscosity or surface tension) (Brown and Lynch 1986; Lederer and Harold 1986). There is a restricted limit in the size of a dose that can be applied to, injected, and tolerated by ocular sites of drug deposition, and in the duration over which an applied dose stays in contact with absorptive surfaces of the eye (whether they are topical or intraocular). From this perspective, it is important to guarantee through proper liquid formulation design that the complete dose is either solubilized in a liquid product or fully available for accurate delivery in the case of solid, semi-solid, or colloidal suspended particulates within a liquid delivery vehicle. The formulation vehicle composition, e.g., pH, ionic content, and strength, as well as the presence of any inactive ingredients, plays a critical role since the allowed practical volumes for ocular delivery of liquid dosage forms lie within 30–100 μL range (depending on the route of administration) (Ghate and Edelhauser 2006; Lee and Robinson 1986). The three main ocular physiological fluids with which liquid ophthalmic formulations come into contact and mix with are tear fluid, aqueous humor, and vitreous humor, while estimations of the ionic content, nature of electrolytes, and pH of these fluids have been of interest from a basic science perspective for nearly a century according to early published records (Meyer and Palmer 1936). In contrary to initial hypothesis that these biological fluids had origins of dialysates (e.g., from blood circulation), their ionic content, presence of hyaluronic acid, and pH which is generally 0.1–0.3 units lower than that of blood suggested more complex biological regulation mechanisms in these ocular compartments and highlighted the importance of understanding their characteristics for drug delivery purposes (Meyer and Palmer 1936).
pH, Buffers, and Buffering Capacity
Furthermore, the pH range of aqueous preparations for ocular administration requires tight control and optimized buffering capacity (β). The latter, e.g., β, has been investigated in several eye-related fluids and displays considerable intersubject variability in ocular biosystems, depending on the methods used, e.g., acid or base titration. For example, local zones of enhanced buffering by human tear fluid across the entire pH spectrum were identified, reflecting multiple endogenous buffering components, primarily bicarbonate and a heterogeneous tear film protein population, among others (Carney et al. 1989). Baseline tear fluid pH values from several reports indicate a range from 7 to 7.5, which is highly dependent on several factors: diurnal fluctuations, e.g., tears are more acidic as sampled from eyes during waking hours of the day (average pH 7.25) than later in the day (pH 7.45) (Carney and Hill 1976); the dynamics attributed to these fluctuations could be related to metabolic byproducts associated with anaerobic conditions during sleep as well as differences in carbon dioxide activity in the eyelids-open vs. eyelids-closed configurations (1 h eyelid patching resulted in a significant acidic shift from pH 7.20 to 7.06 (Coles and Jaros 1984)), and also gender and age, especially in females where tear film pH increases significantly, e.g., 7.06 vs. 7.28, for <40 years of age vs. >40 years of age, respectively (Coles and Jaros 1984). Vitreous humor pH has been estimated in several instances and species, as it is thought to play a role during intraocular hypoxia, acidosis, and optic nerve cell health. Baseline vitreous pH in normotensive eyes is reported to be approximately 7.3, while it can decrease by as much as 0.4 pH units in cases of acute intraocular pressure (IOP) elevation (however, it is reversible if IOP is returned to normal levels within 2 h) (Lu et al. 2001). While the mechanisms of vitreous humor pH regulation are not well known, the influence of liquid intravitreal-injectable ophthalmic formulations for retinal disease treatment on posterior tissue circulation and vitreous pH is of great importance. Within an exploratory context, liquid intravitreal injections of pH 3–8 range have been evaluated and characterized as acceptable or tolerable from a post-hoc histopathological examination perspective (Aguirre et al. 2012). These studies employed specific buffers (at pH 3–4 range with a relatively low β) and counterions to prepare intravitreal liquid vehicles targeted for delivery of new chemical entities (e.g., small-molecule inhibitors of angiogenesis being repurposed from an oral route of delivery in oncology indications for the treatment of wet neovascular age-related macular degeneration (AMD)) (Aguirre et al. 2012; Marra et al. 2011). Specific counterions entertained within this wide pH range included sulfate, maleate, malate, fumarate, citrate, and phosphate; their molar concentrations were maintained in the 10–30 mM range with the intention to allow for rapid pH adjustment in the vitreous chamber microenvironment as the exact buffering mechanism and capacity of the compartment was not well defined (Aguirre et al. 2012, 2018; Marra et al. 2011). The selection of counterions from ionic chemical drug substances, which could subsequently behave as buffers in liquid ophthalmic formulations, or additional buffering agents for setting and controlling final drug product pH, is another important consideration from an ocular safety point of view. While traditional selection and use criteria for pharmaceutical salts can be considered as a starting point (Stahl et al. 2011), there are several physiologically unique principles which may be limitations in an ophthalmological setting. For example, in research formulation development work for a potent, selective vascular endothelial growth factor receptor tyrosine kinase inhibitor, PF-00337210, under consideration for the treatment of age-related macular degeneration, twofold changes were made to maximize safety and ocular delivery properties. Switching from an oral immediate release tablet in an oncology indication, PF-00337210 bismaleate (a rapidly dissolving salt form of the original drug substance) was recrystallized as a stable free-base polymorph to avoid use of maleate counterion intravitreally, thought to elicit retinal tissue toxicity partially through GSH depletion (Aguirre et al. 2012). Furthermore, to optimize the unique physicochemical properties of the drug which would allow for a sterile liquid parenteral injectable product to be developed for early testing (i.e., deliver up to 6 mg of PF-00337210 in a 0.1 mL intravitreal injection), the aqueous solubility was increased to >800 mg/mL using crystalline free base in a safer citrate buffer system at pH 3 with low β (10 mM citrate, β 0.001–0.003) to allow for rapid in situ neutralization of pharmaceutical pH (Marra et al. 2011). Buffering zone offered instantaneous intravitreal neutralization (i.e., from pH 3 to 7) of PF-00337210 doses by the endogenous ampholytes present in vitreous humor allowing for a spontaneous in situ formation of a drug substance precipitate which acted as a dose depot to reduce the frequency of intravitreal injections, expected by virtue of known rapid elimination of small molecules from this intraocular compartment (Aguirre et al. 2012; Raghava et al. 2004).
Liquid ophthalmic formulation preparations whose pH or tonicity is non-physiological are known to stimulate tear turnover, changes in aqueous humor dynamics, and transient ion solute exchange, thereby accelerating drug loss or potential compromise of ocular tissue integrity (Ghate and Edelhauser 2006; Mitra 1993; Shen et al. 2018). Early investigations, however largely based on subjective comfort indices, of appropriate formulation pH for ophthalmic use already suggested that deviating away from eyes’ physiological pH caused non-productive drug losses as opposed to desirable absorption, accompanied by damage to ocular tissues in extreme cases. Furthermore, various buffering agent effects were studied as a function of lacrimation presumably based on human tolerance (Hind and Goyan 1947; Martin and Mims 1950). Plausibly the earliest quantitative approach which utilized dacryoscintigraphy as a method of detecting lacrimation, in direct proportionality to tear drainage rate constant, showed that alkaline and acid pH in liquid formulations decreased ocular bioavailability—for both nonionizable and ionizable drugs (Conrad et al. 1978). Furthermore, changing aspects (diurnal and nocturnal fluctuations) of tear film and ocular surface pH have been explored, and the mechanisms of tear fluid pH regulation have been carefully studied. pH challenges can affect formulation vehicle toleration, drug effectiveness, and clinical signs in disease-related endpoints. Specifically, the buffering capacity of tears shows considerable differences from those seen in the blood, large intrasubject variability, especially toward acidic-range titration. Local ocular zones of enhanced micro-buffering across the pH spectrum have been identified, presumably suggesting the existence of multiple buffering components (bicarbonate, protein, and others) present in ocular fluids (Carney et al. 1989; Coles and Jaros 1984). Perfusion of intraocular aqueous humor containing compartments with solutions of varying pH range revealed that outside of the pH range of 6.5–8.5, morphological and cell-physiology-related alterations occur, including direct cellular damage, as well as disruption of tight-junctional complexes, leading to loss in barrier function integrity within ocular and blood-systemic compartments. Furthermore, analysis of the extent of this breakdown has been shown to be dependent upon the magnitude and the exposure time to altered pH (Gonnering et al. 1979).
Estimations of pH have been performed in tears and aqueous and vitreous humor, reported at 7.25–7.45, 7.5, and 7.32, respectively, and the endogenous buffering capacity of each compartment is estimated to be significantly lower than that of blood in terms of the presence of species which act as buffers and recovery turnover time to baseline pH value following an exogenous stressor (Carney and Hill 1976; Carney et al. 1989; Lu et al. 2001; Paterson et al. 1975). Classical pH-partition hypothesis partially explains the influence of physiological pH (specifically the hydrogen ion concentration normally found in tear fluid or other ocular fluids where liquid dosage forms are deposited) for drugs with an acid dissociation constant (e.g., pKa) on the extent of drug transfer, partitioning, or absorption across the phospholipid bilayer of cells. The concept reasons that when a drug is ionized, it will not be able to get through a lipid membrane, while keeping in mind that the ionized form of a drug is also in a pKa-governed simultaneous equilibrium with its neutral form (Shore et al. 1957). For liquid ophthalmic drug products, the final pH of the formulation has exclusive control over the ratio of drugs’ non-ionized vs. ionized states and therefore has a transient influence on proportion of species with higher lipid solubility. Pioneering reports indicated that the extent of ocular absorption of ionizable drugs must consider pH-dependent lacrimation in addition to the classical pH-partition explanation. Within this context, detailed pharmacokinetic ocular absorption studies of early glaucoma drug, pilocarpine, were able to fully corroborate quantitative estimations illustrating a plateau within the pH-dependent absorption into aqueous humor plot, only by taking into account both lacrimation and pH-partition hypothesis as two opposing effects above physiological pH and pKa of the drug (Conrad et al. 1978). The enhanced delivery of brimonidine is apparent from a comparison of ALPHAGAN® (brimonidine tartrate ophthalmic solution) 0.2% at a pH of 5.6–6.6 (https://www.accessdata.fda.gov/ 1996) vs. ALPHAGAN® P (brimonidine tartrate ophthalmic solution) 0.1% at a pH of 7.4–8.0 (https://www.accessdata.fda.gov/ 2001, 2006), where the 50% lower concentration of brimonidine equivalents in ALPHAGAN® P at a more alkaline pH provides bioequivalence (comparable to aqueous humor, iris ciliary body exposures, and intraocular pressure lowering). By buffering the pH in ALPHAGAN® P to slightly basic and near 7.4–7.8, e.g., at approximately the pKa of brimonidine (Bhagav et al. 2010), the ocular penetration is further enhanced partially due to the tendency of the drug to efficiently diffuse through lipid membranes under such circumstances where dissolved brimonidine species are predominantly unionized in neutral to alkaline formulation environments (Olejnik July 14, 2000). Increasing the pH of vehicles can promote increased corneal penetration for pilocarpine as well in accordance with the pH-partition hypothesis (Shore et al. 1957), while analogous series of experiments with nonionizable drugs and glycerin have been reported to give similar results (Sieg and Robinson 1977). Here, there is additional consideration around an extent of pH-induced lacrimation by the liquid topical ophthalmic vehicle, and the effect on precorneal drug concentration was determined to partially increase pilocarpine absorption at neutral to slightly alkaline pH. Comparisons against neutral, nonionizable controls suggested a primary relationship to pilocarpine’s unique solubility characteristics coupled with less irritation and lacrimation, rather than a direct pH effect on the molecule (Sieg and Robinson 1977). Analogously, previous studies provided support for further development of l-carnosine as a functionally synergistic buffer for topical ophthalmic use, with pharmaceutical compatibility in the context of dosage forms displaying in situ gel-formation properties following eye drop mixing with resident tear fluid. l-Carnosine was shown to have higher buffering capacity (its buffer capacity, b, ranged from 0.002 to 0.01 at 7.5–44 mM of the dipeptide) when compared to tromethamine (e.g., TRIS) at pH values of 6.5–7.6, and superior stability (l-carnosine appeared to be 3–4 times more resistant to thermal acid/base-driven decomposition under most limiting conditions) when assessed against l-histidine (e.g., a common biologic buffer). For ophthalmic pharmacology and therapeutics, where a broad spectrum of topical (or injectable) ophthalmic agents require chronic dosing because of disease etiology or pharmacological mechanism of action, use of l-carnosine as a buffer was proposed to enable applications of emerging sustained delivery technologies which utilize osmotic or ionic in situ gel formation to slow down the clearance of small molecules or biologics from ocular compartments (Singh et al. 2009). Overall, based on the comparatively lower physiological buffering capacity of ocular fluids than that of blood (i.e. blood, plasma, and red blood cells combined—e.g., the typical central compartment for drug distribution—in contrast have virtually unlimited buffering capacity (Salenius 1957)), the final pH and chemical buffer content of liquid ophthalmic products have to be carefully controlled. A global examination of known liquid ophthalmic products (Table 1) indicates that pH is targeted close to neutrality and the concentrations of exogenous buffers used in the product are maintained to a level sufficient to guarantee product quality and not interfere with endogenous ocular physiological pH (which can cause irritation and inter-ocular compartment boundary compromise) (Aguirre et al. 2018; Marra et al. 2011; Younis et al. 2008). Limited examples of drug products displaying a final pH (or range) significantly away from 7 exist, and despite the fact that these come with a strong case from a drug product quality point of view, the adequacy of such digressions from guidance criteria set forth by ocular physiological constraints is contextual, i.e., related to the nature of disease conditions and almost exclusively acute duration of treatment (as opposed to chronic conditions).
Osmolarity and Osmolality
In addition to the pH specification in liquid ophthalmic drug products, the final osmolarity of formulations (typically estimated using freezing point depression approach (Tomlinson et al. 2010)) is another essential biophysical and physiological compatibility attribute. Total solute content has been demonstrated to play a key role in injectable and topical ophthalmic liquid products. Formerly called osmolarity, by definition an osmotic concentration is the product of the osmolality and the mass density of water, in which osmolality is the quotient of the negative natural logarithm of the rational activity of water and the molar mass of water (McNaught and Wilkinson 1997, 2006). Conrad et al. published one of the earliest plausible investigations on the influence of tonicity (in addition to previously discussed pH and local ocular or systemic anesthesia) on lacrimation and topical ocular drug bioavailability. Employing the state-of-the-art microscintigraphy monitoring systems at the time, radiotracer signal dilution was detected in the tear film with hypertonic liquid formulations, suggesting considerable increase in lacrimation. The same was not evident with hypotonic formulations. Furthermore, this relationship of osmolarity and lacrimation had a proxy to ocular pharmacokinetic exposures, in an inverse relationship, where greatest ocular bioavailability was observed with deionized formulations containing a probe/drug, and hypertonicity (up to four times isotonic) giving the lowest (Conrad et al. 1978). Additional influential factors over extents and peak exposures elucidated from these studies were found to depend on precorneal contact time and mixing efficiency with the resident tear film (Conrad et al. 1978; Patton and Robinson 1975; Sieg and Robinson 1975, 1977; Singh et al. 2009). Limiting mechanisms which are apparently exerted by total solute concentration in liquid ophthalmic dosage forms are relative to the tonicity of the blood. While several different explanations exist, in the scenario where formulation osmolarity exceeds physiological tonicity, another phenomenon of rapid fluid extraction from ocular compartments into the vicinity of instilled dose can occur, effectively diluting the total dose in situ and decreasing the driving force for passive diffusive mass transfer to surrounding ocular compartments (Maurice 1971, 1980). From a liquid formulation design perspective, this can have implications on maximal amounts of inactive (esp. solubilizers, co-solvents, buffers, cyclodextrins, surfactants) and active ingredients that can act as solutes or osmolytes, which should be considered during ophthalmic safety and efficacy evaluations. Since excipients often make up a majority of the weight to volume ratio in liquid ophthalmic products, their contribution to osmolarity and final pH is also of paramount importance (Aguirre et al. 2018). Prolonged ocular dosing compartment exposure to hypertonic solutions, e.g., topical or intravitreal ophthalmic delivery, has been shown to be benign on epithelial barrier permeability. However, the opposite is true for hypoosmotic compositions introduced to ocular tissue compartments, which are reported to elicit transient increases in epithelial permeability from a topical delivery perspective, or microscopic findings manifesting themselves as mild retinal degeneration with emergence of eosinophilic bodies from an intravitreal delivery perspective (Aguirre et al. 2018; Maurice 1980).
The ionic content of ocular fluids is known to be modulated on a molecular and cellular level by several endogenous and pharmacological factors of relevance in the eye. Liquid ophthalmic dosage forms which are administered into various compartments of the eye require fine-tuning of their pharmaceutical and pharmacological properties that directly or indirectly influence osmolyte balance to further ensure compatibility, safety, and efficacy. In the anterior segment of the eye, epithelial tissues which line the entire ocular surface and come into full contact with topical liquid ophthalmic dosage forms have been characterized in terms of active and passive net fluid transfer rates across corneal and conjunctival epithelial cells. Chloride is the most abundant physiological anion, and its movement across cell membranes and mucosa/serosa of epithelial tissue layers is known to be tightly coupled to the osmotically driven flux of sodium (an abundant, physiological extracellular cation) (Mobasheri et al. 2005; Pusch and Jentsch 1994). Characterization of active ion transport in the presence and absence of molecules known to affect chloride secretion and sodium absorption in corneal and conjunctival epithelial tissues indicated that the cornea is primarily a sodium absorptive tissue, while the conjunctiva plays a largely chloride secretory role (Chang-Lin et al. 2005; Kompella et al. 1993; Shiue et al. 1998, 2000). This asymmetrical transfer of physiological ions to and from tear fluid by ocular epithelial tissues is thought to modulate composition and concentration of drugs and other solutes within the context of topical ophthalmic liquid dosage forms. While transient perturbation of this osmotic balancing mechanism by extremes in liquid formulation solute content has been shown to result in changes in drug permeability across ocular epithelia (Scholz et al. 2002), the absolute osmolarity of endogenous tear film present on ocular surface is also known to behave as a biomarker for prognosis at various degrees (e.g., mild to moderate) of dry eye disease (Tomlinson et al. 2006; Rocha et al. 2017). Toward addressing the latter, several liquid formulations of secretagogues have been tested in the treatment of ocular surface inflammation relief and tear film dysfunction, most prominent of which maybe diquafosol (Nichols et al. 2004), a purinergic receptor agonist which stimulates chloride coupled net fluid flow into the tear film (Hosoya et al. 2005; Dartt 2002; Shiue et al. 1998; Kompella et al. 1993). Osmotically driven fluid flux also plays a key role in the production of aqueous humor by ciliary epithelial cells. Here, the presence of bicarbonate exchange mechanisms found in the non-pigmented ciliary epithelium has been capitalized pharmaceutically, evidenced by well-documented slowing in the rate of aqueous humor production elicited by carbonic anhydrase inhibitors (e.g., compounds found in liquid ophthalmic drug products like AZOPT® (https://www.accessdata.fda.gov/ 1998) and TRUSOPT® (https://www.accessdata.fda.gov/ 1994)) which reduce the supply of ciliary epithelial cell cytoplasmic bicarbonate (Delamere 2005). Lastly, in the anterior chamber of the eye, fluid (possibly also by virtue of aquaporin water channels (Thiagarajah and Verkman 2002)) coupled anion secretion requires transcorneal endothelial cell net flux of chloride, bicarbonate, and/or lactate, the modulation of which through endogenous factors—such as aging—or exogenous factors which can be introduced through intracameral introduction of various ophthalmic drug products can play a role in cause or therapy for corneal stromal swelling or edema (Bonanno 2012). In the posterior segment of the eye, hypertonicity in liquid injectable ophthalmic preparations has been shown to exert macroscopic changes on a cellular level in retinal tissues in pathology reports (Aguirre et al. 2018). Furthermore, pharmacological findings suggested that INS37217 (a structural analog diquafosol, a secretagogue discussed earlier in the anterior segment setting) was able to stimulate fluid secretion from vitreous-to-choroid direction by activating similar chloride coupled osmotic movement mechanisms in retinal pigmented epithelial cells enhancing the rates of subretinal fluid reabsorption in certain experimentally induced retinal detachments (Maminishkis et al. 2002). Overall, therapeutic usefulness for selective solute control in liquid ophthalmic drug products within the context of treating a variety of retinal diseases that result in fluid accumulation in various posterior segment tissue compartments requires further study to determine if the described osmolarity linked mechanisms could be additive or synergistic in nature.
Inactive Ingredients Found in Liquid Ophthalmic Products
A high-level, global survey of known liquid ophthalmic drug products (Table 1) suggests that the arsenal of excipients available for use in product development is remarkably sparse (e.g., in comparison to other routes of parenteral drug administration). Selection of optimal route for ocular delivery depends on multiple factors, intuitively including the disease condition being treated, ocular tissue physiology (e.g., retina, choroid, and iris-ciliary body) that is targeted for pharmacological intervention, desired treatment modality or duration, as well as patient-disease demographics. Selection of key excipients in liquid ophthalmic drug products involves stratified rationale considerations. Initially choices may be limited from a pragmatic perspective, for example, precedence of use and prior utilization in a reference listed ophthalmic drug product as found in the Inactive Ingredient Search for Approved Drug Products or the Orange Book (https://www.accessdata.fda.gov/ 2019; https://www.hhs.gov/ 2019), or availability of parenteral and pharmaceutical grade excipient bulk from manufacturers which perform compendia testing on the material. However, ultimate restrictions most often come from a lack of basic scientific understanding about the full tolerability and disposition of the preferred inactive ingredients within an ocular context. Secondly, selections of excipients should be driven by a conventional functional role and appropriate requirement within the context of drug product quality, safety, and consistent performance (Rowe et al. 2012). Several existing reports have done a systematic evaluation of various functional excipients from an in vivo veterinary medicine (observational tests, e.g., the Draize eye test) and post-hoc tissue histopathology perspective, although there is limitation to translation from preclinical species to humans (Abraham et al. 2003; Wilhelmus 2001). Emerging research in this specific area of excipient qualification to enable ophthalmic drug delivery and product development could be highly helpful and influential in understanding the safety limits around selection of inactive ingredients in liquid ophthalmic products for development of topical eye drops, intravitreal and sub-tenon injections, or other novel routes of administration into this organ (Aguirre et al. 2012, 2018; Blandford et al. 1992; Younis et al. 2008).
Within this context, a unique and specific consideration among preservatives in liquid ophthalmic products is worthwhile to mention. Although preservatives are technically not inactive ingredients in liquid ophthalmic products, particular basic physiological research reports about additional roles (over those of known bactericidal and bacteriostatic activity) preservatives play in liquid eye products are noteworthy. Benzalkonium chloride has probably one of the most lengthy track records of use in topical eye drop products; however, it is not devoid of limitations in safety and tolerability which have over time resulted in the advent of alternatives like Polyquad, Purite®, and SofZia® (Ammar et al. 2010; Kahook and Noecker 2008; Dong et al. 2004). Furthermore, investigations on the influence benzalkonium chloride and commonly co-employed ethylene diamine tetra-acetic acid on the permeability of several ophthalmic drugs used for management of glaucoma showed a general trend in facilitating drug transport across the cornea and conjunctiva. This was partially attributed to some level of toxic effect that benzalkonium chloride has on ocular epithelial cells, permeabilizing them possibly transiently, however not insignificantly (Ashton et al. 1991; Scholz et al. 2002).
Historical accounts of off-label use of triamcinolone acetonide (a steroidal anti-inflammatory drug substance) within liquid ophthalmic drug product space presented as Kenalog-40® (https://www.accessdata.fda.gov/ 1965) provides a compelling retrospective argument supporting the importance of careful excipient selection within this pharmaceutical development space. Before the advent of TRIESENCE™ (https://www.accessdata.fda.gov/ 2007), Kenalog-40® was widely used via intravitreal and sub-tenon injection routes to treat ocular diseases, such as varieties of noninfectious uveitis and diabetic macular edema (Jonas 2006; Kovacs et al. 2012). As Kenalog-40® evolved into the most widely injected liquid parenteral drug product for triamcinolone acetonide application in various intraocular neovascular and edematous diseases, purification of triamcinolone suspension from this product (designed for intramuscular or intra-articular use only (https://www.accessdata.fda.gov/ 1965)) became important. Once it was clear that the solvent agent was better removed, in order to avoid the potential toxic effects of the vehicle, evaluations of different techniques used to reduce benzyl alcohol (~0.9–1%w/v) from commercially prepared triamcinolone acetonide suspensions were researched and published (Garcia-Arumi et al. 2005; Jonas 2006). Subsequent, more thorough histopathological evaluations of benzyl alcohol showed that the lack of toleration following the excipient’s use in liquid ophthalmic preparations was manifested as conjunctival swelling, corneal and intraglobal opacities, and corneal lesions arising from multiple concentrations and compendia/purity grades available for testing (Younis et al. 2008). Overall, it is important to take a systematic and deliberate approach in the selection and qualification of all inactive ingredients present in liquid ophthalmic drug products, keeping in mind the physiological considerations around the actual, final physiological route of administration into the eye.
Manufacturing Considerations
As introduced earlier, all liquid ophthalmic products—occurring as solutions, suspensions, or more complex dosage forms of small molecules and compounds derived from biological sources—are specialized parenteral dosage forms, e.g., sterile products, that are intended for application to ocular compartments including locations adjacent to the eye and its immediate surrounding periorbital tissues. Ophthalmic routes of administration for liquid products include, but are not limited to: topical drops, subconjunctival, sub-tenon capsule, subretinal, sub- or suprachoroidal, intracorneal, intrascleral, intravitreal, intracameral, juxtascleral, and retrobulbar injection routes (Ghate and Edelhauser 2006). While Table 1 shows a comprehensive list of liquid ophthalmic products, with several off-label used parenterals in an ocular setting, this section succinctly enumerates consolidated, common liquid ophthalmic product preparation and quality test considerations which would apply for manufacturing. The current, electronic, United States Pharmacopeia chapter 771, with encompassed references, is recommended as a helpful resource for obtaining details on new manufacturing guidelines toward de novo development of liquid ophthalmic drug product monographs (United States Pharmacopeial Convention. Committee of Revision, 1979; United States Pharmacopeial Convention).
Sterilization process considerations add one of several important product development boundaries to selected physical, chemical, and formulation attributes for liquid ophthalmic products. Depending on the drug substance, packaging selection for route of administration and final liquid delivery vehicle composition, degradation, and/or morphological changes can occur to liquid suspensions and colloidal systems during sterilization. A particle size cutoff of <0.2 μm is required to consider filtration as a method of terminal sterilization for a liquid ophthalmic drug product. While aseptic processing remains a feasible option, the manufacture of sterile liquid ophthalmic products within class 10 or 100 clean rooms could be limiting to scale and flexibility. Design considerations for the development of steam-in-place sterilization processes, by introduction of pressurized steam into the internal cavities of a vessel used for liquid ophthalmic product manufacturing, have proven to be an effective means of making sure large, stationery processing equipment is compliant with sterility guidelines. While steam-in-place sterilization has several engineering control nuances, it does offer an advantage by potentially eliminating the need for aseptic processing or individual assembly of component parts within a manufacturing line. The latter can still introduce a risk of equipment contamination due to several possible root causes. Many liquid ophthalmic products, which are unit-dose and unpreserved, are manufactured under steam-in-place system procedures which allow the flexibility of non-aseptic fabrication followed by complete sterilization of the closed system carrying the product (Myers and Chrai 1980, 1981, 1982).
Limited aqueous solubility of drug substances is typically the most common consideration leading toward the development of suspension or colloidal-emulsion ophthalmic products (as opposed to aqueous solutions). Emulsion formulation manufacture is within a unique complex drug product category, as establishment of pharmaceutical and bioequivalence between two colloidal liquid ophthalmic products carrying the same drug substance is complicated and challenging (if not, in many cases pragmatically impossible). For such liquid ophthalmic complex drug products (e.g., cyclosporin A containing dosage forms of Restasis® (0.5 mg/mL), Ikervis® (1.0 mg/mL), Papilock mini® (1.0 mg/mL), Modusik-A Ofteno® (1.0 mg/mL), Lacrinmune® (0.5 mg/mL), TJ Cyporin® (0.5 mg/mL), Cyporin® (0.5 mg/mL), and Cyclorin® (0.5 mg/mL) (Lallemand et al. 2017)), it has been documented that “the manufacturing process is the product,” i.e., a well-controlled and well-understood production and scale-up procedure should be engineered to guarantee reproducible product quality, safety, and performance (de Vlieger et al. 2019; Hussaarts et al. 2017). Topical ophthalmic emulsions are generally prepared by dissolving a drug substance into an oil phase, including a suitable emulsifying agent, considering additional suspending excipients, and mixing with the liquid aqueous phase vigorously to homogenize an oil-in-water emulsion. Essentially two macroscopic phases exist, where each phase—the oil and aqueous—is normally sterilized in advance or concurrently with charging into mixing vessel. High-shear homogenization is one approach which can be used to reduce emulsion droplet sizes to (sub)micron distributions, desirable toward improving physical stability of unit micelles by slowing their coalescing rate.
Once prototypical liquid ophthalmic drug products are manufactured, procedures for testing and accepting them need to be developed. Assessment of general quality attributes, e.g., identification, potency, purity (and impurities), sterility, and particulate matter, and in vitro product performance, i.e., dissolution or drug release of the drug substance from a suspension or colloidal drug product, can be found in USP (United States Pharmacopeial Convention. Committee of Revision. 1979; United States Pharmacopeial Convention.). Quality tests assess the integrity of the dosage form, whereas the performance tests assess drug release and other attributes that relate to in vivo drug performance. For example, the aforementioned physicochemical and biophysical considerations around the final pH and solute content, specific to liquid ophthalmic dosage forms, are described in USP 〈pH 791〉 and〈osmolality and osmolarity 785〉. Additionally, liquid ophthalmic drug products are required to be essentially free of visible foreign (extrinsic or intrinsic) particulates and subvisible particles in intra- or extra-ocular injectables. Besides terminal sterilization considerations discussed earlier, further analyses of effectiveness in antimicrobial preservatives (in the case of multidose liquids ophthalmics) and minimization of bacterial endotoxins (e.g., pyrogen-free) are essential (United States Pharmacopeial Convention. Committee of Revision 1979; United States Pharmacopeial Convention).
Design and validation of specific tests is necessary to build a good understanding and proper.
control over the manufacturing process critical for a reproducible, high-quality liquid ophthalmic drug product. For colloidal systems and some suspensions, development of such tests may pose challenges. Active ingredient release testing conducted on complex liquid colloidal ophthalmic drug products or suspensions manufactured under boundary conditions and compared to drug products that are intentionally prepared with meaningful variations in formulation and manufacturing sensitive parameters (i.e., particle size distribution, dose or drug loading, types and/or amounts of inactive ingredients) maybe far from predictive in terms of ophthalmic bioequivalence. The extents and degrees of sensitivity analysis require further discussion and research; although it is pragmatically unachievable to ascertain robust in vitro-in vivo correlations with these assays in an ophthalmic setting, some in vitro release tests and in silico simulations and modeling tools still represent promising avenues for evaluating their ability to distinguish performance (de Vlieger et al. 2019; Gukasyan et al. 2019b; Hussaarts et al. 2017). Several additional specific tests which maybe discriminating from a performance of a manufactured liquid ophthalmic product perspective include those around viscosity, particle size distribution, and inactive ingredients. Inclusion of viscosity evaluations in the specification of liquid ophthalmic products should be based on the types of dosage forms and whether changes in product viscosity will affect the overall performance. For example, in liquid suspensions, depending on the vehicles’ viscosity, if drug particles settle and cake, they must re-disperse promptly in users’ hands to achieve proper dose uniformity and accurate delivery. As mentioned earlier, the opposite is the case for viscosity influence on reliable eye-drop volume dispensing (vs. nozzle engineering) (Brown and Lynch 1986; Lederer and Harold 1986). While particle size and distributions can impact the intensity and duration of ophthalmic pharmacokinetics, the potential for any changes in particle size of ophthalmic suspensions and emulsions also needs to be evaluated. Lastly suitable substances may be added to ophthalmic products to increase stability, provided they are benign in the amounts administered and do not interfere with therapeutic efficacy or with responses to the specified manufacturing-related assays and quality tests (United States Pharmacopeial Convention.; United States Pharmacopeial Convention. Committee of Revision 1979).
In recent years the field of ophthalmic drug discovery and development has witnessed what some experts in the field call a renaissance (Yerxa 2018). With the advent of gene therapies which promise to be thus far the most curative solutions to several genetically inherited retinal diseases, and several new chemical entities being introduced as novel pharmacological mechanisms for management of glaucoma and dry eye disease, the importance of pharmaceutical development of liquid ophthalmic dosage forms remains essential (Gukasyan et al. 2019a). Discovery efforts continue toward treatment of rare genetic ocular diseases, neuroprotection from damage caused by glaucoma at the optic nerve head, and prevention of neovascular wet age-related macular degeneration (AMD) through inhibition and reversal of dry AMD, demand for additional pharmaceutical technology research, and development to support novel drugs in the pipeline. Considerations discussed here for drug substance (any modality), drug product blueprint attributes, and sterile manufacturing guidelines will remain vital and fundamental in clinical testing and commercialization for future progressive liquid ophthalmic drug products.
Abbreviations
- AMD:
-
Age-related macular degeneration
- DCE-GS:
-
S-(1,2-dicarboxyethyl)glutathione
- FDA:
-
Food and Drug Administration
- GSH:
-
Glutathione
- IOP:
-
Intraocular pressure
- K sp :
-
Equilibrium constant for a solubility product
- pI:
-
Isoelectric point
- pKa:
-
Acid dissociation constant
- pKb:
-
Base dissociation constant
- P o/w :
-
Oil/water partition coefficient
- ROS:
-
Reactive oxygen species
- TRIS:
-
Triethanolamine
- USP:
-
United States Pharmacopeia
- UVA:
-
Ultraviolet long-wavelength light radiation
- UVB:
-
Ultraviolet short-wavelength light radiation
- β :
-
Buffering capacity
References
Abraham MH, Hassanisadi M, Jalali-Heravi M, Ghafourian T, Cain WS, Cometto-Muniz JE. Draize rabbit eye test compatibility with eye irritation thresholds in humans: a quantitative structure-activity relationship analysis. Toxicol Sci. 2003;76(2):384–91. https://doi.org/10.1093/toxsci/kfg242.
Aguirre SA, Collette W III, Gukasyan HJ, Huang W. An assessment of the ocular safety of excipient maleic acid following intravitreal injection in rabbits. Toxicol Pathol. 2012;40(5):797–806. https://doi.org/10.1177/0192623312441400.
Aguirre SA, Gukasyan HJ, Younis HS, Huang W. Safety assessment of formulation vehicles following intravitreal administration in rabbits. Pharm Res. 2018;35(9):173. https://doi.org/10.1007/s11095-018-2450-1.
Ahmed I, Gokhale RD, Shah MV, Patton TF. Physicochemical determinants of drug diffusion across the conjunctiva, sclera, and cornea. J Pharm Sci. 1987;76(8):583–6.
Ammar DA, Noecker RJ, Kahook MY. Effects of benzalkonium chloride-preserved, polyquad-preserved, and sofZia-preserved topical glaucoma medications on human ocular epithelial cells. Adv Ther. 2010;27(11):837–45. https://doi.org/10.1007/s12325-010-0070-1.
Ashton P, Podder SK, Lee VH. Formulation influence on conjunctival penetration of four beta blockers in the pigmented rabbit: a comparison with corneal penetration. Pharm Res. 1991;8(9):1166–74.
Bevacizumab (AVASTIN) and age-related macular degeneration. Lower cost does not justify taking risks. Prescrire Int. 2015;24(163):201–4.
Bhagav P, Deshpande P, Pandey S, Chandran S. Development and validation of stability indicating UV spectrophotometric method for the estimation of brimonidine tartrate in pure form, formulations and preformulation studies. Pharm Lett. 2010;2(3):106–22.
Blandford DL, Smith TJ, Brown JD, Pearson PA, Ashton P. Subconjunctival sustained release 5-fluorouracil. Invest Ophthalmol Vis Sci. 1992;33(12):3430–5.
Bonanno JA. Molecular mechanisms underlying the corneal endothelial pump. Exp Eye Res. 2012;95(1):2–7. https://doi.org/10.1016/j.exer.2011.06.004.
Breda SA, Jimenez-Kairuz AF, Manzo RH, Olivera ME. Solubility behavior and biopharmaceutical classification of novel high-solubility ciprofloxacin and norfloxacin pharmaceutical derivatives. Int J Pharm. 2009;371(1–2):106–13. https://doi.org/10.1016/j.ijpharm.2008.12.026.
Brown RH, Lynch MG. Drop size of commercial glaucoma medications. Am J Ophthalmol. 1986;102(5):673–4.
Carney LG, Hill RM. Human tear pH. Diurnal variations. Arch Ophthalmol. 1976;94(5):821–4.
Carney LG, Mauger TF, Hill RM. Buffering in human tears: pH responses to acid and base challenge. Invest Ophthalmol Vis Sci. 1989;30(4):747–54.
Chang-Lin JE, Kim KJ, Lee VH. Characterization of active ion transport across primary rabbit corneal epithelial cell layers (RCrECL) cultured at an air-interface. Exp Eye Res. 2005;80(6):827–36. https://doi.org/10.1016/j.exer.2004.12.012.
Chen SL, Png E, Tong L. Comparison of two artificial tear formulations using aberrometry. Clin Exp Optom. 2009;92(6):519; author reply 519. https://doi.org/10.1111/j.1444-0938.2009.00421.x.
Chien DS, Homsy JJ, Gluchowski C, Tang-Liu DD. Corneal and conjunctival/scleral penetration of p-aminoclonidine, AGN 190342, and clonidine in rabbit eyes. Curr Eye Res. 1990;9(11):1051–9. https://doi.org/10.3109/02713689008997579.
Coles WH, Jaros PA. Dynamics of ocular surface pH. Br J Ophthalmol. 1984;68(8):549–52.
Conrad JM, Reay WA, Polcyn RE, Robinson JR. Influence of tonicity and pH on lacrimation and ocular drug bioavailability. J Parenter Drug Assoc. 1978;32(4):149–61.
Dartt DA. Regulation of mucin and fluid secretion by conjunctival epithelial cells. Prog Retin Eye Res. 2002;21(6):555–76.
Delamere NA. Ciliary body and ciliary epithelium. Adv Organ Biol. 2005;10:127–48. https://doi.org/10.1016/S1569-2590(05)10005-6.
Diehl H, Markuszewski R. Studies on fluorescein-II the solubility and acid dissociation constants of fluorescein in water solution. Talanta. 1985;32(2):159–65.
Dong JQ, Babusis DM, Welty DF, Acheampong AA, Tang-Liu D, Whitcup SM. Effects of the preservative purite on the bioavailability of brimonidine in the aqueous humor of rabbits. J Ocul Pharmacol Ther. 2004;20(4):285–92. https://doi.org/10.1089/1080768041725326.
Edward A, Prausnitz MR. Predicted permeability of the cornea to topical drugs. Pharm Res. 2001;18(11):1497–508.
El-Qutob D. Off-label uses of Omalizumab. Clin Rev Allergy Immunol. 2016;50(1):84–96. https://doi.org/10.1007/s12016-015-8490-y.
Fazelat A, Lashkari K. Off-label use of intravitreal triamcinolone acetonide for diabetic macular edema in a pregnant patient. Clin Ophthalmol. 2011;5:439–41. https://doi.org/10.2147/OPTH.S14584.
FDA. Kenalog®-40 injection. 1965. https://www.accessdata.fda.gov/, https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/014901s042lbledt.pdf.
FDA. PRED FORTE®. 1973. https://www.accessdata.fda.gov/, https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/017011s047lbl.pdf.
FDA. TRUSOPT®. 1994. https://www.accessdata.fda.gov/, https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/020408s047lbl.pdf.
FDA. ALPHAGAN® (brimonidine tartrate ophthalmic solution) 0.2%. 1996. https://www.accessdata.fda.gov/, https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/020613s031lbl.pdf.
FDA. Azopt®. 1998. https://www.accessdata.fda.gov/, https://www.accessdata.fda.gov/drugsatfda_docs/label/2006/020816s009lbl.pdf.
FDA. ALPHAGAN® P (brimonidine tartrate ophthalmic solution) 0.1% or 0.15%. 2001, 2006. https://www.accessdata.fda.gov/, https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021262s020,021770s004lbl.pdf.
FDA. TRIESENCE™. 2007. https://www.accessdata.fda.gov/, https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/022223,022048lbl.pdf.
FDA. (2019) Inactive ingredient search for approved drug products. https://www.accessdata.fda.gov/, https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm.
Fishman GA. Ocular phototoxicity: guidelines for selecting sunglasses. Surv Ophthalmol. 1986;31(2):119–24.
Food and Drug Administration, Center for Drug Evaluation and Research. Orange book: approved drug products with therapeutic equivalence evaluations. 2019. https://www.accessdata.fda.gov/scripts/cder/ob/index.cfm.
Friedrich S, Saville B, Cheng YL. Drug distribution in the vitreous humor of the human eye: the effects of aphakia and changes in retinal permeability and vitreous diffusivity. J Ocul Pharmacol Ther. 1997;13(5):445–59. https://doi.org/10.1089/jop.1997.13.445.
Gan IM, van Dissel JT, Beekhuis WH, Swart W, van Meurs JC. Intravitreal vancomycin and gentamicin concentrations in patients with postoperative endophthalmitis. Br J Ophthalmol. 2001;85(11):1289–93. https://doi.org/10.1136/bjo.85.11.1289.
Garcia-Arumi J, Boixadera A, Giralt J, Martinez-Castillo V, Gomez-Ulla F, Corcostegui B, Garcia-Arumi E. Comparison of different techniques for purification of triamcinolone acetonide suspension for intravitreal use. Br J Ophthalmol. 2005;89(9):1112–4. https://doi.org/10.1136/bjo.2005.067744.
Ghate D, Edelhauser HF. Ocular drug delivery. Expert Opin Drug Deliv. 2006;3(2):275–87. https://doi.org/10.1517/17425247.3.2.275.
Gonnering R, Edelhauser HF, Van Horn DL, Durant W. The pH tolerance of rabbit and human corneal endothelium. Invest Ophthalmol Vis Sci. 1979;18(4):373–90.
Green K, Kearse EC, Yokogaki S, Awata T. Human and rabbit corneal endothelial permeability after different chemical forms of glutathione. Ophthalmic Res. 2001;33(3):151–5. https://doi.org/10.1159/000055662.
Gukasyan HJ, Hailu S, Karami TK. Ophthalmic drug discovery and development. Pharm Res. 2019a;36(5):69. https://doi.org/10.1007/s11095-019-2606-7.
Gukasyan HJ, Hailu S, Karami TK, Graham R. Ocular biopharmaceutics: impact of modeling and simulation on topical ophthalmic formulation development. Drug Discov Today. 2019b; https://doi.org/10.1016/j.drudis.2019.04.002.
Gurbay A, Hincal F. Ciprofloxacin-induced glutathione redox status alterations in rat tissues. Drug Chem Toxicol. 2004;27(3):233–42. https://doi.org/10.1081/DCT-120037504.
Hamalainen KM, Kananen K, Auriola S, Kontturi K, Urtti A. Characterization of paracellular and aqueous penetration routes in cornea, conjunctiva, and sclera. Invest Ophthalmol Vis Sci. 1997;38(3):627–34.
He Y, Li P, Yalkowsky SH. Solubilization of fluasterone in cosolvent/cyclodextrin combinations. Int J Pharm. 2003;264(1–2):25–34.
Hilfiker R, Blatter F, von Raumer M. Relevance of solid-state properties for pharmaceutical products, in polymorphism: in the pharmaceutical industry. Weinheim: Wiley; 2006.
Hind HW, Goyan FM. A new concept of the role of hydrogen ion concentration and buffer system in the preparation of ophthalmic solutions. J Am Pharm Assoc Am Pharm Assoc. 1947;36(2):33–41.
Hogben CA, Tocco DJ, Brodie BB, Schanker LS. On the mechanism of intestinal absorption of drugs. J Pharmacol Exp Ther. 1959;125(4):275–82.
Hosoya K, Lee VH, Kim KJ. Roles of the conjunctiva in ocular drug delivery: a review of conjunctival transport mechanisms and their regulation. Eur J Pharm Biopharm. 2005;60(2):227–40. https://doi.org/10.1016/j.ejpb.2004.12.007.
Hussaarts L, Muhlebach S, Shah VP, McNeil S, Borchard G, Fluhmann B, Weinstein V, Neervannan S, Griffiths E, Jiang W, Wolff-Holz E, Crommelin DJA, de Vlieger JSB. Equivalence of complex drug products: advances in and challenges for current regulatory frameworks. Ann N Y Acad Sci. 2017;1407(1):39–49. https://doi.org/10.1111/nyas.13347.
Jackson TL, Williamson TH. Amikacin retinal toxicity. Br J Ophthalmol. 1999;83(10):1199–200. https://doi.org/10.1136/bjo.83.10.1194f.
Jonas JB. Intravitreal triamcinolone acetonide: a change in a paradigm. Ophthalmic Res. 2006;38(4):218–45. https://doi.org/10.1159/000093796.
Kahook MY, Noecker RJ. Comparison of corneal and conjunctival changes after dosing of travoprost preserved with sofZia, latanoprost with 0.02% benzalkonium chloride, and preservative-free artificial tears. Cornea. 2008;27(3):339–43. https://doi.org/10.1097/ICO.0b013e31815cf651.
Kansy M, Senner F, Gubernator K. Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J Med Chem. 1998;41(7):1007–10. https://doi.org/10.1021/jm970530e.
Kidron H, Vellonen KS, del Amo EM, Tissari A, Urtti A. Prediction of the corneal permeability of drug-like compounds. Pharm Res. 2010;27(7):1398–407. https://doi.org/10.1007/s11095-010-0132-8.
Kompella UB, Kim KJ, Lee VH. Active chloride transport in the pigmented rabbit conjunctiva. Curr Eye Res. 1993;12(12):1041–8.
Kovacs K, Wagley S, Quirk MT, Ceron OM, Silva PA, Singh RJ, Gukasyan HJ, Arroyo JG. Pharmacokinetic study of vitreous and serum concentrations of triamcinolone acetonide after posterior sub-tenon’s injection. Am J Ophthalmol. 2012;153(5):939–48. https://doi.org/10.1016/j.ajo.2011.10.021.
Lallemand F, Schmitt M, Bourges JL, Gurny R, Benita S, Garrigue JS. Cyclosporine A delivery to the eye: a comprehensive review of academic and industrial efforts. Eur J Pharm Biopharm. 2017;117:14–28. https://doi.org/10.1016/j.ejpb.2017.03.006.
Lalwani GA, Berrocal AM, Murray TG, Buch M, Cardone S, Hess D, Johnson RA, Puliafito CA. Off-label use of intravitreal bevacizumab (Avastin) for salvage treatment in progressive threshold retinopathy of prematurity. Retina. 2008;28(3 Suppl):S13–8. https://doi.org/10.1097/IAE.0b013e3181644ad2.
Lederer CM Jr, Harold RE. Drop size of commercial glaucoma medications. Am J Ophthalmol. 1986;101(6):691–4.
Lee VH, Robinson JR. Topical ocular drug delivery: recent developments and future challenges. J Ocul Pharmacol. 1986;2(1):67–108.
Leibowitz HM, Kupferman A, Stewart RH, Kimbrough RL. Evaluation of dexamethasone acetate as a topical ophthalmic formulation. Am J Ophthalmol. 1978;86(3):418–23.
Lexi-Comp Inc., American Pharmacists Association. Pediatric & neonatal dosage handbook. Lexi Comp’s drug reference handbooks. LexiComp; American Pharmacists Association, Hudson, OH, Washington, DC.
Liu Y, Kam WR, Ding J, Sullivan DA. One man’s poison is another man’s meat: using azithromycin-induced phospholipidosis to promote ocular surface health. Toxicology. 2014;320:1–5. https://doi.org/10.1016/j.tox.2014.02.014.
Lu DW, Chang CJ, Wu JN. The changes of vitreous pH values in an acute glaucoma rabbit model. J Ocul Pharmacol Ther. 2001;17(4):343–50. https://doi.org/10.1089/108076801753162753.
Maminishkis A, Jalickee S, Blaug SA, Rymer J, Yerxa BR, Peterson WM, Miller SS. The P2Y(2) receptor agonist INS37217 stimulates RPE fluid transport in vitro and retinal reattachment in rat. Invest Ophthalmol Vis Sci. 2002;43(11):3555–66.
Maren TH, Bar-Ilan A, Conroy CW, Brechue WF. Chemical and pharmacological properties of MK-927, a sulfonamide carbonic anhydrase inhibitor that lowers intraocular pressure by the topical route. Exp Eye Res. 1990;50(1):27–36.
Marra MT, Khamphavong P, Wisniecki P, Gukasyan HJ, Sueda K. Solution formulation development of a VEGF inhibitor for intravitreal injection. AAPS PharmSciTech. 2011;12(1):362–71. https://doi.org/10.1208/s12249-011-9591-4.
Martin FN Jr, Mims JL. Preparation of ophthalmic solutions with special reference to hydrogen ion concentration and tonicity. AMA Arch Ophthalmol. 1950;44(4):561–72.
Masuzawa K, Suzue S, Hirai K, Ishizaki T. 8-Alkoxyquinolonecarboxylic acid and salts thereof. US 5,043,450; 1991.
Matsumoto T, Hara M, Miyashita K, Kato Y. 8-Alkoxyquinolonecarboxylic acid hydrate with excellent stability and process for producing the same. US5880283; 1999.
Maurice DM. The tonicity of an eye drop and its dilution by tears. Exp Eye Res. 1971;11(1):30–3.
Maurice DM. Factors influencing the penetration of topically applied drugs. Int Ophthalmol Clin. 1980;20(3):21–32.
McNaught AD, Wilkinson A. IUPAC. Compendium of chemical terminology. 2nd edn (the “gold book”). Blackwell Scientific Publications, Oxford; 1997, 2006. https://doi.org/10.1351/goldbook.
Meyer K, Palmer JW. On the nature of the ocular fluids. Am J Ophthalmol. 1936;19(10):859–65. https://doi.org/10.1016/S0002-9394(36)92723-X.
Missel PJ, Horner M, Muralikrishnan R. Simulating dissolution of intravitreal triamcinolone acetonide suspensions in an anatomically accurate rabbit eye model. Pharm Res. 2010;27(8):1530–46. https://doi.org/10.1007/s11095-010-0163-1.
Mitra AK. Ophthalmic drug delivery systems. Drugs and the pharmaceutical sciences, vol. 58. New York: Dekker; 1993.
Mobasheri A, Barrett-Jolley R, Shakibaei M, Canessa CM, Martin-Vasallo P. Enigmatic roles of the epithelial sodium channel (ENaC) in articular chondrocytes and osteoblasts: mechanotransduction, sodium transport or extracellular sodium sensing? In: Kamkin A, Kiseleva I, editors. Mechanosensitivity in cells and tissues. Moscow: Academia; 2005.
Mortimer RG, Eyring H. Elementary transition state theory of the Soret and Dufour effects. Proc Natl Acad Sci U S A. 1980;77(4):1728–31.
Myers T, Chrai S. Parenteral fundamentals. Basic principles and applications of bioindicators. J Parenter Drug Assoc. 1980;34(3):234–43.
Myers T, Chrai S. Design considerations for development of steam in-place sterilization processes. J Parenter Sci Technol. 1981;35(1):8–12.
Myers T, Chrai S. Steam-in-place sterilization of cartridge filters in-line with a receiving tank. J Parenter Sci Technol. 1982;36(3):108–12.
Newman A, Wenslow R. Solid form changes during drug development: good, bad, and ugly case studies. AAPS Open. 2016;2(1):2. https://doi.org/10.1186/s41120-016-0003-4.
Nichols KK, Yerxa B, Kellerman DJ. Diquafosol tetrasodium: a novel dry eye therapy. Expert Opin Investig Drugs. 2004;13(1):47–54. https://doi.org/10.1517/13543784.13.1.47.
Olejnik OK, E.D.S. Compositions containing α-2-adrenergic agonist components. USA Patent; 14 July 2000.
Palkama A, Uusitalo H, Raij K, Uusitalo R. Comparison of the effects of adrenergic agonists and alpha-, beta 1-, beta 2-antagonists on the intraocular pressure and adenylate cyclase activity in the ciliary processes of the rabbit. Acta Ophthalmol. 1985;63(1):9–15.
Paterson CA, Pfister RR, Levinson RA. Aqueous humor pH changes after experimental alkali burns. Am J Ophthalmol. 1975;79(3):414–9.
Patton TF, Robinson JR. Influence of topical anesthesia on tear dynamics and ocular drug bioavailability in albino rabbits. J Pharm Sci. 1975;64(2):267–71.
Pawar P, Katara R, Mishra S, Majumdar DK. Topical ocular delivery of fluoroquinolones. Expert Opin Drug Deliv. 2013;10(5):691–711. https://doi.org/10.1517/17425247.2013.772977.
PDR Network LLC. PDR drug information handbook. Montvale: PDR, LLC; 2016.
Photosafety evaluation of pharmaceuticals. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. 2019. https://www.ich.org/products/guidelines/.
Physicians’ desk reference for ophthalmic medicines. Medical Economics, Montvale; 2000.
Pitkanen L, Ranta VP, Moilanen H, Urtti A. Permeability of retinal pigment epithelium: effects of permeant molecular weight and lipophilicity. Invest Ophthalmol Vis Sci. 2005;46(2):641–6. https://doi.org/10.1167/iovs.04-1051.
Prausnitz MR, Noonan JS. Permeability of cornea, sclera, and conjunctiva: a literature analysis for drug delivery to the eye. J Pharm Sci. 1998;87(12):1479–88.
Pusch M, Jentsch TJ. Molecular physiology of voltage-gated chloride channels. Physiol Rev. 1994;74(4):813–27. https://doi.org/10.1152/physrev.1994.74.4.813.
Raghava S, Hammond M, Kompella UB. Periocular routes for retinal drug delivery. Expert Opin Drug Deliv. 2004;1(1):99–114. https://doi.org/10.1517/17425247.1.1.99.
Raghaven KS, Ranadive SA, Gougoutas JZ, DiMarco JD, Parker WL, Davidovich M, Newman A. Gatifloxacin pentahydrate US6413969; 2002.
Ramsay E, Ruponen M, Picardat T, Tengvall U, Tuomainen M, Auriola S, Toropainen E, Urtti A, Del Amo EM. Impact of chemical structure on conjunctival drug permeability: adopting porcine conjunctiva and cassette dosing for construction of in silico model. J Pharm Sci. 2017;106(9):2463–71. https://doi.org/10.1016/j.xphs.2017.04.061.
Ramsay E, Del Amo EM, Toropainen E, Tengvall-Unadike U, Ranta VP, Urtti A, Ruponen M. Corneal and conjunctival drug permeability: systematic comparison and pharmacokinetic impact in the eye. Eur J Pharm Sci. 2018;119:83–9. https://doi.org/10.1016/j.ejps.2018.03.034.
Roberts JE. Screening for ocular phototoxicity. Int J Toxicol. 2002;21(6):491–500. https://doi.org/10.1080/10915810290169918.
Rocha G, Gulliver E, Borovik A, Chan CC. Randomized, masked, in vitro comparison of three commercially available tear film osmometers. Clin Ophthalmol. 2017;11:243–8. https://doi.org/10.2147/OPTH.S127035.
Rowe RC, Sheskey PJ, Cook WG, Fenton ME, American Pharmacists Association. In: Rowe RC, Sheskey PJ, Cook WG, Fenton ME, editors. Handbook of pharmaceutical excipients. 7th ed. London: APhA/Pharmaceutical Press; 2012.
Salenius P. A study of the pH and buffer capacity of blood, plasma and red blood cells. Scand J Clin Lab Invest. 1957;9(2):160–7.
Schoenwald RD, Huang H-S. Corneal penetration behavior of β-blocking agents I: physicochemical factors. J Pharm Sci. 1983a;72(11):1266–72. https://doi.org/10.1002/jps.2600721108.
Schoenwald RD, Huang HS. Corneal penetration behavior of beta-blocking agents I: physiochemical factors. J Pharm Sci. 1983b;72(11):1266–72.
Scholz M, Lin JE, Lee VH, Keipert S. Pilocarpine permeability across ocular tissues and cell cultures: influence of formulation parameters. J Ocul Pharmacol Ther. 2002;18(5):455–68. https://doi.org/10.1089/10807680260362731.
Scozzafava A, Menabuoni L, Mincione F, Briganti F, Mincione G, Supuran CT. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem. 1999;42(14):2641–50. https://doi.org/10.1021/jm9900523.
Shen J, Lu GW, Hughes P. Targeted ocular drug delivery with pharmacokinetic/pharmacodynamic considerations. Pharm Res. 2018;35(11):217. https://doi.org/10.1007/s11095-018-2498-y.
Shirasaki Y. Molecular design for enhancement of ocular penetration. J Pharm Sci. 2008;97(7):2462–96. https://doi.org/10.1002/jps.21200.
Shiue MH, Kim KJ, Lee VH. Modulation of chloride secretion across the pigmented rabbit conjunctiva. Exp Eye Res. 1998;66(3):275–82. https://doi.org/10.1006/exer.1997.0459.
Shiue MH, Kulkarni AA, Gukasyan HJ, Swisher JB, Kim KJ, Lee VH. Pharmacological modulation of fluid secretion in the pigmented rabbit conjunctiva. Life Sci. 2000;66(7):PL105–11.
Shoghi E, Fuguet E, Bosch E, Rafols C. Solubility-pH profiles of some acidic, basic and amphoteric drugs. Eur J Pharm Sci. 2013;48(1–2):291–300. https://doi.org/10.1016/j.ejps.2012.10.028.
Shore PA, Brodie BB, Hogben CA. The gastric secretion of drugs: a pH partition hypothesis. J Pharmacol Exp Ther. 1957;119(3):361–9.
Sieg JW, Robinson JR. Vehicle effects on ocular drug bioavailability i: evaluation of fluorometholone. J Pharm Sci. 1975;64(6):931–6.
Sieg JW, Robinson JR. Vehicle effects on ocular drug bioavailability II: evaluation of pilocarpine. J Pharm Sci. 1977;66(9):1222–8.
Sieg JW, Robinson JR. Vehicle effects on ocular drug bioavailability III: shear-facilitated pilocarpine release from ointments. J Pharm Sci. 1979;68(6):724–8.
Singh SR, Carreiro ST, Chu J, Prasanna G, Niesman MR, Collette Iii WW, Younis HS, Sartnurak S, Gukasyan HJ. L-carnosine: multifunctional dipeptide buffer for sustained-duration topical ophthalmic formulations. J Pharm Pharmacol. 2009;61(6):733–42. https://doi.org/10.1211/jpp/61.06.0005.
Singhal D, Curatolo W. Drug polymorphism and dosage form design: a practical perspective. Adv Drug Deliv Rev. 2004;56(3):335–47. https://doi.org/10.1016/j.addr.2003.10.008.
Stahl PH, Wermuth CG, International Union of Pure and Applied Chemistry. Handbook of pharmaceutical salts: properties, selection, and use. 2nd rev ed. Weinheim: Wiley-VCH; 2011.
Tai MC, Lu DW, Chiang CH. Corneal and scleral permeability of quinolones--a pharmacokinetics study. J Ocul Pharmacol Ther. 2003;19(6):547–54. https://doi.org/10.1089/108076803322660468.
Thiagarajah JR, Verkman AS. Aquaporin deletion in mice reduces corneal water permeability and delays restoration of transparency after swelling. J Biol Chem. 2002;277(21):19139–44. https://doi.org/10.1074/jbc.M202071200.
Thompson AM. Ocular toxicity of fluoroquinolones. Clin Exp Ophthalmol. 2007;35(6):566–77. https://doi.org/10.1111/j.1442-9071.2007.01552.x.
Tomlinson A, Khanal S, Ramaesh K, Diaper C, McFadyen A. Tear film osmolarity: determination of a referent for dry eye diagnosis. Invest Ophthalmol Vis Sci. 2006;47(10):4309–15. https://doi.org/10.1167/iovs.05-1504.
Tomlinson A, McCann LC, Pearce EI. Comparison of human tear film osmolarity measured by electrical impedance and freezing point depression techniques. Cornea. 2010;29(9):1036–41. https://doi.org/10.1097/ICO.0b013e3181cd9a1d.
Tsuboi S, Kobayashi M, Nanba M, Imaoka S, Ohmori S. S-(1,2-dicarboxyethyl)glutathione and activity for its synthesis in rat tissues. J Biochem. 1990a;107(4):539–45. https://doi.org/10.1093/oxfordjournals.jbchem.a123082.
Tsuboi S, Ohnaka M, Ohmori S, Sakaue T, Ogata K, Itano T, Hatase O. Inhibition of platelet aggregation by S-(1,2-dicarboxyethyl)glutathione, intrinsic tripeptide in liver, heart, and lens. Arch Biochem Biophys. 1990b;279(1):146–50.
United States Pharmacopeial Convention. Committee of Revision. The United States pharmacopeia. Rockville: United States Pharmacopeial Convention, Inc.; 1979.
United States Pharmacopeial Convention. USP. NF: the official compendia of drug standards information on disk. Rockville: USPC.
de Vlieger JSB, Crommelin DJA, Tyner K, Drummond DC, Jiang W, McNeil SE, Neervannan S, Crist RM, Shah VP. Report of the AAPS guidance forum on the FDA draft guidance for industry: “drug products, including biological products, that contain nanomaterials”. AAPS J. 2019;21(4):56. https://doi.org/10.1208/s12248-019-0329-7.
Wang W, Sasaki H, Chien DS, Lee VH. Lipophilicity influence on conjunctival drug penetration in the pigmented rabbit: a comparison with corneal penetration. Curr Eye Res. 1991;10(6):571–9.
Wilhelmus KR. The Draize eye test. Surv Ophthalmol. 2001;45(6):493–515.
Yerxa B. Progress in inherited retinal disease drug discovery and development: a foundation’s perspective. Pharm Res. 2018;35(11):239. https://doi.org/10.1007/s11095-018-2514-2.
Yoshida F, Topliss JG. Unified model for the corneal permeability of related and diverse compounds with respect to their physicochemical properties. J Pharm Sci. 1996;85(8):819–23. https://doi.org/10.1021/js960076m.
Young S, Larkin G, Branley M, Lightman S. Safety and efficacy of intravitreal triamcinolone for cystoid macular oedema in uveitis. Clin Exp Ophthalmol. 2001;29(1):2–6.
Younis HS, Shawer M, Palacio K, Gukasyan HJ, Stevens GJ, Evering W. An assessment of the ocular safety of inactive excipients following sub-tenon injection in rabbits. J Ocul Pharmacol Ther. 2008;24(2):206–16. https://doi.org/10.1089/jop.2007.0099.
Zhang Y, Ren K, He Z, Li H, Chen T, Lei Y, Xia S, He G, Xie Y, Zheng Y, Song X. Development of inclusion complex of brinzolamide with hydroxypropyl-beta-cyclodextrin. Carbohydr Polym. 2013;98(1):638–43. https://doi.org/10.1016/j.carbpol.2013.06.052.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 American Association of Pharmaceutical Scientists
About this chapter

Cite this chapter
Gukasyan, H.J., Graham, R. (2021). Liquid Ophthalmic Drug Products: Physicochemical Properties, Formulations, and Manufacturing Considerations. In: Neervannan, S., Kompella, U.B. (eds) Ophthalmic Product Development. AAPS Advances in the Pharmaceutical Sciences Series, vol 37. Springer, Cham. https://doi.org/10.1007/978-3-030-76367-1_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-76367-1_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-76366-4
Online ISBN: 978-3-030-76367-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)