
ABSTRACT
Purpose
To develop a computational model for optimisation of low corneal permeability, which is a key feature in ocular drug development.
Methods
We have used multivariate analysis to build corneal permeability models based on a structurally diverse set of 58 drug-like compounds.
Results
According to the models, the most important parameters for permeability are logD at physiologically relevant pH and the number of hydrogen bonds that can be formed. Combining these descriptors resulted in models with Q 2 and R 2 values ranging from 0.77 to 0.79. The predictive capability of the models was verified by estimating the corneal permeability of an external data set of 11 compounds and by using predicted permeability values to calculate the aqueous humour concentrations in the steady-state of seven compounds. The predicted values correlated well with experimental values.
Conclusion
The developed models are useful in early drug development to predict the corneal permeability and steady-state drug concentration in aqueous humor without experimental data.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
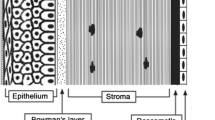
In the treatment of ophthalmic diseases, eye drops are the most convenient, economical and safe way to deliver drugs into the eye compared to invasive delivery routes such as intravitreal or periocular injections. The main route for drug absorption from eye drops is permeation through the cornea (1). However, the cornea is a tight diffusion barrier, which protects the eye against exogenous compounds. It consists of three main layers: the epithelium, stroma and endothelium. The endothelium is a monolayer facing the aqueous humor, and it is considered to have minor significance in drug permeation through the cornea. Tight junctions of the corneal epithelium limit drug permeation through paracellular pores, but lipophilic compounds can pass through the stratified epithelium by the transcellular permeation route. However, the underlying hydrophilic stroma may limit the permeation of very hydrophobic compounds. Accordingly, permeation through cornea has been shown to follow a parabolic relationship with an optimal logP of 2–3 (2). In general, the ocular drug absorption from the tear fluid through the cornea is inefficient. The absolute ocular bioavailability is usually below 5%, while the majority of the drug dose is absorbed rapidly to the systemic blood circulation through the conjunctival and nasal mucosae (3).
Optimisation of the corneal permeability is one of the key features in ocular drug discovery and development. Corneal drug permeability can be determined by diffusion studies with isolated corneas (rabbit, bovine, pig) (2,4) or corneal epithelial cell models (5,6). These are labour intensive, tedious and expensive methods that are not feasible for rapid drug screening. In vivo ocular pharmacokinetic studies are even less suitable for screening, because several animals, usually rabbits, must be sacrificed for each time point in the concentration-time curve. Therefore, new tools are needed for ophthalmic drug discovery to enable rapid evaluation and pre-selection of the compounds before studies with cells, tissues or animals. Such tools would also reduce the number of animal experiments.
Fast and easy-to-use computational methods would be optimal for permeability screening of new molecules before experimental studies. Previously, it has been reported that the absorption of drug-like compounds in the human intestine can be predicted as accurately with computational models as with in vitro experiments (7). Even though quantitative structure-property relationship (QSPR) models for transdermal and intestinal absorption of drugs have been constructed (8–10), QSPR models for the corneal permeability are still at an elementary level. Some computational models for predicting the corneal permeability have been developed (2,11–14), but so many of these models have been constructed for a small set of similar compounds (2,13), and the models are thus not applicable to a broader variety of molecules. Other models use descriptors that are difficult to obtain, like delta logP, the difference between the octanol-water partition coefficient and the alkane-water partition coefficient (11). On the other hand, a more extensive physiological model (15) has been developed, but the use of this model in early ADME context may be limited due to its complexity. The physiological model incorporates the corneal morphology; therefore, it can be used also to investigate the impact of structural changes in the cornea. However, a straightforward QSPR-based algorithm for the prediction of the corneal permeability would be useful in early ocular drug discovery and development.
In this study, we aimed to generate a completely computational model for the corneal permeability in early drug discovery. The model should be easily applicable to a variety of new compounds without the need to produce any prior experimental data. The models of this study are based on a diverse set of 58 drug-like compounds. There are plenty of experimental data available from the literature about the corneal permeability of different drugs or drug-like compounds. However, the permeability values for a compound can differ greatly in different conditions; therefore, we have taken great care to only include experimental data that has been measured in comparable experimental conditions. Since the models are intended for use during the early drug discovery process, we have opted to use only calculated physicochemical properties, and we did not include any experimental descriptors, which can be time-consuming and costly to obtain for new compounds. The resulting models are statistically sound, easy to use, have a broad applicability and can be explained physiologically. The predictive power of the final models was verified by estimating the corneal permeability on an external data set of eleven compounds and by predicting the steady-state concentrations in the aqueous humour in vivo based on the predicted permeability values.
MATERIALS AND METHODS
Compound Data Set
A data set of 58 compounds (Table I and Supplementary Material) was constructed based on an extensive literature search for corneal permeability studies (4,16–24). The reported permeability of the compounds included in the dataset has been measured in vitro in the rabbit cornea. The experiments were performed in Ringer’s buffer during a maximal duration of 6 h, hydration level was below 83%, a mix of O2 and CO2 (95:5) was used to adjust the pH and to mix the buffer, and temperature was set between 34–37°C.
Molecular Descriptors
The molecular descriptors were calculated with the ACDLabs software package version 6.0 (25). Descriptors that are known to affect membrane permeability were selected for this study: molecular weight (MW), molecular volume (MV), polar surface area (PSA), number of hydrogen bond donors (HBD), number of hydrogen bond acceptors (HBA), total number of putative hydrogen bonds, i.e. HBD + HBA (HBtot), the logarithm of the octanol-water partition coefficient of the neutral form (logP) and at pH 7.0, 7.4 and 8.0 (logD7.0, logD7.4 and logD8.0, respectively). The molecular structures of the compounds were imported from ACD/Dictionary, sketched in ACD/ChemDraw version 6.0 (25) or their SMILES codes were extracted from the PubChem database (26) (http://pubchem.ncbi.nlm.nih.gov). All molecular descriptor values of the compounds in the data set are given in Table I.
Multivariate Data Analysis
Multivariate data analyses were performed with Simca-P version 10.5 (27). The diversity of the compounds in the data set was analyzed with principal component analysis (PCA) using all molecular descriptors. The relationship between the logarithm of corneal permeability (logperm) and the molecular descriptors was determined by Partial Least Squares (PLS) analysis (28). Based on their distribution in the PCA plot, the data set was divided into a training set and an internal test set of 29 compounds each. Initial models were calculated from the compounds in the training set. The predictive capability of these models was estimated by cross-validation and by predicting the corneal permeability for the compounds in the internal test set. The cross-validation was performed by dividing the data into seven groups and subsequently leaving out one group, for which compounds the permeability was predicted. The differences between the actual and the predicted permeability values are then calculated and expressed as Q 2 (the cross-validated R 2). A Q 2 value above 0.5 indicates that the model has a better predictive power than chance. The predictive capability of the initial models was validated by predicting the permeability of the compounds in the internal test set. For the final models, the original data set of 58 compounds was used in order to achieve higher accuracy by including more data into the models. The final models were then cross-validated, and the predictive power was tested by predicting the corneal permeability of an external data set of eleven compounds (Table II and Supplementary Material).
The statistical significance of the predictive capability of the final models was estimated by the validate function in Simca-P (27). Here, a PLS model is fitted to randomly shuffled permeability values, and R 2 and Q 2 are calculated. The process was repeated 50 times to get reference distribution of R 2 and Q 2 based on random data. The data was plotted with the Y-axis representing the R 2 and Q 2 values and the X-axis the correlation coefficients between shuffled and original variables. Regression lines were fitted for the R 2 and Q 2 points, and the intercepts on the Y-axis represent “background” by fit to random data. The R 2 and Q 2 intercept for valid models should not exceed 0.3 and 0.05, respectively (27).
Prediction of Aqueous Humour Concentrations Using Permeability Values Calculated with QSPR Models
To further estimate the predictive capability of the developed models, steady-state concentration (Css) in aqueous humour was calculated using permeability values obtained with models 3b and 4b. We selected compounds for which the aqueous humour Css has been studied earlier with the topical infusion method in vivo in the rabbit eye and the ocular clearance reported in the literature (19,22,29,30). In these studies, a constant input rate was achieved by placing a cylinder containing the test solution on the surface of the cornea, and the permeation of test compounds was followed until steady state in aqueous humour concentration was reached.
To calculate Css the following equation was used:
In the equation, Cc is the concentration of the compound applied to the cornea (µg/ml), Papp is the permeability value (cm/s, calculated by model 3b, 4b or measured experimentally), S is the surface area of cornea available for permeation (0.503 cm2) and CL is the clearance from aqueous humour (µL/min).
RESULTS
Molecular Diversity of the Compounds
The ranges of the descriptor values were for MW 167–361 g/mol, MV 95–269 Å3, PSA 3–177 Å2, HBD 0–4, HBA 1–8, HBtot 1–11, logP −2.5 − +4.2, logD7.0 -2.5 − +3.2, logD7.4 −2.6 − +3.0 and logD8.0 −2.9 − +3.1 (Table I). A PCA was performed of the 58 compounds in the data set in order to analyze their molecular diversity. All calculated descriptors were used for the PCA, which resulted in a model with two principal components explaining 43% and 29%, respectively, of the variance in the data set. Fig. 1 shows the PCA score plot of the 58 compounds in the data set. The compounds are quite well separated, with three compounds (5-imino-4-methyl-1,3,4-thiadiazoline-2-sulfonamide, SKF86466 and cinoxacin) outside the elliptic 95% tolerance volume. The three compounds were not excluded, as 5% of the dataset is allowed outside the ellipse. The data set of 58 compounds was at first divided into a training set of 29 compounds and an internal test set of 29 compounds based on their distribution in the PCA score plot (Fig. 1, Table I).

PCA score plot of the 58 compounds in the data set. The black triangles represent compounds in the training set, and the unfilled triangles the compounds in the internal test set. The ellipse depicts the 95% tolerance volume based on hotelling T2 (0.05).
PLS Analysis of Training Set Compounds and Evaluation of Derived Models with Internal Test Set Compounds
A PLS analysis with the ten calculated descriptors and the 29 compounds of the training set resulted in a one-component model with Q 2 = 0.73 and R 2 = 0.80 (model 1a, Table III). In order to construct a simpler model, we then used the variable influence on projection (VIP) function in Simca-P. A VIP value above 1 indicates that the descriptor has a higher than average influence in explaining the corneal permeability. In model 1a, the four lipophilicity variables (logP, logD7.0, logD7.4 and logD8.0) as well as two hydrogen bond variables (HBtot and HBD) had VIP values above 1 (Fig. 2). Using these six descriptors with VIP values above 1 for a new PLS analysis on the training set yielded a one-component model with similar values, Q 2 = 0.73 and R 2 = 0.77 (model 2a, Table III). Finally, when we combined the HBtot descriptor (the hydrogen bond variable with the highest VIP value) with either logD7.4 and logD8.0 (the lipophilicity descriptors with the highest VIP values), we obtained one-component models with Q 2 = 0.75 and R 2 = 0.79 and Q 2 = 0.76 and R 2 = 0.80, respectively (models 3a and 4a, Table III). However, when only the distribution coefficients logD7.4 and logD8.0 were included in the model, the Q 2 value dropped to 0.68 and the R 2 value to 0.69. The models were then evaluated by predicting logPapp of the internal test set compounds. The correlation between experimental and predicted logPapp values of the internal test set compounds for models 3a and 4a are shown in Fig. 3.

VIP plot from PLS analysis using all variables and the training set compounds.

Predicted versus observed logPapp values for the internal test set compounds according to A) model 3a and B) model 4a. To facilitate interpretation, a diagonal line has been drawn. The predicted permeabilities fall inside a factor of 10, as indicated by the dashed lines. The correlation coefficient is 0.780 for model 3a and 0.785 for model 4a.
Final Model Construction
The 58 compounds from the original dataset (both training and internal test sets) were used for the final model calculations. Four models were made: using all ten variables (model 1b, Table III), the HBtot, HBD, logP and the three logD variables (model 2b), HBtot and logD7.4 (model 3b) or HBtot and logD8.0 (model 4b). In order to better understand the importance of each descriptor, we calculated the linear correlation between the logPapp and each of the ten variables using all 58 compounds in the original dataset (Table IV). The best correlations were obtained with HBtot, logD7.4 and logD8.0 (Q 2 and R 2 values > 0.5), whereas no correlation could be found between either MW or MV and logPapp (Q 2 and R 2 values < 0.1). Since the same variables that were found important in the PLS analyses had the best correlation on their own, we excluded the other variables from our final models. The statistical significance of the predictive capability of the final models was evaluated with the validate function in Simca-P. The R 2 and Q 2 Y-intercepts were −0.04 and −0.127 for model 3b and −0.06 and −0.132 for model 4b, respectively, which is well below the upper limits of a statistically valid model. The following equations were obtained for models 3b and 4b (equations for the other models are given in Supplementary Material):
Evaluation of the Predictive Power of the Final Models on an External Test Set
We tested the models 3b and 4b by predicting the corneal permeability of an external test set (Table II). Five compounds (ofloxacin, ciprofloxacin, norfloxacin, timolol and atenolol) were also included in the original data set that was used to build the final models and were removed from the external test set before prediction. The eleven compounds that remained were similar to the compounds used for the model building, as all fit inside the elliptic tolerance volume on the PCA score plot (data not shown). The permeability of the compounds in the external data set had been measured in otherwise similar conditions but in a BSS Plus buffer (Alcon Inc.) (31). The results of the logPapp predictions are given in Table V. Model 4b predicted the corneal permeability for these compounds slightly better than model 3b, which is reflected by the lower RMSEP value: 0.39 for model 4b and 0.42 for model 3b.
Prediction of Aqueous Humour Css
Aqueous humour Css of seven compounds (Table VI) was estimated using permeability values calculated by models 3b and 4b and compared to experimental Css from topical infusion studies. Both models estimated Css values relatively closely to experimental Css measured with constant infusion studies. The difference to the experimental Css values was 0.9–6.7-fold and 0.7–6.3-fold by model 3b and 4b, respectively. When experimental permeability values through isolated rabbit cornea were used to calculate Css, values were 0.7–2.5-fold compared to measured aqueous humour concentrations. For five compounds, namely 2-benzothiazolesulfonamide, clonidine, ethoxzolamide, ibufenac and ibuprofen, the differences between experimental and calculated Css values were only 0.9–3.2-fold for model 3b and 0.7–2.1-fold for 4b. This suggests that steady-state concentration in the intraocular tissues may be estimated by using permeability values based on molecular descriptors as well as experimental permeability values.
DISCUSSION
We have collected corneal permeability data from literature, which has been measured in similar conditions for 58 compounds. In order to make as easily applicable models as possible, we used only calculated descriptors in this study, as experimental descriptors can be hard to obtain for new compounds. The initial models predicted the corneal permeability of the internal test set compounds quite well, as the difference in observed and predicted permeability of the most poorly predicted compounds (bromacetazolamide, labetalol, 3-fluoro-6-sulfonamido-3-substituted-3H-1,3,4,-thiadiozolo-[2,3-C]-1,2,4-thiadiazole and cinoxacin) ranged from 3- to 6-fold. Based on the training set models, only three descriptors were considered for the final models: the total amount of potential hydrogen bonding atoms (both acceptors and donors) HBtot, and a distribution coefficient descriptor, either logD7.4 or logD8.0. Linear correlations between HBtot, logD7.4 or logD8.0 and logPapp supported the importance of these descriptors in explaining corneal permeability. The corneal permeability experiments were performed in the pH range 7.4–7.8, which is likely to be reflected in the high correlation of logD7.4 and logD8.0 to logPapp compared to logD7.0. The final models were constructed using the HBtot, logD7.4 or logD8.0 descriptors and the whole data set of 58 compounds. The final models do not differ statistically from the models with up to ten of the calculated descriptors, with Q 2 and R 2 values > 0.7. Notably, a model including only the two distribution coefficients, logD7.4 and logD8.0, had clearly lower Q 2 and R 2 values, ∼0.6, which indicates that both lipophilicity and hydrogen bonding potential are important for predicting corneal permeability.
The influence of the descriptors in the final models on the corneal permeability can be explained physiologically. The cornea consists of three main barriers: the epithelium, the stroma and the endothelium. The epithelium and the stroma are considered to be the most significant of the three barriers, while the endothelium has a minor impact. The logD value describes the transcellular permeation in the cell layers of the epithelium, with increasing lipophilicity leading to increasing permeability. The negative value of the HBtot descriptor indicates that increasing hydrogen bonding capacity decreases the permeability, which has previously been interpreted to indicate that a lipophilic compound that can penetrate the corneal epithelium could have a decreased permeability rate through the stroma if it forms many hydrogen bonds to the colleagen matrix (32). Our results agree with previous studies that moderate lipophilicity and weak hydrogen bonding potential is favorable for corneal permeability (32) as well as for permeability of other membrane barriers like the intestine (8,9) and the blood-brain barrier(33). It should be noted that at very high lipophilicity values (logP > 4) the corneal permeability decreases, presumably due to the poor desorption from the lipoidal epithelium to the hydrophilic stroma. The QSPR models may not predict such lipophilic compounds, but such substances are not likely to be useful anyway for eyedrop administration, since they have poor water-solubility.
In contrast to some previous studies (12,32), we did not find that molecular weight or volume was an important parameter for the corneal permeability, even though molecular weight is known to affect the permeation across pure lipid bilayers (34). This is explained first by the relatively low variance in molecular weight of the compounds in the data set (167–361 g/mol). However, most of the topically administered ophthalmic drugs fit into this MW range. Drug diffusion across the cornea is affected by various processes making the situation very different from a simple diffusion across a lipid bilayer. Only 1–2 cell layers on the epithelial surface contain tight cell layer structures with tight junctions, whereas even macromolecules can diffuse between the epithelial cells in the deeper layers and in the stroma (35). It is known that large molecular weight limits the corneal permeability of peptides and proteins (36), but for such large molecules, conjunctiva, not cornea, is the main absorption route into the eye (37). Thus, the model is applicable in the molecular weight range 167–361, but it should not be used for macromolecules.
Five compounds (corynanthine, 3-chloro-6-sulfonamido-3-substituted-3H-1,3,4,-thiadiozolo-[2,3-C]-1,2,4-thiadiazole, 4-chloro-6-sulfonamido-3-substituted-3H-1,3,4,-thiadiozolo-[2,3-C]-1,2,4-thiadiazole, 4-dimethylamino-6-sulfonamido-3-substituted-3H-1,3,4,-thiadiozolo-[2,3-C]-1,2,4-thiadiazole and yohimbine) in the data set exceeded the limit we had set for an acceptable hydration level of the corneas measured after the permeability experiment. However, the compounds were not excluded from the data set, since their removal did not significantly improve the obtained models, as the effect was only ∼0.01 to the Q 2, R 2 and RMSE values. The only compound that we excluded from this study was acebutolol, whose corneal permeability has been reported by two sources included in this study (4,18). Previously, it has been identified as an outlier in modeling studies (12,13), and, recently, acebutolol has been reported to be actively transported by MDR-1 in rabbit cornea (38).
The final models performed well on the external data set, with calculated permeability values that were within 2.5-fold difference to observed values for nine of the eleven compounds for model 4b. The observed permeability of nepafenac and moxifloxacin was approximately 8-fold and 4-fold higher than predicted with both models, which could indicate that these compounds are actively transported. A two-fold difference can be considered as excellent, since the influence of experimental variation of measuring corneal permeability is easily of the same scale. For instance, the experimental permeability values for the five excluded compounds (ofloxacin, ciprofloxacin, norfloxacin, timolol and atenolol) varied approximately two-fold, even three-fold for atenolol, between the experiments conducted in our original data set and in the external data set. The models performed almost equally well: thus, the selection of model depends on the available descriptors. For highest accuracy, we recommend that the models are used for compounds with descriptor values inside the range of the descriptor values of the test set compounds (listed in the Results section).
Ocular pharmacokinetics after eye-drop administration is complex due to fast non-productive drug loss from tear fluid by solution drainage and systemic drug absorption through the conjunctiva. These loss processes compete with the trans-corneal drug absorption to the intraocular tissues. To avoid the complexities of the transient eyedrop kinetics, we tested the models 3b and 4b with simplified in vivo experimental set-up from the literature, i.e. the constant drug infusion on the corneal surface and steady-state drug concentration measurements in the aqueous humour. The permeability values that the models predict have been measured experimentally in diffusion chambers during several hours of steady-state permeation. Due to demanding experiments, experimental values of aqueous humour Css and ocular clearance are available for only a few compounds, and only seven compounds could be found for this correlation study. The QSPR models appear useful in the prediction of steady-state aqueous humour concentrations, since Css of aqueous humour predicted with models 3b and 4b were comparable to those calculated with experimentally measured permeability values and fairly close to measured Css for several compounds. Compared to the experimental permeability values, the model 4b predicted Css better in three cases, worse in three cases and equally in one case. The range of Css prediction error was 0.7–6-fold for the computational model and 0.7–3-fold for the experimental permeability experiment. It is worthwhile to notice that long-term medication by eye drops also follows steady-state kinetics, but in that case, the aqueous humour drug concentrations fluctuate around the mean steady-state concentration that is determined by mean daily drug input rate and clearance from the aqueous humour.
CONCLUSION
We have built QSPR models that are applicable to estimate the corneal permeability of passively transported drug-like compounds that fit within the property space of the compounds used to build the model. The models are simple, easy-to use, and statistically sound, and can be explained physiologically. The developed models are useful in the early phase of drug development when molecules are chosen for further studies and experimental data is not yet available, whereas computational descriptors are quick and easy to calculate. The models could also be utilized to estimate intraocular steady-state concentrations.
Abbreviations
- Css :
-
steady-state concentration
- HBA:
-
number of hydrogen bond acceptors
- HBD:
-
number of hydrogen bond donors
- HBtot :
-
total number of putative hydrogen bonds, i.e. HBD + HBA
- logP:
-
the logarithm of the octanol-water partition coefficient of the neutral form
- logperm :
-
the logarithm of the corneal permeability
- logD7.0, logD7.4 and logD8.0 :
-
the logarithm of the octanol-water partition coefficient at pH 7.0, 7.4 and 8.0, respectively
- MV:
-
molecular volume
- MW:
-
molecular weight
- PCA:
-
principal component analysis
- PLS:
-
partial least squares
- PSA:
-
polar surface area
- QSPR:
-
quantitative structure-property relationship
- RMSE:
-
root mean squared error
- RMSEP:
-
root mean squared error of prediction
- VIP:
-
variable importance in the projection
REFERENCES
Maurice DM, Mishima S. In: Sears ML, editor. Ocular pharmacokinetics. Berlin-Heidelberg: Springer Verlag; 1984. p. 16–119.
Schoenwald RD, Ward RL. Relationship between steroid permeability across excised rabbit cornea and octanol-water partition coefficients. J Pharm Sci. 1978;67:786–8.
Urtti A, Salminen L. Minimizing systemic absorption of topically administered ophthalmic drugs. Surv Ophthalmol. 1993;37:435–56.
Huang HS, Schoenwald RD, Lach JL. Corneal penetration behavior of beta-blocking agents II: assessment of barrier contributions. J Pharm Sci. 1983;72:1272–9.
Toropainen E, Ranta VP, Talvitie A, Suhonen P, Urtti A. Culture model of human corneal epithelium for prediction of ocular drug absorption. Invest Ophthalmol Vis Sci. 2001;42:2942–8.
Toropainen E, Ranta VP, Vellonen KS, Palmgren J, Talvitie A, Laavola M et al. Paracellular and passive transcellular permeability in immortalized human corneal epithelial cell culture model. Eur J Pharm Sci. 2003;20:99–106.
Linnankoski J, Ranta VP, Yliperttula M, Urtti A. Passive oral drug absorption can be predicted more reliably by experimental than computational models—fact or myth. Eur J Pharm Sci. 2008;34:129–39.
Linnankoski J, Makela JM, Ranta VP, Urtti A, Yliperttula M. Computational prediction of oral drug absorption based on absorption rate constants in humans. J Med Chem. 2006;49:3674–81.
Winiwarter S, Bonham NM, Ax F, Hallberg A, Lennernas H, Karlen A. Correlation of human jejunal permeability (in vivo) of drugs with experimentally and theoretically derived parameters. A multivariate data analysis approach. J Med Chem. 1998;41:4939–49.
Moss GP, Dearden JC, Patel H, Cronin MT. Quantitative structure-permeability relationships (QSPRs) for percutaneous absorption. Toxicol In Vitro. 2002;16:299–317.
Yoshida F, Topliss JG. Unified model for the corneal permeability of related and diverse compounds with respect to their physicochemical properties. J Pharm Sci. 1996;85:819–23.
Fu XC, Liang WQ. A simple model for the prediction of corneal permeability. Int J Pharm. 2002;232:193–7.
Schoenwald RD, Huang HS. Corneal penetration behavior of beta-blocking agents I: physiochemical factors. J Pharm Sci. 1983;72:1266–72.
Li Y, Liu J, Pan D, Hopfinger AJ. A study of the relationship between cornea permeability and eye irritation using membrane-interaction QSAR analysis. Toxicol Sci. 2005;88:434–46.
Edwards A, Prausnitz MR. Predicted permeability of the cornea to topical drugs. Pharm Res. 2001;18:1497–508.
Duffel MW, Ing IS, Segarra TM, Dixson JA, Barfknecht CF, Schoenwald RD. N-Substituted sulfonamide carbonic anhydrase inhibitors with topical effects on intraocular pressure. J Med Chem. 1986;29:1488–94.
Maren TH, Jankowska L, Sanyal G, Edelhauser HF. The transcorneal permeability of sulfonamide carbonic anhydrase inhibitors and their effect on aqueous humor secretion. Exp Eye Res. 1983;36:457–79.
Wang W, Sasaki H, Chien DS, Lee VH. Lipophilicity influence on conjunctival drug penetration in the pigmented rabbit: a comparison with corneal penetration. Curr Eye Res. 1991;10:571–9.
Chiang CH, Schoenwald RD. Ocular pharmacokinetic models of clonidine-3H hydrochloride. J Pharmacokin Biopharm. 1986;14:175–211.
Chiang CH, Schoenwald RD, Huang HS. Corneal permeability of adrenergic agents potentially useful in glaucoma. J Taiwan Pharmaceut Assoc. 1986;38:67–84.
Schoenwald RD, Houseman JA. Disposition of cyclo-phosphamide in the rabbit and human cornea. Biopharm Drug Dispos. 1982;3:231–41.
Eller MG, Schoenwald RD, Dixson JA, Segarra T, Barfknecht CF. Topical carbonic anhydrase inhibitors. III: optimization model for corneal penetration of ethoxzolamide analogues. J Pharm Sci. 1985;74:155–60.
Wu NC, Chiang CH, Lee AR. Studies of carbonic anhydrase inhibitors: physicochemical properties and bioactivities of new thiadiazole derivatives. J Ocul Pharmacol. 1993;9:97–108.
Jankowska L, Bar-Ilan A, Maren TH. The relations between ionic and nonionic diffusion of sulfonamides across the rabbit cornea. Invest Ophthal Vis Sci. 1986;27:29–37.
Advanced Chemistry Developement, Toronto, Canada.
Bolton EE, Wang Y, Thiessen PA, Bryant SH. PubChem: integrated platform of small molecules and biological activities. Annu Rep Comput Chem. 2008;4:217–41.
Umetrics AB, Box 7960, SE-90719 Umeå, Sweden. Simca-P. 10.5.
Wold S. In: van de Waterbeemd H, editor. PLS for multivariate linear modeling. Weinheim: VCH; 1995. p. 195–218.
Rao CS, Schoenwald RD, Barfknecht CF, Laban SL. Biopharmaceutical evaluation of ibufenac, ibuprofen, and their hydroxyethoxy analogs in the rabbit eye. J Pharmacokinet Biopharm. 1992;20:357–88.
Schoenwald RD, Chien DS. Ocular absorption and disposition of phenylephrine and phenylephrine oxazolidine. Biopharm Drug Dispos. 1988;9:527–38.
Rusinko A, Hellberg MR, May JA, Owen GR. Use of MDCK cell line to predict corneal penetration of drugs. 2007;11114649.
Worth AP, Cronin MTD. Structure-permeability relationship for transcorneal penetration. ATLA. 2000;28:403–13.
Norinder U, Sjoberg P, Osterberg T. Theoretical calculation and prediction of brain-blood partitioning of organic solutes using MolSurf parametrization and PLS statistics. J Pharm Sci. 1998;87:952–9.
Malkia A, Murtomaki L, Urtti A, Kontturi K. Drug permeation in biomembranes: in vitro and in silico prediction and influence of physicochemical properties. Eur J Pharm Sci. 2004;23:13–47.
Tonjum AM. Movement of horseradish peroxidase in the cornea, sclera and the anterior uvea. Acta Ophthalmol (Copenh). 1977;55:771–80.
Urtti A. Challenges and obstacles of ocular pharmacokinetics and drug delivery. Adv Drug Deliv Rev. 2006;58:1131–5.
Ahmed I, Patton TF. Importance of the noncorneal absorption route in topical ophthalmic drug delivery. Invest Ophthalmol Vis Sci. 1985;26:584–7.
Kawazu K, Oshita A, Nakamura T, Nakashima M, Ichikawa N, Sasaki H. Transport of acebutolol through rabbit corneal epithelium. 2006;29:846–9.
ACKNOWLEDGEMENTS
This work was supported by the Academy of Finland. Heikki Käsnänen is thanked for valuable comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Kidron, H., Vellonen, KS., del Amo, E.M. et al. Prediction of the Corneal Permeability of Drug-Like Compounds. Pharm Res 27, 1398–1407 (2010). https://doi.org/10.1007/s11095-010-0132-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-010-0132-8