
Abstract
The growth of a thin film of a high-entropy AlCoCuFeNi alloy on a substrate of silicon (100) was studied using molecular dynamics modeling. The simulation was carried out using the embedded atom model to describe the interaction among Al–Co–Cu–Ni–Fe. Interaction between the atoms of Al, Co, Cu, Fe, Ni, and the Si substrate was modeled using the Lennard–Jones potential, and the interaction between the silicon atoms was described using the Stillinger–Weber potential. Total simulation time has reached 50 ns. It was found that at the first stage of deposition small clusters were formed and the process of crystallization started after ~5 ns of simulation, at the characteristic sizes of clusters of about 2 nm. At the end of the simulation, after the 50 ns of modeling, the simulated film contains a face-centered cubic phase, a body-centered cubic phase, a hexagonal close-packed phase, and an amorphous phase. An analysis of the radial distribution of atoms made it possible to determine the distances between the nearest neighbors and estimate the lattice parameters of these phases.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


1 Introduction
High-entropy alloys (HEAs) are a relatively new class of metallic materials developed by Yeh et al. in 2004. Such alloys usually contain from 5 to 13 basic elements in equiatomic or close to equiatomic concentrations (from 5 to 35%). Due to the high mixing entropy, multicomponent high-entropy alloys typically consist of simple solid solutions with face-centered cubic (FCC) or body-centered cubic (BCC) lattices. Many high-entropy alloys possess unique properties, such as wear resistance, resistance to corrosion and oxidation, radiation resistance, high hardness, and strength [1,2,3,4,5]. Thus, the high-entropy alloys may find use as materials for nuclear reactors applications, medicine, electronics devices, mechanical equipment, rocket casings, and engines. The majority of HEAs were investigated in the as-quenched or homogenized state, whereas much less attention was paid to the study of thin films of high-entropy alloys. However, due to the very high cooling rates required to avoid the formation of complex crystallized phases and freeze the low-ordered structures, HEA is sometimes difficult to synthesize as bulk materials. The study of the growth characteristics of complex multi-element HEA films is an important topic of research considering that thin films can be used in various fields of technology [6]. Several papers [7,8,9] have been devoted to the study of thin film growth processes of one of the earliest and best-studied HEA of the AlCoCrCuFeNi system. However, a five-element AlCoCuFeNi HEA was later developed. This alloy does not contain Cr, which is commonly used in HEAs and helps to improve their performance, but significantly increases the cost of the alloy. In this work, the processes of deposition and growth of thin films of AlCoCuFeNi HEA on the surface of a monocrystalline silicon substrate were investigated by classical molecular dynamics (MD) simulation.
2 Simulation Model and Methods
Classical MD simulation studies of deposition and growth of thin films of AlCoCuFeNi HEA have been performed using Large-Scale Atomic/Molecular Massively Parallel Simulator (LAMMPS, Sandia National Laboratory, USA) [10]. Visualization of snapshots and the coordination analysis was done using open visualization tool software OVITO [11]. A three-dimensional cell with periodic boundary conditions only in two horizontal directions x and y was used. The free boundary condition was used in the vertical z-direction to ensure surface growth. The monocrystalline Si (100) substrate was 10 × 10 × 2.2 nm.
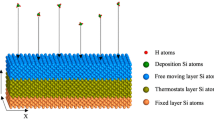
To prevent substrate displacement due to the impact of adatoms on the upper surface of the substrate, the positions of several lower atomic planes were fixed. The intermediate region of the substrate above the stationary atomic planes was controlled by a Berendsen thermostat and maintained at a temperature of 300 K which is similar to the experimental temperature mode to ensure isothermal growth conditions. The atoms of the upper layers of the substrate were not subjected to thermostating and were able to move freely under the impact of the deposited atoms (Fig. 1). The five atoms of metals Al, Co, Cu, Fe, and Ni in the equimolar ratio were directed to a substrate every 10 ps, this leads to the atomic flux density of 5 × 1023 atom/(cm2 s). The rate of 0.5 atom/ps was carefully chosen to allow sufficient time for thermal relaxation with a Berendsen thermostat (i.e., energy relaxation of the substrate after collision). It should be borne in mind that the flux of atoms in the MD simulation is much more than in the experiment, but the obtained data on morphology, structure, and adhesion coefficient are quite adequate [9]. The velocities of the deposited atoms corresponded to the mean kinetic energy of 0.1 eV. The initial positions of the deposited atoms were chosen randomly in the {x, y} plane above substrate so that they were outside the range of the potential that describes the interaction in the z-direction. All of Al, Co, Cu, Fe, and Ni atoms were deposited from an initial position 15.7–16 nm above the substrate.

Schematic picture of the simulation model
The simulation was carried out using the embedded atom model (EAM) [12] to describe the interaction among Al–Co–Cu–Ni–Fe. EAM potentials include empirical formulas that simulate material characteristics such as surface energies, lattice constants, and the heat of solution. The EAM potential is based on the ideas of density functional theory, which generally states that the energy of a solid is a unique function of electron density.
The EAM potential is expressed as a multi-body potential energy function in the following form [12]
where \(f_{i} (\overline{\rho }_{i} )\) is the embedding energy function, \(\phi_{ij} (r_{ij} )\) is a pair interaction potential energy as a function of the distance \(r_{ij}\) between atoms i and j that have chemical sorts a and b, \(\overline{\rho }_{i}\) is the local host electron density. The energy E is the total energy of the atomistic system which comprises the sum of the embedding energy (first term of Eq. (1)) and the short-range pair potential energy (the second term). In the present research, the EAM potential developed by Zhou et al. [13] was used.
The interaction between the atoms of Al, Co, Cu, Fe, Ni, and the Si substrate was modeled using the Lennard–Jones potential
with the parameters summarized in [7, 8], and the interaction between the silicon atoms was described using the Stillinger–Weber potential [14].
3 Results and Discussion
During simulation, 25,000 atoms were deposited to the substrate, which corresponds to the nominal equiatomic AlCoCuFeNi composition, but some of them escaping the surface and the resulting film contained only 24,433 atoms. So the sticking coefficient was ≈ 0.98. The composition of the simulated HEA film and the target composition is shown in Table 1.
Thus, the final composition of the simulated AlCoCuFeNi film was quite close to equiatomic.
According to the results of the simulation, it was found that in the initial stages of growth (0–2 ns) the film represents an islet-like structure formed by small clusters (Fig. 2). Later, the growth of the islands and the formation of continuous coating has begun. Due to the limited number of deposited atoms, it was impossible to obtain a completely continuous film even after 50 ns of simulation time. The film obtained at the end of the simulation is shown in Fig. 3. The maximum thickness of the film has reached 4.3 nm.

Snapshots of AlCoCuFeNi thin film deposited on Si substrate at different times (top view):


AlCoCuFeNi thin film obtained after the 50 ns of simulation time
Following the results of common neighbor analysis (CNA), the process of crystallization started after ~5 ns of simulation, at the characteristic sizes of islands (clusters) of about 2 nm. At the end of the simulation, after the 50 ns of modeling, the simulated film contains a face-centered cubic (FCC) phase (content 20.5%), a body-centered cubic (BCC) phase (content 13.2%), a hexagonal close-packed (HCP) phase (content 39.7%) (Fig. 4), and an indefinite phase (content 26.6%), which, according to the analysis of the radial distribution of atoms (RDF), has an amorphous structure. It should be noted that in bulk AlCoCuFeNi alloys only the presence of FCC and BCC phases was recorded [15], but at the same time in melt-spun films, the formation of the nanotwin structure and stacking faults in the FCC phase was noted [16]. Thus, the specific features of the distribution of the HCP phase atoms (Fig. 4) lead to the conclusion that this phase must be formed mainly of extrinsic (two HCP layers with an FCC layer between them) and intrinsic (two or more adjacent HCP layers) stacking faults in the lattice of the FCC phase. There are also twin boundaries between the FCC twins (a single layer composed of HCP atoms). As for the amorphous phase, its formation is explained by the high cooling rate that occurs during film deposition at which some of the atoms do not have time to rearrange and form a crystalline phase. In the context of the present simulation, this concerns mainly atomic layers located near the film surface, while crystallization processes take place in deeper layers.

Snapshot of AlCoCuFeNi thin-film sliced along the substrate plane (top view). Atoms are colored according to CNA analysis results:

It is well known from the literature, that there are two main criteria by which the high-entropy alloys are usually characterized. This is the entropy of mixing \(\Delta S_{{\rm{mix}}}\) and the enthalpy of mixing \(\Delta H_{{\rm{mix}}}\). However, to predict the phase composition of HEAs, some additional parameters were proposed [1, 2]. These parameters include in particular the valence electron concentration (VEC), the thermodynamic parameter Ω, which takes into account the melting temperature, mixing entropy, and the mixing enthalpy. The important parameter is an atomic-size difference between alloy components which is denoted as δ. Using the data from [17, 18], we calculated \(\Delta S_{{\rm{mix}}}\), \(\Delta H_{{\rm{mix}}}\), δ, VEC, and Ω for the AlCoCuFeNi HEA (Table 2).
According to [1, 2] the HEA alloys for which Ω ≥ 1.1 and δ ≤ 6.6 can form solid solutions without intermetallic compounds. However, simple (not ordered) solid solutions form if −15 kJ/mol < \(\Delta H_{{\rm{mix}}}\) < 5 kJ/mol. The other useful parameter is VEC, which has been proven suitable in determining the phase stability of high-entropy alloys. As pointed in [19] at VEC ≥ 8.0, the sole FCC phase is expected in the alloy; at 6.87 ≤ VEC < 8.0, mixed FCC and BCC phases will coexist and the sole BCC phase is expected at VEC < 6.87. Thus, the simulation results, as well as the experimental results, generally confirm the validity of the above criteria, except for the VEC parameter. But, if the value of VEC is close to the boundary values, predictions of the phase compositions sometimes do not work [5].
Figure 5 presents the total RDF curves after 50 ns of simulation time. RDF patterns are obtained both for the film as a whole and for individual phases separated by CNA analysis. It is clearly visible, that the RDF form of an indefinite phase is intermediate between liquid and crystal, indicating an amorphous structure. The peaks on the RDF curves represent the successive distances between neighbors for different phases in the simulated film. So, from the RDF pattern for whole film these distances are: 1-st neighbor—0.2575 nm, 2-st neighbor—0.3725 nm, 3-st neighbor—0.4525 nm, 4-st neighbor—0.4925 nm.

RDF curves of AlCoCuFeNi thin film: a—whole film, b—amorphous phase, c—FCC phase, d—BCC phase, e—HCP phase
Determining the distances between the nearest neighbors for the BCC and FCC lattices made it possible to estimate the lattice parameters for these phases. Table 3 shows the comparison between estimated parameters of the BCC and FCC lattices from the present MD simulation and the experimental results (Table 3).
As can be seen from Table 3, calculated lattice parameters are in good agreement with the experimental results, taking into account that the estimated average film temperature after 50 ns simulation was above room temperature and was ~350 K.
4 Conclusions
Molecular dynamics simulation was performed to describe the processes of growth and crystallization of thin AlCoCuFeNi HEA films. The structural characteristics and phase composition of films were examined utilizing CNA and RDF. According to simulation results, the growth of AlCoCuFeNi films occurs via a formation of three-dimensional adatom clusters or islands with subsequent coarsening and coalescence. The estimated lattice parameters of the BCC and FCC phases are in good agreement with the lattice parameters of AlCoCuFeNi alloy obtained from the experiment.
References
Murty BS, Yeh JW, Ranganathan S, Bhattacharjee PP (2019) High-entropy alloys. 2nd Edn. Elsevier
Gao MC, Yeh J-W, Liaw PK, Zhang Y (2016) High-entropy alloys: fundamentals and applications. Springer, Berlin
Miracle DB, Senkov ON (2017) A critical review of high entropy alloys and related concepts. Acta Mater 122:448–511. https://doi.org/10.1016/j.actamat.2016.08.081
Bashev VF, Kushnerov OI (2014) Structure and properties of high-entropy CoCrCuFeNiSn x alloys. Phys Met Metallogr 115:692–696. https://doi.org/10.1016/j.jallcom.2016.04.064
Bashev VF, Kushnerov OI (2017) Structure and properties of cast and splat-quenched high-entropy Al–Cu–Fe–Ni–Si alloys. Phys Met Metallogr 118:39–47. https://doi.org/10.1134/S0031918X16100033
Gulivets AN, Zabludovsky VA, Shtapenko EP, Kushnerev AI, Dergachov MP, Baskevich AS (2002) Multilayer compound Co-P films with controlled magnetic properties. Trans IMF 80:154–156. https://doi.org/10.1080/00202967.2002.11871457
Xie L, Brault P, Thomann A-L, Bauchire J-M (2013) AlCoCrCuFeNi high entropy alloy cluster growth and annealing on silicon: a classical molecular dynamics simulation study. Appl Surf Sci 285:810–816. https://doi.org/10.1016/j.apsusc.2013.08.133
Xie L, Brault P, Thomann A, Yang X, Zhang Y, Shang G (2016) Molecular dynamics simulation of Al–Co–Cr–Cu–Fe–Ni high entropy alloy thin film growth. Intermetallics 68:78–86. https://doi.org/10.1016/j.intermet.2015.09.008
Xie L, Brault P, Bauchire J-M, Thomann A-L, Bedra L (2014) Molecular dynamics simulations of clusters and thin film growth in the context of plasma sputtering deposition. J Phys D Appl Phys 47:224004. https://doi.org/10.1088/0022-3727/47/22/224004
Plimpton S (1995) Fast parallel algorithms for short-range molecular dynamics. J Comput Phys 117:1–19. https://doi.org/10.1006/jcph.1995.1039
Stukowski A (2010) Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model Simul Mater Sci Eng 18:015012 . https://doi.org/10.1088/0965-0393/18/1/015012
Daw MS, Foiles SM, Baskes MI (1993) The embedded-atom method: a review of theory and applications. Mater Sci Reports 9:251–310. https://doi.org/10.1016/0920-2307(93)90001-U
Zhou XW, Johnson RA, Wadley HNG (2004) Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers. Phys Rev B 69:144113. https://doi.org/10.1103/PhysRevB.69.144113
Stillinger FH, Weber TA (1985) Computer simulation of local order in condensed phases of silicon. Phys Rev B 31:5262–5271. https://doi.org/10.1103/PhysRevB.31.5262
Liu C, Peng W, Jiang CS, Guo H, Tao J, Deng X, Chen Z (2019) Composition and phase structure dependence of mechanical and magnetic properties for AlCoCuFeNix high entropy alloys. J Mater Sci Technol 35:1175–1183. https://doi.org/10.1016/j.jmst.2018.12.014
Yu PF, Cheng H, Zhang LJ, Zhang H, Ma MZ, Li G, Liaw PK, Liu RP (2016) Nanotwin’s formation and growth in an AlCoCuFeNi high-entropy alloy. Scr Mater 114:31–34. https://doi.org/10.1016/j.scriptamat.2015.11.032
Takeuchi A, Inoue A (2005) Classification of bulk metallic glasses by atomic size difference, heat of mixing and period of constituent elements and its application to characterization of the main alloying element. Mater Trans 46:2817–2829. https://doi.org/10.2320/matertrans.46.2817
Li W-K, Zhou G, Mak TCW (2008) Advanced structural inorganic chemistry. Oxford University Press, Oxford, New York
Guo S, Ng C, Lu J, Liu CT (2011) Effect of valence electron concentration on stability of fcc or bcc phase in high entropy alloys. J Appl Phys 109:103505. https://doi.org/10.1063/1.3587228
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this paper

Cite this paper
Kushnerov, O.I., Bashev, V.F., Ryabtsev, S.I. (2021). Deposition and Growth of the AlCoCuFeNi High-Entropy Alloy Thin Film: Molecular Dynamics Simulation. In: Fesenko, O., Yatsenko, L. (eds) Nanomaterials and Nanocomposites, Nanostructure Surfaces, and Their Applications . NANO 2020. Springer Proceedings in Physics, vol 263. Springer, Cham. https://doi.org/10.1007/978-3-030-74741-1_28
Download citation
DOI: https://doi.org/10.1007/978-3-030-74741-1_28
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-74740-4
Online ISBN: 978-3-030-74741-1
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)