
Abstract
In this chapter, we describe the advances in molecular biology methods and discuss how current methods address the highly heterogeneous nature of human genomic variation. We start presenting a historical perspective of early developments that culminated in our knowledge of human genomes’ enormous diversity. Next, we propose a classification of variants according to these criteria: variant’s length, frequency, and consequence. Variant’s length is critical when choosing between two main groups of methods: fragment-based or sequence-based. We explore these two groups in Sect. 3.3. Two other divisions were considered to describe the methods: if they are targeted or genome-wide approaches and if they focus on common or rare variants. Fluorescence in-situ hybridization (FISH), array comparative genomic hybridization (array-CGH), multiplex ligation-dependent probe amplification (MLPA), and triplet repeat primed PCR (TP-PCR) are discussed in the section of fragment-based methods. Sanger sequencing, genotyping microarrays, and a detailed description of next-generation sequencing are covered in the sequence-based section. In Sect. 3.4, we discuss current applications, including a brief workflow for molecular diagnosis, rare-variant association testing, and polygenic risk scores. Finally, Sect. 3.5 explores perspectives and presents a glimpse of promising methods, such as Cell-free DNA, long-read sequencing, and integrative approaches with other ‘omics’. Although it is still difficult to interrogate the full spectrum of human genome variation using a single method, advances might soon overcome these challenges.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Genomic analysis
- Single nucleotide variants
- Structural variation
- Array-CGH
- MLPA
- Next-generation sequencing
- Target enrichment
- Genome-wide analysis
- Whole-genome sequencing
- Bioinformatics
3.1 Introduction
Human genome variation is highly heterogeneous in scale, distribution across populations, and manifestation (from the molecular level to phenotype). This section will explore current methods that address such heterogeneity, their application regarding the objective of analyses, their advantages and limitations, and, finally, an overview of what is likely to come next. Although a description of historical facts is not the scope of this chapter, a brief reminder of the early developments illustrates the very fast pace rushed by genomic analyses from observations to direct experiments that take place in research and reach medical applications.
The broad variability in scale was empirically stated since the dawn of cytogenetic analyses combined with heredity studies. The rationale behind such a proposal is that variation in the chromosomal scale, observed in microscopy procedures, is relatively rare, and its occurrence is often associated with many clinical conditions (See Chap. 2). Therefore, the heritable phenotypic variability across individuals without major pathologies shall not be explained exclusively by large chromosomal abnormalities but rather from more subtle changes not detected by cytogenetic methods. By the end of the first half of the twentieth century, the determination of species-specific chromosome numbers and the systematic development of analytical methods to study chromosomes, such as karyotyping, naturally led to the comparison of a reference set with samples from patients. Lejeune, in 1959, proposed the correlation of the most common aneuploidy, the trisomy of chromosome 21, with the clinical features typical of Down syndrome [1]. Curiously, a few years earlier, the landmark paper by Watson and Crick describing the DNA structure was published giving rise to modern molecular genetic studies [2]. Across the 1970s, the development and expansion of indirect methods of measuring genetic polymorphisms through electrophoretic patterns in enzymes expressed in blood cells improved our comprehension of the distribution of variation in different populations [3]. Also, during this decade, the use of restriction enzymes, nucleic acid probes, and hybridization had an enormous impact in both detecting variability and pinpointing the genomic context of regions of interest, directly paving steps to genome-wide mapping. Towards the end of the decade, DNA sequencing by chain termination developed by Sanger and colleagues would begin a novel chapter in the sensitive detection of genetic variability [4].
The following decade saw profound advances in molecular biology. In 1986, Kary Mullis developed a method for DNA amplification in vitro, combining a pair of oligonucleotides (of which synthesis had been resolved just a couple of years before), dNTPs, DNA polymerase, and buffer to a series of temperature changes to optimize each step of what became ubiquitously known as PCR (Polymerase-chain Reaction ) [5]. This method allowed precise amplification of specific genomic segments of interest and became an essential tool in most molecular biology protocols. Almost simultaneously, in 1984, the Alta Summit would be the incidental embryo of the largest initiative in human genetics: the Human Genome Project . The ambitious and expensive task would create a definitive reference map from which, to some extent, all projects could rely on and compare their results against [6]. As the project was approved but yet struggled to get funded and to convince the scientific community and society, until the final draft, delivered in 2001, many other advances were published, including the deposition of single-nucleotide polymorphisms and special program reports. One example is the 1993 report, which presented projects to develop and improve mapping, cloning, and assembly protocols, along with computational approaches and ethical implications. Therefore, the Human Genome Project had a pivotal role not only in delivering a reference human genome sequence but also in leveraging an entire ecosystem of research in genomics. Naturally, the next challenge to be tackled would be describing the enormous variant diversity discovered and their role in traits, including disorders with complete or partial genetic etiology.
Even though some current methods promise to approach the full spectrum of variation, it is still challenging to interrogate human genome variation using a single method. This broad range of variant categories also creates a practical problem of how to represent the human genome as a single reference, to which most detected alterations can be compared to, especially when considering population-specific (mostly rare) variation (See Chap. 11). In addition, it implies that for both research and clinical applications, there are several choices to be made which may, in turn, limit the observations and gradually bias the accumulated knowledge on a few classes of variation at the expense of others, due to cost, availability of analytical tools, and interpretation capacities [7].
3.2 Variant Categories
As mentioned before, the heterogeneity of variant types imposes limitations on each technique. Therefore, before choosing an analytical method, it is essential to understand what to expect (and not to expect) of variant categories to be interrogated in each technique. We can classify variants by at least three criteria: size, consequence, and frequency.
Among such criteria, the variant’s length spectrum is key when choosing between two main groups of methods: fragment-based or sequence-based. While at the dawn of genetic analyses, it was a commonplace among scientists to think of variation as major chromosomal rearrangements, currently, due to sequencing techniques, the first type of variant that comes to mind is single nucleotide substitutions or short-ranged insertions and deletions (indels). Although both ends of the spectrum are true and relevant, the amount of variants carried by a population or an individual is likely to be asymmetrically distributed across variant sizes. Very large (over five million base pair—bp) genomic imbalances (insertions and deletions, commonly referred as copy number variants, CNV s; See Chap. 9), translocations and inversions are much less common than single nucleotide substitutions or short-range indels (up to 50 bp). These large events (over 50 bp) are called structural variants (SV) , and there is a substantial range of sizes among them, roughly observed in an inverse correlation with its frequency (it is more common to find shorter than longer SVs) and to its type (it is more common to find CNVs than the other types of SV). As presented in Table 3.1, it is expected that about 4–5 million single nucleotide substitutions can be found on average per diploid genome; about a fifth of that corresponds to short-range indels; 10–100 times less frequently are SVs and, finally inversions , translocations and aneuploidies are the least common types of variants. On the other hand, the total size of the genome affected by the different types of variants presents the inverse trend (Table 3.1) [8,9,10]. Other two types of variants are widespread in the human genome, microsatellites (also known as short tandem repeats, STR ) and mobile element insertions (MEI) . STR consists of stretches of DNA composed of units of 2–15 nucleotides repeated in tandem (See Chap. 6), and because of their highly polymorphic nature they are very useful for DNA profiling easily obtained with standard PCR protocols. Mobile elements are DNA sequences that can change their number of copies or change their location within a genome, eventually affecting genes. MEI constitute approximately 50% of the human genome (See Chap. 8).
Depending on the genomic context, variants can be categorized according to their predicted consequence, which should ideally be validated by subsequent molecular analyses. Each predicted consequence can also be associated with potential changes in the function of the gene products or, alternatively, their direct effect on DNA interaction with regulatory elements.
Annotation tools can cross a variant file and diverse annotation datasets (most based on matched “CPRAs”: chromosome, position, reference allele, and alternate allele) to pinpoint diverse information important to predict variant consequence. If a variant is located within intergenic regions, inferring its consequence can be more challenging since annotations are limited by prediction of sequence-based regulatory motifs or a set of assays that evaluate the evidence of transcription activity, chromatin state, methylation of CpG clusters [11] and, recently, the method of Hi-C sequencing was developed and improved allowing detection of structural interaction of distant regions called ‘TADs’ (Topologically associated domains) and LADs (Lamina-associated domains) [12,13,14]. Such assays hold a promising contribution to genome annotation. The Encode Project Consortium is an effort to systematically improve the understanding of DNA elements and, subsequently, the effect of variants that fall along such regions [15].
When variants fall in regions defined by genes, there are substantially more annotation resources that can be helpful in categorization and inference of predicted consequence. The annotation informs if the variant is noncoding, intronic, UTR regions, or coding. For the latter, annotation informs which amino acid is affected and, if it is synonymous, nonsynonymous, stop gain, stop loss or start loss. Annotation can also flag potential splice sites based on relative location to the exon-intron boundaries and state whether it promotes a frameshift or in-frame (if the variant is an indel).
It is challenging to predict the functional impact of such alterations in their products (proteins or RNA): any given variant may have a neutral consequence; promote a gain of function by either increasing the amount or activity of the product, also known in genetics as hypermorphic ; create a novel function or property (neomorphic ), which could interfere in the other allele (dominant negative effect ) or be expressed in a different tissue or moment (ectopic or heterochronic expression , respectively); finally, a variant can promote a partial or complete loss of function of the original product (hypomorphic or amorphic mutations). Among the latter, it is possible to infer with better precision its consequence, since premature stop codons, loss of start codons, frameshift indels and splicing motifs can be automatically annotated with reasonably high confidence, in addition to the detection of large insertions and deletions that span coding regions of the genes. However, there are pitfalls in automated annotations of potential loss-of-function (pLOF) variants when a frameshift variant has a nearby indel that restores the frame, or if a premature stop codon is located at the last exon (likely to activate nonsense-mediated decay pathway with the affected transcript). Recent algorithms such as LOFTEE (Loss-Of-Function Transcript Effect Estimator) address these putative outcomes [16]. Either way, it is remarkable how such annotations performed on large datasets of variants and allelic frequencies can improve our understanding of a given gene’s intolerance to loss of function by measuring the observed number of pLOFs as compared to expectations based on transcript length and relative position, as a function of mutational saturation in datasets of more than 100 thousand individuals. Such metrics, named pLI (acronym for ‘probability of being loss-of-function intolerant ’) and LOEUF (‘loss-of-function observed/expected upper bound fraction’) after further development of the calculations, are useful resources to estimate haploinsufficiency and the potential impact of variants assuming the gene’s intolerance to inactivation, measured by the observed depletion of pLOF variants, describing genes associated with dominantly inherited disorders caused by hypomorphic function of the gene, which is not always trivial to infer in non-familial (also named sporadic) cases with variants originated by de novo events [16,17,18].
Finally, variants can be categorized by frequency . It is often arbitrary to establish frequency cutoffs and it depends on the application context. In population genomic studies, it is generally accepted that variants above a frequency of 0.5% or 1% in any given population can be considered common. Keep in mind the absolute number of counted alleles: even though the proportion is the same, 1 alternative allele in 200 alleles (0.5%) wouldn’t be confidently tagged as common, as opposed to 0.5% calculated with 100 alternative alleles in 20 thousand alleles (10 thousand individuals, assuming a diploid locus). In molecular diagnosis of monogenic disorders, an upper bound cutoff of 5% can be applied for a stand-alone benign pathogenicity classification [19] and even very low allelic counts in control populations can provide supporting evidence of reduced pathogenic effect in causing Mendelian disorders.
Therefore, very large sequencing-based datasets enabled detection of a wider frequency range, including a set of very rare yet shared variants with allelic frequency as low as 0.005% (result of counting 10 alternative alleles in diploid loci of 100 thousand individuals, or 200 thousand alleles), which are useful in functional inferences such as LOEUF calculations and refinements in pLOF intolerance investigations. In addition, on the extreme of the spectrum, sequencing followed by annotation with large datasets can provide a high number of ultra-rare variants found in a single individual in heterozygous state (termed as singletons ).
As more underrepresented a population is across large public datasets, the larger proportion of singletons can be identified in every sequencing project, given the sampling to avoid small degree relatedness of subjects. As a consequence, as sequencing initiatives containing diverse populations get larger in sample sizes, it is likely that the amount of singletons will eventually reflect a private set of variants shared only in families or lineages, including those that are de novo, that is, present in one individual but not inherited from either parent. Likewise, somatic variants usually detected in sequencing experiments from paired tissues would either fall in the category of mutational hotspots or de novo, besides falling by coincidence on positions that were previously detected in germline experiments. Such frequency spectrum promotes different types of methods for genomic analyses: while common variants can be interrogated in genotyping platforms (containing a selected list of previously known variable loci to be evaluated), rare and ultra-rare variants would only be detected by sequencing methods (without a priori hypotheses on what to find).
3.3 Methods in Genomic Analyses
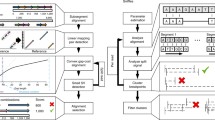
As explained in the previous section, there are genomic alterations of various lengths, a fact that challenges the investigation of the full panorama of variation using a single method. Overall, depending on the length of variants, one method will be optimal over others to detect and describe variation, with high sensitivity and specificity, avoiding false-positives and, particularly, false-negative results. Most current widespread sequencing-based methods begin with random fragmentation of the source DNA in relatively short stretches and all methods rely on sequence alignment. In these cases, detection of duplications and deletions is not trivial, especially in heterozygosity. An alternative strategy, which in fact was developed before sequencing, is to analyze larger fragments of DNA. These methods rely on hybridization or conditional amplification and usually handle longer variants and complex rearrangements better than sequencing-based. We will explore some of these fragment-based methods in the following section and sequencing-based methods right after that. A secondary partition of the methods refers to targeted approaches versus genome-wide approaches, as the former requires some a priori evidence for interrogation of a certain variant, variant list or group of genes, and the latter is exploratory. A decision tree was built to help visualize this rationale (Fig. 3.1). In Sect. 3.4, we discuss some of the current applications, including a workflow for molecular diagnosis, rare-variant association testing, and polygenic risk scores. In Sect. 3.5, we present some promising perspectives, such as, cell-free DNA, long-read sequencing, and omics integration.

Decision tree of selected methods for genomic analyses, based on variant length, frequency, and constitution. Here, the decision was guided by choosing the current gold standard methods for each aim; however, as discussed along with the text, the choice will ultimately favor the best cost-effective method
3.3.1 Fragment-Based Methods
Large SVs are the cause of a significant amount of genetic disorders. In fact, there are more individuals affected by chromosome disorders than for all single-gene diseases . However, as mentioned before, there is a considerable amount of SVs, especially CNVs, per individual genome, indicating neutral or small effects of most variants. As variation grows in length, it becomes less common and more likely to be deleterious. Either way, screening the absence or presence of SVs, and quantifying them (in the case of multiple copies) is relevant in most genomic applications. In this section, we will briefly cover genome-wide and targeted fragment-based methods currently used in genomic analyses. Even though, by definition, whole-chromosome analyses fall within fragment-based methods, traditional karyotype and chromosome banding, observable under a microscope, will not be discussed here. We will cover fluorescence in-situ hybridization (FISH), array comparative genomic hybridization (array-CGH), multiplex ligation-dependent probe amplification (MLPA), and triplet repeat primed PCR (TP-PCR), that are vastly used methods in currently genomic analysis.
3.3.1.1 Fluorescence In-Situ Hybridization (FISH)
The introduction of FISH in the 1980 decade inaugurated the field of molecular cytogenetics that allowed locating specific DNA sequences on chromosomes and greatly expanded the sensitivity of chromosome analysis, becoming a powerful tool used in routine clinical diagnosis [20]. FISH experiment consists of using a fluorescence-labeled DNA or RNA probe capable of hybridizing to a complementary target sequence of a sample DNA. Probes can be labeled indirectly by modified nucleotides containing a hapten or directly by incorporating directly fluorophore-modified nucleotides. Further evaluation of signals under fluorescence microscopy reveals the chromosome location where the labeled probe binds, allowing the detection of various chromosomal abnormalities, including deletions, duplications, inversions, and translocations. The development of FISH came at the same epoch of the advent of the Human Genome Project that made available thousands of clone resources that could be used as probes [21]. One important advantage of FISH is its ability to perform analysis of interphase chromosomes, which allows the analysis of various samples, especially those from solid tumors that do not divide frequently (i.e., do not produce enough analyzable metaphases).
Since its development, many advances have increased the scope and sensitivity of the method. A powerful development was a 24-color karyotyping, called multiplex-FISH (M-FISH) and spectral karyotyping (SKY), in which each chromosome is painted with a different color, allowing a quick scan of all chromosomes to detect large deletions and/or duplications, translocations and complex rearrangements. However, site-specific probes are needed if more detailed information is required [22, 23].
3.3.1.2 Array Comparative Genomic Hybridization (Array-CGH or aCGH )
As explained above, FISH assays are suitable for investigating chromosome imbalances but rely on prior knowledge of which probes to use, one at a time. In contrast, the development of comparative genomic hybridization (CGH) allowed genome-wide screening for CNVs in a single experiment. CGH uses competitive hybridization between a patient and an unaffected control whole-genomic DNA (fluorescently labeled with different colors) to normal metaphase chromosomes. The fluorescence ratio of the patient and control hybridization signals along the chromosomes are then measured, revealing three possible outcomes: an equivalent signal, an overrepresentation or an underrepresentation of the patient’s fluorescent signal [24]. Further development of the technique introduced array-CGH (aCGH), in which microarrays, consisting of a microscope slide with immobilized probes in defined positions, are used as targets instead of metaphase chromosomes [25]. The use of aCGH increased the resolution from 3 to 10 Mb of conventional CGH to 250 kb, and a higher density of probes can be used to increase resolution. Although the use of NGS-sequencing methods is increasingly replacing aCGH for CNV analysis in clinical testing and in research, at present, aCGH is the gold standard method to detect this type of variant and has been particularly useful in studying subtelomeric and pericentromeric rearrangements [26]. However, it is not appropriate for detecting other chromosomal abnormalities, such as inversions and translocation, that can be investigated by M-FISH or WGS.
3.3.1.3 Multiplex Ligation-Dependent Probe Amplification (MLPA)
MLPA is a rapid and cost-effective alternative to diagnose whole-exon CNVs on candidate genes [27]. The MLPA probe consists of two oligonucleotides, both containing the target sequence and a fluorescently labeled universal primer pair, identical for all probes. A stuffer sequence with a different size for each probe is attached to one of the oligonucleotides, giving each probe a unique length. Thus multiple probes can be hybridized simultaneously (multiplex). In the first step, the two oligonucleotides of each probe hybridize to immediately adjacent target DNA sequences. One oligonucleotide contains the binding site recognized by the forward primer; the other contains the binding site recognized by the reverse primer. Then, the pair of probe oligonucleotides that successfully hybridized are ligated, and only the ligated probes are amplified by PCR. Each fragment corresponds to a specific MLPA probe and generates a fluorescent peak that can be detected by capillary electrophoresis. By comparing the peak pattern of the tested sample with the pattern of reference samples, the relative change in copy number can be identified. MLPA can use up to 40 probes in a single reaction, in which each probe is generally used for each exon of a candidate gene. Thus, it is very useful for disorders, such as Duchenne muscular dystrophy, in which a substantial proportion of affected individuals have pathogenic deletions or duplications in a known gene.
3.3.1.4 Triplet Repeat Primed PCR (TP-PCR)
Trinucleotide repeats expansions are the cause of many genetic diseases, particularly neurological and neuromuscular ones (See Chap. 6). Standard PCR protocols are used to detect modest expansions. However, for large expansions (>100 repeats), an alternative method is necessary. Until the development of TP-PCR method in 1996, Southern blotting was the gold standard to analyze this type of variation. However, Southern blot is technically demanding, expensive, and has limited power to detect interrupted alleles, and then encouraged the development of TP-PCR [28]. TP-PCR uses an external primer flanking the repeat plus a primer that can randomly hybridize to multiple possible binding sites within the repeat, resulting in a ladder pattern on the fluorescence trace that enables the identification of expansions compared to samples used as a reference. The method allows identifying large expansions but cannot detect the exact number of repeats if this number is >50. TP-PCR was first developed to scan expanded alleles in myotonic dystrophy, but since then, the technique was validated for many other diseases, such as Friedreich ataxia (FRDA) , Huntington’s disease , and spinocerebellar ataxia type 3 (SCA3) .
3.3.2 Sequence-Based Methods
By definition, methods that evaluate the presence and quantity of DNA fragments, and allow for quantification, irrespective of short-range variations in the sequence itself, were presented in the previous section. On the other hand, sequence-based methods are defined by the ability to interrogate or detect alterations across the sequence of particular DNA stretches. It means that even though fragmentation of DNA itself is often required as an initial processing step, or that the analyzed fragment will physically hybridize with probes, the main outcomes of these methods are the nucleic acid sequences themselves that allow detecting the variation on sequences when compared to a reference. In the following topics, we will cover Sanger sequencing, genotyping microarrays, and detail next-generation sequencing.
3.3.2.1 Sanger Sequencing
Although currently DNA sequencing far surpasses other biomolecules’ sequencing in cost, ease, and, as a consequence, volume of generated data, in early 1970s, methods of protein and RNA sequencing were more advanced, although time-consuming. Nearly in parallel, Maxam and Gilbert’s method of stepwise chemical cleavage of DNA molecule and Sanger and Coulson’s method of DNA extension with chain-terminating nucleotides were successfully implemented in laboratories worldwide, both using fragments separation by electrophoresis [4]. The next development, known as shotgun sequencing, took place in the early 1980s and was extensively used in the Human Genome Project . This method targeted random clones of constructs containing libraries of samples of interest for sequencing and a posteriori computational reassembly of larger DNA fragments. Sanger’s protocol would eventually prevail due to the improvement of the method with fluorescence-based automated machines in 1987.
Sanger sequencing is a reliable method of genomic analyses, targeted for regions of interest which are subjected to amplification or cloning. Therefore, even if a project is designed to cover a library of fragments generated by amplification or fragmentation followed by cloning, individual region of interest analyses will take place. For instance, all exons of a single gene are PCR-amplified or all fragments of a mitochondrial genome from a given tissue are cloned, physically paralleled Sanger reactions (one per plate well or tube) will be performed, generating individual electropherograms, which will be aligned to a reference sequenced or queried across a collection of sequences.
After amplification, products are purified to eliminate non-incorporated nucleotides and primers, and quantified for downstream steps. Sanger sequencing reaction consists of the extension of single strands (one primer is used) by incorporation of standard 2′-deoxyribonucleotides (dNTPs) complementary to the template strand and chain termination after incorporation of fluorescence-labeled 2′,3′-dideoxyribonucleotides (ddNTPs). Reaction parameters such as cycling temperatures, extension times and, especially, dNTP/ddNTP ratios are optimized to produce a library of DNA strands of different lengths with one nucleotide difference each. Each fragment from this library has a fluorescent dye brought by the 3′-end incorporated ddNTP. This reaction is then submitted to a high-density polymer matrix electrophoresis, usually in capillaries, to support the intended separation resolution of one nucleotide. Using a steady voltage, the process of differential migration of the fragments with optimal separation of fragments occurs towards the end of the capillary, where a detector is placed and converts fluorescence to bytes, including intensity parameters. The final result is one electropherogram per reaction, with roughly 800–1000 peaks that can be base-called for further analyses. One standard procedure is to cover the same region at least twice, in two different reactions, one for each strand (namely forward and reverse reactions). Depending on the project, a higher depth of coverage (also known as vertical coverage, meaning how many times a high-quality base is independently sequenced and called) is needed: the draft of the Human Genome Project , which was completed entirely with Sanger sequencing, was 5–10-fold [29].
3.3.2.2 Genotyping Microarrays
The use of hybridization techniques for analyzing nucleic acids started before sequencing technologies and basically consists of exploring the property of complementarity between base pairs of anti-parallel strands of DNA and RNA. As mentioned before for FISH and array-CGH, it is straightforward to observe the results of hybridization between a probe (the sequence we have prior information about) and the region we are interested in detecting/quantifying.
The ability to miniaturize the synthesis of oligonucleotide probes onto a solid phase (usually glass slides, in a process called photolithography), the implementation of improved digital cameras, and the growing knowledge on allelic diversity contributed to the development of ever-higher density genotyping microarrays, often called DNA chips [30]. The overall methodological workflow involves enzymatic fragmentation followed by end repair, adapter ligation, and PCR, enriching the sample in products of less than 1 kb. DNA probes in the chip harbor the selected SNPs in several positions (overlapping probes), and SNPs themselves are selected based on frequency and by location, usually between within two restriction enzyme sites 1 kb apart. Allelic detection by hybridization without this a priori step of size selection by amplification can produce non-specific calls that increase background noise. Although these steps are fairly similar between the two main commercial microarray platforms (Affymetrix and Illumina), each has their own specificities: Illumina uses a probe-linked beadchip embedded in the slides and has a single base extension; Affymetrix uses ligation, and several washes to remove less stringent hybridizations.
These tools were essential to decrease the costs of genome-wide analyses for several applications, from family-based segregation and linkage studies to large sample-sized genome-wide association studies (GWAS) , since automation greatly increases the through-put and reduces experimental variability. Currently, commercially available microarrays include many options of high-density sets of variants (>500 k markers) and enrichment of clinically relevant variants or copy number variants; besides the possibility of some degree of customization. Outside basic research applications, most companies that offer direct-to-consumer testing for ancestry or disease risk alleles are microarray-based. It is important to consider that each microarray chip is designed to interrogate a list of polymorphic alleles previously detected by sequencing projects, which might be biased on their own. Some commercial microarrays were developed to include population-specific variants and to some extent contribute to studies on diverse populations. Many GWAS studies benefit from an increasing density of variants through imputation, in which unobserved genotypes are inferred by using haplotypes from reference panels [31].
3.3.2.3 Next-Generation Sequencing
Pinpointing the large frequency spectrum of genomic variants, from ultra-rare to common, is only achievable by directly sequencing the DNA. As mentioned above, Sanger sequencing method revolutionized genomic science by providing a reliable and reasonably automated protocol that could consistently deliver the nucleic acid sequence of stretches of 700–800 bp. The main limitation of Sanger is parallelization itself. The Human Genome Project public effort overcame this issue using the challenging, costly, and time-consuming solution of distributing the job among hundreds of facilities worldwide, while the private effort did a similar approach, except that the hundreds of machines were centralized in a single facility (improving optimization and reducing costs). Either way, sequencing a whole human genome by the end of the first published draft in 2001 was still priced in the order of magnitude of 100 million dollars.
The (Recent) History of Next-Generation Sequencing
During the late 1990s and early 2000s, emerging sequencing technologies evolved from the combination of microfluidics and molecular assays advances such as emulsion PCR, bridge PCR, and adapter ligation. Three main next-generation sequencing (NGS) platforms were released almost simultaneously in 2005 and 2006: 454 pyrosequencing (later acquired by Roche), Solexa sequencing by synthesis (SBS, later acquired by Illumina) and Agencourt sequencing by oligo ligation detection (SOLiD, later acquired by Applied Biosystems) [32].
Illumina prevailed as the more broadly used method, and its in-depth protocol will be discussed in this section. Before each method is briefly covered, a few important NGS parameters are presented. Considerations about them define the usefulness and cost-effectiveness of each method and quality standards to be observed during analyses. As mentioned previously, depth of coverage is the number of times a base is independently called (i.e., read counts overlapping a single base). Although there is no consensus on an optimum minimum depth, 10-20x is usually the aimed range, even though some applications like somatic mutation detection require deeper coverage. The second parameter is the horizontal coverage, meaning the genomic extension that the sequencing project is aimed at mapping. Both parameters can be planned during the experiment design. The third parameter is the read length, which is usually restricted by the sequencing method. Finally, also limited by the sequencing protocol, the sequence output measured in number of reads and megabases is a value expected by each protocol and sequencing machine. All parameters are useful when designing the experiment, including the ability to multiplex several samples per run, and to expect minimum values for quality control and downstream analyses.
Pyrosequencing protocol provided by 454 involved fragmentation of genomic DNA and ligation to adapters, which would be baited by beads, generating an immobilized library. These beads were then emulsified for optimal isolation (one bead per emulsion compartment), later distributed on a picotiter plate for sequencing cycles (adding polymerase and one dNTP at a time). Incorporated nucleotides would release pyrophosphates, which would cascade a reaction of ATP and luciferase-catalyzed luciferin oxidation, generating visible light. Each well would provide up to 700-bp reads (typically 500 bp), not far from Sanger sequencing and, therefore, minimizing alignment and assembly procedures. Sequencing output of the latest Roche 454 machines was about 14Gb. Homopolymer detection (contiguous nucleotides of a single base) is challenging in most NGS protocols and was particularly critical in 454 chemistry.
Applied Biosystems SOLiD methods also used beads with immobilized oligonucleotides complementary to adapters linked to DNA fragments to be sequenced. Fragments on beads were also amplified and spread onto glass slides in polonies (‘polymerase colonies’). The extension and detection, however, used a unique experimental design where 8-bp long oligonucleotides had four different fluorescent labels to four dinucleotides located at the 3′ end, while the remaining nucleotides of the probe were degenerate. When a dinucleotide is stabilized, a ligase catalyzes the phosphodiester bond, unextended strands are capped and the fluorophore removed with 3 bp cleaved at the 5′ end of the probe, allowing the next cycle. The dinucleotide color code can be decoded by repeating the cycles offsetting the initiation primer by 1 base pair (n-1, n-2, n-3, and n-4), tiling the probe-fragment complementarity and generating overlaps between each sequence of colors. Although a complex procedure, this dinucleotide color-coded overlaps allows each base to be covered twice per read, increasing accuracy without substantially increasing cost. Each SOLiD output per run as for the last available machine was 90Gb. Two major drawbacks for this method probably caused its resistance to use and later discontinuation: very short read length of 50–60 bp, promoting a reduction in alignment quality and computational complexity increase; and problems in sequencing palindromic regions, which form hairpins and reduces ligation of probes to a critical level.
Illumina’s Sequencing by Synthesis
Illumina has established the main adopted technology by overall markets using the sequence-by-synthesis (SBS) method developed and improved from a combination of the original patents by Solexa and Lynx technologies. SBS consists of fragmentation of DNA in regular-sized segments to be ligated with adapters. Usually, fragments are around 500 bp of length obtained from native DNA, but mate-pair libraries aim at 2–5 kb long inserts and linked-read assays start with high molecular weight DNA that is isolated prior to fragmentation (Chromium technology, 10X Genomics). In all cases, only the extremes are sequenced (paired-end method), but regular spacing (short and long-range) improves de novo assembly and the ability to detect SVs. Indexing the fragments during library prep consists of ligating oligonucleotides to the ends of the fragment, before adapters’ ligation. Indices play a barcoding role allowing libraries to be pooled together, therefore multiplexing the run with several samples. After sequencing, computational scripts for demultiplexing will reassign reads to individual sample identifiers. Both adapters have complementary surface-bound oligonucleotides in a structure called a flow cell. Fragments are denatured and each strand is annealed with the fixed oligonucleotides for extension, and complementary strands are synthesized, now covalently attached (with phosphodiester bonds) to the flow cell. Clusters are generated by bridge amplification, populating the surroundings of the initial fragment with copies. As sequencing cycles begin , these clusters will be detected and monitored by a camera to register the incorporation of each dNTP to the fragment. The key for each added dNTP is that they have a reversible property, blocking the incorporation of more than one dNTP per cycle and unblocking after washing for further extension (Fig. 3.2). Reads range from 50 to 300 bp, but for genomic purposes are usually set to 150 bp. The paired-end sequencing allows for another round of dNTP incorporation cycles (also 150 bp), extending the complementary strands and providing a total of 2×150 bp long separated by the remaining unsequenced spacer of the original sample. It is relevant to stress that the paired reads are not reverse complements of each other, but rather the extremities of each fragment. The redundant representation of a region more than once (measured by depth of coverage) is, therefore, a result of independently sequenced reads from different fragments [32]. Output of Illumina SBS relies on a few parameters, some fixed and some adjustable: flow cell load capacity (there are different flow cell designs for each machine), cluster density (there is an optimal value), choice of single-end versus paired-end and sample multiplexing. Currently, Illumina offers a range of systems with outputs from 2 Gb to 6 Tb, which can be chosen in accordance with the project. In the clinical routine, it is a common practice to confirm findings using a different method, Sanger sequencing if the variant has short length. Sanger sequencing is commonly used to confirm or exclude the parent’s carrier status.

Overview of NGS protocol (Illumina’s sequencing by synthesis). Genomic DNA is fragmented in regular sizes (around 500 bp), which are ligated to adapters (lilac and light yellow) with indices (in this example, light blue for one individual and light red for another individual). Samples are pooled and optionally target enriched (in this example, carried out by probe capture attached to beads). Libraries are loaded onto flow cells containing oligonucleotide probes complementary to adapters. In any given probe, a fragment will be used as template for extension, and the original fragment is washed away. In a series of bridge amplifications, a cluster of fragments attached by both strands is generated. Sequencing by synthesis is performed in cycles, including incorporation of dNTPs that function as reversible terminators, and clusters are monitored for dNTP incorporation. Cycles restart to the reverse fragment within the same cluster, providing a paired-end read
Target Enrichment
As we have previously stated, the growing capacity of parallel sequencing millions to billions reads provides an opportunity to either go very deep on vertical coverage or very broad on horizontal coverage. In latest Illumina models (NovaSeq 6000), one can choose the higher output system of dual flow cells with 6 Tb output per run (and 20 billion reads). That means one human whole-genome (diploid genome of 6.4 Gb) could be sequenced at over 900x of coverage, or 900 whole-genomes could be sequenced at 1x. Although possible, both situations are not economically feasible, or actually desirable, for most applications. As we will see in the next section, whole-genome sequencing aimed at 30x is an accepted standard. But how would we make use of such a large output to make it both useful and cost-effective?
Besides multiplexing, which allows for pooling multiple samples along the same sequencing run, there are methods designed to enrich libraries with regions of interest that can be prioritized in sequencing experiments. Prior to the distribution of the libraries onto flow cells for cluster generation and sequencing, target enrichment methods can be performed. Two main strategies can be chosen to enrich libraries: probe-based capture or amplicon generation. In the first strategy, single-stranded DNA or RNA oligonucleotides are designed for the chosen regions of interest, synthesized, and attached to a solid phase surface such as a glass slide (resembling microarrays), or, more commonly, beads, followed by hybridization steps. Oligonucleotides probes must be long enough to allow for some mismatching, including indels, otherwise, allele dropout could be an issue (when one allele is not captured with equivalent success of the other allele). This also can be achieved by designing multiple overlapping probes spanning the region of interest (in a tiling setup), as some commercial options emphasize to be an improvement in enrichment by capture efficiency. The second strategy is to use multiplex amplification, generating amplicons of the regions of interest, using either primers or molecular inversion probes. There are advantages and disadvantages to amplicon-based enrichment. Customization and processing are easier and simpler than capture by hybridization of probes, and overall costs can be lower. However, there is a limitation on the number of amplicons that can be generated (amplicons that enrich whole-exomes were developed later and are usually more expensive than probes counterparts). Comparisons generally indicate that even with higher coverage, amplicon-based enrichment can be less uniform and provide a higher proportion of false-positives and false-negative results. However, for smaller panels and applications such as microbiome profiling, amplicon-based enrichment is widely considered [33].
Probably the most used application of NGS so far is target-enriched sequencing of human samples, specifically whole-exomes and gene panels. The combination of technological advances , reduction in equipment and reagent costs, multiplexing samples and target-enrichment led to an explosion of NGS data generation from the 2010s, both for academic purposes and clinical setups. Noteworthy is that the management and analysis of the incredible amount of data generated since then were only possible with the parallel development and advances of bioinformatics. Laboratories were able to standardize and streamline protocols to offer gene panels directed to groups of disorders such as hereditary cancer, neuromuscular and developmental disorders, costing no more than any complex health-related exam. Whole-exome sequencing (WES) had a pivotal role in the identification of genes associated with monogenic diseases, many of which required few family members to achieve probable candidates, a task that used to take time-consuming steps of STR profiling and linkage analyses. WES varies in terms of horizontal coverage, depending on how far into UTRs and introns the probes are designed to capture the target, but 120 Mb per diploid exome is a general value to consider. When aimed at 100x, the above-mentioned Illumina NovaSeq output could produce 500 WES per run, at a cost (reagents only) of about 100USD. Even adding other essential costs of equipment, computational resources, and, especially, high-skilled staff for producing and analyzing the data, WES will certainly stay as the gold standard for molecular diagnostics for a while [34, 35].
Whole-Genome Sequencing
Skipping the step of target enrichment and loading onto the flow cell the library of fragments ligated with adaptors and indices will produce the once holy grail of the scientific community worldwide: sequencing the whole human genome. The 1000 Genomes Project was launched in 2008 with the ambitious effort of sequencing thousands of individuals from 26 populations. Phase 1 included low-coverage whole-genome sequencing (WGS) of 179 individuals, WES of nearly 700 individuals and high-coverage WGS of two trios. The project was able to deposit a large number of variants previously unknown and paved the way for several other initiatives [36]. Now, there are many countries aiming at 100 thousand WGS along with extensive clinical data to improve precision medicine initiatives by providing both reference datasets and research substrates for the discovery of novel genes and loci associated with traits.
It is important to mention the advantages and disadvantages of WGS over other methods. As compared to WES, sequencing the entire genome allows interrogation of both common and rare variants within and outside coding regions. The high-density microarrays are useful when researchers are agnostically detecting association signals across the genome (when performing GWAS), and more often than not, signals fall within intergenic or intronic regions. If the association is truly a proxy of causal variants nearby, WGS would be useful in both steps, allowing fine-mapping to pinpoint candidates of causality within variants of lower frequency. WGS uniformity in horizontal coverage allows an improvement in phasing (or haplotyping) estimates (the attribution of the relative position of two or more alleles in cis or trans configurations). By chance, paired-end reads can harbor informative variants that aid algorithms to keep track of the phase, which can be useful in classification pathogenicity of variants in recessively inherited disorders. Phasing software can take advantage of this information together with estimated haplotype frequencies to infer haplotypes throughout the genome. The alternative to that is applying the gold-standard phasing procedure, which analyzes trios and duos; however, this strategy is often disregarded in favor of sampling more unrelated individuals. In addition, both properties of long-range horizontal coverage combined with uniform vertical coverage facilitate detection, mapping, and description of SVs, which will be covered in the next section.
An important disadvantage of WGS over WES or targeted panels is the cost, not only of equipment and reagents but also of computational resources to process and store generated data. While the wet lab steps (including sequencing runs) of WGS had just breached down the 1000USD barriers, storage alone can represent 5% of this value per year. In addition to that, annotation of noncoding regions is still challenging and the gain in diagnostic capacities from WES to WGS is not yet clear. For research purposes, on the other hand, WGS is an excellent tool that embraces many analytical possibilities [37].
NGS Analyses
The extensive use of NGS-based tests and NGS for research gave rise to a whole community of users, composed of wet-lab researchers and technicians, bioinformaticians, programmers, and analysts. Two interesting things came as a result of this: standardized protocols and recommended guidelines were developed and improved over time, building confidence and reproducibility of results; and a vast universe of alternative methods were tested allowing researchers to apply NGS in several different manners. This section will focus on the basic pipeline and comment on workflows that support the most common applications.
As previously described, the sequencer captures the position and intensities of clusters across flow cells, cycle after cycle, with a camera-like sensor. To reduce the volume of data generated at this process, a Real-Time Analyzer software (Illumina proprietary resource) converts data to BCL format, a binary file that contains raw information on each cluster output. This file must be exported to a server where the primary bioinformatics pipeline will take place. The first step involves demultiplexing and conversion of BCL to FASTQ files, a text-based file that stores the sequence ID (including the cluster position, useful for paired-end sequencing), the sequence itself, and per-base sequencing quality.
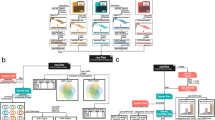
At this point, mapping can proceed using alignment software. Two strategies can take place: de novo assembly or alignment to a genome reference. Keep in mind that the original Human Genome Project had precisely this challenge (besides generating all raw data): assembly is computationally costly since all reads must query themselves to build up contigs based on overlapping stretches. Since then, updates on the human reference genome were made, and interesting discussions on how a reference should be represented to account for diversity became common. Alignment algorithms can be tuned in several parameters: if they are too strict, reads containing true alternate alleles might not be considered; if they are too permissive, mapping quality will decrease, since reads will align to many loci. Also, indels and larger SVs tend to penalize alignments and depending on the size of the variant, the read length itself, and the genomic context, some of those variants will not be aligned and, therefore, will not be called. BWA-MEM is the more commonly used free aligner and outputs a raw BAM file (the binary version of a SAM file) which provides information on the position of the reads relative to the reference genome of choice and mapping quality. Then, a few intermediate steps that involve marking and removing PCR duplicates, local realignment for improved indel detection, and recalibrating base scores to the local and overall sample context are performed to obtain an analysis-ready BAM file . BAM files are ready for visualization by a genome browser such as IGV, providing an image of piled-up reads aligned to a reference genome sequence (Fig. 3.3a). Base mismatches of the aligned data could only mean three things: true variants, sequencing errors or alignment errors. The following step is to integrate the mismatches across the reads, effectively calling variants. HaplotypeCaller is the variant calling tool recommended by the Genome Analysis Toolkit (GATK) , a recommended general protocol provided and maintained by the Broad Institute of Harvard and MIT and used worldwide [38]. It outputs a (very large) file, named gVCF, that has the same format of a standard VCF , except it contains the genotypes of all called positions, whether it is a variant call or not (Fig. 3.3b).

Schematic representation of BAM file from one individual (a), gVCF files from two individuals (b), and combined VCF file with two individuals (c). In the BAM file (a) we can see an example of paired-end reads containing variants in colors (all grey portions of the reads are matching the reference). Genomic position is represented as a ruler above, and depth of coverage as the light grey graph below. Below the reads, the reference genome is shown along with a basic gene/transcript annotation in blue (in this case, representing the two first exons, where the first has a 5’UTR portion and direction of gene in the genome). Note that paired-end brought evidence of phasing between the first green variant (in homozygous state) with the second blue variant (in heterozygous state), which in turn is also in cis with the third red variant. gVCF files contain all called positions (b), and variant based quality (which is also included in the INFO field, along with several other metrics of sequencing, alignment, base calling, and variant calling). VCF file summarizes positions with variants (c) and includes site quality and information (now as an aggregate of all individuals included in this combined file). This is ready for annotation and downstream analyses
The gVCF can also block information whenever a sample has reference alleles for consecutive stretches (indicating the end of each block in the INFO column), as well as an indication of the spot for a non-reference allele. The FORMAT column will contain guidance to read the genotypes, which generally contain the inferred genotype itself referring to REF and ALT status, depth of coverage for each allele (AD), depth of coverage at the site (DP) and genotype quality score (GQ). Some files can contain a strand-specific allele depth, which can be useful to evaluate strand bias. The ID column represents the only place with “outside” information (an annotation, by definition), and should be completed with rsID from dbSNP. INFO column is usually populated with several quality statistics: in gVCF refers to the site and individual genotypes, but in combined VCF may include the overall site quality and other metrics such as allele count (AC), allele number (AN), inbreeding coefficient, and Hardy-Weinberg statistics . The same for QUAL, which individually represents the site and genotype quality but in conjunction with other samples, provides a flag with confidence levels for the site (Fig. 3.3c).
Next, this individual gVCFs can be combined with other samples in a joint cohort to a joint-call step that will result in a standard VCF file containing only positions in which there is at least one alternate allele. The header of VCF files can store a number of meta-information, including the description of the entries present in the body of the VCF file and the commands used to obtain the VCF. The steps presented here compose the pipeline recommended by the GATK Best Practices to identify germline short-range variants, which is used by most bioinformaticians and very appraised by the scientific community. The GATK Best Practices validated pipelines with recommended software, quality parameters, and continued improvements for different types of variants.
The steps presented here compose the pipeline recommended by the Genome Analysis Toolkit (GATK) Best Practices to identify germline short-range variants, which is used by most bioinformaticians and very appraised by the scientific community. The GATK Best Practices is a product of the collaborative effort provided by the Broad Institute (Boston, MA) which thoroughly validated pipelines and provided a workflow with recommended software, quality parameters, and continued improvements for different types of variants.
Either an individual VCF or a combined VCF (with multiple individuals) can now be annotated to provide context to the findings. As mentioned above, annotation is a procedure that will systematically intersect findings with previous knowledge stored in datasets and includes straightforward basic annotations such as rsID, gene, and transcripts. There are several annotations that can be relevant for various analyses such as the frequency of variants across different datasets, the association of genes to disorders, pathogenicity assertions, prediction of deleteriousness by different algorithms, context of protein domains. There are several annotators in use, most freely available such as ANNOVAR [39] and Ensembl Variant Effect Predictor (VEP) [40], but it is common that laboratories add in-house scripts for specific annotations.
In the previous sections, we have stated that NGS-based analyses are not the gold-standard method for the detection of large SVs. One reason is that uniformity of reads (both in vertical and horizontal coverage) is not always predictable and varies within the individual sample and across samples. For instance, although the peaks of depth surrounding an exon in a WES sample can reach over 200x (in a sample aimed at 100x), it is not trivial to infer if that particular exon was better captured than the others or if it represented a duplication. The same goes for heterozygous deletions: a drop in depth of coverage can be caused by a deletion or by a lower capture performance. However, there are several algorithms and workflows that use read-depth measures to successfully detect a high proportion of CNVs from NGS data, most of the time through exome or gene panels. In fact, NGS-based CNV analysis is increasing in both clinical and research contexts as a cost-effective choice to study a broader range of variants [41]. The optimum choice for short-range NGS-based CNV analysis would be to use paired-end deep-coverage (>30x) WGS, which has the main advantage of more coverage uniformity (i.e. less variation of depth along chromosomes and among individuals). This characteristic facilitates the definition of a reference depth to which deviations can be tested and allow the extensive use of read-pair (RP) and split-read (SR) methods [42]. RP detects discordant pairs in which the span and/or the orientation of read-pairs are inconsistent with the expected insert size. If a deletion spans a well-covered region, paired-end reads will align to the boundaries of the deletion, and will appear to be more distant from each other than expected (Fig. 3.4). On the other hand, read-pairs closer than expected indicate insertions. SR identifies split sequence-read signatures breaking the alignment to the reference (gaps indicate deletions and stretches indicate insertions), detecting the precise boundaries of the variation (breakpoints). RP and SR are useful to identify other types of SVs, including inversions and translocations [43].

BAM file with an example of paired-end clustering and split reads. One of the chromosomes had a deletion, which dropped local depth of coverage and promoted paired-end alignments to be more spaced than average. Also, a few reads were able to align to the reference after splitting. This example was adapted from a patient case from HUG-CELL (USP, São Paulo, Brazil)
3.4 Analysis of Rare and Common Variants to Understand Diseases and Traits
3.4.1 Workflow for Molecular Diagnosis
Molecular diagnosis for patients affected by rare genetic disorders with monogenic patterns of inheritance is straightforward [35]. It begins with a deep clinical evaluation of the patients and family members, which will provide clues for diagnostic hypotheses. Family history of disorders or related phenotypes, occurrence of consanguineous marriages in the family, age of manifestation and clinical progression, and age of parents help the physician narrow down possible candidates. Further access to public databases such as OMIM , Orphanet , and GeneReviews , indicate one or more genes previously associated with the condition or part of the phenotypes that can be prioritized and interrogated with methods described in this chapter. Choosing the best method to start the diagnostic investigation is not trivial: previous knowledge on the disease and genes is critical for establishing a rational stepwise set of tests. Once the test is performed, a complete pipeline for analysis, including access to databases of variants and related literature, will provide a report to be returned in genetic counseling consultation. In the case the test is NGS-based, such as targeted panels or WES, there are general recommendations provided by the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) that support the workflow, pathogenicity classification, and handling of secondary findings [19]. There are several ethical concerns to this process, which are not the scope of this chapter.
There are, however, concerns regarding the clinical level of evidence and penetrance of variants that must be addressed. Monogenic disorders are generally caused by one or two variants of large effect size, meaning that the presence of such variants greatly increases the risk of manifestation (up to complete penetrance in some cases such as Huntington’s disease). Most common disorders, however, have multifactorial etiologies, with an environmental component and a genetic component. Given that the genetic component is usually polygenic, causative variants have, individually, low to moderate effect sizes and are distributed across dozens to hundreds of loci. In research, large GWAS efforts identified many loci associated with multifactorial traits, indeed improving the knowledge on the architecture of such traits and unraveling part of molecular and cellular mechanisms involved in these conditions . Some alleles in genes such as APOE were robustly associated with Alzheimer’s disease and cognitive impairment with a relatively high odds ratio (3–15, depending on the study and zygosity), but as any single susceptibility allele of a multifactorial disorder, it explains only partially the phenotypic variability [44]. Even though companies offer tests and reports with variants of reduced penetrance in direct-to-consumer tests, the clinical validity of such associations is still under discussion by scientific and medical societies.
3.4.2 Rare-Variant Association Testing
Although successfully discovering tens of thousands of variants robustly associated with diverse traits and better understanding their genetic architecture, GWAS hits explain only a small proportion of phenotypic variability (a problem referred to as the missing heritability). GWAS is, by design, focused on common variants (usually defined as variants with minor allele frequency > 0.01) obtained from genotyping are interrogated using microarrays. Part of the missing heritability arises because many common variants have very small effect sizes that could be detected only with increasing sample sizes. The ever-increasing sample sizes of recent GWAS and meta-analyses intend to address this problem. Another part of the missing heritability arises from not considering rare variants, which were shown to collectively reach significant effect sizes. However, even if GWAS included rare variants in its analysis, detecting association with standard GWAS protocols would require enormous sample sizes. To address this issue, methods for collapsing rare variants per gene, per genomic region, or per pathway have been developed and improved overtime. The rationale is quite straightforward: case and control groups of individuals are sequenced (ideally WES or WGS) and rare variants are computed per group, within candidate genes, regions, or pathways, or alternatively, multiple combinations of genes are tested. Variant annotations can be used in weighing each aggregate, and some tests are prepared to combine common variants as well. Many publicly available algorithms perform these tests, also known as burden and nonburden tests, that can well complement GWAS or be used when larger sample sizes are not available [45, 46].
3.4.3 Polygenic Risk Scores
GWAS usually identifies individual association signals for each variant and strict thresholds are applied to ensure the exclusion of false-positive results. However, as mentioned, there are many loci truly associated with the traits that do not reach statistical significance due to several reasons: reduced effect size, low frequency, population-specific linkage disequilibrium patterns, and epistasis with more loci. With the recent availability of very large cohorts with genotyping data and comprehensive phenotypic information, such as the UK BioBank , testing combinations of variants based on GWAS summary statistics (effect sizes measured as odds ratio or regression betas) could be performed [47]. Using a protocol for reducing interdependence of variant’s signal by pruning blocks under linkage disequilibrium and thresholding p-values, many researchers are exploring UK BioBank’s large sample sizes (about 500 thousand genotyped individuals) to identify combinations of variants that successfully stratify individuals by disease risk (or trait levels). These profiles can be interrogated in a validation set, providing a distribution of a polygenic risk score (PRS). Both the scientific communities and medical societies are enthusiastic about the application of PRS in several traits and disorders, since these profiles would eventually anticipate clinical interventions for individuals at higher risks, at ultimately low costs (microarrays). The full extent of rare variant contributions to PRS is yet to be elucidated. However, there are already studies on breast cancer and hypercholesterolemia showing the combined effect of different PRS risks and carrier status of high effect size monogenic variants, providing a good perspective on the clinical applications of PRS and that WGS might be the ultimate test to embrace all dimensions [48].
One important drawback, currently under discussion, is that source samples used in GWAS are still biased towards Europeans, and PRS transferability to other populations is challenging, with significant reductions of predictive power (See Chap. 11). Holding the same caution alert on direct-to-consumer testing, it is noteworthy that admixed individuals or individuals from diverse populations different from the original sourced in large GWAS might not benefit from such PRS-based tests and may receive reports with reduced clinical validity [49].
3.5 Perspectives
A brief glimpse of the rapid development and improvements in genomic analysis methods was presented in the previous sections. The consolidation of NGS as the gold-standard sequencing method does not mean that all challenges imposed by genomic complexity have been fully addressed. In this section, our intention is to introduce selected methods under implementation that are likely to complement or eventually replace the current protocols.
3.5.1 Cell-Free DNA
Cell-free DNA (cfDNA) refers to any degraded DNA fragment present in the circulation and other biological fluids. They were first detected in 1948 and since then many studies investigating their possible association with different diseases were conducted. Currently, detection and analyses of cfDNA originated from tumors and from the fetus in a pregnant woman are widely used. The rationale is to detect somatic mutations from tumor and either chromosomal imbalances (such as aneuploidies) or de novo mutations from the fetus (absent from mother and father). Plasma cfDNA concentration is usually low, and DNA is very fragmented, so the depth of coverage usually is aimed higher and paired comparisons are performed: plasma from patient vs buffy coat (blood fraction containing mononucleated cells) from the same patient, or plasma from mother vs buffy coat or saliva from mother [50].
The investigation of cfDNA from tumors is known as liquid biopsy, a noninvasive procedure that allows routine clinical screening to detect resistance mechanisms to inform treatment, and to monitor the response to treatment and residual disease. Besides, evaluating the use of liquid biopsy as a clinical tool for early cancer detection is currently an active area of research.
Another widely application of this type of sample is to obtain cfDNA from the fetus and screen for the most common aneuploidies through NGS sequencing. This test, known as noninvasive prenatal testing (NIPT) or noninvasive prenatal screening (NIPS) , has the advantage of being noninvasive, in contrast to invasive procedures to obtain fetal DNA which brings risks to pregnancy, and of being more accurate. The exam can be performed from the ninth week of gestation and although it is highly accurate, it is important to keep in mind that the accuracy is not high enough to be considered a diagnostic test. Although the focus is on detecting common aneuploidies, other important known SVs, particularly microdeletion syndromes can be investigated. Besides, methods to investigate monogenic diseases are currently being developed and validated [51].
3.5.2 Long-Read Sequencing
NGS-based methods use relatively short-reads that challenge the determination of several types of genomic variation, such as SVs, pseudogenes, and highly similar genes, highly repetitive regions (including disease-related repeat expansions), and highly diverse haplotypes (such as HLA regions). Besides, short-reads complicate inference of phasing information (for compound heterozygosity determination), particularly for very rare variants and singletons. Some methodological improvements address these issues by modifying NGS libraries preparation using mate-pair (Illumina), linked-reads (10X), and Hi-C assays, that capture three-dimensional chromatin conformation and provide evidence on structural interactions. However, all-new technologies arose in the past decade to fill this gap, with continuous fragments sequenced from few kb to megabases (named long-reads), directly obtained from native DNA. We will briefly explore two main platforms for long-read sequencing, Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) [52].
PacBio developed a protocol named SMRT sequencing (single-molecule, real-time) which uses kilobase-long fragments (up to more than 100 kb) with adapters connected to their ends forming hairpins (leaving the double-strand capped, able to circularize in complementary single strands). This structure is then assembled with DNA polymerase for loading into a SMRT Cell, a much reduced flow cell with a nanophotonic surface containing zero-mode waveguides (ZMW), which works as chamber reactions and photons. The incorporation of fluorescently labeled dNTPs excites the dye and a camera sensor, which detects the incorporation in real time for each ZMW. Fluorophore is removed before each light pulse to prevent spectral interference. Each forward and reverse strands keep circulating for some rounds, providing a measure of depth of coverage. Median read length in PacBio is around 10–60 kb and although read accuracy averages 90%, there are high fidelity protocols that reach over 99%, with a 10–30 Gb throughput per flow cell. In this protocol, cost per Gb reaches about 86USD (over twice Illumina’s NovaSeq current pricing).
ONT begins with a linear DNA fragmented in long stretches (from 1 kb to a few megabases long) that are in double-strand form and are ligated to a sequence adapter attachable to a motor protein. The flow cells are composed of membranes embedded with thousands of engineered nanopores, with which motor proteins will interact and unwind DNA into the pores, transiently disrupting the electric current of the membrane. The current changes are base-pair specific and since the rate of translocation is controlled, there is enough resolution for detection of individual base pairs and homopolymers. Detection occurs in real-time and although there are larger platforms for loading multiple flow cells, ONT became famous for providing the smallest sequencer ever created to this day (MinION), with the size of a smartphone and the ability to be transported. The ability to consistently sequence reads of dozens of kb and ease of transport allowed MinION to become very popular among microbiologists during field trips, and more recently, to sequence the entire viral genomes of Zika and SARS-CoV-2, in many locations [53, 54]. With variable accuracy of 87–98% depending on the platform, ONT reads range from 10 to 200 kb (but reads over 1 Mb were obtained and replicated), with outputs from 2 to 100Gb per flow cell and costs approaching Illumina’s NovaSeq.
Several important accomplishments were already achieved by long-read sequencing, including the ability to distinguish modified bases such as methylation state of cytosines, relevant in epigenomic studies (See Chap. 4). In addition to that, native RNA sequencing has been reported with ONT and replicated and promises an interesting future in identifying full-length transcript isoforms. Modification in RNA bases (epitranscriptomics) is also being explored using ONT. Accuracy of long-read sequencing is still behind as compared to NGS, but combining both methods have demonstrated a significant gain in de novo genome assembly and confirmation of structural variation. There are novel methods for target enrichment of DNA loci using CRISPR-Cas9 that, in theory, improve accuracy of long-read sequencing by increasing depth of coverage. Adoption of long-read sequencing may improve our ability to detect, catalogue, and interpret haplotypes directly inferred from genomes and transcriptomes.
3.5.3 Omics Integration
The studies on genetic variation are often descriptive, as providing evidence on the consequences of this type of findings is challenging. While there is not a truly systematic strategy of defining functional consequences to molecules, cells, tissues, organs, and clinical manifestations of all variants found in one individual, a group of patients, or a population, there are orthogonal methods that help drawing a larger picture.
The ENCODE Project Consortium has the objective of deeply annotating DNA elements of the genome by integrating research groups and methods to describe and validate regions of the genome that interact with transcription factors, chromatin structure, and methylation sites [15]. The Genotype-Tissue Expression (GTEx) project , on the other hand, intended the creation of a resource on gene expression and its regulation in dozens of human tissues, providing a full description of variation, expression conditions, and transcriptional outcomes [55]. More recently, the Human Cell Atlas project was launched to integrate research groups involved in cellular models, including genomics, transcriptomics, proteomics, and metabolomics. Novel technologies such as single-cell transcriptomics , allows a deep description of cellular states under different conditions and mapping signatures involved in pathology can give insight in gene function [56, 57].
In addition, large initiatives that intend to collect and follow up clinical data and other traits in a population-level scale along with biological sampling, as an example of the UK BioBank , are already contributing for our understanding of the association between genomic variability and outcomes. Combining data from hundreds of thousands of individuals improves detection of small effect variants and polygenic profiles. Several biobanks also include other levels of biomedically relevant experiments such as RNA-Seq, epigenomics, proteomics, and metabolomics, all of which can be integrated using both agnostic approaches such as deep learning or candidate-driven by piling up individual-level information (“thick-data”) [58].
In this chapter, we presented how genomic methods have constantly been evolving over the past decades, and with all the new technologies and the enormous population samples being analyzed, we can expect that the coming years will continue to bring significant advances to genomic science, ultimately making precision medicine a reality in clinical routine.
References
Lejeune J, Gautier M, Turpin R. Study of somatic chromosomes from 9 mongoloid children. C R Hebd Seances Acad Sci. 1959;248(11):1721–2.
Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737–8. https://doi.org/10.1038/171737a0.
O'Brien SJ, MacIntyre RJ. An analysis of gene-enzyme variability in natural populations of Drosophila melanogaster and D. simulans. Am Nat. 1969;103(930):97–113.
Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–7. https://doi.org/10.1073/pnas.74.12.5463.
Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol. 1986;51:263–73. https://doi.org/10.1101/sqb.1986.051.01.032.
Cook-Deegan RM. The Alta summit, December 1984. Genomics. 1989;5(3):661–3. https://doi.org/10.1016/0888-7543(89)90042-6.
Eichler EE. Genetic variation, comparative genomics, and the diagnosis of disease. N Engl J Med. 2019;381(1):64–74. https://doi.org/10.1056/NEJMra1809315.
Abel HJ, Larson DE, Regier AA, Chiang C, Das I, Kanchi KL, Layer RM, Neale BM, Salerno WJ, Reeves C, Buyske S, Matise TC, Muzny DM, Zody MC, Lander ES, Dutcher SK, Stitziel NO, Hall IM. Mapping and characterization of structural variation in 17,795 human genomes. Nature. 2020;583(7814):83–9. https://doi.org/10.1038/s41586-020-2371-0.
Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, Gardner EJ, Rodriguez OL, Guo L, Collins RL, Fan X, Wen J, Handsaker RE, Fairley S, Kronenberg ZN, Kong X, Hormozdiari F, Lee D, Wenger AM, Hastie AR, Antaki D, Anantharaman T, Audano PA, Brand H, Cantsilieris S, Cao H, Cerveira E, Chen C, Chen X, Chin CS, Chong Z, Chuang NT, Lambert CC, Church DM, Clarke L, Farrell A, Flores J, Galeev T, Gorkin DU, Gujral M, Guryev V, Heaton WH, Korlach J, Kumar S, Kwon JY, Lam ET, Lee JE, Lee J, Lee WP, Lee SP, Li S, Marks P, Viaud-Martinez K, Meiers S, Munson KM, Navarro FCP, Nelson BJ, Nodzak C, Noor A, Kyriazopoulou-Panagiotopoulou S, Pang AWC, Qiu Y, Rosanio G, Ryan M, Stutz A, Spierings DCJ, Ward A, Welch AE, Xiao M, Xu W, Zhang C, Zhu Q, Zheng-Bradley X, Lowy E, Yakneen S, McCarroll S, Jun G, Ding L, Koh CL, Ren B, Flicek P, Chen K, Gerstein MB, Kwok PY, Lansdorp PM, Marth GT, Sebat J, Shi X, Bashir A, Ye K, Devine SE, Talkowski ME, Mills RE, Marschall T, Korbel JO, Eichler EE, Lee C. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun. 2019;10(1):1784. https://doi.org/10.1038/s41467-018-08148-z.
Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, Pitsillides AN, LeFaive J, Lee S-b, Tian X, Browning BL, Das S, Emde A-K, Clarke WE, Loesch DP, Shetty AC, Blackwell TW, Wong Q, Aguet F, Albert C, Alonso A, Ardlie KG, Aslibekyan S, Auer PL, Barnard J, Barr RG, Becker LC, Beer RL, Benjamin EJ, Bielak LF, Blangero J, Boehnke M, Bowden DW, Brody JA, Burchard EG, Cade BE, Casella JF, Chalazan B, Chen Y-DI, Cho MH, Choi SH, Chung MK, Clish CB, Correa A, Curran JE, Custer B, Darbar D, Daya M, Andrade Md, DeMeo DL, Dutcher SK, Ellinor PT, Emery LS, Fatkin D, Forer L, Fornage M, Franceschini N, Fuchsberger C, Fullerton SM, Germer S, Gladwin MT, Gottlieb DJ, Guo X, Hall ME, He J, Heard-Costa NL, Heckbert SR, Irvin MR, Johnsen JM, Johnson AD, Kardia SLR, Kelly T, Kelly S, Kenny EE, Kiel DP, Klemmer R, Konkle BA, Kooperberg C, Köttgen A, Lange LA, Lasky-Su J, Levy D, Lin X, Lin K-H, Liu C, Loos RJF, Garman L, Gerszten R, Lubitz SA, Lunetta KL, Mak ACY, Manichaikul A, Manning AK, Mathias RA, McManus DD, McGarvey ST, Meigs JB, Meyers DA, Mikulla JL, Minear MA, Mitchell B, Mohanty S, Montasser ME, Montgomery C, Morrison AC, Murabito JM, Natale A, Natarajan P, Nelson SC, North KE, O’Connell JR, Palmer ND, Pankratz N, Peloso GM, Peyser PA, Post WS, Psaty BM, Rao DC, Redline S, Reiner AP, Roden D, Rotter JI, Ruczinski I, Sarnowski C, Schoenherr S, Seo J-S, Seshadri S, Sheehan VA, Shoemaker MB, Smith AV, Smith NL, Smith JA, Sotoodehnia N, Stilp AM, Tang W, Taylor KD, Telen M, Thornton TA, Tracy RP, Berg DJVD, Vasan RS, Viaud-Martinez KA, Vrieze S, Weeks DE, Weir BS, Weiss ST, Weng L-C, Willer CJ, Zhang Y, Zhao X, Arnett DK, Ashley-Koch AE, Barnes KC, Boerwinkle E, Gabriel S, Gibbs R, Rice KM, Rich SS, Silverman E, Qasba P, Gan W, Papanicolaou GJ, Nickerson DA, Browning SR, Zody MC, Zöllner S, Wilson JG, Cupples LA, Laurie CC, Jaquish CE, Hernandez RD, O’Connor TD, Abecasis GR (2019) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. bioRxiv.
Lloyd JP, Tsai ZT-Y, Sowers RP, Panchy NL, Shiu S-H, Gojobori J. A model-based approach for identifying functional intergenic transcribed regions and noncoding RNAs. Mol Biol Evol. 2018;35(6):1422–36. https://doi.org/10.1093/molbev/msy035.
Gonzalez-Sandoval A, Gasser SM. On TADs and LADs: spatial control over gene expression. Trends Genet. 2016;32(8):485–95. https://doi.org/10.1016/j.tig.2016.05.004.
Melo US, Schöpflin R, Acuna-Hidalgo R, Mensah MA, Fischer-Zirnsak B, Holtgrewe M, Klever M-K, Türkmen S, Heinrich V, Pluym ID, Matoso E, Bernardo de Sousa S, Louro P, Hülsemann W, Cohen M, Dufke A, Latos-Bieleńska A, Vingron M, Kalscheuer V, Quintero-Rivera F, Spielmann M, Mundlos S. Hi-C identifies complex genomic rearrangements and TAD-shuffling in developmental diseases. Am J Hum Genet. 2020;106(6):872–84. https://doi.org/10.1016/j.ajhg.2020.04.016.
Szabo Q, Bantignies F, Cavalli G. Principles of genome folding into topologically associating domains. Sci Adv. 2019;5(4):eaaw1668. https://doi.org/10.1126/sciadv.aaw1668.
The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. https://doi.org/10.1038/nature11247.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O'Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Genome Aggregation Database C, Neale BM, Daly MJ, MacArthur DG. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43. https://doi.org/10.1038/s41586-020-2308-7.
Antonarakis SE. Carrier screening for recessive disorders. Nat Rev Genet. 2019;20(9):549–61. https://doi.org/10.1038/s41576-019-0134-2.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation C. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. https://doi.org/10.1038/nature19057.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Van Prooijen-Knegt AC, Van Hoek JFM, Bauman JGJ, Van Duijn P, Wool IG, Van der Ploeg M. In situ hybridization of DNA sequences in human metaphase chromosomes visualized by an indirect fluorescent immunocytochemical procedure. Exp Cell Res. 1982;141(2):397–407. https://doi.org/10.1016/0014-4827(82)90228-2.
The BAC Resource Consortium, Cheung VG, Nowak N, Jang W, Kirsch IR, Zhao S, Chen XN, Furey TS, Kim UJ, Kuo WL, Olivier M, Conroy J, Kasprzyk A, Massa H, Yonescu R, Sait S, Thoreen C, Snijders A, Lemyre E, Bailey JA, Bruzel A, Burrill WD, Clegg SM, Collins S, Dhami P, Friedman C, Han CS, Herrick S, Lee J, Ligon AH, Lowry S, Morley M, Narasimhan S, Osoegawa K, Peng Z, Plajzer-Frick I, Quade BJ, Scott D, Sirotkin K, Thorpe AA, Gray JW, Hudson J, Pinkel D, Ried T, Rowen L, Shen-Ong GL, Strausberg RL, Birney E, Callen DF, Cheng JF, Cox DR, Doggett NA, Carter NP, Eichler EE, Haussler D, Korenberg JR, Morton CC, Albertson D, Schuler G, de Jong PJ, Trask BJ. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature. 2001;409(6822):953–8. https://doi.org/10.1038/35057192.
Schrock E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith MA, Ning Y, Ledbetter DH, Bar-Am I, Soenksen D, Garini Y, Ried T. Multicolor spectral karyotyping of human chromosomes. Science. 1996;273(5274):494–7. https://doi.org/10.1126/science.273.5274.494.
Speicher MR, Ballard SG, Ward DC. Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat Genet. 1996;12(4):368–75. https://doi.org/10.1038/ng0496-368.
Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258(5083):818–21. https://doi.org/10.1126/science.1359641.
Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20(2):207–11. https://doi.org/10.1038/2524.
Shaffer LG, Bejjani BA, Torchia B, Kirkpatrick S, Coppinger J, Ballif BC. The identification of microdeletion syndromes and other chromosome abnormalities: cytogenetic methods of the past, new technologies for the future. Am J Med Genet C: Semin Med Genet. 2007;145C(4):335–45. https://doi.org/10.1002/ajmg.c.30152.
Schouten JP. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic acids Res. 2002;30(12):57e–57. https://doi.org/10.1093/nar/gnf056.
Warner JP, Barron LH, Goudie D, Kelly K, Dow D, Fitzpatrick DR, Brock DJ. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet. 1996;33(12):1022–6. https://doi.org/10.1136/jmg.33.12.1022.
Shendure J, Balasubramanian S, Church GM, Gilbert W, Rogers J, Schloss JA, Waterston RH. DNA sequencing at 40: past, present and future. Nature. 2017;550(7676):345–53. https://doi.org/10.1038/nature24286.
Bumgarner R. Overview of DNA microarrays: types, applications, and their future. Curr Protoc Mol Biol. 2013; https://doi.org/10.1002/0471142727.mb2201s101.
Tam V, Patel N, Turcotte M, Bosse Y, Pare G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20(8):467–84. https://doi.org/10.1038/s41576-019-0127-1.
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012;2012:1–11. https://doi.org/10.1155/2012/251364.
Samorodnitsky E, Jewell BM, Hagopian R, Miya J, Wing MR, Lyon E, Damodaran S, Bhatt D, Reeser JW, Datta J, Roychowdhury S. Evaluation of hybridization capture versus amplicon-based methods for whole-Exome sequencing. Hum Mutat. 2015;36(9):903–14. https://doi.org/10.1002/humu.22825.
Weiss MM, Van der Zwaag B, Jongbloed JDH, Vogel MJ, Brüggenwirth HT, Lekanne Deprez RH, Mook O, Ruivenkamp CAL, van Slegtenhorst MA, van den Wijngaard A, Waisfisz Q, Nelen MR, van der Stoep N. Best practice guidelines for the use of next-generation sequencing applications in genome diagnostics: a National Collaborative Study of Dutch genome diagnostic laboratories. Hum Mutat. 2013;34(10):1313–21. https://doi.org/10.1002/humu.22368.
Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet. 2018;19:253. https://doi.org/10.1038/nrg.2017.116.
The 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. https://doi.org/10.1038/nature15393.
Koboldt Daniel C, Steinberg Karyn M, Larson David E, Wilson Richard K, Mardis ER. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155(1):27–38. https://doi.org/10.1016/j.cell.2013.09.006.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. https://doi.org/10.1101/gr.107524.110.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. https://doi.org/10.1093/nar/gkq603.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The Ensembl variant effect predictor. Genome Biol. 2016;17(1):122. https://doi.org/10.1186/s13059-016-0974-4.
Tattini L, D’Aurizio R, Magi A. Detection of genomic structural variants from next-generation sequencing data. Front Bioeng Biotechnol. 2015;3:92. https://doi.org/10.3389/fbioe.2015.00092.
Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):i333–9. https://doi.org/10.1093/bioinformatics/bts378.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, Pineda-Alvarez D, Aradhya S, Martin CL. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the clinical genome resource (ClinGen). Genet Med. 2019;22(2):245–57. https://doi.org/10.1038/s41436-019-0686-8.
Ridge PG, Hoyt KB, Boehme K, Mukherjee S, Crane PK, Haines JL, Mayeux R, Farrer LA, Pericak-Vance MA, Schellenberg GD, Kauwe JSK, Adams PM, Albert MS, Albin RL, Apostolova LG, Arnold SE, Asthana S, Atwood CS, Baldwin CT, Barber RC, Barmada MM, Barnes LL, Barral S, Beach TG, Becker JT, Beecham GW, Beekly D, Bennett DA, Bigio EH, Bird TD, Blacker D, Boeve BF, Bowen JD, Boxer A, Burke JR, Burns JM, Buxbaum JD, Cairns NJ, Cantwell LB, Cao C, Carlson CS, Carlsson CM, Carney RM, Carrasquillo MM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Crane PK, Cribbs DH, Crocco EA, Cruchaga C, De Jager PL, DeCarli C, Demirci FY, Dick M, Dickson DW, Doody RS, Duara R, Ertekin-Taner N, Evans DA, Faber KM, Fairchild TJ, Fallon KB, Fardo DW, Farlow MR, Ferris S, Foroud TM, Frosch MP, Galasko DR, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Goate AM, Graff-Radford NR, Green RC, Growdon JH, Hakonarson H, Hamilton RL, Hamilton-Nelson KL, Hardy J, Harrell LE, Honig LS, Huebinger RM, Huentelman MJ, Hulette CM, Hyman BT, Jarvik GP, Jicha GA, Jin L-W, Jun G, Kamboh MI, Karydas A, Katz MJ, Kauwe JSK, Kaye JA, Kim R, Kowall NW, Kramer JH, Kukull WA, Kunkle BW, LaFerla FM, Lah JJ, Larson EB, Leverenz JB, Levey AI, Li G, Lieberman AP, Lin C-F, Lipton RB, Lopez OL, Lunetta KL, Lyketsos CG, Mack WJ, Marson DC, Martin ER, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Montine TJ, Morris JC, Mukherjee S, Murrell JR, Myers AJ, Naj AC, O'Bryant S, Olichney JM, Pankratz VS, Parisi JE, Partch A, Paulson HL, Perry W, Peskind E, Petersen RC, Pierce A, Poon WW, Potter H, Quinn JF, Raj A, Raskind M, Reiman EM, Reisberg B, Reisch JS, Reitz C, Ringman JM, Roberson ED, Rogaeva E, Rosen HJ, Rosenberg RN, Royall DR, Sager MA, Sano M, Saykin AJ, Schneider JA, Schneider LS, Seeley WW, Smith AG, Sonnen JA, Spina S, St George-Hyslop P, Stern RA, Swerdlow RH, Tanzi RE, Thornton-Wells TA, Trojanowski JQ, Troncoso JC, Tsuang DW, Valladares O, Van Deerlin VM, Van Eldik LJ, Vardarajan BN, Vinters HV, Vonsattel JP, Wang L-S, Weintraub S, Welsh-Bohmer KA, Wendland JR, Wilhelmsen KC, Williamson J, Wingo TS, Winslow AR, Wishnek S, Woltjer RL, Wright CB, Wu C-K, Younkin SG, Yu C-E, Yu L. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging. 2016;41:200.e213–20. https://doi.org/10.1016/j.neurobiolaging.2016.02.024.
Peloso GM, Rader DJ, Gabriel S, Kathiresan S, Daly MJ, Neale BM. Phenotypic extremes in rare variant study designs. Eur J Hum Genet. 2015;24(6):924–30. https://doi.org/10.1038/ejhg.2015.197.
Sazonovs A, Barrett JC. Rare-variant studies to complement genome-wide association studies. Annu Rev Genomics Hum Genet. 2018;19(1):97–112. https://doi.org/10.1146/annurev-genom-083117-021641.
Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, Kathiresan S. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–24. https://doi.org/10.1038/s41588-018-0183-z.
Fahed AC, Wang M, Homburger JR, Patel AP, Bick AG, Neben CL, Lai C, Brockman D, Philippakis A, Ellinor PT, Cassa CA, Lebo M, Ng K, Lander ES, Zhou AY, Kathiresan S, Khera AV. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun. 2020;11(1):3635. https://doi.org/10.1038/s41467-020-17374-3.
Martin AR, Gignoux CR, Walters RK, Wojcik GL, Neale BM, Gravel S, Daly MJ, Bustamante CD, Kenny EE. Human demographic history impacts genetic risk prediction across diverse populations. Am J Hum Genet. 2017;100(4):635–49. https://doi.org/10.1016/j.ajhg.2017.03.004.
Bronkhorst AJ, Ungerer V, Holdenrieder S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol Detect Quantif. 2019;17:100087. https://doi.org/10.1016/j.bdq.2019.100087.
Breveglieri G, D’Aversa E, Finotti A, Borgatti M. Non-invasive prenatal testing using fetal DNA. Mol Diagn Ther. 2019;23(2):291–9. https://doi.org/10.1007/s40291-019-00385-2.
Logsdon GA, Vollger MR, Eichler EE. Long-read human genome sequencing and its applications. Nat Rev Genet. 2020;21(10):597–614. https://doi.org/10.1038/s41576-020-0236-x.
Candido DS, Claro IM, de Jesus JG, Souza WM, Moreira FRR, Dellicour S, Mellan TA, du Plessis L, Pereira RHM, Sales FCS, Manuli ER, Thézé J, Almeida L, Menezes MT, Voloch CM, Fumagalli MJ, Coletti TM, da Silva CAM, Ramundo MS, Amorim MR, Hoeltgebaum HH, Mishra S, Gill MS, Carvalho LM, Buss LF, Prete CA, Ashworth J, Nakaya HI, Peixoto PS, Brady OJ, Nicholls SM, Tanuri A, Rossi ÁD, Braga CKV, Gerber AL, de C. Guimarães AP, Gaburo N, Alencar CS, ACS F, Lima CX, Levi JE, Granato C, Ferreira GM, Francisco RS, Granja F, Garcia MT, Moretti ML, Perroud MW, TMPP C, Lazari CS, Hill SC, de Souza Santos AA, Simeoni CL, Forato J, Sposito AC, Schreiber AZ, MNN S, de Sá CZ, Souza RP, Resende-Moreira LC, Teixeira MM, Hubner J, PAF L, Moreira RG, Nogueira ML, Ferguson NM, Costa SF, Proenca-Modena JL, ATR V, Bhatt S, Lemey P, Wu C-H, Rambaut A, Loman NJ, Aguiar RS, Pybus OG, Sabino EC, Faria NR. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science. 2020;369(6508):1255–60. https://doi.org/10.1126/science.abd2161.
Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, Oliveira G, Robles-Sikisaka R, Rogers TF, Beutler NA, Burton DR, Lewis-Ximenez LL, de Jesus JG, Giovanetti M, Hill SC, Black A, Bedford T, Carroll MW, Nunes M, Alcantara LC, Sabino EC, Baylis SA, Faria NR, Loose M, Simpson JT, Pybus OG, Andersen KG, Loman NJ. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc. 2017;12(6):1261–76. https://doi.org/10.1038/nprot.2017.066.
Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, Gillard B, Ramsey K, Sullivan S, Bridge J, Magazine H, Syron J, Fleming J, Siminoff L, Traino H, Mosavel M, Barker L, Jewell S, Rohrer D, Maxim D, Filkins D, Harbach P, Cortadillo E, Berghuis B, Turner L, Hudson E, Feenstra K, Sobin L, Robb J, Branton P, Korzeniewski G, Shive C, Tabor D, Qi L, Groch K, Nampally S, Buia S, Zimmerman A, Smith A, Burges R, Robinson K, Valentino K, Bradbury D, Cosentino M, Diaz-Mayoral N, Kennedy M, Engel T, Williams P, Erickson K, Ardlie K, Winckler W, Getz G, DeLuca D, MacArthur D, Kellis M, Thomson A, Young T, Gelfand E, Donovan M, Meng Y, Grant G, Mash D, Marcus Y, Basile M, Liu J, Zhu J, Tu Z, Cox NJ, Nicolae DL, Gamazon ER, Im HK, Konkashbaev A, Pritchard J, Stevens M, Flutre T, Wen X, Dermitzakis ET, Lappalainen T, Guigo R, Monlong J, Sammeth M, Koller D, Battle A, Mostafavi S, McCarthy M, Rivas M, Maller J, Rusyn I, Nobel A, Wright F, Shabalin A, Feolo M, Sharopova N, Sturcke A, Paschal J, Anderson JM, Wilder EL, Derr LK, Green ED, Struewing JP, Temple G, Volpi S, Boyer JT, Thomson EJ, Guyer MS, Ng C, Abdallah A, Colantuoni D, Insel TR, Koester SE, Little AR, Bender PK, Lehner T, Yao Y, Compton CC, Vaught JB, Sawyer S, Lockhart NC, Demchok J, Moore HF. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45(6):580–5. https://doi.org/10.1038/ng.2653.
Han X, Zhou Z, Fei L, Sun H, Wang R, Chen Y, Chen H, Wang J, Tang H, Ge W, Zhou Y, Ye F, Jiang M, Wu J, Xiao Y, Jia X, Zhang T, Ma X, Zhang Q, Bai X, Lai S, Yu C, Zhu L, Lin R, Gao Y, Wang M, Wu Y, Zhang J, Zhan R, Zhu S, Hu H, Wang C, Chen M, Huang H, Liang T, Chen J, Wang W, Zhang D, Guo G. Construction of a human cell landscape at single-cell level. Nature. 2020;581(7808):303–9. https://doi.org/10.1038/s41586-020-2157-4.
Rozenblatt-Rosen O, Stubbington MJT, Regev A, Teichmann SA. The human cell atlas: from vision to reality. Nature. 2017;550(7677):451–3. https://doi.org/10.1038/550451a.
Misra BB, Langefeld C, Olivier M, Cox LA. Integrated omics: tools, advances and future approaches. J Mol Endocrinol. 2019;62(1):R21–45. https://doi.org/10.1530/jme-18-0055.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Naslavsky, M.S., de Oliveira Scliar, M. (2021). Methods to Study Genomic DNA Sequence Variation. In: Haddad, L.A. (eds) Human Genome Structure, Function and Clinical Considerations. Springer, Cham. https://doi.org/10.1007/978-3-030-73151-9_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-73151-9_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-73150-2
Online ISBN: 978-3-030-73151-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)