
Abstract
In this contribution we analyzed the Vanadium speciation and structural local environment in two V-bearing Na-phosphate glasses by means of V K-edge X-ray Absorption Spectroscopy. The two glasses have starting molar proportions Na2O:V2O5:P2O5 equal to 45:10:45 (NaPV10) and 10:80:10 (NaPV80). Accurate analysis of the pre-edge peak of the two glasses allowed to determine the presence of [4]V5+ and [5]V5+ in NaPV80 and of [5]V4+ and [6]V4+ in NaPV10. EXAFS derived V–O distances for the two glasses are in agreement with literature data for the above mentioned V species. The determined V local geometries and the EXAFS derived bond distances have been used to build four structural models for [4]V5+, [5]V5+, [5]V4+ and [6]V4+ in the studied glasses; these models have been in turn used to calculate theoretical XANES spectra by means of the MXAN code. The partial contributions of each V species have been summed in order to fit the experimental XANES spectra of the two glasses. Iterative fitting of the structural parameters of each structural model resulted in a good agreement between experimental and theoretical XANES data. The analysis strategy used in the present work allowed to gain information of V speciation and on the structural local environment of the [4]V5+, [5]V5+, [5]V4+ and [6]V4+ species present in the analyzed glasses. Combined use of different spectral regions and data analysis routines allowed to successfully simulate XANES spectra and to gain an in depth knowledge of V local environment even in a complex system like amorphous glass containing different V species.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
17.1 Introduction
Glasses containing transition metals find applications in technological fields like power cells [1] and high power laser devices [2] because of their peculiar optical, electronic and magnetic properties. The semiconductive behavior and properties of glasses containing transition elements, and in particular Vanadium phosphate glasses, have long been known [3], but it was only after the theoretical studies by Sir Nevil Francis Mott [4,5,6], that the scientific community started to be interested in the possibility to exploit the properties of those kind of glasses for technological applications.
Among several glass matrices, phosphate glasses are interesting due to their easy preparation at low temperatures, strong glass forming role, high ionic conductivities and several applications [7,8,9,10,11]. The properties of phosphate glasses can be change by the addition of modifier halides or oxides of alkali, alkaline earth and transition metals into the glassy network [12,13,14,15,16]. Moreover glasses containing transition metal (TM) such as V2O5 are especially attractive because of their applications as cathode materials in batteries, memory switching, electrical threshold and optical switching devices [17,18,19].
The electrical conductivity of these glasses containing different amounts of V2O5 depends on the presence of two different valence states (V5+ and V4+) [20, 21]. The electronic conductivity is due to by small polaron hopping between such ions while the alkali oxide (Li+ or Na+) when added into a glass, can contribute to the charge transport and mixed conductivity to be observed. Generally, ionic conductivity depends on alkali concentration and ion mobility and if we assume that the motion of alkali ions and polarons are independent, the electrical conductivity can increase with increasing alkali content. Glasses containing transition metals and alkali, however, exhibit highly variable behavior—from strong conductivity anomalies to conductivity only slightly dependent on alkali content in the glass [22, 23].
17.2 Materials and Methods
17.2.1 Glass Synthesis
Two Na-V phosphate glasses have been synthetized with molar proportions Na2O:V2O5:P2O5 equal to 45:10:45 (NaPV10) and 10:80:10 (NaPV80) by mixing appropriate amounts of ammonium metavanadate (NH4VO3), ammonium dihydrogen orthophosphate (NH4H2PO4) and sodium carbonate (Na2CO3), and heating the mixture in a muffle furnace up to 450 °C. The powders were maintained at this temperature for 3 h in order to decompose NH4H2PO4 and NH4VO3. After that the mixtures were melted at 1000 °C for 30 min. The melt was then quickly quenched on a graphite mold at room temperature and annealed for 2 h near the Glass Transition (Tg) temperature (300 and 230 °C for NaPV10 and NaPV80 respectively) in order to relieve mechanical stresses.
17.2.2 XAS Data Collection and Analysis
XAS spectra of the two glass samples have been collected at the XAFS beamline of the ELETTRA storage ring, operated at 2.4 GeV in top-up mode with a typical current of 160 mA and a maximum current of 300 mA. The monochromator was equipped with a pair of Si(111) crystals. Harmonic were rejected using a pair of Pt-coated mirrors. Spectra were collected in transmission mode and the intensities of the incident and transmitted beams have been measured with two ionization chambers.
The raw XAS spectra have been subject to background subtraction and to normalization of the edge jump to one by using the Athena package [24].
Pre-edge peak data of the XANES spectra have been examined according to the procedure already used in [25,26,27] in order to determine V oxidation state and local coordination geometry. EXAFS data have been analyzed by means of the Horae package, which gives a graphical interface to the IFEFFIT procedure.
Calculation of theoretical XANES spectra have been performed thanks to the MXAN package [28, 29], that uses a Real Space Multiple Scattering (RSMS) method in the Green function formalism to calculate the final states of the Dyson equation in the framework of the muffin tin approximation for the Coulomb part of the potential. The RSMS is the one contained in the CONTINUUM code [30], the self energy can be chosen according to the Hedin-Lundqist and the Xα; the damping of the XAFS oscillations is taken into account using a plasmon resonance model. No thermal displacement model is used.
A remarkable advantage of this code is the possibility to fit structural parameters (e.g. bond distances and bond angles) together with non-structural parameters, like muffin-tin spheres overlap, and fermi energy, with a least square method in order to obtain a best fit between experimental and theoretical spectra. However, this package does not use a Self Consistent Field method to calculate the potentials and charge densities, so that variables like interstitial potentials must be input. This choice (explained in details in [29]) has been made in order to save computational time and perform a fit in a reasonable time without losing physical rigor.
This procedure has already been successfully used to calculate XANES theoretical spectra in a set of vanadium model compounds with different V oxidation states and coordination geometries [31], resulting in good agreement between experimental and theoretical spectra.
17.3 Results
17.3.1 V K-Edge XANES Pre-edge Peak
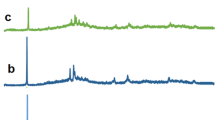
The XANES spectra of the two glasses are shown in Fig. 17.1: clear differences are visible in the raw intensity of the pre-edge peak and in its energy position, indicating the predominance of different V oxidation states and coordination geometries in these two samples. Also marked differences in the EXAFS oscillation are evidence different V–O distances.

V K-edge XAS spectra of the two glasses studied. The two spectra have been vertically offset for clarity. In the inset, the EXAFS signal is shown as a function of the photoelectron wave vector
In order to determine quantitatively the importance of different V species in the samples, the pre-edge peak has been compared to those of V model compounds with different oxidation states and coordination geometries according to the procedure described in [25, 27]. The pre-edge peak of sample NaPV80 can be adequately modeled by a linear combination of the pre-edge peak of Vanadinite and V2O5 (containing V5+ respectively in tetrahedral coordination and in square pyramidal coordination). It is thus evident, that this glass contains dominantly V5+ with two different coordination geometry in almost similar amounts: 40 mol % [4]V5+ and 60 mol % [5]V5+ (where the term in square brackets represents the coordination number).
On the other hand, the pre-edge peak of sample NAPV10 displays normalized Intensity and centroid Energy intermediate between those of cavansite and V2O4 (containing V4+ respectively in square pyramidal and octahedral coordination). This samples thus displays predominantly the presence of V4+ with 2 different coordination geometries in very similar proportions: 45 mol % [6]V4+ and 55 mol % [5]V4+ (Fig. 17.2).

Background subtracted pre-edge peak of the 2 glass samples, compared with selected V model compounds (left panel); IN the right panel are shown the pre-edge peak normalized intensity and centroid energy of the 2 studied glasses, compared to those of V model compounds with known V oxidation state and coordination environment. NaPV80 displays the presence of penta-valent Vanadium in tetrahedral and square pyramidal coordination, whereas NaPV10 displays the presence of tetravalent V in square pyramidal and in octahedral coordination
17.3.2 V K-Edge EXAFS
From the data obtained, we built 4 different clusters with V bonded to 4, 5 or 6 oxygens, choosing appropriate V–O distances according to literature data for [4]V5+, [5]V5+, [5]V4+ and [6]V4+ species. These starting clusters were used in order to calculate single- and multiple-scattering paths to be used for EXAFS data analysis.
The two clusters used for NaPV10 consist of:
-
(i)
V in fivefold coordination, with a vanadyl V–O bond at 1.6 Å, 4 basal V–O bonds at 1.95 Å with 4 O-P bonds at 1.53 Å (so at 3.48 Å from the central V) and an unbonded oxygen at 2.9 Å;
-
(ii)
V in a regular sixfold coordination with 6 symmetric V–O bonds at 1.95 Å (average value from literature data) and 6 O–P bonds at 1.53 Å (at 3.48 Å from the central V).
The two clusters used for NaPV80 consist of:
-
(i)
V in fivefold coordination, with a vanadyl V–O bond at 1.58 Å, 4 basal V–O bonds at distances ranging from 1.78 to 2.05 Å with 4 O–P bonds at 1.53 Å;
-
(ii)
V in a regular tetrahedral coordination with 4 V–O bonds at 1.71 Å and 6 O–P bonds at 1.53 Å (at 3.48 Å from the central V).
Single scattering amplitude and phase have been calculated for every V–O, V–V, and V–P distance. EXAFS data analysis included fitting thee distances, a global amplitude factor, the ratio between [4]V5+ and [5]V5+ (for NaPV80) or [5]V4+ and [6]V4+ species (for NaPV10), and two different edge energies (each relative to a different V species) for each spectrum.
The results of the fit are shown in Fig. 17.3, whereas the fitted distances are reported in Table 17.1.

Fit results for the V K-edge EXAFS spectra of the NaPV10 (left) and NaPV80 (right) glasses studied
17.3.3 Theoretical V K-Edge XANES Spectra
The results of the EXAFS fit have been used in order to modify the initial structural model used to calculate EXAFS contribution; these, in turns have been used in order to calculate XANES theoretical spectra. Follows the description of the structural model used for the calculation of the theoretical XANES spectra.
NaPV10 V4+ in octahedral coordination
According to literature data [32, 33] [6]V4+ commonly has a shorter bond length (not necessarily a vanadyl, i.e. a double V=O bond), a long trans bond opposed to it and 4 equatorial bonds with intermediate bond lengths. In particular the vanadyl bond is in the range 1.48–1.74 Å, the trans bond in the range 2–2.6 Å and the equatorial bonds in the range 1.86–2.16 Å. Our choice was thus to refine 3 individual distances: a vanadyl distance, a trans distance and one individual distance for the 4 equatorial bonds. The bond angles were optimized in couples using the same fit variable for opposite couples of atoms. The P atoms were linked to the respective O atoms. Since the refinement of V2O4 in [31] was not accurate enough, the muffin-tin fit parameters, i.e. the overlap and the interstitial potential, were let to move almost freely.
NaPV10 V4+ in square pyramidal coordination
In this case from literature data [33, 34] we know that V has a short vanadyl double bond V=O, 4 basal bonds and sometimes a trans bond that can be considered unbonded. The vanadyl bond distance is in the range 1.48–1.66 Å, the basal bond distances in the range 1.86–2.12 Å and the trans oxygen distance (to the opposite site of the vanadyl bond) is at about 2.9 Å. The fit variables were thus chosen as in the case of the octahedral part: one distance for the vanadyl bond, one for the trans bond, one for all the 4 basal bonds. Again, the bond angles were optimised coupling opposite atoms. The P atoms were linked to the respective O atoms and the MT variables were again chosen from the values found in [31] and let free to slightly vary (a few percent) around the starting value.
NaPV10 optimization of the theoretical XANES spectrum
The fit of the experimental spectrum was performed by summing two contributions, so that the structural and non structural parameters of one contribution are fitted every time, keeping fixed the other contribution. The relative importance of each contribution (i.e. the value [5]V5+/([4]V5+ + [5]V5+)) was initially chosen according to the values determined by the pre-edge peak (i.e. 0.5). This value has been then refined during each optimization cycle. Since there is no possibility to refine the relative quantity of the two contributions and the relative energy shift, to estimate those quantities we adopted this strategy.
MXAN allows to add a previously calculated spectrum and fit the difference; If we want to fit a spectrum in which two components are predominant so that S = νA + (1 − ν)B, we can consider the case 0.5 < ν < 1 (i.e., the case with a dominant A species). We can first calculate the theoretical spectra for each V species (A0 and B0), then we can fix B to B0 (because, the less important contribution is thought to influence the least the spectrum) and refine A; the fit result (A1), can be then fixed and we can do a second cycle of refinements, this time fixing A1 and refining B0. This procedure can be repeated several times until the residual of the fit does not change significantly.
We performed 10 optimization cycles, checking the value of the residual until we got a stable value of the disagreement index. We will not report all of the procedure, but only show the final results. The best fit is shown in Fig. 4a, while the best fit parameters are displayed in Table 17.2. The calculated spectrum is in very good agreement with the experimental one. All the features of the experimental spectrum are reproduced in the theoretical spectrum, with the correct energy position and intensity. In particular the resonance peaks “a” and “b” are present and accurately reproduced. This is an indication that the MS is accurately calculated. The small discrepancy in the position and intensity of peak “c” can be due to (i) a badly estimated broadening of the spectrum and (ii) the presence of Na which is not included in the structural model used for the calculation of the theoretical spectrum. The refined bond distances are in good agreement with those determined by the EXAFS analysis and so an overall agreement with bond distances is found in tetravalent V. For the octahedrally coordinated V we have found that a better fit is obtained if the 6 distances are not all regular, but distorting the polyhedron. This is in good agreement also with literature data, both for crystalline structures [26, 33] and for V phosphate glasses [35].

Fit results for the V K-edge XANES spectra of the NaPV10 (left) and NaPV80 (right) glasses. The black dotted lines represent the experimental spectra, whereas the black continuous lines the weighted sum of the theoretical spectra calculated for each Vanadium species (colored dotted lines)
The result of this procedure is reported in Table 17.2.
From the calculations we found out that the ratio [6]V/[5]V + [6]V is around 0.3, i.e. closer to the value found in the EXAFS refinement (0.27) than that found in the pre-edge peak analysis (0.45). We believe that the actual value is so somewhere intermediate between 0.3 and 0.45, i.e. intermediate between the pre-edge peak determination, the EXAFS fit and the XANES fit.
NaPV80 optimization of the theoretical XANES spectrum
The fit procedure was the same of NaPV10, and a convergence was achieved after 6 cycles, even though, as exposed before, for the fivefold coordinated V some further refinements were necessary. The best fit, as determined by the residual, is shown in Fig. 4b, while the fit parameters are displayed in Table 17.2.
The calculated spectrum is in very good agreement with the experimental one. For clarity, in Fig. 4b we also displayed a calculation without the plasmon broadening (the energy dependent part of the broadening, see Sect. 17.2.2). This has been shown because some features (“c” and “d”) are a bit less prominent after the broadening.
It is interesting to notice that those peaks have analogues in the V2O5 spectrum, in particular the peaks “d” and “f” in [31]. MXAN slightly underestimated the same peaks in V2O5 as in NaPV80. This can be an indication that using a similar atomic configuration for the two samples was a good choice. The “a” and “b” peaks, that correspond to the strongest MS contribution, are less accurately determined for NaPV80 than for NaPV10. This could possibly happen due to the presence of V in the second coordination shell, which leads to stronger multiple scattering that is less accurately calculated by MXAN at low energy [30]. The V–O bond distances in the fourfold coordinated contribution are shorter than those calculated with the EXAFS refinement, but closer to the average of the V–O distances in the literature (1.72 Å [33]). All of the bond distances found in the square pyramidal contribution were slightly longer with respect to those found in EXAFS, with the exception of the trans V–O bond, that we found to be shorter (2.52 Å vs. 2.86 Å of EXAFS). The XANES distances are anyway compatible to the EXAFS distances within the error bars.
As for NaPV10, after the fit we tried to optimize the energy shift and the ratio a = [5]V/[5]V + [4]V by calculating the residual as a function of a and Energy shift. The results are summarized in Table 17.2. In this case the lower residual for the a parameter (0.7) was found to be closer to that calculated with the pre-edge peak analysis (0.60) than to that determined by the EXAFS analysis. Anyway, the XANES value falls in the limit of the error bar, so probably the actual value is somewhere intermediate between those obtained from the EXAFS and the XANES calculations. The determined relative energy shift was found to be 1.5 eV.
NaPV80 V5+ in tetrahedral coordination
From the EXAFS analysis we were able to determine only the first coordination shell of atoms, i.e. the 4 neighbouring oxygens. For the XANES analysis we kept as starting parameters this configuration, according to literature data [36]. We also considered the tetrahedron to be regular for the bond distances, using a single fit variable for the 4 V–O distances. We refined the 4 bond angles independently.
The MT parameters were chosen from the model compounds analysis and let free to slightly vary around the values found for [4]V5+ in [31] i.e. 0.2 for the MT overlap and −13 eV for the interstitial potential.
NaPV80 V5+ in square pyramidal coordination
In this case the EXAFS analysis did not give us a clear answer about the V local structure, so we performed several trials with different atomic configurations to determine the most suitable one according to the residual. The details will not be reported here, but the trials involved (i) the removal of the P atom with an eventual substitution of a V atom at opportune distance determined by the V2O5 structure (the three configurations being thus with a P atom in the second coordination shell, a V atom in the second coordination shell and no atoms in the second coordination shell); (ii) the removal of some of the V atoms in the second coordination shell. The best configuration was found to be: (i) a vanadyl bond; (ii) a trans bond opposed to it; (iii) a basal bond with two V atoms in the corner (a configuration already found in V2O5, see [37] and [31]); (iv) a basal bond with a P atom opposed to it; (v) two basal bonds bended towards the 2 V atoms in the second coordination shell. The fit was performed in different steps:
first we varied the structural parameters in groups (basal oxygens, second shell atoms, vanadyl and trans oxygens) to check the behaviour of those parameters on the theoretical spectrum; after reaching a configuration that was satisfactory we have released all the variables as previously done for [5]V4+ in NaPV10: one common fit parameter for the basal oxygen atoms, one for the vanadyl and one for the trans oxygen. The bond angles were varied in couples, the P atom was linked again to the corresponding O atom. The V atoms coordinates in the second coordination shell were independent and free to move.
17.4 Conclusions
In both cases we have shown how the combined analysis of all the regions of the absorption spectrum can lead to a deep knowledge on the V local structure of phosphate glasses, even in complex cases when several different Vanadium species are present. The pre-edge peak analysis was able to give us precious information about (i) the coordination geometries and (ii) oxidation states of the glasses. In systems like V-bearing glasses, in which V can be in a variety of coordination geometries and oxidation states, this procedure provided important information: in fact, by determining the V coordination geometries we were able to build the starting cluster, while by determining the V oxidation state we were able to estimate if the bond distances determined by ab-initio calculations were reasonable. Through EXAFS analysis we were able to refine the bond distances obtaining a radial distribution function that at the end we geometrically refined by means of MS geometrical fits of the XANES region.
The analysis performed on the different parts of the spectra converged to the same results, even though some questions, like the Na structural role in these glasses and the long V–O bond distances in the [5]V5+ component of NaPV80, are still unsolved.
References
Y. Sakurai, J. Yamaki, Correlations between microstructure and electrochemical behaviour of amorphous V2O5-P2O5 in lithium cells. J. Electrochem. Soc. 135(4), 791–796 (1988)
J. Campbell, T. Suratwala, Nd-doped phosphate glasses for high-energy/high-peak-power lasers. J. Non-Cryst. Solids 263–264, 318–341 (2000)
M. Munakata, Electrical conductivity of high vanadium phosphate glass. Solid-State Electron. 1, 159–163 (1960)
N. Mott, Electrons in disordered structures. Adv. Phys. 16(61), 49–144 (1967)
N. Mott, Conduction in glasses containing transition metal ions. J. Non-Cryst. Solids 1(1), 1–17 (1968)
I. Austin, N. Mott, Polarons in crystalline and non-crystalline materials. Adv. Phys. 18(71), 41–102 (1969)
P.A. Tick, Water durable glasses with ultra low melting temperatures. Phys. Chem. Glasses 25(6), 149–154 (1984)
H.S. Liu, P.Y. Shih, T.S. Chin, Low melting PbO-ZnO-P2O5 glasses. Phys. Chem. Glasses 37(6), 227–235 (1996)
R.K. Brow, Review: the structure of simple phosphate glasses. J. Non-Cryst Solids 263–264, 1–28 (2000)
H. Yung, P.Y. Shih, H.S. Liu, T.S. Chin, Nitridation effect on properties of stannous-lead phosphate glasses. J. Am. Ceram. Soc. 80, 2213 (1997)
C.M. Shaw, J.E. Shelby, The effect of stannous oxide on the properties of stannous fluorophosphate glasses. Phys. Chem. Glasses 29, 87 (1988)
R.K. Brow, Nature of alumina in phosphate glass: I, properties of sodium aluminophosphate glass. J. Am. Ceram. Soc. 76, 91 (1993)
S. Kabi, A. Ghosh, Dynamics of Ag+ ions and immobile salt effect in CdI2 doped silver phosphate glasses. Solid State Ion. 187, 39 (2011)
A. Shaw, A. Ghosh, Influence of immobile ions on the length scale of ion transport in conducting phosphate glasses. J. Phys. Chem. C 116, 24255 (2012)
C. Mugoni, M. Montorsi, C. Siligardi, H. Jain, J. Non-Cryst. Solids 383, 137–140 (2014)
C. Mugoni, H. Jain, M. Montorsi, M. Montecchi, A. Kovalskiy, C. Siligardi, Structural origin of electrical conductivity of copper lithium metaphosphate glasses. J. Non-Cryst. Solids 447, 91–97 (2016)
A. Ghosh, Memory switching in bismuth-vanadate glasses. J. Appl. Phys. 64, 2652 (1988)
J. Livage, J.P. Jollivet, E. Tronc, Electronic properties of mixed valence oxide gels. J. Non-Cryst. Solids 121, 35 (1990)
Y. Sakuri, J. Yamaki, V2O5-P2O5 glasses as cathode for lithium secondary battery. J. Electrochem. Soc. 132, 512 (1985)
S. Jayaseelan, P. Muralidharan, M. Venkateswarlu, N. Satyanarayan, Ion transport and relaxation studies of silvervanadotellurite glasses at low temperatures. Mater. Chem. Phys. 87(2–3), 370–377 (2004)
Y. Dimitriev, V. Dimitrov, M. Arnaudov, D. Topalov, IR-spectral study of vanadate vitreous systems. J. Non Cryst. Solids 57(1), 147–156 (1983)
G.D.L.K. Jayasinghe, M.A.K.L. Dissanayake, P.W.S.K. Bandaranayake, J.L. Souquet, D. Foscallo, Solid State Ionics 93, 291 (1997)
J.R. Macdonald D.R. Franceschetti, Impedance spectroscopy emphasising solid materials and systems, in ed. by JR Macdonald (Wiley, New York, 1987), p. 98.
B. Ravel, M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12(4), 537–541 (2005)
G. Giuli, E. Paris, J. Mungall, C. Romano, D. Dingwell, V oxidation state and coordination number in silicate glasses by XAS. Am. Miner. 89(11–12), 1640–1646 (2004)
J. Wong, F.W. Lytle, R.P. Messmer, D.H. Maylotte, K-edge absorption spectra of selected vanadium compounds. Phys. Rev. B 30, 5596–5610 (1984)
A. Moretti, G. Giuli, F. Nobili, A. Trapananti, G. Aquilanti, R. Tossici, R. Marassi, Structural and electrochemical characterization of vanadium-doped LiFePO4 cathodes for lithium-ion batteries. J. Electrochem. Soc. 160(6), A940–A949 (2013)
M. Benfatto, S. Della Longa, Geometrical fitting of experimental XANES spectra by a full multiple-scattering procedure. J. Synchrotron Rad. 8(4), 1087–1094 (2001).
M. Benfatto, S. Della Longa MXAN: new improvements for potential and structural refinement. J. Phys.: Conf. Ser. 190(1), 012031, (2009)
C. Natoli, M. Benfatto, A unifying scheme of interpretation of x-ray absorption spectra based on the multiple scattering theory. J. Phys. Colloques 47(C8), 11–23 (1986)
F. Benzi, G. Giuli, S. Della Longa, E. Paris, Vanadium K-edge XANES in vanadium-bearing model compounds: a full multiple scattering study. J. Synchrotron Rad. 23(4), 947–952 (2016)
K.D. Rogers, An x-ray diffraction study of semiconductor and metallic vanadium dioxide. Powder Diffr. 8, 240–244 (1993)
M. Schindler, F.C. Hawthorne, W.H. Baur, Crystal chemical aspects of vanadium: polyhedral geometries, characteristic bond valences, and polymerization of (VOn) polyhedra. Chem. Mater. 12(5), 1248–1259 (2000)
R. Danisi, T. Armbruster, B. Lazic, In situ dehydration behavior of zeolite-like cavansite: a single-crystal X-ray study. Am. Miner. 97, 1874–1880 (2012)
U. Hoppe, N. Wycko, M. Schmitt, R. Brow, A. Schöps, A. Hannon, Structure of V2O5-P2O5 glasses by X-ray and neutron diffraction. J. Non-Cryst. Solids 358(2), 328–336 (2012)
M. Nagashima, T. Armbruster, Palenzonaite, berzeliite, and manganberzeliite: (As5+, V5+, Si4+)O4 tetrahedra in garnet structures. Mineral. Mag. 76(5), 1081–1097 (2012)
V. Shklover, T. Haibach, F. Ried, R. Nesper, P. Novak, Crystal structure of the product of Mg2+ insertion into V2O5 single crystals. J. Solid State Chem. 123(2), 317–323 (1996)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this paper

Cite this paper
Benzi, F., Paris, E., Della Longa, S., Mugoni, C., Siligardi, C., Giuli, G. (2021). V K-Edge XANES Full Multiple Scattering Study of V-Bearing Phosphate Glasses. In: Di Cicco, A., Giuli, G., Trapananti, A. (eds) Synchrotron Radiation Science and Applications. Springer Proceedings in Physics, vol 220. Springer, Cham. https://doi.org/10.1007/978-3-030-72005-6_17
Download citation
DOI: https://doi.org/10.1007/978-3-030-72005-6_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-72004-9
Online ISBN: 978-3-030-72005-6
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)