
Abstract
Currently, the world’s requirement for energy and chemicals is satisfied by tapping petroleum, coal, and natural gas resources. The continuously escalating energy demands for the betterment of life necessitate the search for alternate sources of energy. Sustainability, probably the “word of this century,” will be a prime force in stimulating scientists and technologists to look for alternate options in making fuels, chemicals and polymers that are irreplaceable in our lives. Renewable biomass, having useful carbon atoms, could be explored with significant potential to produce chemicals and materials that include polymers. The approach also has an intrinsic advantage of balancing CO2 emission, thereby aiding our environment. Researchers worldwide have made considerable advancements on biomass value addition for producing fuels, chemicals and materials. Broadly, biomass are categorized as lignocellulose and lipid-based wherein the former offers humungous scope of reaction chemistries that are deployable in a biorefinery akin to petro-refinery. Among them, levulinic acid (LA), 5-hydroxymethylfurfural (HMF) and 2,5-furandicarboxylic acid (FDCA) and their related products render multifunctional properties with diverse opportunities for applications. In the last decade, intense research has been carried out on these molecules. In this chapter, we shall discuss the background of these platform chemicals, multifarious catalytic approaches made and process tools deployed, in particular for γ-valerolactone, LA-based plasticizers, HMF and FDCA. Challenges on these approaches and possible strategies to overcome them will also be discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Biomass valorization
- Levulinic acid derivatives
- γ-Valerolactone
- Furan compounds
- 5-Hydroxymethylfurfural
- 2,5-Furandicarboxylic acid
1 Introduction
At present, the world’s energy and chemical requirements are largely met through fossilized sources like petroleum, coal and natural gas. The improvement in the quality of life and, consequently, the increasing demand for energy necessitate the search for alternate and sustainable solutions to energy crisis. Sustainability, arguably the “word of this century,” has been instrumental in stimulating scientists and technologists to explore alternative options in making fuels, chemicals and polymers that facilitate our daily lives. Carbon-rich biomass is the only renewable source that could be explored with significant potential for producing chemicals including polymers. This is de facto an attractive option considering the balance between the availability and consumption patterns. The approach also has an intrinsic advantage of reducing indispensability on fossil fuels to an extent besides reducing CO2 emission, thereby benefitting our environment. Researchers all over the world have enthusiastically contributed to the value addition of biomass for producing chemicals. Many countries with strong roots in agriculture, availability of extensive forest/agro-resources and vast coastline for marine macro−/micro-algae are likely to generate non-edible/waste biomass which can potentially be harnessed to produce chemicals sustainably in the years to come.
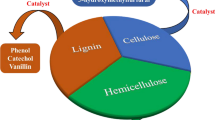
Generally, lignocellulosic biomass consists of 38–50% cellulose, 23–32% hemicellulose and 15–25% lignin, which constitute about 80–90% of the total biomass. Cellulose is a non-branched water-insoluble polysaccharide consisting of several hundreds to tens of thousands of glucose units linked through 1,4-β-glycosidic ether bonds. Cellulose is the most abundant biopolymer synthesized by nature; its amount is estimated at approximately 2 × 109 tons/year [1]. Hemicellulose is a polymeric network connecting lignin and cellulose. Although lower in molecular weight than cellulose, hemicellulose consists of C6 sugars (glucose, mannose and galactose) and C5 sugars (arabinose, xylose and so on). Lignin is a complex three-dimensional cross-linked amorphous polymer consisting of three major methoxylated phenylpropanoid units (coumaryl alcohol, coniferyl alcohol and sinapyl alcohol) connected by strong C–O (~60–70%) and C–C (~25–35%) linkages [2, 3]. Thus, the primary components have great potential for the production of diverse chemicals [4].
The Department of Energy (USA) with NREL (National Renewable Energy Laboratory) and PNNL (Pacific Northwest National Laboratory) have conducted an extensive study to identify valuable sugar-based building blocks [5]. Of the 300 initially selected candidates, a list of 30 interesting chemicals was obtained through an iterative process. The list was further reduced to 12 building blocks considering potential markets, complexity of synthetic pathway, functionality of molecule and abundance. Of these 12 platform molecules, some are obtained through biochemical methods and some others via thermochemical pathway. 1,4-Diacids (succinic, fumaric and malic), 3-hydroxypropionic acid, aspartic acid, glutamic acid and itaconic acid were obtained from biochemical pathway. Xylitol, 2,5-furandicarboxylic acid, levulinic acid, glucaric acid, sorbitol and 3-hydroxybutyrolactone were reported through thermochemical pathway.
From the last decade, our research group is associated with biomass conversion, working on heterogeneous catalytic approaches for the conversion and value addition of the above-mentioned platform molecules including the preparation of xylitol, sorbitol, succinic acid, 5-hydroxymethylfurfural (HMF) and 2,5-furandicarboxylic acid (FDCA). We have also focused on the valorization of levulinic acid into various value-added products by finding efficient catalysts and synthetic protocols. In this chapter, we have covered a brief outline of levulinic acid synthesis, state of the art on catalytic interventions for preparation of levulinic acid-based products such as γ-valerolactone, arylated-γ-lactones and 4,4-diaryl-substituted pentanoic acid/esters. A summarized report on HMF preparation and its selective conversion into FDCA using ruthenium-based catalysts has also been discussed here. The end of the chapter discusses the challenges of the mentioned conversions.
2 Levulinic Acid
Levulinic acid (LA), a keto carboxylic acid from biomass, is an important building block for fuels and chemicals harnessed by catalytic transformations. LA (C5H8O3) is a white crystalline solid (<30 °C), soluble in aqueous and in common organic solvents including polar solvents. LA is a potential intermediate for making alkyl/aryl-substituted cyclic lactones, aliphatic alcohols including diols, cyclic/acyclic amides, ketals, esters, acid halides, diketones, amino/halo-substituted keto carboxylic acids, alkane/alkenes, valeric acid/esters, cyclic ethers, etc.
2.1 Preparation of Levulinic Acid
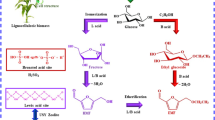
LA is mainly prepared from cellulose-derived glucose by involving two steps. In the first step of the reaction, glucose isomerizes to fructose and is subsequently dehydrated to HMF. In the second step, HMF under hydrolysis in aqueous medium forms LA along with formic acid as a by-product (Scheme 1) [6]. The conversion in the presence of alcoholic medium forms alkyl levulinates and alkyl formates [7]. Many reports reveal the direct conversion of lignocellulose biomass (wheat straw, pulp slurry, sorghum grain, corn starch, water hyacinth, bagasse and rice husk) to LA using homogeneous acid catalysts [6, 8,9,10,11,12,13]. Reports of LA preparation are also available from cellulose, glucose and HMF individually, and these conversions mainly used homogeneous catalysts including mineral acids, super acids, metal halides and acidic ionic liquids. In the course of the past few years, researchers have also developed many solid acid catalysts [14,15,16,17,18,19,20,21,22,23,24,25].

Simplified reaction mechanism for the production of LA via HMF path
The hemicellulose-based xylose is also a precursor for the preparation of LA and its esters through furfural and furfuryl alcohol as intermediates (Scheme 2). Furfural undergoes selective hydrogenation using metal-supported catalysts with furfuryl alcohol as the product. Furfuryl alcohol in the presence of water results in LA and LA esters (alkyl levulinates) in alcoholic medium. Furfuryl alcohol conversion to LA involves acid catalysts including homogeneous acid catalysts (mineral acids), acidic ionic liquids and solid acid catalysts [26,27,28,29,30].

Simplified reaction mechanism for the production of LA from furfural via furfuryl alcohol as intermediate
2.2 Applications of Levulinic Acid
Multiple functionalities of LA including carboxyl, carbonyl and so on facilitate its participation in various organic transformations including oxidation, hydrogenation, hydrocyclization, reductive amination, condensation, etc. (Scheme 3) [31]. We are extensively working on the preparation of γ-valerolactone, arylated-γ-lactones and 4,4-disubstituted pentanoic acid/esters. A detailed discussion pertaining to these chemical transformations has been covered in the following sections.

Catalytic organic transformation of LA to industrially valuable products
2.2.1 Hydrocyclization of Levulinic Acid to γ-Valerolactone
LA under H2 environment primarily results in useful products such as γ-valerolactone (Gvl), 1,4-pentanediol (1,4-PDO), 2-methyltetrahydrofuran (2-MTHF) and pentanoic acid, depending on the catalytic system and reaction parameters [32,33,34,35]. All these LA derivatives are industrially relevant; for example, Gvl is an attractive platform chemical and has various applications in the production of biofuels, i.e. valeric esters [36], aromatic hydrocarbons [37], liquid alkanes [38, 39], polymer intermediates, i.e. α-methylene-γ-valerolactone, 1,4-pentanediol and adipic acid [40,41,42]. In addition, Gvl itself is a green solvent for organic transformations including the conversion of lignocellulose components (Fig. 1) [43,44,45,46,47,48,49,50].

Applications of Gvl in the preparation of polymers and fuel intermediates
Generally, the mechanistic pathway for the conversion of LA to Gvl involves two routes [51]. Path-a culminates in the hydrogenation of LA to γ-hydroxypentanoic acid prior to an intramolecular esterification to obtain Gvl, and path-b follows the esterification of the enol form of LA to α-angelica lactone (α-AL) and its subsequent hydrogenation to Gvl (Scheme 4). Over-hydrogenation results in the ring opening of Gvl forming 1,4-pentanediol as its product, under acidic conditions. At higher temperatures, diol undergoes cyclization to 2-methyltetrahydrofuran (2-MTHF).

Reaction mechanism for hydrocyclization of LA to Gvl
Various research groups explored the catalytic hydrocyclization of LA and levulinates to Gvl using heterogeneous and homogeneous catalytic routes under batch and continuous flow conditions with molecular hydrogen, alcohols and formic acid as hydrogen sources [35, 52,53,54,55,56,57]. The homogeneous catalysts such as complexes of Ru/Ir/Pd, etc. were found to be quite active for this conversion. However, inherent problem of separation and catalyst disposal are bottlenecks in homogeneous catalytic systems. Often expensive chemicals and multistep reaction protocols are required for catalyst preparation, thus limiting their applicability in large scales.
Non-noble metal-supported heterogeneous catalysts including Fe, Ni, Cu, Cr, Co and Mo were reported towards this hydrocyclization. Raney Ni® and Cu-/Ni- metal supported and combination of these two are exclusively reported using three hydrogen sources (H2 gas, isopropanol and formic acid) in either static or continuous vapour phase. The Zr-based catalysts were mainly reported for Meerwein-Ponndorf-Verley (MPV) of levulinic acid followed by cyclization to Gvl using alcohol as hydrogen source. Heterogeneous catalytic systems based on supported noble metals such as Ru, Pd, Pt and Au are active towards LA to Gvl conversion. Among these, Ru catalysts are most active under comparatively milder reaction conditions.
Layered double hydroxide (LDH) materials were found to be attractive in many catalytic applications because of its multifunctionality and tunable nature. Pristine LDH, calcined LDH (CLDH), LDH-derived ex situ and in situ catalysts and metal supported on LDH materials were extensively reported for levulinic acid conversion to Gvl. The following section reviewed the prior art of this conversion using LDH materials including the contribution from our group.
Hydrocyclization of LA to Gvl Using Layered Double Hydroxide-Based Materials
2.2.1.1 LDH-Derived Catalysts for Hydrocyclization of LA to Gvl
The LDH-derived catalysts for LA to Gvl conversion, were first reported by Yan et al. who introduced Cu-Cr, Cu-Al and Cu-Fe CLDH catalyst precursors (derived from LDH by calcination). All the mentioned CLDH catalyst precursors showed good catalytic activity for Gvl (yield 90.7% for Cu-Cr, 87.3% for Cu-Al and 87.3% for Cu-Fe) (Table 1, entries 1–4). The Cu2+ oxide of CLDH generates Cu(0) under reaction conditions (200 °C, 70 bar H2 for 10 h in water) confirmed by PXRD, which is the active catalyst for hydrogenation. The authors observed a decrease in the catalytic activity (in the case of Cu-Cr) upon reuse because of carbon deposits on the surface of the catalyst during the reaction. To avoid this problem, they reactivated the catalyst by calcination (550 °C for 3 h) and was rewarded with similar catalytic activity up to monitored three reaction cycles [58,59,60].
Li research group demonstrated a series of Al-LDH-based catalyst precursors using different metals such as Fe, Cu, Ni and Co. From these M-Al LDH (M = metal) precursors, the active catalyst species was prepared by reduction under H2 (50 mL/min) at 700 °C for 2.5 h, and obtained catalysts are denoted as M(0)/Al2O3. Among the catalysts screened by the Li group, Co/Al2O3 (derived from Co-Al LDH) showed excellent activity towards LA to Gvl conversion with 100% conversion and >99% selectivity at 180 °C, 50 bar H2 for 3 h in 1,4-dioxane medium (Table 1, entries 5–9). The LDH precursor-derived Co/Al2O3 is more active than Co/γ-Al2O3 (prepared by Co impregnation on γ-Al2O3 followed by reduction) and Co/Al2O3-CR (derived from Co-Al LDH through calcination followed by pre-reduction), suggesting that the synthesis protocol of Co/Al2O3 (derived from Co-Al LDH) greatly affected the catalytic performance. The used Co/Al2O3 catalyst was recovered from the reaction mixture by magnetic separation. The authors observed that, after reaction, the Co nanoparticle surface underwent oxidation to cobalt oxide in air during catalyst separation. Thus, prior to each run (while reusability), the used catalyst was washed with water and re-reduced under H2 flow (50 mL/min) at 700 °C for 1 h. The activity of the Co/Al2O3 was maintained up to four reaction cycles following this pretreatment procedure [61].
Magnetic catalyst (Ni(0)-Cu(0)/MgAlFe LDH) was reported by Chen research group for Gvl synthesis in methanol medium with 98.1% yield and full conversion of LA (Table 1, entries 10 and 11). The magnetically active catalyst (Ni(0) and Cu(0)) is prepared from LDH precursor by prior or external reduction in H2 atmosphere at 500 °C for 3 h. The prior reduction temperature of LDH affected the catalytic activity; increased yield of Gvl was observed by increasing the reduction temperature from 350 to 500 °C, and further increment (>500 °C) resulted in a decrease in the selectivity of Gvl because of particle sintering at high temperature. Decrease in catalytic activity of the material (Ni(0)-Cu(0)/MgAlFe LDH) was observed in the reusability studies with increase in the number of cycles. Thus, to maintain the catalyst activity, the used catalyst was reactivated by prior reduction (500 °C for 3 h) for each cycle, and the methodology showed good reusability [62].
Kantam research group reported LDH-derived active catalyst Cu(0)-Ni(0)/MgAl LDH for this conversion with 100% yield of Gvl at relatively mild reaction conditions (140 °C, 30 bar H2 pressure for 3 h) in 1,4-dioxane solvent (Table 1, entry 12). Calcination (600 °C for 6 h) followed by prior reduction (140 °C, 30 bar H2 for 2 h) procedure was used to generate the active catalyst (Ni(0) and Cu(0)) from its LDH precursor via intermediate metal oxides (CLDH). In this case also, gradual decrease in catalytic activity of LDH-derived material was observed upon reuse (66% yield at fourth cycle). Hence, to improve reusability, the catalyst was reactivated at 140 °C, 30 bar H2 for 2 h, and it showed better catalytic activity (fourth cycle 92%) as compared with un-reactivated catalyst [63].
Li and co-researchers disclosed Zr containing Ni catalyst such as Ni(0)/Zr-Al CLDH for vapor-phase continuous production of Gvl from neat LA at 250 °C and ambient H2 pressure (Table 1, entries 13 and 14). The active catalyst is prepared from Ni-Zr-Al LDH catalyst precursor by calcination (500 °C for 4 h in static air) followed by reduction (600 °C for 2 h in H2/Ar atmosphere). The active LDH-derived catalyst (Ni(0)/Zr-Al LDH) exhibited the highest selectivity of Gvl (97.7%) as compared to Zr-free catalyst such as Ni/Al2O3 (85.3%), and the acidity of Zr enhanced the cyclization of reaction intermediate (γ-hydroxy pentanoic acid) to Gvl via path-a mechanism (Scheme 4) [64].
Chary et al. reported in situ hydrogen transfer (from formic acid) for Gvl preparation from LA in continuous mode. In this methodology, it was observed that Mg-Al CLDH (derived from Mg-Al LDH calcination at 500 °C for 6 h) was an efficient catalytic material by considering stability and activity as compared with physical mixture of MgO-Al2O3 and as-synthesized Mg-Al LDH (without calcination). Under the reaction conditions (270 °C and N2 flow), Mg-Al CLDH showed complete LA conversion with 98% Gvl selectivity (Table 1, entries 15–17). Reactivated (calcination at 450 °C for 1 h) MgO-Al2O3 CLDH showed good results during reusability studies as compared to the spent catalyst [65].
Fu and co-authors studied several Ni-supported catalysts for this conversion by varying the supports such as MgO, Al2O3 and Mg-Al CLDH. Among these supports, Ni(0) on Mg-Al CLDH (derived from Ni-Mg-Al LDH through calcination followed by reduction) showed the highest yield (99.7%) of Gvl in 1,4-dioxane medium at 160 °C, 30 bar H2 for 1 h. High surface area and uniform dispersion of Ni on Mg-Al CLDH was the reason for better catalytic activity as compared to other examined catalysts (Table 1, entries 18–20). They also conducted reusability of CLDH-derived active catalyst (Ni/Mg-Al CLDH) and showed good recyclability even in the absence of reactivation [66].
The above-mentioned synthetic protocol for the generation of active catalyst from LDH precursor consumes a significant amount of energy. To avoid these drawbacks, we recently reported Ni-Al LDH catalyst precursor (without calcination or pre-reduction) for this conversion in water. Interestingly, Ni(0)/boehmite (Ni-Al LDH-derived) was obtained during the reaction and confirmed by PXRD and TEM. The PXRD of NiAl-LDH showed the presence of (003), (006), (012), (015), (018), (110) and (113) planes that are characteristic of hydrotalcite (Fig. 2), which completely converted to (020), (120), (140) and (051) planes of boehmite (JCPDS Card No. 01-074-290), and (111) with (200) for Ni (0) (JCPDS Card No.: 00-004-0850). Platelet-like morphology of NiAl-LDH underwent a change to hexagonal rods of boehmite with embedded spherical Ni(0) particles having <20 nm size as shown in Fig. 3. The generated active catalyst (Ni(0)/boehmite) showed superior catalytic activity towards the reaction with 100% yield of Gvl (Table 1, entry 21). The active catalyst was recyclable up to four cycles without any activation and only a minor decrease in LA conversion in each cycle, maintaining the Gvl selectivity [67].

Powder XRD pattern for NiAl-LDH (catalyst precursor) and Ni(0)@boehmite (catalyst obtained after the reaction). The XRD pattern for Ni(0)@boehmite matched well with nickel (JCPDS Card No: 004-0850; stick pattern included) and boehmite (JCPDS Card No: 01-074-290)

TEM images showing the morphology and crystallinity of NiAl-LDH catalyst precursor and Ni(0)@boehmite catalyst at different magnifications (catalyst obtained after the reaction)
Ma et al. disclosed in situ-reduced nano-Cu/AlOOH catalyst for alkyl levulinates to Gvl using Cu2(OH)2CO3/AlOOH-LDH precursor using 2-propanol as hydrogen source [68]. Under optimized condition (180 °C for 5 h), the in situ-reduced catalyst showed 90.5% yield of Gvl (96% conversion of methyl levulinate) with Cu/Al molar ratio 3/1 (Table 1, entry 22). The pre-reduced catalyst Cu/Al2O3 has lower activity towards the reaction and resulted in 78% yield of Gvl because of the agglutination of active catalytic Cu(0) metal particles on Al2O3 (Table 1, entry 23). The in situ Cu/AlOOH catalyst has strong acidic sites (obtained from TPD analysis) and could reasonably enhance the reaction. Various alcohols as hydrogen sources including primary (methanol, ethanol, 1-propanol and 1-butanol), secondary (2-propanol and 2-butanol) and tertiary (t-butanol) alcohols were studied for the reaction using Cu2(OH)2CO3/AlOOH-LDH precursor. It was observed that primary and tertiary alcohols are inefficient for hydrogen generation and cannot participate in the reaction, wherein secondary alcohols such as 2-propanol and 2-butanol showed remarkable activity to obtain Gvl. Using the in situ catalyst, authors identified significant loss in Gvl yield while increasing the number of catalytic cycles, and they rationalized this from the increase (50 nm from 35 nm) in Cu(0) metal particles size by the TEM analysis of the spent catalyst.
Aytam research group reported ex situ-reduced Ni(0)/Al2O3-TiO2 catalyst , derived from Ni-Al-Ti LDH precursor [69]. The mixed metal catalyst has high catalytic activity for vapor-phase hydrocyclization of LA to Gvl as compared with Ni(0)/TiO2 and Ni(0)/Al2O3 (Table 1, entries 24–26). The high concentration of Lewis acidic sites (confirmed by DRIFT) along with metallic Ni active sites on the surface (confirmed by CO pulse chemisorption) in Ni(0)/Al2O3-TiO2 supports the higher activity of the material. The Lewis acidity of support (Al2O3-TiO2), i.e. peaks at 1630–1600/cm and 1450/cm, influences the dehydration of LA for the formation of angelica lactone (path-b mechanism in Scheme 4) which underwent subsequent hydrogenation to Gvl using active metallic Ni(0). The catalyst was also tested for transfer hydrocyclization of LA with formic acid as hydrogen donor, resulting in higher angelica lactone yield compared to desired Gvl which may be due to insufficient hydrogen (obtained from decomposition formic acid) or deactivation/leaching of active Ni metal under such acidic conditions. The catalyst is active up to 17 h time-on-stream using molecular hydrogen, and a further increase in time and/or increase in reaction cycles, a decrease in the conversion of LA is observed due to carbon (coke formation) deposition on the catalyst.
Transfer hydrocyclization of ethyl levulinate to Gvl using hierarchical multilevel-supported bimetallic catalyst (derived from Al2O3@NiCuAl-LDH precursor) was reported [70]. The bimetallic NiCu catalyst with 0.5 Cu/Ni molar ratio resulted in high activity (89% yield of Gvl) in the presence of isopropanol as hydrogen source (Table 1, entry 27). Alumina-supported bimetallic catalyst (NiCu) has highest density of acidic and basic sites. Lewis acidic sites of catalytic material can interact with carbonyl group of ethyl levulinate through the electron pair on the oxygen atom and are active for C=O hydrogenation. The basic sites of the catalyst are also responsible for MPV reduction and facilitate ethyl levulinate activation. As-fabricated bimetallic NiCu catalyst has good stability and activity for recycling studies. The bimetallic catalyst shows no metal leaching and no particle agglomeration during the reaction.
2.2.1.2 LDH-Supported Metal Catalysts for Hydrocyclization of LA to Gvl
LDH materials as catalytic support were also reported for hydrocyclization of LA. Venugopal group reported Ru(0)/Mg-La CLDH prepared by impregnation procedure using RuCl3 with Mg-La CLDH in aqueous medium. The Ru(0)-supported CLDH catalyst was the first basic supported catalyst for this reaction which showed good catalytic activity (92% LA conversion with >99% Gvl selectivity) in toluene medium at 80 °C, 5 bar H2 for 4 h (Table 1, entry 28) [71]. Furthermore, Rajaram group also reported Pt-supported Mg-Al CLDH for LA to Gvl conversion in aqueous medium at room temperature, 30 bar H2 for 24 h with >99% yield of Gvl (Table 1, entry 29) [72].
Our group reported in situ-generated Ru(0)/MgAl-LDH catalyst from Ru(O/OH)/LDH and hydrous ruthenium oxide (HRO)/MgAl LDH during the reaction. The in situ-regenerated catalysts showed remarkable catalytic activity with >99% yield of Gvl under mild reaction conditions (80 °C, 10 bar H2 for 30 min) in water (Table 1, entries 30 and 31). The Ru(0)/Mg-Al LDH material is prepared by wet impregnation (Mg-Al LDH with RuCl3 solution) followed by in situ reduction during the reaction [73]. The main advantage of this invention is to avoid prior reduction for preparation of Ru(0) from ruthenium precursor. Prior reduction of catalysts mostly consumes more energy and sometimes may even be higher than the input energy used for the reaction.
Cu-supported MgAl-LDH catalyst (prepared from colloidal deposition method) was reported by Asiri research group for hydrocyclization of LA to Gvl under vapour phase using fixed-bed reactor [74]. At optimized conditions, i.e. 260 °C, 10 bar H2, and 30 mL/min H2 flow rate, the 3% Cu/MgAl-LDH catalyst (0.5 g) showed 87.5% conversion of LA (WHSV = 0.456/h) with 95% selectivity of Gvl (Table 1, entry 32). Three per cent Cu loading enhances the overall acidity of the material which initiates the intramolecular esterification of γ-hydroxy pentanoic acid to obtain Gvl. Increasing the temperature to >260 °C decreased the selectivity of Gvl because of the onset of competing reactions such as the dehydration of LA to angelica lactone and ring opening of obtained Gvl into valeric acid. The spent Cu/MgAl-LDH catalyst shows decrease in activity towards the reaction and is associated with Cu particle size increment (5.3 nm from 2 nm) due to agglomeration as well as decrease in the surface area and acidity.
2.2.2 Arylated-γ-Lactones from Levulinic Acid and Aromatics
Preparation of arylated-γ-lactones (Agvls) was demonstrated by Yonezawa research group using homogeneous catalysts such as methanesulfonic acid (MsOH), triflic acid (TfOH) and polyphosphoric acid (PPA) [75]. Among these, triflic acid catalyst showed the highest yield of corresponding lactones. m-Br anisole, o-Br anisole and unsubstituted anisole ended with 84, 46, and 81% yields of desired Agvls at room temperature for 8 h (Scheme 5). However, homogeneous catalysts used are difficult to remove from the product mixture and pollute the environment while disposing. Homogeneous acid catalyst needs to be neutralized after reaction with large amount of bases which demands additional energy and chemical input and increases the overall process cost.

Preparation of Agvls from levulinic acid with aromatics using triflic acid
A patent by Hattori et al., in JP 2011201847-A, disclosed the preparation of Agvls from levulinic acid with aromatics using various solid acid catalysts [76]. The reactions were performed under microwave irradiation in the presence of solvents. The drawback of this invention is the formation of multiple products under microwave irradiation, rendering poor selectivity for the desired product. Another drawback of this invention is the use of microwave irradiation that poses difficulty and challenges for scale-up operations as these are to be prepared in bulk scale (in tonnage) where conventional heating in batch or continuous reactors would be more feasible technologically. Also, various chlorinated high boiling solvents such as 1,2-dichlorobenzene were used that are not environmentally friendly and have recyclability issues.
We reported this transformation under solvent-free conditions using conventional heating. Various zeolites were identified for preliminary screening owing to their properties, i.e. high crystallinity, thermal stability, tunable acidity, reusability and shape selectivity, which find use in industrially relevant transformations especially in petrochemicals. Among zeolites screened, Na/H-β, Na-Y, and Na-ZSM-5, Na-β and H-β were found active for the mentioned conversion. The high activity of β-zeolite is attributed to its unique pore topology and the presence of local structural defects through octahedral extra-framework aluminium (AlO6) that render Lewis acidic properties in the material, resulting in the observed conversion and product selectivity. H-β zeolite fared better compared to Na-β. Under optimized condition, the highest yield of γ-lactone was obtained with anisole: LA 4:1 molar ratio in the absence of solvent at 150 °C using 50 wt% catalyst loading after 12 h (Table 2, entry 1).
Substrate scope was extended from ethoxy to butoxy benzenes. Increasing the alkyl chain length had a positive effect on LA conversion with a selectivity for para-substituted γ-lactones (Table 2, entries 2–4). Aromatic hydrocarbon cumene did not participate in the reaction. Inability of the relatively less electron-rich aromatic ring in cumene to take part in the nucleophilic attack on the carbonyl carbon of levulinic acid could be a reason for its inactive nature (Scheme 6). The ring electron density was increased by the lone pair of electron present on the heteroatom and in the substrates with electron-donating substituents. Thioanisole showed 83% selectivity of para-isomeric γ-lactone (Table 2, entry 5). This synthetic protocol was further explored with 2-methylthiophene and thiophene, which resulted in the selective formation of γ-lactone (90% and 80%) with 93% and 96% LA conversion (Table 2, entries 6 and 7).

Mechanistic pathway for Agvls formation from LA with anisole using β-zeolite
Recyclability of H-β catalyst was also studied. Organic carbon that adhered to the used catalyst (UH-β) surface was removed by calcination in air at 550 °C for 3 h. The used calcined catalyst (UH-βC) and fresh H-β catalyst showed comparable yield and selectivity for the synthesis of γ-lactone. A scalability study at 10 g of LA with anisole showed an increase in γ-lactone yield with time. After 6, 12, 18 and 24 h, 38, 62, 80 and 90% yield of para-substituted γ-lactone was obtained with 95, 94, 92 and 90% selectivity, respectively.
Mechanistic investigation for the formation of Agvls (Scheme 6) suggests that the lone pairs of the keto carbonyl group of LA interact with Lewis acidic sites of aluminium in zeolitic framework, present due to local defects [77, 78]. A partial positive charge generated on the keto carbon is responsible for the nucleophilic attack resulting in the formation of γ-hydroxy pentatonic acid intermediate. This unstable species underwent intramolecular esterification to γ-lactone on removal of water [79]. The higher activity of H-β zeolite compared to Na-β is possibly due to the presence of Bronsted acidic protons on H-β zeolite.
2.2.3 Synthesis of 4,4-Diryl-Substituted Pentanoic Acid/Esters from Levulinic Acid and Aromatics
2.2.3.1 Preparation of Diphenolic Acid
Bisphenol A (BPA), a popular plasticizer prepared from the acid-catalysed condensation of phenol and acetone (2:1 ratio), is a commercial polymer precursor, used for thermoplastic polymers, polycarbonates and epoxy resins and is also used in the making of paper currencies by several countries [80, 81]. Diphenolic acid (DPA), a structural analogue and a potential alternate to BPA, is a lignocellulose-derived bisphenol having diaromatic-substituted moiety as 4,4-bis(4-hydroxyphenyl)valeric acid, prepared on condensation of LA with phenol under acid-catalysed reaction (Scheme 7) [82]. Many catalysts are reported for this conversion. Initially, the reaction was reported with Bronsted mineral acids (H2SO4 and HCl) [83]. However, these are toxic and corrosive, and hence homogeneous mineral acids were replaced by insoluble heterogeneous catalysts.

Preparation of diphenolic acid from levulinic acid with phenol
The insoluble cesium-substituted Keggin-type heteropolyacid such as Cs1.5H4.5P2W18O62 showed 70.5% yield of DPA with 88% selectivity of p,p′-DPA isomer at 150 °C, molar ratio of phenol to LA 4:1 for 10 h [84]. The heteropolyacid catalyst revealed good reusability for three repetitive runs.
A water-tolerant solid acid catalyst , i.e. mesoporous H3PW12O40/SBA-15 (obtained from sol-gel co-condensation), has been reported for DPA synthesis [85,86,87]. The H3PW12O40/SBA-15 has high surface area, well-defined pore size and uniform distributed catalytic sites. The SBA with 15.7% loading of H3PW12O40 showed better activity towards the reaction, 80% conversion of levulinic acid with 2.9:1 ratio of DPA (p,p′) isomer to DPA (o,p′) isomer. The high pore size of catalytic material facilitates the accessibility of the reactive sites in the framework for the reactants. The H3PW12O40/SBA-15 catalyst is regenerated by calcination (420 °C) and was recycled for five reaction cycles with almost similar catalytic activity.
Sels research group reported a new class of acid catalyst, i.e. sulfonated hyperbranched poly(arylene oxindoles) (SHPAOs) in combination with thiol co-catalyst. Using SHPAO catalyst, various thiol compounds, e.g. benzylthiol, ethanethiol, 1-propanethiol, 1-butanethiol, 2-propanethiol and 2-methyl-2-propanethiol, were explored at 200 °C for 16 h under inert atmosphere and absence of solvent [88, 89]. The less sterically hindered co-catalyst/additive ethanethiol showed high yield of DPA (52.9%) with high selectivity of p,p′ isomer (19.5 ratio of the p,p′-DPA to the o,p′-DPA isomer). The role of thiol promoter in catalytic mechanism cycle is mentioned in Scheme 8.

Mechanism for formation of diphenolic acid using sulfonic acid-functionalized catalyst with thiol promoter
High yield of DPA has been reported using Bronsted acidic ionic liquids (BAILs) as catalyst [90]. The combination of ionic liquid and propanethiol showed 91% yield of DPA including high selectivity of p,p′-DPA isomer (ratio of p,p′/o,p′ is >100) at 60 °C for 48 h (Scheme 9). The catalytic system is also applied for the preparation of diphenolic esters by reacting alkyl levulinates with phenol. The alkyl levulinates, methyl, ethyl and n-butyl, gave 57%, 58% and 75% yield of corresponding diphenolic esters, and diphenolic acid was observed as the side product. Interestingly, high yield of diphenolic esters was observed using levulinic acid as the reactant in a suitable alcoholic medium. Methanol, ethanol and n-butanol resulted in 87%, 83% and 86% yield of the corresponding diphenolic esters, and a minor yield of diphenolic acid (2–6%) was reported. The catalytic system had good stability and was recyclable for four reaction cycles, with a minor decrease in the product yield.

Ionic liquid-catalysed diphenolic acid formation using propanethiol
Recently, Shen et al. reported different acid catalysts for this conversion such as [BSMim]OAc, as [BSMim]CF3SO3, [BSMim]HSO4, [Bpy]HSO4, [AMim]Br, [BMin]Cl, NH3SO3H, p-TSA and HCl at 60 °C, 24 h, LA to phenol (1:4) ratio, 50 mol% of catalyst and 1 mol% ethanethiol w.r.t LA. Among them, [BSMim]CF3SO3 and [BSMim]HSO4 resulted in 100% selectivity for p,p′-DPA with 81 and 75% conversion of LA. At optimized condition, [BSMim]HSO4 showed 93% conversion with desired p,p′-DPA as the only product [91].
2.2.3.2 Preparation of 4,4-Bis(5-Methylfuran-2-yl) Pentanoic Acid and Esters
Zhang research group disclosed the acid-catalysed preparation of γ,γ-di-2-methylfuran pentanoic acid (4,4-bis(5-methylfuran-2-yl) pentanoic acid) and its esters from levulinic acid and its derivatives (angelica lactone and ethyl levulinate) with 2-methylfuran by hydroxyalkylation-alkylation (HAA) (Schemes 10 and 11) [92, 93]. Nafion-212 resin showed good catalytic activity towards the desired product. At reaction conditions (50 °C for 1 h), angelica lactone showed high yield of HAA product with 81.3%, wherein 12.0% and 4.8% yield of HAA product was reported from ethyl levulinate and levulinic acid. The reusability of Nafion-212 resin was investigated for six reaction cycles with marginal decrease after the third run, attributed to handling loss of the catalyst.

Hydroxyalkylation-alkylation of ethyl levulinate with 2-methylfuran

Hydroxyalkylation-alkylation of angelica lactone with 2-methylfuran
The product from HAA reaction, i.e. 4,4-bis(5-methylfuran-2-yl)pentanoic acid, is converted into 4,4-bis(5-methyl-tetrahydrofuran-2-yl)pentan-1-ol intermediate by hydrogenation over 5% Pd/C catalyst. The substituted pentanol is converted into liquid alkanes ((C9–C15 diesel range alkanes (81–82%), C5–C8 gasoline range alkenes (10–11%) and C1–C4 alkanes (1–2%)) through hydrodeoxygenation using again 5% Pd/C catalyst under continuous flow (Scheme 12) [92].

Liquid alkanes from obtained 4,4-bis(5-methylfuran-2-yl)pentanoic acid through hydrogenation followed hydrodeoxygenation
2.2.3.3 Preparation of (4,4-Bis(5-Methylthiophen-2-yl)pentanoic Acid
Our group reported the preparation of (4,4-bis(5-methylthiophen-2-yl)pentanoic acid denoted as bisthiophenic acid-methyl (BTA-M)) from levulinic acid with 2-methylthiophene for the first time using solid acid catalysts (Scheme 13). Among the catalysts used such as zeolites, clay and resins, the resin catalysts (Indion-190 and Amberlyst-15) were more active. Indion-190, an inexpensive resin catalyst, showed 98% selectivity and 99% conversion (based on LA) of BTA-M under optimized solvent-free reaction conditions, i.e. levulinic acid to 2-methylthiophene molar ratio is 2.5, 30 wt% of Indion-190 catalyst w.r.t LA, 110 °C for 6 h. The recyclability of Indion-190 for the preparation of BTA-M was demonstrated for three reaction cycles with a slight decrease in the conversion of reactants.

Preparation of BT(A/E)-M from levulinic acid and its esters with 2-Met using acid catalysts
Preparation of alkyl 4,4-bis(5-methylthiophen-2-yl)pentanoate (bisthiophenic alkyl esters-methyl (BTAE-M)) from levulinic esters (methyl, ethyl and butyl) with 2-methylthiophene using Indion-190 catalyst was also attempted (Scheme 13). In the presence of methyl and ethyl levulinates, high conversion (99 and 94%) and good selectivities of related products methyl 4,4-bis(5-methylthiophen-2-yl)pentanoate (96%) and ethyl 4,4-bis(5-methylthiophen-2-yl)pentanoate (95%) were observed. Under similar conditions, butyl levulinate showed less conversion (13%) with poor selectivity of butyl 4,4-bis(5-methylthiophen-2-yl)pentanoate (38%). This poor conversion may be due to the bigger size of butyl levulinate, which is sterically precluded from interacting with the acidic active sites of the catalyst.
The applications of BTA-M and BTAE-M were explored in various domains. We tested BTA-M compound as lubricant, plasticizer and antioxidant and antibacterial agent. Surprisingly, the molecule showed good applicability in the field of polymers as plasticizing agent. Moreover, the compound has good antioxidant property.
3 5-Hydroxymethylfurfural
5-Hydroxymethylfurfural (HMF) is an organic compound which was first identified in 1875 as an intermediate product during the synthesis of LA from sugar under sulphuric acid treatment. Mainly, HMF is considered as a platform chemical for the synthesis of bio-based fuel additives and polyester building blocks.
3.1 Preparation of 5-Hydroxymethylfurfural
In principle, HMF preparation Fructose dehydration to HMF conversion mechanism is simple, i.e. removal of three water molecules from hexose using an acid catalyst. However, practically synthesizing HMF is quite complicated because of the side reactions under acidic medium, i.e. hydration of HMF to LA with formic acid as a byproduct and homo-polymerization of HMF into poly-furanics which result in poor yield of HMF. According to the literature, cellulose or starch can be converted to HMF in three steps as follows:
-
1.
Acid-catalysed hydrolysis of cellulose or starch into glucose monomeric units.
-
2.
Acid-catalysed isomerization of glucose to fructose.
-
3.
Acid-catalysed dehydration of fructose to HMF.
The key to achieve good yields of HMF is to choose the best catalyst, and the reaction condition simultaneously should suppress the formation of byproducts.
3.1.1 Catalytic systems reported for Fructose Dehydration to HMF
According to the literature, there are two dehydration mechanistic pathways, resulting in cyclic and acyclic intermediates [94]. However, cyclic pathway is well accepted around the scientific community (Scheme 14). Many catalysts were reported for this conversion including mineral/organic acids, metal salts/oxides, heteropolyacids, functionalized carbon materials, ion-exchange resins, clay and zeolites. The summarized reports of each type of catalytic materials are discussed in the following sections.

Proposed mechanism for the dehydration of fructose to HMF using acid catalyst
3.1.1.1 Mineral and Organic Acids
Mineral acid (HCl, H3PO4, H2SO4)-catalysed homogenepus catalytic reactions are reported in batch and continuous flow conditions (Table 3, entries 1–5) [95,96,97,98,99,100]. High HMF conversion rate was observed regardless of the nature of the acid, and it is well known that H+ ions are responsible for the dehydration reaction. Using mineral acid catalysts, the difference in the selectivity of HMF is observed by varying medium and anions associated with the acids [95].
In continuous micro-reactor system using HCl catalyst, 96% conversion of fructose with 85% selectivity of HMF in mixed solvents (MIBK/2-butanol + H2O + DMSO) was reported (Table 3, entry 1). H2SO4 and HCl showed excellent yield of HMF, i.e. 95 and 97% in the presence of ionic liquid (Table 3, entries 2 and 3). 67% yield of HMF was observed with H3PO4 as catalyst in [BMIM]Cl ionic liquid (Table 3, entry 4). Fructose in DMSO medium using formic acid catalyst afforded maximum yield of HMF (99%) (Table 3, entry 5). Practical problems observed in making HMF using mineral acids as catalyst are corrosion of reactor vessel and product separation.
3.1.1.2 Metal Salts
Lewis acidic metal chlorides were found to be excellent materials for acid-catalysed dehydration of fructose for HMF production. AlCl3, catalysts from lanthanide group, chromium chloride, zinc chloride, lithium chloride, niobium chloride, indium chloride and cupric chloride were also employed as catalysts in DMSO, water, sulfolane and ionic liquids like [BMIM]Cl and [EMIM]Cl solvents (Table 4, entries 1–9) [101,102,103,104,105,106,107,108,109]. The highest yield of HMF was observed with LaCl3 and CrCl3 (92.6 and 96%). Other metal salts afforded good to moderate yield of HMF and some of them were recyclable, e.g. AlCl3 and InCl3.
In a noticeable study, our group reported that the synergism among alumina, metal salt and solvent played an important role in the direct conversion of glucose to HMF. Alumina helped in the isomerization of glucose to fructose, while copper chloride and solvent promoted fructose dehydration to HMF (Scheme 15). Using this catalytic system, several carbohydrates were chosen for the preparation of HMF such as glucose, fructose, galactose, cellobiose, sucrose and starch (Table 4, entries 10 and 11) [110]. Reaction at higher glucose concentration (50 wt% w.r.t to DMSO) was carried out at 140 °C. The HMF yield was 52% with the retention of efficiency even at high substrate concentration.

Conversion of glucose to HMF using combination of Al2O3 and CuCl2 in DMSO medium
3.1.1.3 Metal Oxides
Inexpensive metal oxides were extensively studied for fructose dehydration in different solvent systems (Table 5). Armaroli et al. reported niobium phosphate as heterogeneous catalyst for this dehydration reaction in water as solvent medium. Although the conversion level is low, selectivity (100%) towards HMF was excellent, and the catalyst was reusable for four cycles (Table 5, entry 1) [111]. Carniti group demonstrated fructose to HMF dehydration reaction in a continuous flow reactor system using silica-niobia mixed metal oxide catalyst (Table 5, entry 2) [112, 113]. The mentioned fixed-bed reactor system was operational up to several hundred hours, but the selectivity and yields were very low. At high temperature (175 °C), silicoaluminophosphate catalyst yielded 80% HMF at 1 h and was reusable up to five cycles (Table 5, entry 3) [114]. Binary mixtures of solvents (water +2-butanol) was reported using tantalum hydroxide (treated with 1 M phosphoric acid) as a catalyst at 160 °C and afforded 90% yield of HMF (Table 5, entry 4). Catalyst recyclability experiments were done by washing the used catalyst several times with water followed by oven drying overnight and used for further reaction cycles [115]. Saha research group discussed self-assembled mesoporous titania microspheres for this dehydration reaction in binary solvent system, i.e. dimethylacetamide and 10% lithium chloride with 74.2% yield of HMF (Table 5, entry 5). Lewis acidic TiO2 is responsible for the glucose to fructose conversion, and lithium chloride was responsible for fructose dehydration [116]. Sulphated zirconia (prepared by impregnation with H2SO4) reported moderate HMF yield (72.8%) from fructose in acetone-DMSO medium under microwave irradiation (Table 5, entry 6) [117].
3.1.1.4 Heteropolyacid (HPA)-Based Catalysts
Heteropolyacids are a class of strong acids having certain metal or non-metal-based oxo-anionic clusters in the system. Due to their excellent physicochemical properties, Keggin structure, thermal stability and high acidity, they have been extensively reported as catalysts for acid-catalysed organic transformations including dehydration reactions [118,119,120]. Xiao et al. demonstrated that H3PW12O40 and H4SiW12O40 are highly efficient catalysts for this dehydration reaction using 1-butyl-3-methylimidazolium chloride [BMIM]Cl as a solvent. HMF was obtained with 99% yield in only 5 min at 80 °C under microwave irradiation condition (Table 6, entry 1) [121].
Jiang group reported a novel water-tolerant C16H3PW11CrO39 catalyst, prepared from cetyltrimethyl ammonium salt of transition metal (Cr(III))-substituted polyoxometalate (C16-cetyltrimethyl ammonium). The novel catalyst showed moderate performance even at high concentration of fructose (30 wt%) (Table 6, entry 2) [122]. The high catalytic activity at concentrated fructose solution is due to the coexistence of Lewis and Bronsted acidic sites and also the hydrophobic groups present in the catalyst.
Wang research group found an economic and environmental friendly method for obtaining HMF from fructose using Cs2.5H0.5PW12O40 catalyst in a biphasic system (water + MIBK). Seventy-four per cent yield of HMF with 94.75% conversion of fructose at 115 °C for 1 h was reported, and the catalyst was reported to be effective up to high concentrations of fructose (50 wt%) as well (Table 6, entry 3) [123]. Silver ion exchanged phosphotungstic acid (Ag3PW12O40) also proved to be an excellent catalytic system for fructose dehydration to HMF (Table 6, entry 4) [124]. To overcome the difficulty in separation and large usage of ionic liquids (ILs), HPA-ILs composites were developed for this acid-catalysed dehydration reaction. Such a system was reusable at least five times (Table 6, entry 5) [125]. Ganji recently reported tin-loaded silicotungstic acid (Sn-STA-2) by hydrothermal microwave treatment which showed good conversion and yield of HMF in DMSO medium . The surface acidity of the catalyst (Sn-STA-2) plays a crucial role for optimum catalytic activity (Table 6, entry 6) [126].
3.1.1.5 Ion-Exchange Resins
Ion-exchange resins are insoluble solid matrix/support normally in the form of 0.5–1 nm size beads or balls. The appearance varies from yellow, brown and faded black colour, made from organic polymeric substrate. Predominantly, two main classes of ion-exchange resins are available, styrene-based sulfonic acid resins and Dow-type resins [127, 128]. The resin catalysts show high catalytic activity towards many acid-catalysed reactions. Hence, the researchers explored them for the dehydration of fructose to HMF. A comparative study was done by Nijhuis group with acidic heterogeneous catalysts like alumina, aluminosilicate, zirconium phosphate, niobic acid, ion-exchange resin, Amberlyst-15 and mordenite (MOR)-zeolite for fructose dehydration to 5-hydroxymethylfurfural (HMF) in aqueous medium. It was reported that the order of Bronsted acidity contributing to the dehydration reaction is Amberlyst-15 > MOR>ZrPO4 > SiO2-Al2O3 > Nb2O5 > Al2O3. The authors concluded that HMF selectivity correlates with the presence of Bronsted acidity in Amberlyst-15 and zeolite-MOR catalysts, whereas Lewis acidity is responsible for the decreased HMF selectivity due to the initial condensation of fructose to humins over Lewis acidity (Table 7, entry 1) [129].
Mu group extensively studied fructose dehydration in low boiling solvents using Amberlyst-15 acidic resin catalyst and yielded approximately 50% HMF (Table 7, entry 2) in THF medium and in binary mixture of solvents (THF and methanol). HMF along with 5-methoxymethylfurfural (MMF) were obtained with an increase in the total yield to 65%. Using methanol as solvent, etherification was initiated as HMF was converted into MMF with 37% yield (Table 7, entry 2) [130]. Commercially attractive process for the production of HMF from high fructose corn syrup was developed by Jeong et al. They found 1,4-dioxane to be a promising solvent media after screening various solvents using Amberlyst-15 a solid acid catalyst. HMF yield was 80% with the reusability of the catalyst up to five times and solvent recycled after simple distillation (Table 7, entry 3) [131]. Morales group reported high sulfonic acid-loaded acidic resin as a heterogeneous catalyst for the dehydration of fructose to HMF in DMSO, yielding 93% of HMF in 1 h (Table 7, entry 4). The authors also studied this reaction with glucose as a starting material, which yielded 33% HMF in 24 h. In this case, DMSO as a solvent played an important role in dehydrating glucose to anhydroglucose which facilitated the production of HMF by reducing side reaction [132].
A study done by our group showed the influence of DMSO and dimethylformamide (DMF) on the recyclability of Amberlyst-15 catalyst for the dehydration of fructose to HMF. In case of DMSO as a solvent, stable activity of the catalyst is noted, whereas in DMF, significant loss of activity is observed up on recycling. The loss of activity in DMF is due to the neutralization of acidic sites by the formation of ammonium ions which was inferred from FT-IR and CHNS studies. To regenerate the catalytic activity, dilute acid treatment was required (Table 7, entry 5) [133].
3.1.1.6 Functionalized Carbon Materials
High stability of carbon materials and complementary properties on hydrophobicity have resulted in the use of mainly sulfonated carbonaceous materials as solid catalysts for fructose dehydration to HMF. Wang group reported a carbon-based solid acid, prepared from glucose and p-toluenesulfonic acid (TsOH), and used for catalytic dehydration of fructose into HMF (91.2% yield). The catalyst was active in DMSO at 130 °C for 1.5 h (Table 8, entry 1). Moreover, this catalyst showed a good reusability for five reaction cycles [134]. Huang et al. reported cellulose-sulphuric acid as catalyst, prepared from cellulose treated with chlorosulfonic acid. The recyclable solid acid catalyst revealed high yield (93.6%) of HMF in DMSO for 45 min (Table 8, entry 2) [135].
A series of sulfonic acid-functionalized carbon materials (C-SO3H), including poly(p-styrenesulfonic acid)-grafted carbon nanotubes (CNT-PSSA), poly(p-styrenesulfonic acid)-grafted carbon nanofibers (CNF-PSSA), benzenesulfonic acid-grafted CMK-5 (CMK-5-BSA) and benzenesulfonic acid-grafted carbon nanotubes (CNT-BSA), was studied for this conversion. Among these functionalized carbons, CNT-PSSA showed exceptional catalytic activity and afforded 89% HMF yield (Table 8, entry 3). This excellent catalytic activity is due to the high Bronsted acid strength which is confirmed by potentiometric acid-base titration and ion chromatography [136].
Russo et al. adopted a simple synthetic procedure for the preparation of carbon-silica composite activated with SO3H groups by acid treatment with different acid concentration. The reaction carried out in the binary solvent mixture (ethanol-water) at 170 °C for 1 h resulted in fructose conversion up to 78% with 43% yield of HMF (Table 8, entry 4) [137]. The authors claimed that increase in the ratio of surface activating agent and mass of carbon precursor eventually increases the acid strength of the catalyst, and this is also accompanied by the creation of mesoporosity which increases the accessibility of the reactants towards the active sites. Zhu research group reported graphene oxide as a catalyst for this conversion. In this work, a series of co-solvents were employed with DMSO. 20 vol% 2-proponal (with DMSO 80 vol%) binary solvent system yielded 90% HMF (Table 8, entry 5). The authors revealed that a small number of sulfonic groups and abundance of oxygen-containing groups (alcohols, epoxides, carboxylates) play a synergic role in maintaining the high performance of graphene oxide [138].
3.1.1.7 Clay and Zeolitic Materials
Zeolites are microporous aluminosilicates widely used as commercial adsorbents, more particularly in gas adsorption and bearing worldwide application as industrial catalysts. Zeolites have large open cage-like structure that form channels, having large pore size and high surface area which greatly allow the reactants into the zeolite framework and result in good catalytic activity. This property of zeolites culminated in their classification under the materials known as “molecular sieves”. Zeolites are more advantageous over other solid acid catalysts, having advantages like easy separation compared to homogenous catalysts, catalyst regeneration upon high temperature treatment and stable at high temperature in aqueous condition compared to ion-exchange resin catalysts. Some highlighted results of fructose dehydration investigated using zeolite catalysts and listed in Table 9.
Moreau et al. performed dehydration of fructose into 5-hydroxymethylfurfural in a batch mode in the presence of H-form of zeolites as catalysts at 165 °C and in a binary solvent mixture consisting of water and methylisobutylketone (1:5 by volume). The catalytic activity observed depended on both the acidic sites and structural properties of the catalyst used. Good selectivity towards HMF was achieved using mordenite-zeolite catalyst with the Si/Al ratio—11 (Table 9, entries 1 and 2). The high selectivity obtained was correlated by shape-selective properties of catalyst which can be reused for several runs after thermal treatment without considerable loss in activity [139, 140].
Zhang group discussed the preparation of novel zeolite microspheres for the dehydration of fructose to HMF. They adopted a simple synthetic procedure named polymerization-induced colloid aggregation (im-PICA) for the preparation of zeolite microspheres (ZMSs) with a hierarchical porous structure and uniform particle size. As expected, these zeolite microspheres afforded good yield of HMF (77%) (Table 9, entry 3) [141]. The catalytic activity of Sn-montmorillonite (mont) was explored towards this conversion in DMSO-THF binary mixture of solvents, and the HMF yield was 78% in 1 h at 160 °C from fructose (Table 9, entry 4) [142]. The aluminium-exchanged montmorillonite-K-10 clay was found to be an efficient catalyst for the dehydration of fructose to HMF in DMSO solvent with 93.2% yield at 120 °C in 3 h (Table 9, entry 5) [143]. Yang et al. provided a green and economic pathway for the conversion of fructose to HMF using natural clay attapulgite (ATP) in a 2-butanol-water system. The highest activity was observed with phosphoric acid-treated attapulgite (ATP-P) catalyst. This excellent activity was attributed to the appropriate amount of Bronsted acidic sites which brought the HMF yield of 96.3% (Table 9, entry 6) [144].
3.2 Applications of 5-Hydroxymethylfurfural
HMF is a multifunctional molecule which can undergo many organic transformations, for example, oxidation, hydrogenation, etherification, hydrodeoxygenation, hydration, decarboxylation, hydrogenation-hydrogenolysis and esterification, with several high value-added products as mentioned in Scheme 16. Thus, HMF is known as the “sleeping giant” in the field of biomass and is a common building platform reported in the survey by DOE and EU.

Organic transformations of HMF
Among the applications FDCA conversion of HMF, the selective oxidation product FDCA is attractive in the present scenario as it is a precursor for polyethylene furanoate (PEF), a suitable alternative for the replacement of fossil-based terephthalic acid (TPA)-derived polyethylene terephthalate (PET) for making plastics (Scheme 17) [145]. Unfortunately, HMF is not stable for long periods under ambient conditions, though pure HMF remains stable up to 8 months under freezing conditions. Presence of small impurity promotes the formation of dimer and oligomers.

Preparation of PET (from terephthalic acid) and PEF (from FDCA)
3.2.1 History of FDCA Synthesis
FDCA was first produced from the dehydration of mucic acid in the presence of strong acid (48% HBr) by Fitting et al. This conversion is industrially un-attractive due to high cost of starting material, long reaction time and relatively high reaction temperature. Further, a process was developed from xylose-derived furfural. Unfortunately, this pathway was also aborted due to the multistep synthesis including a number of intermediates (Scheme 18). Thus, the researchers strived to look for the alternative, efficient and economical pathway for the production of FDCA [145].

Preparation of FDCA from furfural
3.2.2 Selective Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid
Several furanic derivatives can be obtained via catalytic oxidation of HMF, including 2,5-diformylfuran (DFF), 5-formyl-2-furancarboxylic acid (FFCA), 5-hydroxymethyl-2-furancarboxylic acid (HFCA) and 2,5-furandicarboxylic acid (FDCA). Among these multiple oxidation products, an efficient and selective preparation of FDCA is a challenging task. The reaction scheme of HMF to FDCA is mentioned in Scheme 19.

Oxidation reaction network of HMF to possible products
Selective oxidation of HMF to FDCA is advantageous because it is a one-pot synthesis. Conversion of HMF to FDCA was demonstrated using several homogeneous catalysts such as KMnO4, HNO3, Co(OAc)2, Mn(OAc)2, Zn(OAc)2, CuCl2, etc. [145]. Working with homogeneous catalyst has some drawbacks like reduced FDCA yield, side products, cumbersome separation and purification of product from reaction mixture, use of additives and reusability of the catalyst.
Oxidation of HMF to FDCA was demonstrated extensively using various heterogeneous catalysts with molecular oxygen (O2), air, H2O2, t-BuOOH and KMnO4 as oxidants, but researchers mostly prefer molecular oxygen due to its high oxidation potential, low cost and environmentally benign nature. Non-noble mono-metal or its mixed metal oxide heterogeneous catalysts from Mn, Co, Fe, Ce, Cu and Li are reported for this oxidation reaction with base or under base-free conditions (Table 10, entry 1-15). Among the non-noble metal-containing catalysts, Fe, Mn and Ce metal oxides were exclusively reported for this conversion in organic, aqueous and ionic liquid medium with moderate to good yields of FDCA. For further improvement in FDCA selectivity, researchers developed many noble metal (Au, Rh, Pt, Pd and Ru) catalytic systems under base or base-free aqueous medium using O2/air oxidant (Table 10, entry 16-19 and Table 11). Even though good yields were achieved using Rh and Pt, these expensive materials increased the cost of the end product. Au, Pd and a combination of these metals as catalysts showed high activity using base additive at mild reaction conditions and short time as compared with base-free reaction set-ups. Moreover, according to the recent analysis, the Pd metal price raised by 400%, and this precious metal is now $1351.40 for 25 g, and hence more expensive than platinum ($792.30).
Recently, Ru-based catalysts are becoming attractive towards this oxidation, as there are several advantages behind it: stability in aqueous medium, high selectivity of FDCA at relatively milder reaction conditions (preferably low O2/air pressure), base-free and availability of multiple Ru species such as Ru(0), oxide and hydroxide. Moreover, the price of Ru is comparatively less than other noble metals. Important findings for the HMF oxidation to FDCA using Ru metal-supported catalysts preferably under aqueous base-free medium are covered in the following section.
3.2.2.1 3.2.2.1 Preparation of FDCA from Selective Oxidation of HMF over Supported Ru Catalysts and its activity comparison with other metal catalysts
Gorbanev et al. studied Ru(OH)x catalyst on different oxide supports such as TiO2, Al2O3, Fe3O4, ZrO2, CeO2, MgO, La2O3, hydrotalcite, hydroxyapatite and MgO. La2O3 for conversion of HMF into FDCA in aqueous medium, under base-free condition with low to moderate oxygen pressure [164]. ZrO2- and Al2O3-supported catalysts induced the formation of formic acid as side product. Fe3O4 and hydroxyapatite supports showed good selectivity of the desired product; however, in both cases, the observed mass balance of the reaction was low, due to the formation of solid humins in the course of the reaction. Even though good selectivity of FDCA was achieved using basic Mg oxide supports such as MgO, MgO.La2O3 and hydrotalcite, the authors found leaching of Mg in the reaction medium. Compared with TiO2, CeO2 support showed high conversion and selectivity; at 140 °C, 2.5 bar O2 for 18 h, Ru(OH)x/CeO2 afforded 60% yield of FDCA and 10% of 5-hydroxymethyl-2-furan-carboxylic acid (Table 11, entry 1), and no other side products (levulinic acid and formic acid) were detected using CeO2 support. Recyclability of Ru(OH)x/CeO2 system was also demonstrated for three cycles by the authors.
Palkovits research group introduced Ru supported on covalent triazine framework (CTF) catalysts for this oxidation. The catalytic activity of Ru/CTF was compared with conventional catalysts such as Ru/C, Pt/C, Ru/γ-Al2O3 and Pd/C under base-free aqueous medium using synthetic air as the oxidant (20 bar) (Table 11, entries 2–6) [165]. Ru/γ-Al2O3 and Pd/C exhibited low conversion of HMF and no discernable yield of FDCA. Pt/C catalyst was very active for FDCA formation (56%) but less mass balance was reported for the reaction; formation of polymeric/oligomeric products of FDCA could be the reason behind. In the case of Ru-based catalysts, mass balance is generally good, and side products DFF and FFCA are formed in addition to FDCA. Ru/CTF showed 41.4% yield of FDCA which is higher than Ru/C (23.7%) catalyst under similar reaction conditions. High surface area (2071 m2/g) and high polarity related to hydrophilicity seem to have positive effects in this conversion. Interestingly, the authors observed high yield of FDCA (77%) by treating Ru/CTF catalyst with DMSO before reaction with a reaction time of 3 h (Table 11, entry 7). Ru/CTF has good reusability and was found to be stable compared to conventional Ru/C.
Yi et al. investigated HMF to FDCA over commercially available Ru/C catalyst [166]. Initially, the reaction was conducted with strong and weak base additives, and the authors observed that weak base such as CaCO3 is attractive and showed high yield (95%) of FDCA under O2 pressure (2 bar) (Table 11, entries 8–12). Moreover, a clear solution was obtained in the presence of weak bases, while a brownish coloured solution was obtained for strong bases (NaOH or KOH) owing to the degradation of HMF and promotion of side reactions. Ru/C catalyst was applied in the absence of base as well. At optimized reaction condition (120 °C, 5 bar O2 for 10 h), the catalyst afforded 88% yield of FDCA with complete conversion of HMF (Table 11, entry 13). Recyclability for Ru/C was also examined, and after each reaction, the catalyst was washed with methanol before being used, that showed good activity and stability for three reaction cycles. From the control experiments, the authors proposed the reaction path, following the Route-A mechanism (Scheme 16) via intermediate DFF and FFCA.
Mishra et al. discussed supported Ru catalysts for air oxidation of HMF to FDCA in aqueous medium and absence of base. In this work, supports were chosen from various Mg- and Co-based oxides such as MnCo2O4, CoMn2O4, MnCo2CO3, MnO2 and CoO [167]. Among them, Ru/MnCo2O4 exhibited the highest selectivity for FDCA (99.1% yield) at relatively milder reaction conditions (120 °C, 24 bar air for 10 h) (Table 11, entries 14–18). The appropriate acidic sites in Ru/MnCo2O4 catalyst was confirmed by NH3-TPD, Bronsted acid sites (10.7 mmol/g) and Lewis acid-coordinated NH3 (7.3 mmol/g), i.e. a total of 18 mmol/g aided in the selective formation of FDCA. Moreover, MnCo2O4 support is a crystalline cubic system, whereas others have distorted spinel structures (e.g. CoMn2O4 is body-centred tetragonal phase). Generally, the spinel system has more adsorption affinity of oxygen. Additionally, using MnCo2O4 support had high surface area (151.1 m2/g) and high Ru dispersion % (39.2) which were beneficial for its higher activity. Ru/MnCo2O4 was reused for five cycles with similar conversion and selectivity. ICP analysis precluded even traces of leached Ru metal in the solution.
Yang et al. investigated the effect of reaction parameters and solvents towards HMF oxidation using RuIII-incorporated zirconium phosphate (ZrP-Ru) catalyst [168]. Various solvents were used for this study including H2O, MeCN, ethanol, methanol, DMSO, MIBK, toluene and p-chlorotoluene. Among these, DMSO, toluene and p-chlorotoluene resulted in high selectivity of FDCA (Table 11, entries 19–25). In the case of alcohol solvents, etherified product was predominant which could be attributed to the Lewis acidity of Zr. Addition of base and increasing the temperature of the reaction improved the selectivity of FDCA (Table 11, entry 26). The decrease in selectivity of FDCA may be associated with the feed pattern of O2 pressure, as the authors flushed the reaction set-up with oxygen under atmospheric pressure at a flow rate of 20 mL/min.
Pichler et al. demonstrated different ZrO2-supported ruthenium catalysts for this conversion in aqueous basic medium using O2 gas as the oxidant (10 bar) [169]. The high surface area-containing catalysts such as Ru/ZrO2 H-SBA and Ru/ZrO2 H-aero showed high catalytic activity and 97% and 100% conversion of HMF with 87% and 97% selectivity of FDCA (Table 11, entries 27 and 28). The high activity of these supports is associated with high surface area; for Ru/ZrO2 H-SBA the surface area is 256 m2/g and 239 m2/g for Ru/ZrO2 H-aero. The experiments show that the size of the ruthenium particles is crucial for the catalytic performance; larger surface area leads to the formation of small Ru particles (0.8 nm for Ru/ZrO2 H-aero, 1.0 nm for Ru/ZrO2 H-SBA) which were found to be more active than bigger Ru particles observed in other synthesized and commercial (monoclinic and tertragonal) ZrO2 supports. Ru/ZrO2 H-aero catalyst was tested for reusability studies, and a decrease in activity was observed with increasing reaction cycles, but no loss in the carbon balance and no leaching of Ru metal was observed.
Antonyraj et al. studied different oxide supports such as MgO, MgAlO, La2O3, CeO2 and ZrO2 [170]. Among these supports, MgAlO (prepared by co-precipitation under low supersaturation followed by calcination)-supported Ru catalyst showed better activity and afforded 99% yield of FDCA which is higher as compared with other supported Ru catalysts (Table 11, entries 29–33). Because of the high surface area (200 m2/g) and appropriate basic property (6.09 mmol/g) of MgAlO, it was found to be highly active. In the case of other supports including MgO, their observed surface area was less than 100 m2/g. Even though La2O3 support has strong basic sites, the catalyst showed poor conversion due to the less surface area (13 m2/g) and surface acidity. In the case of CeO2 and ZrO2, less surface area and no basic property resulted in poor selectivity towards FDCA. The authors also studied the reusability of both catalysts, i.e. Ru/MgO and Ru/MgAlO supports were found to be dissolved during the reaction, but Ru/MgO showed good catalytic activity/stability for five reaction cycles.
Gao et al. studied highly dispersed ruthenium nanoparticles on hydroxyapatite (HAP) solid support for the aerobic oxidation of HMF to FDCA in aqueous medium under base-free condition [171]. A systematic study done by the authors includes the effect of HMF/Ru molar ratio, oxygen pressure, reaction temperature, kinetic study, catalyst stability upon reuse, surface acidity and basicity, morphology and dispersion and valence state of surface Ru. At optimized condition (HMF/Ru molar ratio: 25, 120 °C, 10 bar O2 for 1 h), Ru/HAP showed >99% yield of FDCA (Table 11, entry 34). The temperature was varied from 60 to 120 °C. At 60 °C, only FFCA intermediate was observed, which clearly showed that Ru/HAP catalyst is capable of converting other oxidized intermediates such as DFF and HMFCA to FFCA at this temperature. Upon increasing the temperature to 120 °C, >99 yield of FDCA from FFCA intermediate was reported. The authors disclosed that well-dispersed Ru0 and acidic-basic sites located in the HAP support were essential to drive this base-free oxidation process. The catalyst was reusable up to five cycles, and after five cycles, slight deactivation was observed which can be reversed by reducing the catalyst under H2 at 350 °C.
Sharma et al. demonstrated metal (Pt/Pd/Ru/Co/Ni)-functionalized carbon nanotubes (CNT) for base-free aerobic oxidation of HMF to FDCA in aqueous medium [172]. The noble metal-based catalysts such as Pt/CNT, Pd/CNT and Ru/CNT showed high yield, i.e. 98, 96 and 95%, respectively, of FDCA under relatively mild reaction condition, i.e. 100 °C, 30 bar O2 for 12 h (Table 11, entries 35–37). The screened non-noble metals such as Ni and Co-loaded CNT were selective towards DFF (Table 11, entries 38 and 39). In the absence of metal, simple-functionalized CNT material gave 86% yield of FFCA (Table 11, entry 40). In this article, the authors clearly explained the mechanism of HMF to FDCA using metal-supported functionalized CNT (M/CNT) catalyst. Initially, the hydroxyl group of HMF is adsorbed on the surface of M/CNT catalyst, followed by C–H activation. Two hydrogen atoms (H of CH2 and OH) were abstracted by the support and removed by oxygen molecule favouring the formation of DFF. The functional groups on the CNT interacted with the aldehyde group (of DFF) which undergo hydrolysis and form FFCA intermediate. Finally, FFCA easily converts into FDCA with similar hydrolysis mechanism as mentioned above. This work also successfully investigated the adsorption behaviour of substrate and product on CNT surface. Among these catalysts, noble metals have more selectivity and activity towards FDCA formation because of their excellent ability to attract the β-H atom from the methyl side chain of HMF. The catalyst ability for the attraction of β-H group is in the order Pt > Pd > Ru on CNT; accordingly, FDCA product selectivity also varied.
4 Conclusion and Challenges
4.1 Conclusions
The importance of biomass conversion and primary building blocks of carbohydrates is discussed at the beginning of this chapter. The application and importance of levulinic and furan-based molecules were explained with pertinent examples. A brief outline of levulinic acid synthesis and its catalytic conversion to various valuable products such as γ-valerolactone, arylated-γ-lactones and 4,4-disubstituted pentanoic acid/esters was covered. Prior art on hydrocyclization of levulinic acid to γ-valerolactone in the presence of different hydrogen sources using layered double hydroxide-based catalysts is discussed along with their catalytic performances. Preparation of arylated-γ-lactones (levulinic acid and aromatics) was introduced in this chapter using various homogeneous and heterogeneous (zeolite) catalysts. The acid-catalysed conversion of levulinic acid with aromatics into 4,4-disubstituted pentanoic acid/esters is disclosed such as diphenolic acid, γ,γ-di-2-methylfuran pentanoic acid (4,4-bis(5-methylfuran-2-yl)pentanoic acid) and (4,4-bis(5-methylthiophen-2-yl)pentanoic acid. The possible applications of 4,4-disubstituted pentanoic acid/esters were also mentioned.
A brief discussion on 5-hydroxymethylfurfural (HMF) synthesis preferably from fructose dehydration has been covered using various acid catalysts including mineral/organic acids, metal salts, metal oxides, heteropolyacids, ion-exchange resins, functionalized carbon materials, clay and zeolitic materials. We focused on the performance of mentioned catalysts by considering conversion of fructose, yield of HMF, stability, reusability, etc.. Finally, brief outline of the results of selective oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid is discussed with emphasis on Ru-based catalyst for this conversion under base-free aqueous medium using O2/air as the oxidant.
4.2 Challenging Tasks
-
Several processes have been demonstrated for the synthesis of γ-valerolactone. However, the problem lies with its applicability. It is studied as fuel blender and precursor for polymers. But the high cost of Gvl starting material such as levulinic acid (2–3$/kg) is the bottleneck for its use in the abovementioned fields. So, finding the application of Gvl, as a solvent in agrochemical formulations, as an electrolyte in electrochemical cells and as alternative high boiling liquids, may be economically viable and might result in impactful research.
-
Finding the suitable application for novel arylated-γ-lactones and 4,4-disubstituted pentanoic acid/esters, which are emerging areas in biorefinery, is also relevant but challenging.
-
Even though maximum possible selectivity of HMF has been achieved from fructose, HMF separation from the reaction medium and obtaining the desired purity are still challenging tasks for researchers. Also, the relatively higher cost for fructose compared to other sugar starting materials creates a problem in its exploration for bulk scale synthesis of HMF. HMF production directly from cellulose is highly desirable and has been presently reported in moderate yields. Its implementation requires a cost-competitive process. While using cellulose as a starting material for HMF synthesis, the side reactions, i.e. the formation of levulinic acid and poly-furanics (humins), has to be suppressed at optimal condition and catalyst. Thus, there is a scope for development in this area.
-
Selective oxidation of HMF to FDCA was reported at a maximum concentration of 3–5 wt% in aqueous medium which is not viable at the industrial scale. Thus, for this conversion, high concentration of HMF has to be studied with high selectivity of FDCA by implementing technical/reaction tools.
References
Sasaki M, Adschiri T, Arai K (2003) Production of cellulose II from native cellulose by near- and supercritical water solubilization. J Agric Food Chem 51:5376–5381
Li C, Zhao X, Wang A, Huber GW, Zhang T (2015) Catalytic transformation of lignin for the production of chemicals and fuels. Chem Rev 115:11559–11624
Achyuthan KE, Achyuthan AM, Adams PD, Dirk SM, Harper JC, Simmons BA, Singh AK (2010) Supramolecular self-assembled chaos: polyphenolic lignin’s barrier to cost-effective lignocellulosic biofuels. Molecules 15:8641–8688
Delidovich I, Hausoul PJ, Deng L, Pfutzenreuter R, Rose M, Palkovits R (2016) Alternative monomers based on lignocellulose and their use for polymer production. Chem Rev 116:1540–1599
Gauthier C, Chiche B, Finiels A, Geneste P (1989) Influence of acidity in friedel-crafts acylation catalyzed by zeolites. J Mol Catal 50:219–229
Rackemann DW, Doherty WOS (2011) The conversion of lignocellulosics to levulinic acid. Biofuels Bioprod Biorefin 5:198–214
Stocker M (2008) Biofuels and biomass-to-liquid fuels in the biorefinery: catalytic conversion of lignocellulosic biomass using porous materials. Angew Chem 47:9200–9211
Bevilaqua DB, Rambo MKD, Rizzetti TM, Cardoso AL, Martins AF (2013) Cleaner production: levulinic acid from rice husks. J Clean Prod 47:96–101
Yang Z, Kang H, Guo Y, Zhuang G, Bai Z, Zhang H, Feng C, Dong Y (2013) Dilute-acid conversion of cotton straw to sugars and levulinic acid via 2-stage hydrolysis. Ind Crop Prod 46:205–209
Victor A, Pulidindi IN, Gedanken A (2014) Levulinic acid production from Cicer arietinum, cotton, Pinus radiata and sugarcane bagasse. RSC Adv 4:44706–44711
Morone A, Apte M, Pandey RA (2015) Levulinic acid production from renewable waste resources: bottlenecks, potential remedies, advancements and applications. Renew Sust Energ Rev 51:548–565
Tabasso S, Montoneri E, Carnaroglio D, Caporaso M, Cravotto G (2014) Microwave-assisted flash conversion of non-edible polysaccharides and post-harvest tomato plant waste to levulinic acid. Green Chem 16:73–76
Li J, Jiang Z, Hu L, Hu C (2014) Selective conversion of cellulose in corncob residue to levulinic acid in an aluminum trichloride–sodium chloride system. ChemSusChem 7:2482–2488
Peng L, Lin L, Zhang J, Zhuang J, Zhang B, Gong Y (2010) Catalytic conversion of cellulose to levulinic acid by metal chlorides. Molecules 15:5258–5272
Upare PP, Yoon J-W, Kim MY, Kang H-Y, Hwang DW, Hwang YK, Kung HH, Chang J-S (2013) Chemical conversion of biomass-derived hexose sugars to levulinic acid over sulfonic acid-functionalized graphene oxide catalysts. Green Chem 15:2935–2943
Hegner J, Pereira KC, DeBoef B, Lucht BL (2010) Conversion of cellulose to glucose and levulinic acid via solid-supported acid catalysis. Tetrahedron Lett 51:2356–2358
Weingarten R, Kim YT, Tompsett GA, Fernández A, Han KS, Hagaman EW, Conner WC Jr, Dumesic JA, Huber GW (2013) Conversion of glucose into levulinic acid with solid metal(IV) phosphate catalysts. J Catal 304:123–134
Ramli NAS, Amin NAS (2015) Fe/HY zeolite as an effective catalyst for levulinic acid production from glucose: characterization and catalytic performance. Appl Catal B Environ 163:487–498
Suacharoen S, Tungasmita DN (2013) Hydrothermolysis of carbohydrates to levulinic acid using metal supported on porous aluminosilicate. J Chem Technol Biotechnol 88:1538–1544
Szabolcs A, Molnar M, Dibo G, Mika LT (2013) Microwave-assisted conversion of carbohydrates to levulinic acid: an essential step in biomass conversion. Green Chem 15:439–445
Shen Y, Sun J-K, Yi Y-X, Wang B, Xu F, Sun RC (2015) One-pot synthesis of levulinic acid from cellulose in ionic liquids. Bioresour Technol 192:812–816
Ren H, Girisuta B, Zhou Y, Liu L (2015) Selective and recyclable depolymerization of cellulose to levulinic acid catalyzed by acidic ionic liquid. Carbohydr Polym 117:569–576
Ren H, Zhou Y, Liu L (2013) Selective conversion of cellulose to levulinic acid via microwave-assisted synthesis in ionic liquids. Bioresour Technol 129:616–619
Sun Z, Xue L, Wang S, Wang X, Shi J (2016) Single step conversion of cellulose to levulinic acid using temperature-responsive dodeca-aluminotungstic acid catalysts. Green Chem 18:742–752
Saravanamurugan S, Riisager A (2013) Zeolite catalyzed transformation of carbohydrates to alkyl levulinates. ChemCatChem 5:1754–1757
Wang G, Zhang Z, Song L (2014) Efficient and selective alcoholysis of furfuryl alcohol to alkyl levulinates catalyzed by double SO3H-functionalized ionic liquids. Green Chem 16:1436–1443
Zhang Z, Dong K, Zhao ZK (2011) Efficient conversion of furfuryl alcohol into alkyl levulinates catalyzed by an organic-inorganic hybrid solid acid catalyst. ChemSusChem 4:112–118
Demma Carà P, Ciriminna R, Shiju NR, Rothenberg G, Pagliaro M (2014) Enhanced heterogeneous catalytic conversion of furfuryl alcohol into butyl levulinate. ChemSusChem 7:835–840
Huang YB, Yang T, Zhou MC, Pan H, Fu Y (2016) Microwave-assisted alcoholysis of furfural alcohol into alkyl levulinates catalyzed by metal salts. Green Chem 18:1516–1523
Zhu SH, Cen YL, Guo J, Chai JC, Wang JG, Fan WB (2016) One-pot conversion of furfural to alkyl levulinate over bifunctional Au-H4SiW12O40/ZrO2 without external H2. Green Chem 18:5667–5675
Timokhin BV, Baransky VA, Eliseeva GD (1999) Levulinic acid in organic synthesis. Russ Chem Rev 68:73–84
Luo W, Bruijnincx PCA, Weckhuysen BM (2014) Selective, one-pot catalytic conversion of levulinic acid to pentanoic acid over Ru/H-ZSM5. J Catal 320:33–41
Upare PP, Lee J-M, Hwang YK, Hwang DW, Lee J-H, Halligudi SB, Hwang J-S, Chang J-S (2011) Direct hydrocyclization of biomass-derived levulinic acid to 2-methyltetrahydrofuran over nanocomposite copper/silica catalysts. ChemSusChem 4:1749–1752
Mizugaki T, Nagatsu Y, Togo K, Maeno Z, Mitsudome T, Jitsukawa K, Kaneda K (2015) Selective hydrogenation of levulinic acid to 1,4-pentanediol in water using a hydroxyapatite-supported Pt-Mo bimetallic catalyst. Green Chem 17:5136–5139
Wright WRH, Palkovits R (2012) Development of heterogeneous catalysts for the conversion of levulinic acid to gamma-valerolactone. ChemSusChem 5:1657–1667
Lange J-P, Price R, Ayoub PM, Louis J, Petrus L, Clarke L, Gosselink H (2010) Valeric biofuels: a platform of cellulosic transportation fuels. Angew Chem Int Ed 49:4479–4483
Zhao Y, Fu Y, Guo QX (2012) Production of aromatic hydrocarbons through catalytic pyrolysis of gamma-valerolactone from biomass. Bioresour Technol 114:740–744
Bond JQ, Alonso DM, Wang D, West RM, Dumesic JA (2010) Integrated catalytic conversion of gamma-valerolactone to liquid alkenes for transportation fuels. Science 327:1110–1114
Alonso DM, Bond JQ, Dumesic JA (2010) Catalytic conversion of biomass to biofuels. Green Chem 12:1493–1513
Manzer LE (2004) Catalytic synthesis of α-methylene-γ-valerolactone: a biomass-derived acrylic monomer. Appl Catal A Gen 272:249–256
Van de Vyver S, Roman-Leshkov Y (2013) Emerging catalytic processes for the production of adipic acid. Cat Sci Technol 3:1465–1479
Lange J-P, Vestering JZ, Haan RJ (2007) Towards ‘bio-based’ nylon: conversion of [gamma]-valerolactone to methyl pentenoate under catalytic distillation conditions. Chem Commun 33:3488–3490
Alonso DM, Wettstein SG, Mellmer MA, Gurbuz EI, Dumesic JA (2013) Integrated conversion of hemicellulose and cellulose from lignocellulosic biomass. Energy Environ Sci 6:76–80
Horváth IT (2008) Solvents from nature. Green Chem 10:1024–1028
Duan Z-Q, Hu F (2012) Highly efficient synthesis of phosphatidylserine in the eco-friendly solvent γ-valerolactone. Green Chem 14:1581–1583
Stradi A, Molnar M, Ovari M, Dibo G, Richter FU, Mika LT (2013) Rhodium-catalyzed hydrogenation of olefins in γ-valerolactone-based ionic liquids. Green Chem 15:1857–1862
Strappaveccia G, Ismalaj E, Petrucci C, Lanari D, Marrocchi A, Drees M, Facchetti A, Vaccaro L (2015) A biomass-derived safe medium to replace toxic dipolar solvents and access cleaner heck coupling reactions. Green Chem 17:365–372
Strappaveccia G, Luciani L, Bartollini E, Marrocchi A, Pizzo F, Vaccaro L (2015) γ-Valerolactone as an alternative biomass-derived medium for the Sonogashira reaction. Green Chem 17:1071–1076
Pongrácz P, Bartal B, Kollár L, Mika LT (2017) Rhodium-catalyzed hydroformylation in γ-valerolactone as a biomass-derived solvent. J Organomet Chem 847:140–145
Fegyverneki D, Orha L, Láng G, Horváth IT (2010) Gamma-valerolactone-based solvents. Tetrahedron 66:1078–1081
Gundekari S, Srinivasan K (2019) Screening of solvents, hydrogen source, and investigation of reaction mechanism for the hydrocyclisation of levulinic acid to γ-valerolactone using Ni/SiO2-Al2O3 catalyst. Catal Lett 149:215–227
Osatiashtiani A, Lee AF, Wilson K (2017) Recent advances in the production of gamma-valerolactone from biomass-derived feedstocks via heterogeneous catalytic transfer hydrogenation. J Chem Technol Biotechnol 92:1125–1135
Liguori F, Moreno-Marrodan C, Barbaro P (2015) Environmentally friendly synthesis of gamma-valerolactone by direct catalytic conversion of renewable sources. ACS Catal 5:1882–1894
Tang X, Zeng X, Li Z, Hu L, Sun Y, Liu S, Lei T, Lin L (2014) Production of γ-valerolactone from lignocellulosic biomass for sustainable fuels and chemicals supply. Renew Sust Energ Rev 40:608–620
Dutta S, Yu IKM, Tsang DCW, Ng YH, Ok YS, Sherwood J, Clark JH (2019) Green synthesis of gamma-valerolactone (GVL) through hydrogenation of biomass-derived levulinic acid using non-noble metal catalysts: a critical review. Chem Eng J 372:992–1006
Xue ZM, Liu QL, Wang JF, Mu TC (2018) Valorization of levulinic acid over non-noble metal catalysts: challenges and opportunities. Green Chem 20:4391–4408
Yu ZH, Lu XB, Liu C, Han YW, Ji N (2019) Synthesis of gamma-valerolactone from different biomass-derived feedstocks: recent advances on reaction mechanisms and catalytic systems. Renew Sust Energ Rev 112:140–157
Yan K, Chen A (2013) Efficient hydrogenation of biomass-derived furfural and levulinic acid on the facilely synthesized noble-metal-free Cu–Cr catalyst. Energy 58:357–363
Yan K, Liao J, Wu X, Xie X (2013) A noble-metal free Cu-catalyst derived from hydrotalcite for highly efficient hydrogenation of biomass-derived furfural and levulinic acid. RSC Adv 3:3853–3856
Yan K, Chen A (2014) Selective hydrogenation of furfural and levulinic acid to biofuels on the ecofriendly Cu–Fe catalyst. Fuel 115:101–108
Long X, Sun P, Li Z, Lang R, Xia C, Li F (2015) Magnetic Co/Al2O3 catalyst derived from hydrotalcite for hydrogenation of levulinic acid to γ-valerolactone. Chin J Catal 36:1512–1518
Zhang J, Chen J, Guo Y, Chen L (2015) Effective upgrade of Levulinic acid into γ-valerolactone over an inexpensive and magnetic catalyst derived from hydrotalcite precursor. ACS Sustain Chem Eng 3:1708–1714
Gupta SSR, Kantam ML (2018) Selective hydrogenation of levulinic acid into γ-valerolactone over Cu/Ni hydrotalcite-derived catalyst. Catal Today 309:189–194
Li W, Fan GL, Yang L, Li F (2016) Highly efficient vapor-phase hydrogenation of biomass-derived Levulinic acid over structured nanowall-like nickel-based catalyst. ChemCatChem 8:2724–2733
Hussain SK, Kumar VV, Pethan RN, Putra KB, Chary KVR (2018) Synthesis of γ-Valerolactone from Levulinic acid and formic acid over Mg-Al Hydrotalcite like compound. ChemistrySelect 3:6186–6194
Jiang K, Sheng D, Zhang ZH, Fu J, Hou ZY, Liu XY (2016) Hydrogenation of levulinic acid to γ-valerolactone in dioxane over mixed MgO-Al2O3 supported Ni catalyst. Catal Today 274:55–59
Gundekari S, Srinivasan K (2017) In situ generated Ni(0)@boehmite from NiAl-LDH: an efficient catalyst for selective hydrogenation of biomass derived levulinic acid to γ-valerolactone. Catal Commun 102:40–43
Ma MW, Liu H, Cao JJ, Hou P, Huang JH, Xu XL, Yue HJ, Tian G, Feng SH (2019) A highly efficient Cu/AlOOH catalyst obtained by in situ reduction: catalytic transfer hydrogenation of ML into γ-GVL. Mol Catal 467:52–60
Gundeboina R, Gadasandula S, Velisoju VK, Gutta N, Kotha LR, Aytam HP (2019) Ni-Al-Ti hydrotalcite based catalyst for the selective hydrogenation of biomass-derived levulinic acid to γ-valerolactone. Chemistryselect 4:202–210
Liu M, Li S, Fan G, Yang L, Li F (2019) Hierarchical flower-like bimetallic NiCu catalysts for catalytic transfer hydrogenation of ethyl levulinate into γ-valerolactone. Ind Eng Chem Res 58:10317–10327
Swarna Jaya V, Sudhakar M, Naveen Kumar S, Venugopal A (2015) Selective hydrogenation of levulinic acid to γ-valerolactone over a Ru/Mg-LaO catalyst. RSC Adv 5:9044–9049
Bal R, Pendem C, Bordolol A, Konthala L, Narayan S, Manoj K, Saran S (2018) Process for the production of γ-Valerolactone. US patent 10,005,747 B2
Kannan Srinivasan SG (2019) Process for the preparation of γ-Valerolactone by catalytic hydrogenation of levulinic acid using Ru-based catalysts. US patent 10,221,149 B2
Mitta H, Seelam PK, Chary KVR, Mutyala S, Boddula R, Inamuddin AAM (2018) Efficient vapor-phase selective hydrogenolysis of bio-Levulinic acid to γ-valerolactone using Cu supported on hydrotalcite catalysts. Glob Challenges 2:1800028
Yonezawa N, Koike M, Kameda A, Naito S, Hino T, Maeyama K, Ikeda T (2002) Chemospecificity in arylations of δ- and γ-ketocarboxylic acids with P2O5–MsOH, TfOH, and related acidic media. Synth Commun 32:3169–3180
Makiko H, Yumi M, Hiroshi Y (2011) Oxygen-containing cyclic compound and method for producing the same. JP 2011201847 A
Jansen JC, Creyghton EJ, Njo SL, van Koningsveld H, van Bekkum H (1997) On the remarkable behaviour of zeolite Beta in acid catalysis. Catal Today 38:205–212
Zhao Z, Xu S, Hu MY, Bao X, Peden CHF, Hu J (2015) Investigation of aluminum site changes of dehydrated zeolite H-Beta during a rehydration process by high-field solid-state NMR. J Phys Chem C 119:1410–1417
Galletti AMR, Antonetti C, De Luise V, Martinelli M (2012) A sustainable process for the production of γ-valerolactone by hydrogenation of biomass-derived levulinic acid. Green Chem 14:688–694
Singh AP (1992) Preparation of bisphenol-a over zeolite catalysts. Catal Lett 16:431–435
Darbre PD (2015) Chapter 1 – what are endocrine disrupters and where are they found? In: Darbre PD (ed) Endocrine disruption and human health. Academic Press, Boston, pp 3–26
Bozell JJ, Moens L, Elliott DC, Wang Y, Neuenscwander GG, Fitzpatrick SW, Bilski RJ, Jarnefeld JL (2000) Production of levulinic acid and use as a platform chemical for derived products. Resour Conserv Recycling 28:227–239
Bader AR, Kontowicz AD (1954) γ,γ-Bis-(p-hydroxyphenyl)-valeric acid. J Am Chem Soc 76:4465–4466
Yu X, Guo Y, Li K, Yang X, Xu L, Guo Y, Hu J (2008) Catalytic synthesis of diphenolic acid from levulinic acid over cesium partly substituted wells–dawson type heteropolyacid. J Mol Catal A Chem 290:44–53
Guo Y, Li K, Yu X, Clark JH (2008) Mesoporous H3PW12O40-silica composite: efficient and reusable solid acid catalyst for the synthesis of diphenolic acid from levulinic acid. Appl Catal B Environ 81:182–191
Guo Y, Li K, Clark JH (2007) The synthesis of diphenolic acid using the periodic mesoporous H3PW12O40-silica composite catalysed reaction of levulinic acid. Green Chem 9:839–841
Li K, Hu J, Li W, Ma F, Xu L, Guo Y (2009) Design of mesostructured H3PW12O40-silica materials with controllable ordered and disordered pore geometries and their application for the synthesis of diphenolic acid. J Mater Chem 19:8628–8638
Van de Vyver S, Helsen S, Geboers J, Yu F, Thomas J, Smet M, Dehaen W, Román-Leshkov Y, Hermans I, Sels BF (2012) Mechanistic insights into the kinetic and regiochemical control of the thiol-promoted catalytic synthesis of diphenolic acid. ACS Catal 2:2700–2704
Van de Vyver S, Geboers J, Helsen S, Yu F, Thomas J, Smet M, Dehaen W, Sels BF (2012) Thiol-promoted catalytic synthesis of diphenolic acid with sulfonated hyperbranched poly(arylene oxindole)s. Chem Commun 48:3497–3499
Liu H-F, Zeng F-X, Deng L, Liao B, Pang H, Guo Q-X (2013) Bronsted acidic ionic liquids catalyze the high-yield production of diphenolic acid/esters from renewable levulinic acid. Green Chem 15:81–84
Shen Y, Sun J, Wang B, Xu F, Sun R (2014) Catalytic synthesis of diphenolic acid from levulinic acid over Bronsted acidic ionic liquids. Bioresources 9:3264–3275
Wang W, Li N, Li S, Li G, Chen F, Sheng X, Wang A, Wang X, Cong Y, Zhang T (2016) Synthesis of renewable diesel with 2-methylfuran and angelica lactone derived from carbohydrates. Green Chem 18:1218–1223
Li G, Li N, Wang Z, Li C, Wang A, Wang X, Cong Y, Zhang T (2012) Synthesis of high-quality diesel with furfural and 2-methylfuran from hemicellulose. ChemSusChem 5:1958–1966
De Melo FC, De Souzaa RF, Coutinhob PLA, De Souza MO (2014) Synthesis of 5-hydroxymethylfurfural from dehydration of fructose and glucose using ionic liquids. J Braz Chem Soc 25(12)
Cinlar B, Wang T, Shanks BH (2013) Kinetics of monosaccharide conversion in the presence of homogeneous Bronsted acids. Appl Catal A Gen 450:237–242
Tuercke T, Panic S, Loebbecke S (2009) Microreactor process for the optimized synthesis of 5-hydroxymethylfurfural: a promising building block obtained by catalytic dehydration of fructose. Chem Eng Technol 32:1815–1822
Sievers C, Musin I, Marzialetti T, Olarte MB, Agrawal PK, Jones CW (2009) Acid-catalyzed conversion of sugars and furfurals in an ionic-liquid phase. ChemSusChem 2:665–671
Li C, Zhao ZK, Wang A, Zheng M, Zhang T (2010) Production of 5-hydroxymethylfurfural in ionic liquids under high fructose concentration conditions. Carbohydr Res 345:1846–1850
Ray D, Mittal N, Chung WJ (2011) Phosphorous pentoxide mediated synthesis of 5-HMF in ionic liquid at low temperature. Carbohydr Res 346:2145–2148
Thananatthanachon T, Rauchfuss TB (2010) Efficient route to hydroxymethylfurans from sugars via transfer hydrogenation. ChemSusChem 3:1139–1141
De S, Dutta S, Saha B (2011) Microwave assisted conversion of carbohydrates and biopolymers to 5-hydroxymethylfurfural with aluminium chloride catalyst in water. Green Chem 13:2859–2868
Seri K, Inoue Y, Ishida H (2000) Highly efficient catalytic activity of lanthanide(III) ions for conversion of saccharides to 5-hydroxymethyl-2-furfural in organic solvents. Chem Lett 29:22
Deng T, Cui X, Qi Y, Wang Y, Hou X, Zhu Y (2012) Conversion of carbohydrates into 5-hydroxymethylfurfural catalyzed by ZnCl2 in water. Chem Commun 48:5494–5496
Caes BR, Raines RT (2011) Conversion of fructose into 5-(hydroxymethyl)furfural in sulfolane. ChemSusChem 4:353–356
Shen Y, Sun J, Yi Y, Wang B, Xu F, Sun R (2014) 5-Hydroxymethylfurfural and levulinic acid derived from monosaccharides dehydration promoted by InCl3 in aqueous medium. J Mol Catal A Chem 394:114–120
Shen Y, Xu YF, Sun JK, Wang B, Xu F, Sun RC (2014) Efficient conversion of monosaccharides into 5-hydroxymethylfurfural and levulinic acid in InCl3-H2O medium. Catal Commun 50:17–20
Mittal N, Nisola GM, Chung W-J (2012) Facile catalytic dehydration of fructose to 5-hydroxymethylfurfural by niobium pentachloride. Tetrahedron Lett 53:3149–3155
Pidko EA, Degirmenci V, Hensen EJM (2012) On the mechanism of lewis acid catalyzed glucose transformations in ionic liquids. ChemCatChem 4:1263–1271
Eminov S, Wilton-Ely JDET, Hallett JP (2014) Highly selective and near-quantitative conversion of fructose to 5-hydroxymethylfurfural using mildly acidic ionic liquids. ACS Sustain Chem Eng 2:978–981
Sampath G, Srinivasan K (2017) Remarkable catalytic synergism of alumina, metal salt and solvent for conversion of biomass sugars to furan compounds. Appl Catal A Gen 533:75–80
Tiziana Armaroli GB, Carlini C, Giuttari M, Sbrana G, Galletti AMR (2000) Acid sites characterization of niobium phosphate catalysts and their activity in fructose dehydration to 5-hydroxymethyl-2-furaldehyde. J Mol Catal A Chem 151:233–243
Carniti P, Gervasini A, Marzo M (2010) Silica–niobia oxides as viable acid catalysts in water: effective vs. intrinsic acidity. Catal Today 152:42–47
Carniti P, Gervasini A, Marzo M (2011) Absence of expected side-reactions in the dehydration reaction of fructose to HMF in water over niobic acid catalyst. Catal Commun 12:1122–1126
Bhaumik P, Dhepe PL (2013) Influence of properties of SAPO’s on the one-pot conversion of mono-, di- and poly-saccharides into 5-hydroxymethylfurfural. RSC Adv 3:17156
Yang F, Liu Q, Yue M, Bai X, Du Y (2011) Tantalum compounds as heterogeneous catalysts for saccharide dehydration to 5-hydroxymethylfurfural. Chem Commun 47:4469–4471
De S, Dutta S, Patra AK, Bhaumik A, Saha B (2011) Self-assembly of mesoporous TiO2 nanospheres via aspartic acid templating pathway and its catalytic application for 5-hydroxymethyl-furfural synthesis. J Mater Chem 21:17505
Qi X, Watanabe M, Aida TM, Smith RL Jr (2009) Sulfated zirconia as a solid acid catalyst for the dehydration of fructose to 5-hydroxymethylfurfural. Catal Commun 10:1771–1775
Timofeeva MN (2003) Acid catalysis by heteropoly acids. Appl Catal A Gen 256:19–35
Kozhevnikov IV (1998) Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem Rev 98:171–198
Yunxiang Qiao ZH (2009) Polyoxometalate–based solid and liquid salts for catalysis. Curr Org Chem 13:1347–1365
Xiao Y, Song Y-F (2014) Efficient catalytic conversion of the fructose into 5-hydroxymethylfurfural by heteropolyacids in the ionic liquid of 1-butyl-3-methyl imidazolium chloride. Appl Catal A Gen 484:74–78
Zheng H, Sun Z, Yi X, Wang S, Li J, Wang X, Jiang Z (2013) A water-tolerant C16H3PW11CrO39 catalyst for the efficient conversion of monosaccharides into 5-hydroxymethylfurfural in a micellar system. RSC Adv 3:23051
Zhao Q, Wang L, Zhao S, Wang X, Wang S (2011) High selective production of 5-hydroymethylfurfural from fructose by a solid heteropolyacid catalyst. Fuel 90:2289–2293
Fan C, Guan H, Zhang H, Wang J, Wang S, Wang X (2011) Conversion of fructose and glucose into 5-hydroxymethylfurfural catalyzed by a solid heteropolyacid salt. Biomass Bioenergy 35:2659–2665
Qu Y, Huang C, Zhang J, Chen B (2012) Efficient dehydration of fructose to 5-hydroxymethylfurfural catalyzed by a recyclable sulfonated organic heteropolyacid salt. Bioresour Technol 106:170–172
Ganji P (2020) Synthesis and catalytic performance of SnxSTA by microwave-assisted hydrothermal synthesis for fructose to HMF. Biomass Conv Bioref 10:823–830
Harmer MA, Sun Q (2001) Solid acid catalysis using ion-exchange resins. Appl Catal A Gen 221:45–62
Barbaro P, Liguori F (2009) Ion exchange resins: catalyst recovery and recycle. Chem Rev 109:515–529
Ordomsky VV, van der Schaaf J, Schouten JC, Nijhuis TA (2012) Fructose dehydration to 5-hydroxymethylfurfural over solid acid catalysts in a biphasic system. ChemSusChem 5:1812–1819
Zhu H, Cao Q, Li C, Mu X (2011) Acidic resin-catalysed conversion of fructose into furan derivatives in low boiling point solvents. Carbohydr Res 346:2016–2018
Jeong J, Antonyraj CA, Shin S, Kim S, Kim B, Lee K-Y, Cho JK (2013) Commercially attractive process for production of 5-hydroxymethyl-2-furfural from high fructose corn syrup. J Ind Eng Chem 19:1106–1111
Morales G, Melero JA, Paniagua M, Iglesias J, Hernández B, Sanz M (2014) Sulfonic acid heterogeneous catalysts for dehydration of C6-monosaccharides to 5-hydroxymethylfurfural in dimethyl sulfoxide. Chin J Catal 35:644–655
Sampath G, Kannan S (2013) Fructose dehydration to 5-hydroxymethylfurfural: remarkable solvent influence on recyclability of Amberlyst-15 catalyst and regeneration studies. Catal Commun 37:41–44
Wang J, Xu W, Ren J, Liu X, Lu G, Wang Y (2011) Efficient catalytic conversion of fructose into hydroxymethylfurfural by a novel carbon-based solid acid. Green Chem 13:2678–2681
Liu B, Zhang Z, Huang K (2013) Cellulose sulfuric acid as a bio-supported and recyclable solid acid catalyst for the synthesis of 5-hydroxymethylfurfural and 5-ethoxymethylfurfural from fructose. Cellulose 20:2081–2089
Liu R, Chen J, Huang X, Chen L, Ma L, Li X (2013) Conversion of fructose into 5-hydroxymethylfurfural and alkyl levulinates catalyzed by sulfonic acid-functionalized carbon materials. Green Chem 15:2895–2903
Russo PA, Antunes MM, Neves P, Wiper PV, Fazio E, Neri F, Barreca F, Mafra L, Pillinger M, Pinna N, Valente AA (2014) Solid acids with SO3H groups and tunable surface properties: versatile catalysts for biomass conversion. J Mater Chem A 2:11813–11824
Wang H, Kong Q, Wang Y, Deng T, Chen C, Hou X, Zhu Y (2014) Graphene oxide catalyzed dehydration of fructose into 5-hydroxymethylfurfural with isopropanol as cosolvent. ChemCatChem 6:728–732
Moreau C, Durand R, Pourcheron C, Razigade S (1994) Preparation of 5-hydroxymethylfurfural from fructose and precursors over H-form zeolites. Ind Crop Prod 3:85–90
Moreau C, Durand R, Razigade S, Duhamet J, Faugeras P, Rivalier P, Ros P, Avignon G (1996) Dehydration of fructose to 5-hydroxymethylfurfural over H-mordenites. Appl Catal A Gen 145:211–224
Shi Y, Li X, Hu J, Lu J, Ma Y, Zhang Y, Tang Y (2011) Zeolite microspheres with hierarchical structures: formation, mechanism and catalytic performance. J Mater Chem 21:16223–16230
Wang J, Ren J, Liu X, Xi J, Xia Q, Zu Y, Lu G, Wang Y (2012) Direct conversion of carbohydrates to 5-hydroxymethylfurfural using Sn-Mont catalyst. Green Chem 14:2506–2512
Liu B, Gou Z, Liu A, Zhang Z (2015) Synthesis of furan compounds from HMF and fructose catalyzed by aluminum-exchanged K-10 clay. J Ind Eng Chem 21:338–339
Yang F, Weng J, Ding J, Zhao Z, Qin L, Xia F (2019) Effective conversion of saccharides into hydroxymethylfurfural catalyzed by a natural clay. Attapulgite Renew Energy 151:829–836
Sajid M, Zhao X, Liu D (2018) Production of 2,5-furandicarboxylic acid (FDCA) from 5-hydroxymethylfurfural (HMF): recent progress focusing on the chemical-catalytic routes. Green Chem 20:5427–5453
Tong X, Yu L, Chen H, Zhuang X, Liao S, Cui H (2017) Highly efficient and selective oxidation of 5-hydroxymethylfurfural by molecular oxygen in the presence of Cu-MnO2 catalyst. Catal Commun 90:91–94
Krivtsov I, García-López EI, Marcì G, Palmisano L, Amghouz Z, García JR, Ordóñez S, Díaz E (2017) Selective photocatalytic oxidation of 5-hydroxymethyl-2-furfural to 2,5-furandicarboxyaldehyde in aqueous suspension of g-C3N4. Appl Catal B Environ 204:430–439
Hayashi E, Komanoya T, Kamata K, Hara M (2017) Heterogeneously-catalyzed aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid with MnO2. ChemSusChem 10:654–658
Han X, Li C, Liu X, Xia Q, Wang Y (2017) Selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over MnOx–CeO2 composite catalysts. Green Chem 19:996–1004
Gui Z, Saravanamurugan S, Cao W, Schill L, Chen L, Qi Z, Riisager A (2017) Highly selective aerobic oxidation of 5-hydroxymethyl furfural into 2,5-diformylfuran over Mn-Co binary oxides. ChemistrySelect 2:6632–6639
Cui M, Huang R, Qi W, Su R, He Z (2017) Cascade catalysis via dehydration and oxidation: one-pot synthesis of 2,5-diformylfuran from fructose using acid and V2O5/ceramic catalysts. RSC Adv 7:7560–7566
Zuo X, Venkitasubramanian P, Busch DH, Subramaniam B (2016) Optimization of Co/Mn/Br-Catalyzed oxidation of 5-Hydroxymethylfurfural to enhance 2,5-furandicarboxylic acid yield and minimize substrate burning. ACS Sustain Chem Eng 4:3659–3668
Zhang Y, Xue Z, Wang J, Zhao X, Deng Y, Zhao W, Mu T (2016) Controlled deposition of Pt nanoparticles on Fe3O4@carbon microspheres for efficient oxidation of 5-hydroxymethylfurfural. RSC Adv 6:51229–51237
Ventura M, Aresta M, Dibenedetto A (2016) Selective aerobic oxidation of 5-(Hydroxymethyl)furfural to 5-formyl-2-furancarboxylic acid in water. ChemSusChem 9:1096–1100
Nguyen CV, Liao Y-T, Kang T-C, Chen JE, Yoshikawa T, Nakasaka Y, Masuda T, Wu KCW (2016) A metal-free, high nitrogen-doped nanoporous graphitic carbon catalyst for an effective aerobic HMF-to-FDCA conversion. Green Chem 18:5957–5961
Neațu F, Marin RS, Florea M, Petrea N, Pavel OD, Pârvulescu VI (2016) Selective oxidation of 5-hydroxymethyl furfural over non-precious metal heterogeneous catalysts. Appl Catal B Environ 180:751–757
Wang S, Zhang Z, Liu B (2015) Catalytic conversion of fructose and 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid over a recyclable Fe3O4–CoOx magnetite nanocatalyst. ACS Sustain Chem Eng 3:406–412
Saha B, Gupta D, Abu-Omar MM, Modak A, Bhaumik A (2013) Porphyrin-based porous organic polymer-supported iron(III) catalyst for efficient aerobic oxidation of 5-hydroxymethyl-furfural into 2,5-furandicarboxylic acid. J Catal 299:316–320
Sádaba I, Gorbanev YY, Kegnaes S, Putluru SSR, Berg RW, Riisager A (2013) Catalytic performance of zeolite-supported vanadia in the aerobic oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran. ChemCatChem 5:284–293
Bao L, Sun F-Z, Zhang G-Y, Hu T-L (2020) Aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over holey 2 D Mn2O3 nanoflakes from a Mn-based MOF. ChemSusChem 13:548–555
Ait Rass H, Essayem N, Besson M (2013) Selective aqueous phase oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over Pt/C catalysts: influence of the base and effect of bismuth promotion. Green Chem 15:2240–2251
Siyo B, Schneider M, Pohl M-M, Langer P, Steinfeldt N (2014) Synthesis, characterization, and application of PVP-Pd NP in the aerobic oxidation of 5-hydroxymethylfurfural (HMF). Catal Lett 144:498–506
Xu J, Liu J, Ma J, Cai J, Du Z, Ma H (2015) Advances in catalytic synthesis of bio-based dicarboxylic acid. Sci Sin Chim 45:526–532
Gorbanev YY, Kegnæs S, Riisager A (2011) Effect of support in heterogeneous ruthenium catalysts used for the selective aerobic oxidation of HMF in water. Top Catal 54:1318–1324
Artz J, Palkovits R (2015) Base-free aqueous-phase oxidation of 5-hydroxymethylfurfural over ruthenium catalysts supported on covalent triazine frameworks. ChemSusChem 8:3832–3838
Yi G, Teong SP, Zhang Y (2016) Base-free conversion of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over a Ru/C catalyst. Green Chem 18:979–983
Mishra DK, Lee HJ, Kim J, Lee H-S, Cho JK, Suh Y-W, Yi Y, Kim YJ (2017) MnCo2O4 spinel supported ruthenium catalyst for air-oxidation of HMF to FDCA under aqueous phase and base-free conditions. Green Chem 19:1619–1623
Wang F, Yuan Z, Liu B, Chen S, Zhang Z (2016) Catalytic oxidation of biomass derived 5-hydroxymethylfurfural (HMF) over Ru III -incorporated zirconium phosphate catalyst. J Ind Eng Chem 38:181–185
Pichler CM, Al-Shaal MG, Gu D, Joshi H, Ciptonugroho W, Schuth F (2018) Ruthenium supported on high-surface-area zirconia as an efficient catalyst for the base-free oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid. ChemSusChem 11:2083–2090
Antonyraj CA, Huynh NTT, Lee KW, Kim YJ, Shin S, Shin JS, Cho JK (2018) Base-free oxidation of 5-hydroxymethyl-2-furfural to 2,5-furan dicarboxylic acid over basic metal oxide-supported ruthenium catalysts under aqueous conditions. J Chem Sci 130:156
Gao T, Yin Y, Fang W, Cao Q (2018) Highly dispersed ruthenium nanoparticles on hydroxyapatite as selective and reusable catalyst for aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid under base-free conditions. Mol Catal 450:55–64
Sharma P, Solanki M, Sharma RK (2019) Metal-functionalized carbon nanotubes for biomass conversion: base-free highly efficient and recyclable catalysts for aerobic oxidation of 5-hydroxymethylfurfural. New J Chem 43:10601–10609
Acknowledgments
CSIR-CSMCRI Communication No. 4/2021. The authors thank CSIR, New Delhi for financial support under the projects MLP-0028 and CSC-0123.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gundekari, S., Kalusulingam, R., Dakhara, B., Mani, M., Mitra, J., Srinivasan, K. (2021). Levulinic Acid- and Furan-Based Multifunctional Materials: Opportunities and Challenges. In: Pant, K.K., Gupta, S.K., Ahmad, E. (eds) Catalysis for Clean Energy and Environmental Sustainability. Springer, Cham. https://doi.org/10.1007/978-3-030-65017-9_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-65017-9_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65016-2
Online ISBN: 978-3-030-65017-9
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)