
Abstract
Pulmonary artery hypertension (PAH) is a devastating cardiopulmonary disease characterized by vascular remodeling and obliteration of the precapillary pulmonary arterioles. Alterations in the structure and function of pulmonary vessels result in the resistance of blood flow and can progress to right-sided heart failure, causing significant morbidity and mortality. There are several types of PAH, and the disease can be familial or secondary to an underlying medical condition such as a connective tissue disorder or infection. Regardless of the cause, the exact pathophysiology and cellular interactions responsible for disease development and progression are largely unknown.
There is significant evidence to suggest altered immune and vascular cells directly participate in disease progression. Inflammation has long been hypothesized to play a vital role in the development of PAH, as an altered or skewed immune response favoring a proinflammatory environment that can lead to the infiltration of cells such as lymphocytes, macrophages, and neutrophils. Current treatment strategies focus on the dilation of partially occluded vessels; however, such techniques have not resulted in an effective strategy to reverse or prevent vascular remodeling. Therefore, current studies in human and animal models have attempted to understand the underlying pathophysiology of pulmonary hypertension (PH), specifically focusing on the inflammatory cascade predisposing patients to disease so that better therapeutic targets can be developed to potentially reverse or prevent disease progression.
The purpose of this chapter is to provide a comprehensive review of the expanding literature on the inflammatory process that participates in PH development while highlighting important and current studies in both animal and human models. While our primary focus will be on cells found in the adaptive and innate immune system, we will review all potential causes of PAH, including cells of the endothelium, pulmonary lymphatics, and genetic mutations predisposing patients. In addition, we will discuss current therapeutic options while highlighting potential future treatments and the questions that still remain unanswered.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
19.1 Introduction
In this chapter, we will review the literature in animal and human models supporting the role of immune cells and how they interact with vascular cells in the development of pulmonary arterial hypertension (PAH) and provide future perspectives on therapeutic application.
Pulmonary arterial hypertension (PAH) is a devastating cardiopulmonary disorder characterized by narrowing and/or disorganization of the terminal pulmonary arterioles. If untreated, PAH can progress to right ventricular failure, causing significant morbidity and mortality. Pathogenic drivers contributing to the extensive obliterative changes seen in PAH throughout the vasculature are still under investigation, but there is evidence to suggest that inflammation is a major contributor.
Immune cells such as T and B lymphocytes have been hypothesized to play a role in PAH development for more than 50 years. Inflammatory cell aggregates composed of T and B lymphocytes, macrophages, dendritic cells, and mast cells have been documented to surround pulmonary vasculature in both animal and human models with PAH [1]. There is ongoing debate whether inflammatory processes are the cause and propagation of vascular remodeling or just a consequence. Regardless of the etiology, several animal and human models have revealed that high levels of inflammatory mediators are predictive of worse clinical outcomes, highlighting their clinical significance and therapeutic potential [2, 3].
Intriguingly, idiopathic pulmonary arterial hypertension (IPAH)-associated tertiary lymphoid tissues (organized ectopic lymphoid follicles) suggest that impaired or hyperpermeable infiltration of the lymphatic collection system is associated with elevated immune cell recruitment. Animal models of PH, which partially recapitulate human pathology with a variable degree of vascular remodeling, have dissected various inflammatory pathways and identify a variety of proinflammatory mediators. PAH is associated with several infectious and autoimmune diseases, including HIV, HHV-8, HCV, and schistosomiasis. These associations suggest that an off-target inflammatory response can inadvertently damage the pulmonary vasculature [4]. Inflammatory dysregulation and PAH are also well-documented in patients with autoimmune disorders. Ten percent of patients with systemic sclerosis have diagnosed PH. A lower incidence is seen in patients with other connective tissue disorders, such as systemic lupus erythematosus and Sjogren’s syndrome [5, 6].
Additionally, IPAH patients can present with Raynaud’s phenomena and scleroderma. Increased levels of antinuclear/endothelial cell antibodies and autoantibodies can also be detected in IPAH serum [6,7,8,9]. While it has been recognized that autoimmunity is highly associated with PAH, we still lack definitive evidence to show that autoimmunity itself is a direct cause of the cellular changes leading to PAH development in animal or human models. For instance, sex, age, functional class, duration of symptoms, or hemodynamic status has no significant impact on the expression level of autoimmune-related antibodies in patients with PAH.
19.2 T Cells
T cells are essential components of the adaptive immune response. In general, three subsets, T helper cells (Th, CD4+), T-regulatory cells (Treg, CD4 + CD25highFoxP3+), and cytotoxic T (Tc, CD8+) cells, are required for equilibrium and homeostasis [10]. Th cells are further subdivided into Th1, Th2, and Th17 cells, named after the proinflammatory cytokines they secrete. Each subtype has a specific role and response in the inflammatory cascade. Th and Tc cells produce a proinflammatory response, while Treg cells exert a balancing response, for self-tolerance and preventing autoimmunity. When an imbalance between the T-cell subtypes occurs, an exaggerated response can be seen secondary to the overexpression of proinflammatory cytokines released by Th cells or the anti-inflammatory cytokines due to Treg cell products. Numerous animal and human models have investigated the relationship between T-cell dysregulation and PAH . The balance and homeostasis of T cells along with their cytokines are vital to prevent the loss of self-tolerance, which may predispose patients to inflammation and the development of PAH [11] (Table 19.1).
19.3 Treg Cells
Treg cells play a vital role in regulating the inflammatory response of Th cells to self and foreign antigens. The downregulation of Tregs and their associated anti-inflammatory cytokines can lead to increased proliferation of Th17 cells. Tregs maintain immune homeostasis by suppressing CD4, CD8, and NK Cells [12]. They can accomplish this by interfering with the major histocompatibility complex II (MCHII) complex on T cells via interleukin 10 (IL-10), inhibiting dendritic cells antigen recognition process, and suppressing T-cell expansion [13, 14]. The Treg cell surface molecule, galectin-1, can also bind to effector T cells and dendritic cells, causing cell cycle arrest and apoptosis [1].
Regulating immune dysfunction is critical to preventing the progression of PAH. Human and animal models show that the imbalance of the Treg/Th17 ratio correlates with PAH disease severity in those with idiopathic, genetic, and PAH secondary to connective tissue disease, highlighting the role of Treg in PAH [15]. Athymic rats have a T-cell immunodeficiency that renders them particularly sensitive to developing severe PH. To some extent, these animals can recapitulate severe human pulmonary arterial hypertension [16].
Athymic rats injected with SU5416, a vascular endothelial growth factor-2 (VEGF-2) inhibitor, have been shown to develop significant right ventricle (RV) remodeling, perivascular inflammation, smooth muscle hypertrophy, and occlusive arteriolar lesions [16]. However, when CD4 + CD25hi Treg populations were restored in these inbred models prior to vascular injury by SU5416 injection, the development of pulmonary disease was prevented and revealed reduced tumore necrosis factor a (TNF-a)/IL-6. This finding suggests that Tregs may be important immune regulators preventing the propagation of vascular injury by limiting inflammation [17]. Intriguingly, the same inbred athymic female rats lacking normal Treg activity exhibited greater inflammation and developed more significant PH than their male counterparts, revealing that the Treg estrogen receptor isoform associated with inflammation activation pathways [18]. In the hypoxic mouse model of PH, injection with Treg cells significantly reduces right ventricular systolic pressure (RVSP) and proinflammatory cytokine expression compared to controls [15].
While the immune imbalance between Treg and Th has been documented in PAH, it is unclear if the development of PAH is due to the T-cell subset imbalance itself or the inflammation from vascular damage. Further studies are needed to identify the exact role of Treg cells in PAH development. Furthermore, the process of CD4 + CD25- conversion to CD4 + CD25+ subsets, as well as its origin and function still need more characterization. The protective effects against PAH seen in Treg-restored animal models suggest a potential for therapeutic strategies.
Treg cells and their anti-inflammatory byproducts play a vital role in vascular remodeling, via (1) inhibiting smooth muscle proliferation, (2) protecting endothelium function, and (3) preventing extracellular matrix proteins deposition. Numerous studies in human and animal models have demonstrated the regulatory effects that Treg cells exhibit on smooth muscle proliferation. Levels of proinflammatory cytokines, such as IL-6 and IL-1b, in smooth muscle cells from humans with PAH decrease with recombinant Treg cell injection compared to controls [19]. Human smooth muscle cells incubated with Treg cells in hypoxic conditions have also been shown to have reduced densities when compared to control groups. This further suggests that Treg cells regulate growth and hypertrophy of the vasculature and smooth muscle cells under inflammatory conditions.
The proposed mechanism by which Tregs control pulmonary artery smooth muscle cells is via suppressing Akt/ERK, the pathway responsible for triggering cell growth, survival, and motility [19]. The inhibition of the Rho1/RhoA kinase pathway has shown the potential to correct the imbalance between Th17 and Treg cells. The correction of this ratio and re-establishing immune cell homeostasis could lead to decreased smooth muscle growth and inflammation [20]. Regardless of the exact mechanism, it is clear from experimental studies that Tregs may prevent the hypertrophy of smooth muscle cells in pulmonary arterioles.
Treg dysfunction also has direct interaction with endothelial cells and can decrease inflammatory effects known to aggravate PH progression [21]. Circulating Treg cell function is downregulated in patients with idiopathic PAH, favoring inflammatory proprieties and resulting in the development of PAH in an endothelium leptin-dependent manner [22]. IL-10, an anti-inflammatory cytokine produced by Treg cells, is protective against the development of PAH by enhancing nitric oxide synthase phosphorylation in pulmonary arteriole endothelial cells.
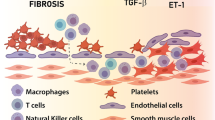
Treg cells have suppressive effects on collagen accumulation by suppressing TGF-b1 and fibroblast growth factor 9 (Fgf-9) secretion [23]. These cytokines directly inhibit the excessive activation of fibroblasts, decreasing collagen and extracellular matrix proteins deposited within arterioles [23,24,25]. In addition to regulating fibroblasts in pulmonary arterioles, Treg cells downregulate cardiac fibroblasts via the secretion of IL-10, contributing to the control of ventricular modeling and the development of right ventricular hypertrophy (RVH) seen in PAH [26] (Fig. 19.1).

The role of the inate and adaptive response leading to PAH and vascular remodeling
It remains unclear how immune reconstitution regulates endothelial bone morphogenic protein receptor 2 (BMPR2) and by what mechanism endothelial injury is attenuated. Of note, several reports have described increased Tregs in the peripheral circulation of idiopathic PH patients [27, 28]. Thus, Treg abnormalities in certain PH patient groups should be further investigated.
In summary, these findings suggest that Tregs may reverse vasculature changes during the progression of PAH . Thus, restoring of Treg function may provide a path for future therapeutic strategies.
19.4 TH17 Cells
Treg cell suppression induces the generation of TH17 cells from naÏve T cells, leading to increased inflammatory response. This response is secondary to the release of proinflammatory cytokines, such as IL-17 IL-21 and IL-22, which provide defense against foreign pathogens. Most studies have investigated and described the relationship between Treg cells and PAH, and TH17 overexpression has also been described in both animal and human models of PH.
The TH17 dominant proinflammatory state has been documented in several diseases to predispose patients to PAH. Patients with PAH secondary to a connective tissue disorder have an increase in peripheral TH17 cells, cytokines, and mRNA levels [29]. Furthermore, using immunophenotyping on monocyte-driven dendritic cells, IPAH patients were noted to have higher levels of the T-cell activating molecules CD86 and CD40 after dexamethasone pretreatment [30]. These studies suggest an increase in TH17 cell number in patients with PAH. When immune dysfunction leads to a skewed TH17 reaction , increased cytokines, such as IL-6 and TNF-a, stimulate the release of granulocyte macrophage colony stimulating factor receptor(GM-CSFR). GM-CSFR release results in the recruitment of macrophages. Studies show that leukotriene B4 (LTB4) signaling elicits increased GM-CSF levels [31]. The increased macrophages can directly induce vascular injury and remodeling via endothelial cell apoptosis and smooth muscle proliferation.
Cytokines derived from TH17 cells, including IL-6 and IL-21, potentially play a role in the pathophysiology of PAH and provide possible therapeutic targets for patients. IL-6 blockade through the receptor antibody MR16-1 ameliorated hypoxia-induced PH in mice models and prevented accumulating of macrophages and TH17 cells in lungs [32]. In animal models, mice depleted of CD4 cells and those treated with SR10001, a TH17 cell inhibitor, prevented vascular remodeling when exposed to chronic hypoxia [33].Overall, experimental data appear to be associated with a dysregulated immune response and increased TH17 expression . However, it is unclear if this overexpression is a cause or result of PAH.
19.5 Th2 Cells
Th2 cells interact with the innate immune system to activate a humoral response to pathogens. They play a major role in the promotion of eosinophilic responses, IgE production, atopy, and combating extracellular pathogens, such as parasites and worms. Th2 cells produce IL-4 and IL-13, which stimulate B-cell proliferation and antibody class switching. Originally described as being an antagonist to the development of autoimmune pathology, Th2 cells and their related cytokines have been shown to exacerbate PH progression in certain diseases, such as Schistosomiasis. In experimental models, mice with insufficient or inhibited CD4+ Th2 cells were protected against PH development after Schistosoma exposure. CD4+ Th2 cells are recruited from the circulation of Schistosoma egg-sensitized mouse model and necessary for the development of PH [34].
Experimental models have suggested type 2 inflammatory cytokines released by TH2 cells, specifically IL-4 and IL-13, which participate in PH development. This is demonstrated by reduced inflammation and alleviated RVSP in mouse models after inhibition of IL-4 and IL-13. Human and rodent models have increased expression of chemoattractant receptor homologous molecule expressed on Th2 cell (CRTH2) in circulating CD3 + CD4+ T cells. The disruption of CRTH2 was further shown to suppress Th2 cells , including IL-4 and IL-13. Thus, results in improved vascular remodeling and PH suggest that Th2 cells directly participated in the progression and development of IPAH [35]. While these models suggest that Th2 cells are involved with PAH development, more clinical studies are needed before causation is proven.
19.6 T Cytotoxic Cells
CD8+ T lymphocytes , also known as cytotoxic T cells, identify cells marked for destruction via interacting with antigens bound to class 1 MHC molecules and promote cell death in response to the release of perforin, granzymes, and other molecules. Decreased levels of cytotoxic T cells have been well documented as a consequence of aging, as well as various chronic diseases.
A decreased number of circulating CD8 cells have been recognized as an adverse prognostic marker in cancer and chronic viral infections such as HIV [36]. This response is known as cytotoxic T-cell depletion, which has also been documented in patients with PAH. It is hypothesized that the abnormalities are in response to a self or foreign antigen, causing depletion of cytotoxic T cells or T-cell exhaustion. Studies have demonstrated that deficiencies in cytotoxic T cells and NK cells may be independently associated with death and a decreased 6 min walk test in patients with PAH [37]. Increased percentage of Treg cells (CD25high FoxP3+CD4+) along with decreased CD8+ T cells in peripheral blood has been detected in IPAH patients [38].
While it is thought the imbalance of cytotoxic T cells described is in response to surrounding immune dysregulation, immunohistochemistry of lung tissue from PAH patients revealed increased perivascular CD8+ compared to controls, suggesting that CD8+ T cells potentially have an active role in vascular remodeling [39]. However, phenotype and functional analyses of CD8 cells through the different stages of PAH are needed to further understand the mechanism and contribution of these cells in disease progression to determine their exact role.
19.7 Neutrophils
Neutrophils are the predominant circulating leukocyte in the body and a staple of the immune system. They act as first responders to infection and inflammation through phagocytosis to clear debris and recruit cells of the innate and adaptive immunity. While few studies have explored the role of neutrophils in the development of PAH, some have found an increased neutrophil presence and activity in animal and human models.
Increased neutrophil accumulation in the lungs has been seen in monocrotaline (MCT)-induced rats and hypoxic mice [40,41,42]. Furthermore, an increased neutrophil-to-lymphocyte ratio has also positively correlated with the New York Heart Association PAH functional class and predicted survival, suggesting neutrophils have prognostic capabilities of disease severity [43].
While it is not well understood why the increase in neutrophils is associated with PAH progression, a wide range of neutrophil byproducts may theoretically contribute. Neutrophils produce neutrophil elastase (NE), which is one of the primary proteolytic enzymes responsible for the release of neutrophil extracellular traps (NETs). A relationship between neutrophil elastance activity and vascular changes in PAH experimental rat models has been documented [44]. There are anecdotal documents that showed augmented NE release both from neutrophils isolated from PAH patients and PAH smooth muscle cells (SMCs) and in rat models of PH [45,46,47,48]. NETs assist in microbial trapping and elimination as its normal function [49], but the emergence of excessive NETs has been identified in vascular pathologies in PAH, which promotes SMC proliferation, induces EC apoptosis, and increases matrix deposition [50, 51]. The degradation of the extracellular membrane leads to the release of growth factors and proinflammatory cytokines, promoting extracellular membrane remodeling and disease progression. In addition, markers of NETs were identified in proximity to plexiform lesions in PAH lung tissues.
The numerous mechanisms of neutrophils contribute to PAH that has led to studies to look into therapeutic options. Interestingly, the inhibition of neutrophil elastase has been noted to reverse disease in experimental models, specifically in monocrotaline mice and the Sugen/hypoxia rat model following treatment with elafin, an endogenous elastance inhibitor [40]. These findings have been replicated in human models. Explanted lung tissue with PAH treated with elafin displayed regression of intimal changes and increased vascular lumen size [52]. In addition to tissue destruction leading to PH, neutrophil elastase activates IL-1b, a proinflammatory cytokine released by endothelial cells, which can reciprocally promote the neutrophil survival and increase matrix deposition.
The role of intrinsic neutrophil abnormalities and alterations in NET and NE release in different stages of PH and during progressive vascular remodeling remain elusive. Nevertheless, a treatment targeting neutrophil elastance is a challenging therapeutic strategy to treat PAH, as they have a vital role in host defense and would leave recipients exposed to infection. More clinical research is needed to determine the exact role and mechanisms of neutrophils in PAH development and progression.
19.8 Macrophages
Macrophages are white blood cells whose primary role is to engulf pathogens and foreign particles found throughout the body via phagocytosis. They are a key component of the innate immune system, presenting manufactured antigens to T cells for differentiation and activation of the adaptive immune system. The infiltration of macrophages is prominent in the inflammatory infiltrate of plexiform lesions in experimental and different forms of clinical PAH [39, 41, 53, 54]. Additionally, depletion of macrophage prevents experimental PH and portopulmonary hypertension [55, 56].
Elevated levels of monocyte-recruiting chemokines in the lung, along with increased peripheral blood monocytes, have been reported in human and animal models with PH. After migration of the monocytes into the pulmonary vasculature, these cells may differentiate into perivascular macrophages through Ccl2 and Cx3cl1 activation [57]. Inhibition of Cx3cI1/Cx3cr1 signaling leads to decreased interstitial macrophage expansion and reduced pulmonary vasculature remodeling and inflammation in rodent models with PH [57].
Without Treg cell suppression, macrophages are activated after an insult and participate in vascular remodeling leading to PAH. This has been suggested by the protective effects seen against PAH development in animal models lacking alveolar macrophages [54]. The depletion of alveolar macrophages in rats exposed to chronic hypoxia has a protective effect against pulmonary artery pressure changes, suggesting macrophages participate directly in the development of PH. Rats exposed to chronic hypoxia with decreased alveolar macrophages had no change in serum monocyte-chemoattractant protein-1 (MCP-1), suggesting that MCP-1 is likely not involved in the recruitment and differentiation of macrophages leading to PAH development [58]. Regardless of how macrophage recruitment occurs, their presence in the distal arterioles of both human and animal models with PH is more sophisticatedly documented.
Humans and SU5416 rat models have shown LTB4, a proinflammatory molecule derived from arachidonic acid and released from macrophages is a key component in PH progression. It was found that LTB4, through inhibition of the endothelial nitric oxide synthetase, causes endothelial cell apoptosis leading to proliferation and hypertrophy of human pulmonary artery smooth muscle cells. Blocking macrophage-derived LTB4 biosynthesis or signal transduction reverses experimental PH, and depleting CD68+ macrophages prevents PH from developing in Sugen-treated athymic rats [56]. Activation of macrophages is closely linked to epigenetic changes that stimulate fibroblast proliferation in PAH patients and experimental models [59].
Although the exact mechanisms of how macrophage participation in vascular remodeling and how their interaction with vascular cells are unknown, targeting macrophages to decrease PAH progression may be promising therapeutic strategies.
19.9 Dendritic Cells
Dendritic cells (DCs) act as professional antigen-presenting cells and are key players in the activation of naïve T cells. Their main function is to act as an intermediate between the innate and adaptive immune system, processing foreign antigens for T-cell presentation and differentiation [60]. They have recently been found to be a key modulator in many disorders, including asthma, autoimmunity, and tumorigenesis [61,62,63].
In experimental PH and clinical PAH, immature dendritic cells accumulate in remodeled pulmonary vessels, suggesting their involvement in the immunopathology of pulmonary hypertension [64]. Besides their T-cell activating function, DCs are crucial for the presence and preservation of tertiary lymphoid organs (TLOs) seen near pulmonary blood vessels, which consist of other myeloid cells, T-, B cells, and monocytes [65, 66].
Intriguingly, multiple DC subsets can be found in steady states, such as conventional DCs (cDCs) and plasmacytoid DCs (pDCs). Under inflammatory conditions, monocytes can differentiate into monocyte-derived DCs (mo-DCs). In IPAH patients, the proportion of circulating cDCs is decreased compared to controls, potentially as a result of an increased cDC migration toward lung TLOs [67]. In IPAH lungs, pDC numbers are enhanced and pDCs are specifically located around the pulmonary vessels, while circulating pDC numbers are unaltered [68]. During the inflammation, pDCs produce type-I IFN and chemokine secretion such as CXCL10 and promote activation of immune cells [69]. Inflammation and chemokines can also attract monocytes to lung of IPAH and CTD-PAH patients, and gave rise to mo-DCs, further exacerbate inflammation and trigger influx of inflammatory cells [70].
In summary, DC subset distribution and activation status play important roles in the pathobiology of autoimmune diseases and most likely in the development of IPAH and CTD-PAH. However, little is known about DC subset distribution and function in IPAH, CTD-PAH, and autoimmune diseases.
19.10 Mast Cells
Classically, mast cells are identified in tissues by their unique products of chymase and tryptase. These products were originally identified during tumor angiogenesis and later reported to increase and correlate with the severity of pulmonary hypertension and pulmonary vascular remodeling [71,72,73,74].
Rats with mutations in mast cell growth factor receptor c-kit develop less PH and vascular remodeling when exposed to MCT [75]. Rats with flow associated PAH that treated with mast cell stabilization attenuated pulmonary vascular remodeling and had a lower chymase activity, correlating with more favorable hemodynamics and pulmonary vascular remodeling [76]. Human tissues with IPAH have shown elevated levels of chymase-positive mast cells, with its product chymase to convert angiotensin 1 to angiotensin 2 and activate cytokines TGF-b and IL-18. These cytokines are noted to directly contribute to the development of hypertension and atherosclerosis [77]. In addition, tryptase was shown to induce pulmonary artery smooth muscle cell proliferation and migration, as well as the synthesis of matrix protein deposition [78]. Although these findings strongly indicate the role of mast cell in pulmonary hypertension, the underlying molecular mechanism is not yet understood.
Interestingly, after intervention with mast cell inhibitors , cromolyn and fexofenadine, PAH patients showed a decrease in tryptase/leukotriene LTE4/VEGF, along with increase in exhaled nitric oxide, a commonly used vasodilator [74]. In a small clinical study, imatinib, a tyrosine kinase inhibitor that targets c-Kit and some subtypes of PH revealed decreased circulating progenitor cells/mast cells and in parallel a decrease in pulmonary vascular resistance [79].
These findings suggest that potential therapies targeting mast cells may show promise for future treatment strategies. Recent studies have also indicated that mast cells directly participate in the formation of bronchus-associated lymphoid tissue (BALT), and c-Kit+ cells were found locally surrounding these structures, suggesting mast cells contribute to their formation and development [66, 80]. Lymphoid structures found in patients with chronic lung disease provide a structure and base for inflammatory cells to congregate and accelerate the progression of vascular remodeling [81]. Mast cells can recruit and activate B cells by the release of IL-6, promoting the formation of this tertiary lymphoid tissue. Additionally, mast cells can induce T-cell activation, proliferation, and cytokine secretion [82]. Conversely, mast cells can suppress Tregs that were reported to protect against hypoxia-induced PH19.
Although mast cell therapy may present a testable and promising strategy, further studies are needed to determine if preventing mast cell migration early in the disease course can prevent vascular remodeling or in the late disease stage.
19.11 Cytokines
Cytokines represent a large group of signaling proteins that have important roles mediating systemic biological responses, including inflammation and immunity. These molecules are typically produced by cells of the immune system, but can also be released from endothelium and vasculature in response to stressful environments and triggers [83]. Specific cytokines are elevated in PAH patients and are known to correlate with disease severity [84]. Identifying the temporal relationship and effects cytokines have on the development of PAH is essential to understanding pathophysiology and disease progression. Therapies targeting specific cytokine responses and pathways are a high yield area for potential treatments and therapeutic strategies.
19.12 IL-6
IL-6 is a potent cytokine with a wide range of proinflammatory properties affecting immune regulation, inflammation, and metabolic pathways. It plays a vital role in regulating the balance between TH17 cells and Treg cells. Oversecretion can cause an immune response favoring Th17 cells and place subjects at risk for PAH [85].
The exact mechanism that IL-6 contributes to PH development is unclear. Studies suggest that unopposed mitogen-activated protein kinase (MAPK) intracellular signaling and decreased TGF-b expression result in proliferative and antiapoptotic signaling pathways [86]. IL-6 also has prognostic value in patients with PH. Serum levels have been shown to be an independent predictor of survival in patients with PH, and in conjunction with other markers of disease severity, can predict patient outcome [87]. Animal models have demonstrated the direct effect IL-6 has on the development of PH. Rats injected with recombinant human IL-6 display increased RVH and under normoxic conditions develop PH [88]. Conversely, IL-6 knockout mice exposed to hypoxia were found to be resistant to the development of increased RVP [89]. The most convincing study for IL-6 inducing PH revealed that transgenic mice overexpressing IL-6 developed muscularization of the proximal arterial tree, distal arteriolar vessels, and were found to have occlusive proliferative lesions consisting of endothelial cells and T lymphocytes [90].
Potential therapies targeting IL-6 and its signaling pathways may help decrease progression or even reverse the vascular remodeling seen in PH. However, more research is needed to determine how to target specific pathways activated by IL-6 contributing to PH development and to identify which patients would benefit.
19.13 IL-1b
IL-1b is a proinflammatory cytokine that, like IL-6, is shown to correlate with worse outcomes in PH patients. Levels of IL-1b decrease prostacyclin PGI2, a metabolite of arachidonic acid, which possesses vasodilatory and antiproliferative effects protective against the development of PH.
Murine models of PH also demonstrate high serum levels of IL-1b, and the initiation of an IL-1b receptor antagonist has been shown to decrease PH and RVH [91]. In addition, pulmonary artery smooth muscle cells treated with IL-1b display increased levels of COX-2 mRNA, a key enzyme in prostacyclin synthesis [90].
While IL-1b itself has properties leading to PAH, it can also activate other proinflammatory cytokines and molecules. Cleavage of IL-18 by IL-1b converting enzyme generates the biologically active IL-18, which is elevated in patients with pulmonary vascular disease. Vascular injury releases IL-18 from smooth muscle cells and causes the local proliferation and recruitment of smooth muscle cells , leading to hypertrophy and PAH progression [92].
19.14 Other Secreted Factors
Other cytokines , such as IL-4, IL-5, IL-8, IL-10, IL-13, VEGF, and TNF-a, have been shown to be abnormal in human and animal models of PH. IL-8 plays a role in the early vascular remodeling and development of PH via its proangiogenic and antiapoptotic properties that act as a growth factor for endothelial cells [93]. Injections of TNF-a into rat models can cause increased vascular activity and remodeling [94].
CXCL12a , a potent proangiogenic chemokine, is also elevated in patients with PH compared to those without. The chemoattractant has been shown to cause endothelial proliferation through the CXCR7 and CXCR4 receptors on endothelial cells [95]. Levels also correlate with disease severity, as they have been found to be an independent risk factor for earlier death and correlate with mean pulmonary arterial pressure [96]. IL-10, a potent anti-inflammatory cytokine released by T cells, is increased in patients with PAH, likely due to counter-regulatory mechanisms as levels have been found to inversely correlate with prostacyclin therapy and be decreased in patients following cardiopulmonary bypass [97].
Abnormal levels of other biological markers, such as osteoprotegerin, also known as tumor necrosis factor receptor superfamily member 11B (TNFSRF11B), have been noted in patients with PAH. Increased expression and secretion of TNFSRF11B have also been identified in patients with PH. Multiple studies have also found that elevated serum osteoprotegerin levels correlate with hemodynamic markers predictive of disease severity [98]. It is hypothesized that multiple different pathways contributing to PAH development can cause increased osteoprotegerin, including increased BMPR2 expression [99].
Whether over or under expression of specific cytokines leads to the development of PH or if the abnormalities detected occur secondary to disease progression remain unclear. Growing evidence suggests that levels of cytokines can distinguish PAH into four distinct immune phenotypes [100]. Independent of underlying etiology, these four immune phenotypes may play a role in the development of future therapies, as patients can be categorized into an immune phenotype and treated with appropriate therapy. Immune clusters found that groups had different signals for various pathways contributing to PAH development. Some groups skewed toward a TH1 response while others toward a TH17 response or adaptive immunity.
Immune phenotyping could offer a framework for therapies in clinical settings. Therapies targeting immunity would be beneficial in patients who lack an increased inflammatory response based on their phenotype and vice versa. The potential of combined treatments based on a patient’s phenotype is a potential future therapeutic option. Targeted treatment toward cells, cytokines, and other molecules overexpressed in an immune phenotype could provide multitargeted treatment strategies personalized for a patient’s inflammatory phenotype.
19.15 Other Cell Types
BMPR2 is a serine/threonine receptor kinase that binds to bone morphogenic proteins, which are a type of TGF-b ligand. BMPR2 is involved in various cellular functions, including osteogenesis, cell growth, and differentiation. It functions to inhibit the proliferation of vascular endothelial and smooth muscle cells, preventing arterial damage and local inflammatory response. Inhibition of BMPR2 gene expression leads to unregulated proliferation and survival of endothelial cells through disordered TGF-b signaling, contributing to vascular remodeling [101]. BMPR2 mutations are the most common genetic mutations associated with PAH. Roughly 80% of patients with HPAH and 30% of IPAH patients have a mutation within the BMPR2 gene [102]. In response to a reduction in BMPR2 function in the endothelium, it is hypothesized that the integrity of the endothelium barrier may be compromised leading to apoptosis, the release of TGF-b , and the development of apoptosis-resistant clones [103]. In contrast, smooth muscle cells proliferate due to TGF-b signaling and undergo an exaggerated growth response leading to vascular remodeling [104]. It is reasonable to speculate that BMPR2 deficiency can increase vascular–immune interaction, with increased immune cells infiltration to the subintimal blood or lymphatic vascular structures.
19.16 Lymphatics
The lymphatic vascular system transports fluid, immune cells, and wastes through the body to help prevent edema within tissues, facilitate an immune response, and remove harmful toxins. Lymphatic vessels are found abundantly in the lung, help maintain a fluid balance, and precipitate an immune response. Small lymphatic vessels around distal bronchus drain into larger vessels and eventually into the right and left lymphatic ducts. Lymph nodes, the major site of T and B cells, filter foreign particles and house an immune response to pathogens [105].
Disrupted lymphatic function and flow can lead to an inflammatory state and alveolar damage characterized by the formation of tertiary lymphoid organs (TLOs) in chronic lung disease [106]. Thus, perivascular lymphatic infiltrates may be a major source to form TLOs seen in severe PAH. After lung transplantation, interrupted lymphatic vessels are associated with the induction of allograft tolerance as well as rejection [107]. There is very little pulmonary research focusing on lymphatic circulation. The characterization and examination of the lymphatic system in PH need more investigation .
19.17 Endothelial Cells
Endothelial cells (ECs) play a key role in maintaining vascular homeostasis in response to various stimuli and regulate inflammation through mediators such as nitric oxide (NO), endothelin-1 (ET1), cell adhesion molecules, cytokines, and chemokines. Endothelial cell dysfunction has been shown to contribute to the development of multiple cardiac and vascular diseases, including PAH and heart failure [108].
Under pathological conditions such as inflammation and hypoxia, pulmonary artery endothelial cells decrease the production of vasodilators such as nitric oxide and vascular growth factors, favoring vasoconstriction of the distal pulmonary arteries [109]. Unregulated endothelial cell proliferation and neoangiogenesis can result in the formation of plexiform lesions (PLs), glomerular like vascular structures, seen in severe PAH [108, 110]. PLs consist of disorganized endothelium channels as well as a uniform myogenic origin cells and immune cells. Rat model exposed to chronic hypoxia and SU5416 has been shown to form PL-like structures that partially resemble human pathology [111, 112]. Bioinformatics analysis further validates the mixture of cell identities. The cellular contribution to the process of PL formation and EC recruitment of inflammatory cells need more clarification.
There is evidence that pulmonary arteries also have a permeability defect in animal models of PH [113, 114]. The pulmonary endothelium aids in the passage of circulating immune cells through alveolar capillaries and closely associated with distal air sacs. The inflammatory and immune cells that migrate into the lung parenchyma of PH can be from multiple resources, such as the pulmonary arterioles, the vasa vasorum, perivascular capillary network, or even the lymphatic vasculature. Following initial tethering at the endothelial cell surface (as known as the classic paradigm of the leukocyte adhesion cascade [115]), leukocytes start to roll and firmly arrest on the vessel surface to ultimately migrate into the subendothelial space, typically via a paracellular route but occasionally also via a transcellular route.
Some studies have analyzed circulation soluble adhesion molecules and their role in cell migration into the lung. For example, one of circulating soluble adhesion molecules P-selectin was shown to be elevated in the plasma of PAH patients or CTEPH, as well as in animal models [116,117,118]. A few other studies have addressed the expression of endothelial adhesion molecules, ICAM-1, VCAM-1, and E-selectin, in PAH tissues that could be related to BMPR2 mutations [119, 120]. Taken together, adhesion molecule expression correlates with leukocyte interaction within the pulmonary endothelium in PH.
However, the mechanisms of these molecules impact on endothelium permeability are still lacking. Circulating endothelial cells (CECs) may participate in processes of vascular injury and tumorigenesis or interaction with immune cells. Additionally, endothelial progenitor cells (EPCs) are bone marrow-derived cells involved in homeostasis, but also physiological and pathological angiogenesis. The increase in proinflammatory cytokines also favors platelet adherence and activation of coagulation cascades, leading to further arteriole occlusion [121].
The role of these cells in the pathobiology of PH is yet to be elucidated. Although bone marrow-derived endothelial progenitor cells have been tested as a therapeutic option in animal and human models with promising results, with much of the measured benefit attributed to gene manipulation [122,123,124]. Regardless, inhibiting endothelial cell apoptosis and migration may stop a necessary initial key step in PAH pathogenesis and provide future therapeutic options.
19.18 Pericytes
Pericytes are mesenchymal-derived mural cells that wrap around endothelial cells throughout the entire capillary vasculature in all organs. Due to its controversy and lack of unique cellular markers, pericytes are largely ignored and under investigation.
Our groups recently discovered that impaired endothelial–pericyte interaction contributed to small vessel loss in PAH [125]. Intriguingly, SDF1 (aka CXCL12), an inflammatory cytokine, regulated pericyte migration and lineage and potentially associated with pulmonary arterial muscularization [126, 127]. In addition to their vascular functions, pericytes regulate different aspects of immune responses, though most of our understanding of pericyte-related immune responses was elucidated from brain or placenta pericytes.
Some studies suggested that central nervous system microvascular pericytes may display macrophage-like/nonprofessional antigen-presenting cell characteristics and involve in several possible immune responses [128]. A clear distinction of pericytes versus macrophages was lacking and the conclusions need to be extensively tested on multiple lineages tracing models. Whether lung pericytes represent the same phenotype seen in the brain and participate in inflammatory processes has remained entirely unclear.
In rat lung, pericytes were demonstrated to upregulate TLR4, increase vessel permeability, and produce of IL-1b upon LPS exposure [129,130,131]. LPS has been shown to generate NO in pericytes, leading to vasodilation. In addition, an iNOS-independent pathway was associated with lung pericyte contractility [132]. Vascular endothelial growth factor (VEGF) may relate to the induction of endothelial nitric oxide, inducing vascular leakage and inflammation. VEGF also modifies the contractile response of lung pericytes. This mechanism may play a role in the increased permeability demonstrated in inflammatory conditions [133].
In the capillary, dynamic changes in response to proinflammatory signals are necessary for the efficient recruitment of leukocytes. Sequential interactions of endothelium-expressed cytokines with circulating immune cells can initiate extravasation during a series of processes, as discussed above, known as the leukocyte adhesion cascade [134].
Endothelium was extensively studied, much more so than pericytes, during acute inflammation. For example, in distal microvessels, the regions with partial coverage of mural cells appear to be preferential inflammatory sites for the transmigration of neutrophils [135]. A follow-up study further characterized the low matrix expression region and identified pericyte partial coverage that could be preferred sites for monocytes and neutrophil migration [136]. Intriguingly, increased neutrophil recruitment and transmigration into extravascular tissue are more associated with EC–pericytes bilayers than a monolayer, suggesting the cytokine released by pericytes could promote vascular inflammation [137].
Subsequent studies showed that pericyte generated basement membrane as a cellular matrix composite model could be the front line barrier to affect leucocyte recruitment upon TNF-a exposure [138]. Pericytes triggered the chemotactic migration of interstitial neutrophils and macrophages after extravasation from capillary [139]. Thus, pericytes can be crucial for the efficient navigation of cells of the innate immune system, which can execute their effector functions at the local inflammation. Using confocal intravital microscopy, pericytes facilitated leukocyte trafficking into sites of inflammation in vivo [140]. Increased PDGFRb signaling induced a panel of immune response genes in pericytes. Lung pericyte-like cells release proinflammatory molecules following epithelial injury and promote acute inflammatory responses by recruiting leukocytes [141].
All studies support the vital role of pericytes in mediating inflammatory and immune signaling, whether lung specific pericytes can recapitulate the same physiological function still require further characterization.
19.19 Other Vascular Mural Cells
In response to hypoxia and other stimuli, the pulmonary vessels undergo proliferation and hypertrophy secondary to enhanced cytokine production. This complex remodeling of the vasculature includes all layers of the pulmonary vasculature but especially affects the medial layer. The increased medial thickness of the pulmonary arteries is well documented in both human and animal models. The process is driven by increased numbers and hypertrophy of its principal cellular constituent, smooth muscle cells (SMCs).
It was proposed that inflammation could recruit SMC populations and enhance their contribution to pulmonary vascular remodeling because hyperproliferative SM-like cells are observed in local occluded vessels. Several studies have also shown that sustained hypoxia induces the recruitment of mesenchymal progenitor cells in the perivasculature. The recruitment of these cells is critical to the development of PAH [142, 143]. It is possible that the inflammatory vasculature changes seen are due to these migrating cells, as well as resident smooth muscle cells, which resume the capabilities needed for vascular remodeling.
While the exact timing and role of smooth muscle cells in PAH development is unclear, therapies targeting the proliferation of smooth muscle cells in animal models have shown success. This includes inhibiting the receptors tyrosine kinase, mTOR, p38 and CDK4/6 [144]. Imatinib, discussed previously, has shown promising results in improving exercise capacity and hemodynamic in patients with advances PAH; however, its clinical use is limited by adverse effects [145]. Future studies are needed to determine if therapies targeting different phenotypes of smooth muscle cells will be of benefit.
Therapeutic strategies targeting dendritic cell migration have also shown some promise in the treatment of PAH. Imatinib, a platelet-derived growth factor receptor antagonist STI571, has been demonstrated to reverse vascular remodeling in monocrotaline exposed rats through effects on smooth muscle cells [146]. However, imatinib has multiple other mechanisms of action, including targeting the common Ras/MAPK and Jak/STAT pathway. Before the effects of imatinib on dendritic cells are confirmed, more studies in human models are needed to better understand the exact roles of dendritic cells have in the progression of PAH.
Fibroblasts, the most common cell found in connective tissue, are responsible for providing the framework and structure of the extracellular matrix. They play a critical role in wound healing and the surrounding environment of pulmonary vessels, reacting to local inflammation and stress, and in return, activating the innate immune system. The release of proinflammatory cytokines and growth factors from stimulated fibroblasts is a potential contributor to the development of PAH, and has been shown in animal and human models to potentially contribute to disease progression.
Activated fibroblasts found in the pulmonary artery adventitia have been documented in experimental and human models with PAH to display antiapoptotic, hyperproliferative, and proinflammatory features [59, 147, 148]. They have also been shown to recruit cells of the innate immune system to participate in disease progression. Fibroblasts of PH patients were shown to recruit and activate naïve macrophages [149]. The exact mechanism of fibroblast hyperactivity and resistance to apoptosis is unclear. Recent studies have shown that hyperactive fibroblasts may be regulated by a pro-oxidase status secondary to complex I deficiency in mitochondrial oxidase phosphorylation [150].
Furthermore, experimental results in mice exposed to hypoxia with mutations in FGFR1 and FGFR2 are consistent with PH compared to controls. This suggests that endothelial receptor activation leads to endothelial cell survival and decreased apoptosis protecting against the development of PH [151]. More studies are needed to confirm that cells exhibiting this mitochondrial abnormality exist during the development of PH before they are targeted for potential therapeutic strategies.
Until recently, it was thought that smooth muscle-like cells expressing a-SMactin accumulate in arterioles due to expansion of resident cells. New literature suggests that circulating progenitor cells, such as fibrocytes from the bone marrow, migrate to the pulmonary vasculature and are responsible for vascular remodeling. These cells continually produce extracellular membrane component-modifying enzymes that alter the structural composition of the lung [152]. These cells can differentiate into myofibroblasts in the presence of TGFb. Activated myofibroblasts are included in the organized thrombotic tissues of Group 4 CTEPH [153]. The differentiation between fibroblast and myofibroblast can mediate adventitial remodeling that found in larger sized pulmonary artery (Fig. 19.2).

The role of Treg cells in PAH development
19.20 Conclusions
There is increasing evidence that immune dysregulation plays an important role in the pathogenesis and progression of PAH. Inflammatory cell recruitment, cytokine and autoantibody production, and enhanced vascular wall remodeling lead to the abnormal interplay between the immune system and the pulmonary vasculature. Current therapies for PAH mainly target vasodilators NO and prostacyclin, or vasoconstrictors ET-1. Other combined strategies include optimizing cardiac function, such as the use of diuretics and calcium channel blockers. While these medications have transformed PAH management in the past few decades, new studies and a better understanding of PAH pathophysiology will improve therapies by targeting immune cells and inflammation, preventing cellular and vascular changes. Targeting inflammatory cascades before cellular changes are seen can not only treat PAH, but also prevent its progression altogether.
Abbreviations
- CRTH2:
-
Chemoattractant receptor homologous molecule expressed on Th2 cell
- CTD-PAH:
-
Connective tissue disease-associated pulmonary artery hypertension
- CTEPH:
-
Chronic thromboembolic pulmonary hypertension
- DC:
-
Dendritic cell
- ET1:
-
Endothelin-1
- GM-CSFR:
-
Granulocyte macrophage colony stimulating factor receptor
- HCV:
-
Hepatitis C virus
- HHV:
-
Human Herpes virus 6
- IPAH:
-
Idiopathic pulmonary arterial hypertension
- LTB4:
-
Leukotriene B4
- MAPK:
-
Mitogen-activated protein kinase
- MCP-1:
-
Monocyte-chemoattractant protein-1
- NE:
-
Neutrophil elastase
- NET:
-
Neutrophil extracellular trap
- NO:
-
Nitric oxide
- PAH:
-
Pulmonary arterial hypertension
- PH:
-
Pulmonary hypertension
- PL:
-
Plexiform lesion
- RV:
-
Right ventricle
- RVH:
-
Right ventricular hypertrophy
- RVSP:
-
Right ventricular systolic pressure
- SU5416:
-
Sugen 5416 selective inhibitor of the vascular endothelial growth factor receptor
- Tc:
-
Cytotoxic T cells
- Th:
-
T helper cells
- TLO:
-
Tertiary lymphoid organ
- TNFSRF11B:
-
Tumor necrosis factor receptor superfamily member 11B
- Treg:
-
T-regulatory cells
- VEGF-2:
-
Vascular endothelial growth factor-2
References
Tamosiuniene R, Nicolls MR. Regulatory T cells and pulmonary hypertension. Trends Cardiovasc Med. 2011;21:166–71.
Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, Walker C, Budd DC, Pepke-Zaba J, Morrell NW. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–7.
Cracowski JL, Chabot F, Labarere J, Faure P, Degano B, Schwebel C, Chaouat A, Reynaud-Gaubert M, Cracowski C, Sitbon O, Yaici A, Simonneau G, Humbert M. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur Respir J. 2014;43:915–7.
McKinley L, Logar AJ, McAllister F, Zheng M, Steele C, Kolls JK. Regulatory T cells dampen pulmonary inflammation and lung injury in an animal model of pneumocystis pneumonia. J Immunol. 2006;177:6215–26.
Mathai SC, Hassoun PM. Pulmonary arterial hypertension in connective tissue diseases. Heart Fail Clin. 2012;8:413–25.
Dib H, Tamby MC, Bussone G, Regent A, Berezne A, Lafine C, Broussard C, Simonneau G, Guillevin L, Witko-Sarsat V, Humbert M, Mouthon L. Targets of anti-endothelial cell antibodies in pulmonary hypertension and scleroderma. Eur Respir J. 2012;39:1405–14.
Rawson AJ, Woske HM. A study of etiologic factors in so-called primary pulmonary hypertension. Arch Intern Med. 1960;105:233–43.
Isern RA, Yaneva M, Weiner E, Parke A, Rothfield N, Dantzker D, Rich S, Arnett FC. Autoantibodies in patients with primary pulmonary hypertension: association with anti-Ku. Am J Med. 1992;93:307–12.
Rich S, Kieras K, Hart K, Groves BM, Stobo JD, Brundage BH. Antinuclear antibodies in primary pulmonary hypertension. J Am Coll Cardiol. 1986;8:1307–11.
Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–52.
Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res. 2014;115:189–202.
Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, Ley TJ. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–46.
Sziksz E, Pap D, Lippai R, Beres NJ, Fekete A, Szabo AJ, Vannay A. Fibrosis related inflammatory mediators: role of the IL-10 cytokine family. Mediat Inflamm. 2015;2015:764641.
Perry JSA, Lio CJ, Kau AL, Nutsch K, Yang Z, Gordon JI, Murphy KM, Hsieh CS. Distinct contributions of Aire and antigen-presenting-cell subsets to the generation of self-tolerance in the thymus. Immunity. 2014;41:414–26.
Qiu H, He Y, Ouyang F, Jiang P, Guo S, Guo Y. The role of regulatory T cells in pulmonary arterial hypertension. J Am Heart Assoc. 2019;8:e014201.
Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med. 2007;175:1280–9.
Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, Long CS, Voelkel NF, Nicolls MR. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res. 2011;109:867–79.
Tamosiuniene R, Manouvakhova O, Mesange P, Saito T, Qian J, Sanyal M, Lin YC, Nguyen LP, Luria A, Tu AB, Sante JM, Rabinovitch M, Fitzgerald DJ, Graham BB, Habtezion A, Voelkel NF, Aurelian L, Nicolls MR. Dominant role for regulatory T cells in protecting females against pulmonary hypertension. Circ Res. 2018;122:1689–702.
Chu Y, Xiangli X, Xiao W. Regulatory T cells protect against hypoxia-induced pulmonary arterial hypertension in mice. Mol Med Rep. 2015;11:3181–7.
Li C, Liu PP, Tang DD, Song R, Zhang YQ, Lei S, Wu SJ. Targeting the RhoA-ROCK pathway to regulate T-cell homeostasis in hypoxia-induced pulmonary arterial hypertension. Pulm Pharmacol Ther. 2018;50:111–22.
Voelkel NF, Tamosiuniene R, Nicolls MR. Challenges and opportunities in treating inflammation associated with pulmonary hypertension. Expert Rev Cardiovasc Ther. 2016;14:939–51.
Huertas A, Tu L, Gambaryan N, Girerd B, Perros F, Montani D, Fabre D, Fadel E, Eddahibi S, Cohen-Kaminsky S, Guignabert C, Humbert M. Leptin and regulatory T-lymphocytes in idiopathic pulmonary arterial hypertension. Eur Respir J. 2012;40:895–904.
Peng X, Moore MW, Peng H, Sun H, Gan Y, Homer RJ, Herzog EL. CD4+CD25+FoxP3+ regulatory Tregs inhibit fibrocyte recruitment and fibrosis via suppression of FGF-9 production in the TGF-beta1 exposed murine lung. Front Pharmacol. 2014;5:80.
MacDonald KP, Blazar BR, Hill GR. Cytokine mediators of chronic graft-versus-host disease. J Clin Invest. 2017;127:2452–63.
Garibaldi BT, D'Alessio FR, Mock JR, Files DC, Chau E, Eto Y, Drummond MB, Aggarwal NR, Sidhaye V, King LS. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol. 2013;48:35–43.
Cao Y, Xu W, Xiong S. Adoptive transfer of regulatory T cells protects against Coxsackievirus B3-induced cardiac fibrosis. PLoS One. 2013;8:e74955.
Austin ED, Rock MT, Mosse CA, Vnencak-Jones CL, Yoder SM, Robbins IM, Loyd JE, Meyrick BO. T lymphocyte subset abnormalities in the blood and lung in pulmonary arterial hypertension. Respir Med. 2010;104:454–62.
Ulrich S, Nicolls MR, Taraseviciene L, Speich R, Voelkel N. Increased regulatory and decreased CD8+ cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration. 2008;75:272–80.
Gaowa S, Zhou W, Yu L, Zhou X, Liao K, Yang K, Lu Z, Jiang H, Chen X. Effect of Th17 and Treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediat Inflamm. 2014;2014:247372.
Hautefort A, Girerd B, Montani D, Cohen-Kaminsky S, Price L, Lambrecht BN, Humbert M, Perros F. T-helper 17 cell polarization in pulmonary arterial hypertension. Chest. 2015;147:1610–20.
Serezani CH, Kane S, Collins L, Morato-Marques M, Osterholzer JJ, Peters-Golden M. Macrophage dectin-1 expression is controlled by leukotriene B4 via a GM-CSF/PU.1 axis. J Immunol. 2012;189:906–15.
Hashimoto-Kataoka T, Hosen N, Sonobe T, Arita Y, Yasui T, Masaki T, Minami M, Inagaki T, Miyagawa S, Sawa Y, Murakami M, Kumanogoh A, Yamauchi-Takihara K, Okumura M, Kishimoto T, Komuro I, Shirai M, Sakata Y, Nakaoka Y. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci USA. 2015;112:E2677–86.
Maston LD, Jones DT, Giermakowska W, Howard TA, Cannon JL, Wang W, Wei Y, Xuan W, Resta TC, Gonzalez Bosc LV. Central role of T helper 17 cells in chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2017;312:L609–L24.
Kumar R, Mickael C, Kassa B, Sanders L, Koyanagi D, Hernandez-Saavedra D, Freeman S, Morales-Cano D, Cogolludo A, McKee AS, Fontenot AP, Butrous G, Tuder RM, Graham BB. Th2 CD4(+) T cells are necessary and sufficient for Schistosoma-pulmonary hypertension. J Am Heart Assoc. 2019;8:e013111.
Chen G, Zuo S, Tang J, Zuo C, Jia D, Liu Q, Liu G, Zhu Q, Wang Y, Zhang J, Shen Y, Chen D, Yuan P, Qin Z, Ruan C, Ye J, Wang X-J, Zhou Y, Gao P, Zhang P, Liu J, Jing Z-C, Lu A, Yu Y. Inhibition of CRTH2-mediated Th2 activation attenuates pulmonary hypertension in mice. J Exp Med. 2018;215:2175–95.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–9.
Edwards AL, Gunningham SP, Clare GC, Hayman MW, Smith M, Frampton CM, Robinson BA, Troughton RW, Beckert LE. Professional killer cell deficiencies and decreased survival in pulmonary arterial hypertension. Respirology. 2013;18:1271–7.
Ulrich S, Nicolls MR, Taraseviciene L, Speich R, Voelkel N. Increased regulatory and decreased CD8+ cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration. 2008;75:272–80.
Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, Scheed A, Ritter C, Dahal BK, Vater A, Klussmann S, Ghofrani HA, Weissmann N, Klepetko W, Banat GA, Seeger W, Grimminger F, Schermuly RT. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:897–908.
Taylor S, Dirir O, Zamanian RT, Rabinovitch M, Thompson AAR. The role of neutrophils and neutrophil elastase in pulmonary arterial hypertension. Front Med (Lausanne). 2018;5:217.
Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N, Stenmark KR. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–69.
Schultze AE, Wagner JG, White SM, Roth RA. Early indications of monocrotaline pyrrole-induced lung injury in rats. Toxicol Appl Pharmacol. 1991;109:41–50.
Yildiz A, Kaya H, Ertas F, Oylumlu M, Bilik MZ, Yuksel M, Polat N, Akil MA, Atilgan Z, Ulgen MS. Association between neutrophil to lymphocyte ratio and pulmonary arterial hypertension. Turk Kardiyol Dern Ars. 2013;41:604–9.
Zhu L, Wigle D, Hinek A, Kobayashi J, Ye C, Zuker M, Dodo H, Keeley FW, Rabinovitch M. The endogenous vascular elastase that governs development and progression of monocrotaline-induced pulmonary hypertension in rats is a novel enzyme related to the serine proteinase adipsin. J Clin Invest. 1994;94:1163–71.
Rose F, Hattar K, Gakisch S, Grimminger F, Olschewski H, Seeger W, Tschuschner A, Schermuly RT, Weissmann N, Hanze J, Sibelius U, Ghofrani HA. Increased neutrophil mediator release in patients with pulmonary hypertension−suppression by inhaled iloprost. Thromb Haemost. 2003;90:1141–9.
Kim YM, Haghighat L, Spiekerkoetter E, Sawada H, Alvira CM, Wang L, Acharya S, Rodriguez-Colon G, Orton A, Zhao M, Rabinovitch M. Neutrophil elastase is produced by pulmonary artery smooth muscle cells and is linked to neointimal lesions. Am J Pathol. 2011;179:1560–72.
Spiekerkoetter E, Alvira CM, Kim YM, Bruneau A, Pricola KL, Wang L, Ambartsumian N, Rabinovitch M. Reactivation of gammaHV68 induces neointimal lesions in pulmonary arteries of S100A4/Mts1-overexpressing mice in association with degradation of elastin. Am J Physiol Lung Cell Mol Physiol. 2008;294:L276–89.
Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6:698–702.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5.
Aldabbous L, Abdul-Salam V, McKinnon T, Duluc L, Pepke-Zaba J, Southwood M, Ainscough AJ, Hadinnapola C, Wilkins MR, Toshner M, Wojciak-Stothard B. Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2016;36:2078–87.
Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, Gallant M, Martinod K, Ten Cate H, Hofstra L, Crijns HJ, Wagner DD, Kietselaer B. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol. 2013;33:2032–40.
Nickel NP, Spiekerkoetter E, Gu M, Li CG, Li H, Kaschwich M, Diebold I, Hennigs JK, Kim KY, Miyagawa K, Wang L, Cao A, Sa S, Jiang X, Stockstill RW, Nicolls MR, Zamanian RT, Bland RD, Rabinovitch M. Elafin reverses pulmonary hypertension via Caveolin-1-dependent bone morphogenetic protein signaling. Am J Respir Crit Care Med. 2015;191:1273–86.
Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–85.
Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, Mitsialis SA, Kourembanas S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation. 2011;123:1986–95.
Thenappan T, Goel A, Marsboom G, Fang YH, Toth PT, Zhang HJ, Kajimoto H, Hong Z, Paul J, Wietholt C, Pogoriler J, Piao L, Rehman J, Archer SL. A central role for CD68(+) macrophages in hepatopulmonary syndrome. Reversal by macrophage depletion. Am J Respir Crit Care Med. 2011;183:1080–91.
Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, Gera L, Farkas L, Rabinovitch M, Zamanian RT, Inayathullah M, Fridlib M, Rajadas J, Peters-Golden M, Voelkel NF, Nicolls MR. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med. 2013;5:200ra117.
Florentin J, Coppin E, Vasamsetti SB, Zhao J, Tai Y-Y, Tang Y, Zhang Y, Watson A, Sembrat J, Rojas M, Vargas SO, Chan SY, Dutta P. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J immunol. 2018;1950(200):3612–25.
Zaloudikova M, Vytasek R, Vajnerova O, Hnilickova O, Vizek M, Hampl V, Herget J. Depletion of alveolar macrophages attenuates hypoxic pulmonary hypertension but not hypoxia-induced increase in serum concentration of MCP-1. Physiol Res. 2016;65:763–8.
Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, Sokol RJ, Strassheim D, Meyrick B, Yeager ME, Flockton AR, McKeon BA, Lemon DD, Horn TR, Anwar A, Barajas C, Stenmark KR. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol. 2011;187:2711–22.
Li C, Liu P, Song R, Zhang Y, Lei S, Wu S. Immune cells and autoantibodies in pulmonary arterial hypertension. Acta Biochim Biophys Sin. 2017;49:1047–57.
van Rijt LS, Lambrecht BN. Dendritic cells in asthma: a function beyond sensitization. Clin Exp Allergy. 2005;35:1125–34.
Palucka AK, Blanck J-P, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFN-α in autoimmune diseases. Proc Natl Acad Sci USA. 2005;102:3372–7.
Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–66.
Perros F, Dorfmuller P, Souza R, Durand-Gasselin I, Mussot S, Mazmanian M, Herve P, Emilie D, Simonneau G, Humbert M. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur Respir J. 2007;29:462–8.
Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum Pathol. 1997;28:434–42.
Perros F, Dorfmuller P, Montani D, Hammad H, Waelput W, Girerd B, Raymond N, Mercier O, Mussot S, Cohen-Kaminsky S, Humbert M, Lambrecht BN. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:311–21.
Wang W, Yan H, Zhu W, Cui Y, Chen J, Wang X, Li S, Zhu J. Impairment of monocyte-derived dendritic cells in idiopathic pulmonary arterial hypertension. J Clin Immunol. 2009;29:705–13.
Marsh LM, Jandl K, Grunig G, Foris V, Bashir M, Ghanim B, Klepetko W, Olschewski H, Olschewski A, Kwapiszewska G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2018;51
Yang T, Li ZN, Chen G, Gu Q, Ni XH, Zhao ZH, Ye J, Meng XM, Liu ZH, Xiong CM, He JG. Increased levels of plasma CXC-chemokine ligand 10, 12 and 16 are associated with right ventricular function in patients with idiopathic pulmonary arterial hypertension. Heart Lung. 2014;43:322–7.
Itoh T, Nagaya N, Ishibashi-Ueda H, Kyotani S, Oya H, Sakamaki F, Kimura H, Nakanishi N. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology. 2006;11:158–63.
Ribatti D, Vacca A, Nico B, Crivellato E, Roncali L, Dammacco F. The role of mast cells in tumour angiogenesis. Br J Haematol. 2001;115:514–21.
Mitani Y, Ueda M, Maruyama K, Shimpo H, Kojima A, Matsumura M, Aoki K, Sakurai M. Mast cell chymase in pulmonary hypertension. Thorax. 1999;54:88–90.
Hamada H, Terai M, Kimura H, Hirano K, Oana S, Niimi H. Increased expression of mast cell chymase in the lungs of patients with congenital heart disease associated with early pulmonary vascular disease. Am J Respir Crit Care Med. 1999;160:1303–8.
Farha S, Sharp J, Asosingh K, Park M, Comhair SA, Tang WH, Thomas J, Farver C, Hsieh F, Loyd JE, Erzurum SC. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm Circ. 2012;2:220–8.
Gilfillan AM, Rivera J. The tyrosine kinase network regulating mast cell activation. Immunol Rev. 2009;228:149–69.
Bartelds B, van Loon RLE, Mohaupt S, Wijnberg H, Dickinson MG, Boersma B, Takens J, van Albada M, Berger RMF. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest. 2012;141:651–60.
Caughey GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev. 2007;217:141–54.
Kwapiszewska G, Markart P, Dahal BK, Kojonazarov B, Marsh LM, Schermuly RT, Taube C, Meinhardt A, Ghofrani HA, Steinhoff M, Seeger W, Preissner KT, Olschewski A, Weissmann N, Wygrecka M. PAR-2 inhibition reverses experimental pulmonary hypertension. Circ Res. 2012;110:1179–91.
Farha S, Dweik R, Rahaghi F, Benza R, Hassoun P, Frantz R, Torres F, Quinn DA, Comhair S, Erzurum S, Asosingh K. Imatinib in pulmonary arterial hypertension: c-Kit inhibition. Pulm Circ. 2014;4:452–5.
Colvin KL, Cripe PJ, Ivy DD, Stenmark KR, Yeager ME. Bronchus-associated lymphoid tissue in pulmonary hypertension produces pathologic autoantibodies. Am J Respir Crit Care Med. 2013;188:1126–36.
Breitling S, Hui Z, Zabini D, Hu Y, Hoffmann J, Goldenberg NM, Tabuchi A, Buelow R, Dos Santos C, Kuebler WM. The mast cell-B cell axis in lung vascular remodeling and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2017;312:L710–L21.
Bulfone-Paus S, Bahri R. Mast cells as regulators of T cell responses. Front Immunol. 2015;6:394.
Price LC, Wort SJ, Perros F, Dorfmuller P, Huertas A, Montani D, Cohen-Kaminsky S, Humbert M. Inflammation in pulmonary arterial hypertension. Chest. 2012;141:210–21.
Groth A, Vrugt B, Brock M, Speich R, Ulrich S, Huber LC. Inflammatory cytokines in pulmonary hypertension. Respir Res. 2014;15:47.
Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40:1830–5.
Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–44, 28p following 44.
Heresi GA, Aytekin M, Hammel JP, Wang S, Chatterjee S, Dweik RA. Plasma interleukin-6 adds prognostic information in pulmonary arterial hypertension. Eur Respir J. 2014;43:912–4.
Golembeski SM, West J, Tada Y, Fagan KA. Interleukin-6 causes mild pulmonary hypertension and augments hypoxia-induced pulmonary hypertension in mice. Chest. 2005;128:572S–3S.
Miyata M, Sakuma F, Yoshimura A, Ishikawa H, Nishimaki T, Kasukawa R. Pulmonary hypertension in rats. 2. Role of interleukin-6. Int Arch Allergy Immunol. 1995;108:287–91.
Itoh A, Nishihira J, Makita H, Miyamoto K, Yamaguchi E, Nishimura M. Effects of IL-1beta, TNF-alpha, and macrophage migration inhibitory factor on prostacyclin synthesis in rat pulmonary artery smooth muscle cells. Respirology. 2003;8:467–72.
Voelkel NF, Tuder RM, Bridges J, Arend WP. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol. 1994;11:664–75.
Ross DJ, Strieter RM, Fishbein MC, Ardehali A, Belperio JA. Type I immune response cytokine-chemokine cascade is associated with pulmonary arterial hypertension. J Heart Lung Transplant. 2012;31:865–73.
Li A, Varney ML, Valasek J, Godfrey M, Dave BJ, Singh RK. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis. 2005;8:63–71.
Stevens T, Janssen PL, Tucker A. Acute and long-term TNF-alpha administration increases pulmonary vascular reactivity in isolated rat lungs. J Appl Physiol. 1985;1992(73):708–12.
Costello CM, McCullagh B, Howell K, Sands M, Belperio JA, Keane MP, Gaine S, McLoughlin P. A role for the CXCL12 receptor, CXCR7, in the pathogenesis of human pulmonary vascular disease. Eur Respir J. 2012;39:1415–24.
McCullagh BN, Costello CM, Li L, O’Connell C, Codd M, Lawrie A, Morton A, Kiely DG, Condliffe R, Elliot C, McLoughlin P, Gaine S. Elevated plasma CXCL12alpha is associated with a poorer prognosis in pulmonary arterial hypertension. PLoS One. 2015;10:e0123709.
Lei Y, Zhen J, Ming XL, Jian HK. Induction of higher expression of IL-beta and TNF-alpha, lower expression of IL-10 and cyclic guanosine monophosphate by pulmonary arterial hypertension following cardiopulmonary bypass. Asian J Surg. 2002;25:203–8.
Condliffe R, Pickworth JA, Hopkinson K, Walker SJ, Hameed AG, Suntharaligam J, Soon E, Treacy C, Pepke-Zaba J, Francis SE, Crossman DC, Newman CM, Elliot CA, Morton AC, Morrell NW, Kiely DG, Lawrie A. Serum osteoprotegerin is increased and predicts survival in idiopathic pulmonary arterial hypertension. Pulm Circ. 2012;2:21–7.
Lawrie A, Waterman E, Southwood M, Evans D, Suntharalingam J, Francis S, Crossman D, Croucher P, Morrell N, Newman C. Evidence of a role for osteoprotegerin in the pathogenesis of pulmonary arterial hypertension. Am J Pathol. 2008;172:256–64.
Sweatt AJ, Hedlin HK, Balasubramanian V, Hsi A, Blum LK, Robinson WH, Haddad F, Hickey PM, Condliffe R, Lawrie A, Nicolls MR, Rabinovitch M, Khatri P, Zamanian RT. Discovery of distinct immune phenotypes using machine learning in pulmonary arterial hypertension. Circ Res. 2019;124:904–19.
Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, Granton J, Stewart DJ. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98:209–17.
Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, Trembath RC, Loyd JE. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J. 2019;53:1801899.
McDonald PP, Fadok VA, Bratton D, Henson PM. Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF-beta in macrophages that have ingested apoptotic cells. J Immunol. 1999;163:6164–72.
Morrell NW. Pulmonary hypertension due to BMPR2 mutation: a new paradigm for tissue remodeling? Proc Am Thorac Soc. 2006;3:680–6.
Schraufnagel DE. Lung lymphatic anatomy and correlates. Pathophysiology. 2010;17:337–43.
Reed HO, Wang L, Sonett J, Chen M, Yang J, Li L, Aradi P, Jakus Z, D'Armiento J, Hancock WW, Kahn ML. Lymphatic impairment leads to pulmonary tertiary lymphoid organ formation and alveolar damage. J Clin Invest. 2019;129:2514–26.
Cui Y, Liu K, Lamattina AM, Visner G, El-Chemaly S. Lymphatic vessels: the next frontier in lung transplant. Ann Am Thorac Soc. 2017;14:S226–S32.
Sakao S, Tatsumi K, Voelkel NF. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res. 2009;10:95.
Perros F, Ranchoux B, Izikki M, Bentebbal S, Happe C, Antigny F, Jourdon P, Dorfmuller P, Lecerf F, Fadel E, Simonneau G, Humbert M, Bogaard HJ, Eddahibi S. Nebivolol for improving endothelial dysfunction, pulmonary vascular remodeling, and right heart function in pulmonary hypertension. J Am Coll Cardiol. 2015;65:668–80.
Pietra GG, Edwards WD, Kay JM, Rich S, Kernis J, Schloo B, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Histopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, primary pulmonary hypertension registry. Circulation. 1989;80:1198–206.
Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–54.
Jonigk D, Golpon H, Bockmeyer CL, Maegel L, Hoeper MM, Gottlieb J, Nickel N, Hussein K, Maus U, Lehmann U, Janciauskiene S, Welte T, Haverich A, Rische J, Kreipe H, Laenger F. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am J Pathol. 2011;179:167–79.
Zhou C, Townsley MI, Alexeyev M, Voelkel NF, Stevens T. Endothelial hyperpermeability in severe pulmonary arterial hypertension: role of store-operated calcium entry. Am J Physiol Lung Cell Mol Physiol. 2016;311:L560–9.
Francis M, Xu N, Zhou C, Stevens T. Transient receptor potential channel 4 encodes a vascular permeability defect and high-frequency Ca(2+) transients in severe pulmonary arterial hypertension. Am J Pathol. 2016;186:1701–9.
Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–89.
Yaoita N, Shirakawa R, Fukumoto Y, Sugimura K, Miyata S, Miura Y, Nochioka K, Miura M, Tatebe S, Aoki T, Yamamoto S, Satoh K, Kimura T, Shimokawa H, Horiuchi H. Platelets are highly activated in patients of chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2014;34:2486–94.
Sakamaki F, Kyotani S, Nagaya N, Sato N, Oya H, Satoh T, Nakanishi N. Increased plasma P-selectin and decreased thrombomodulin in pulmonary arterial hypertension were improved by continuous prostacyclin therapy. Circulation. 2000;102:2720–5.
Hironaka E, Hongo M, Sakai A, Mawatari E, Terasawa F, Okumura N, Yamazaki A, Ushiyama Y, Yazaki Y, Kinoshita O. Serotonin receptor antagonist inhibits monocrotaline-induced pulmonary hypertension and prolongs survival in rats. Cardiovasc Res. 2003;60:692–9.
Vengethasamy L, Hautefort A, Tielemans B, Belge C, Perros F, Verleden S, Fadel E, Van Raemdonck D, Delcroix M, Quarck R. BMPRII influences the response of pulmonary microvascular endothelial cells to inflammatory mediators. Pflugers Arch. 2016;468:1969–83.
Le Hiress M, Tu L, Ricard N, Phan C, Thuillet R, Fadel E, Dorfmuller P, Montani D, de Man F, Humbert M, Huertas A, Guignabert C. Proinflammatory signature of the dysfunctional endothelium in pulmonary hypertension. Role of the macrophage migration inhibitory factor/CD74 complex. Am J Respir Crit Care Med. 2015;192:983–97.
Diller GP, Thum T, Wilkins MR, Wharton J. Endothelial progenitor cells in pulmonary arterial hypertension. Trends Cardiovasc Med. 2010;20:22–9.
Spees JL, Whitney MJ, Sullivan DE, Lasky JA, Laboy M, Ylostalo J, Prockop DJ. Bone marrow progenitor cells contribute to repair and remodeling of the lung and heart in a rat model of progressive pulmonary hypertension. FASEB J. 2008;22:1226–36.
Stewart DJ, Zhao YD, Courtman DW. Cell therapy for pulmonary hypertension: what is the true potential of endothelial progenitor cells? Circulation 2004;109:e172–3. Author reply e-3.
Zhao YD, Courtman DW, Deng Y, Kugathasan L, Zhang Q, Stewart DJ. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ Res. 2005;96:442–50.
Yuan K, Orcholski ME, Panaroni C, Shuffle EM, Huang NF, Jiang X, Tian W, Vladar EK, Wang L, Nicolls MR, Wu JY, de Jesus Perez VA. Activation of the Wnt/planar cell polarity pathway is required for pericyte recruitment during pulmonary angiogenesis. Am J Pathol. 2015;185:69–84.
Yuan K, Liu Y, Zhang Y, Nathan A, Tian W, Yu J, Sweatt AJ, Shamshou EA, Condon D, Chakraborty A, Agarwal S, Auer N, Zhang S, Wu JC, Zamanian RT, Nicolls MR, de Jesus Perez VA. Mural cell SDF1 signaling is associated with the pathogenesis of pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2020;62:747–59.
Yuan K, Shamskhou EA, Orcholski ME, Nathan A, Reddy S, Honda H, Mani V, Zeng Y, Ozen MO, Wang L, Demirci U, Tian W, Nicolls MR, de Jesus Perez VA. Loss of endothelium-derived Wnt5a is associated with reduced Pericyte recruitment and small vessel loss in pulmonary arterial hypertension. Circulation. 2019;139:1710–24.
Balabanov R, Washington R, Wagnerova J, Dore-Duffy P. CNS microvascular pericytes express macrophage-like function, cell surface integrin alpha M, and macrophage marker ED-2. Microvasc Res. 1996;52:127–42.
Edelman DA, Jiang Y, Tyburski J, Wilson RF, Steffes C. Toll-like receptor-4 message is up-regulated in lipopolysaccharide-exposed rat lung pericytes. J Surg Res. 2006;134:22–7.
Edelman DA, Jiang Y, Tyburski JG, Wilson RF, Steffes CP. Lipopolysaccharide up-regulates heat shock protein expression in rat lung pericytes. J Surg Res. 2007;140:171–6.
Edelman DA, Jiang Y, Tyburski JG, Wilson RF, Steffes CP. Cytokine production in lipopolysaccharide-exposed rat lung pericytes. J Trauma. 2007;62:89–93.
Speyer CL, Steffes CP, Tyburski JG, Ram JL. Lipopolysaccharide induces relaxation in lung pericytes by an iNOS-independent mechanism. Am J Physiol Lung Cell Mol Physiol. 2000;278:L880–7.
Donoghue L, Tyburski JG, Steffes CP, Wilson RF. Vascular endothelial growth factor modulates contractile response in microvascular lung pericytes. Am J Surg. 2006;191:349–52.
Muller WA. Mechanisms of transendothelial migration of leukocytes. Circ Res. 2009;105:223–30.
Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M, Maxwell PH, Sorokin L, Nourshargh S. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. 2006;203:1519–32.
Voisin MB, Woodfin A, Nourshargh S. Monocytes and neutrophils exhibit both distinct and common mechanisms in penetrating the vascular basement membrane in vivo. Arterioscler Thromb Vasc Biol. 2009;29:1193–9.
Ayres-Sander CE, Lauridsen H, Maier CL, Sava P, Pober JS, Gonzalez AL. Transendothelial migration enables subsequent transmigration of neutrophils through underlying pericytes. PLoS One. 2013;8:e60025.
Lauridsen HM, Pober JS, Gonzalez AL. A composite model of the human postcapillary venule for investigation of microvascular leukocyte recruitment. FASEB J. 2014;28:1166–80.
Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Bruhl ML, Gartner F, Khandoga AG, Legate KR, Pless R, Hepper I, Lauber K, Walzog B, Massberg S. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and 'instruct' them with pattern-recognition and motility programs. Nat Immunol. 2013;14:41–51.
Proebstl D, Voisin MB, Woodfin A, Whiteford J, D'Acquisto F, Jones GE, Rowe D, Nourshargh S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. 2012;209:1219–34.
Hung CF, Mittelsteadt KL, Brauer R, McKinney BL, Hallstrand TS, Parks WC, Chen P, Schnapp LM, Liles WC, Duffield JS, Altemeier WA. Lung pericyte-like cells are functional interstitial immune sentinel cells. Am J Physiol Lung Cell Mol Physiol. 2017;312:L556–L67.
Burke DL, Frid MG, Kunrath CL, Karoor V, Anwar A, Wagner BD, Strassheim D, Stenmark KR. Sustained hypoxia promotes the development of a pulmonary artery-specific chronic inflammatory microenvironment. Am J Physiol Lung Cell Mol Physiol. 2009;297:L238–50.
Farha S, Asosingh K, Xu W, Sharp J, George D, Comhair S, Park M, Tang WH, Loyd JE, Theil K, Tubbs R, Hsi E, Lichtin A, Erzurum SC. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood. 2011;117:3485–93.
Stenmark KR, Frid MG, Graham BB, Tuder RM. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc Res. 2018;114:551–64.
Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galie N, Gomez-Sanchez MA, Grimminger F, Grunig E, Hassoun PM, Morrell NW, Peacock AJ, Satoh T, Simonneau G, Tapson VF, Torres F, Lawrence D, Quinn DA, Ghofrani HA. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127:1128–38.
Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–21.
Panzhinskiy E, Zawada WM, Stenmark KR, Das M. Hypoxia induces unique proliferative response in adventitial fibroblasts by activating PDGFbeta receptor-JNK1 signalling. Cardiovasc Res. 2012;95:356–65.
Das M, Burns N, Wilson SJ, Zawada WM, Stenmark KR. Hypoxia exposure induces the emergence of fibroblasts lacking replication repressor signals of PKCzeta in the pulmonary artery adventitia. Cardiovasc Res. 2008;78:440–8.
El Kasmi KC, Pugliese SC, Riddle SR, Poth JM, Anderson AL, Frid MG, Li M, Pullamsetti SS, Savai R, Nagel MA, Fini MA, Graham BB, Tuder RM, Friedman JE, Eltzschig HK, Sokol RJ, Stenmark KR. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J Immunol. 2014;193:597–609.
Plecitá-Hlavatá L, Tauber J, Li M, Zhang H, Flockton AR, Pullamsetti SS, Chelladurai P, D'Alessandro A, El Kasmi KC, Ježek P, Stenmark KR. Constitutive reprogramming of fibroblast mitochondrial metabolism in pulmonary hypertension. Am J Respir Cell Mol Biol. 2016;55:47–57.
Woo KV, Weinheimer C, Kovacs A, Ornitz D. Impact of endothelial fibroblast growth factors on pulmonary hypertension. J Am Coll Cardiol. 2017;69:1901.
Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–16.
Maruoka M, Sakao S, Kantake M, Tanabe N, Kasahara Y, Kurosu K, Takiguchi Y, Masuda M, Yoshino I, Voelkel NF, Tatsumi K. Characterization of myofibroblasts in chronic thromboembolic pulmonary hypertension. Int J Cardiol. 2012;159:119–27.
Qiu H, He Y, Ouyang F, Jiang P, Guo S, Guo Y. The role of regulatory T cells in pulmonary arterial hypertension. J Am Heart Assoc. 2019;8:e014201.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Klouda, T., Yuan, K. (2021). Inflammation in Pulmonary Arterial Hypertension. In: Wang, YX. (eds) Lung Inflammation in Health and Disease, Volume I. Advances in Experimental Medicine and Biology, vol 1303. Springer, Cham. https://doi.org/10.1007/978-3-030-63046-1_19
Download citation
DOI: https://doi.org/10.1007/978-3-030-63046-1_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-63045-4
Online ISBN: 978-3-030-63046-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)