
Abstract
Molecular neurocytogenetic (neurocytogenomic) studies have shown the human brain to demonstrate somatic genome variability (mosaic aneuploidy, subchromosomal rearrangements). Chromosomal mosaicism and instability rates vary during ontogeny in the human brain: dramatic increase of the rates in the early brain development follows by a significant decrease in the postnatal period. It is highly likely that rates of mosaicism and instability increase in the aging brain. Alternatively, chromosome-specific instability (aneuploidy and interphase chromosome breaks) and increased levels of chromosomal mosaicism confined to the brain are associated with a wide spectrum of neurodevelopmental and neurodegenerative diseases. Neurocytogenetic/neurocytogenomic analyses may provide further insights into genome organization at the chromosomal level in cells of such a high-functioning system as the human brain. Here, we review studies of interphase chromosomes in the human brain. In this instance, the role of molecular neurocytogenetics and neurocytogenomics in current genetics, genomics, and cell biology of the human brain is discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Human brain
- Interphase chromosomes
- Molecular cytogenetics
- Cytogenomics
- Chromosome instability
- Genome instability
- Disease
Introduction
The availability of interphase molecular cytogenetic techniques (e.g., fluorescence in situ hybridization (FISH) with chromosome- and site-specific DNA probes) has made possible to analyze chromosomes in almost all cellular populations in humans (Soloviev et al. 1995; Yurov et al. 1996, 2013; Vorsanova et al. 2010c; Hu et al. 2020). Neural chromosomes have been found to demonstrate high rates of variations manifesting as aneuploidy (gain/loss of chromosomes in a cell), which has been hypothesized to mediate neuronal diversity and brain diseases. Currently, chromosomal variation in the human brain has shown to represent a mechanism for a variety of neurodegenerative and psychiatric diseases (Yurov et al. 2001, 2018b; Iourov et al. 2006c; Kingsbury et al. 2006; Arendt et al. 2009; Jourdon et al. 2020). Actually, one can distinguish two main directions of studying interphase chromosomes in the human brain: (I) analysis of numerical and structural chromosomal changes (i.e., aneuploidy, structural abnormalities, copy number variations (CNV), chromosome instability, etc.) and (II) uncovering genome organization at the chromosomal level. The former has been the focus of numerous molecular neurocytogenetic and neurocytogenomic studies, whereas the latter is likely to become a purpose of further neurocytogenetic research.
In the present chapter, we review the latest advances in studying chromosomes in the human brain at microscopic, submicroscopic, and molecular levels. Theoretical and practical issues of brain-specific cytogenomic analyses are considered.
Interphase Chromosomes and Brain Ontogeny: Natural Chromosomal Variations
The complexity , plasticity, and intercellular variability of the human brain are likely to be generated during early ontogenetic stages and to be mediated by genomic content of neural progenitor cells (Muotri and Gage 2006; Rohrback et al. 2018b). The developing mammalian brain is characterized by high levels of chromosomal variations affecting ~30% of cells (Rehen et al. 2001; Yurov et al. 2005, 2007a). More precisely, the developing human brain is demonstrated to possess 30–35% of aneuploid cells (1.25–1.45% per chromosome) revealed by methods based on fluorescence in situ hybridization (FISH). These are multiprobe FISH, quantitative FISH (QFISH), and interphase chromosome-specific multicolor banding (ICS-MCB) (Yurov et al. 2005, 2007a; Iourov et al. 2010a, 2019a) (Fig. 4.1). Additionally, the developing human brain is the only embryonic tissue so far, which has demonstrated confined chromosomal mosaicism in contrast to confined placental mosaicism (Yurov et al. 2007a). At the subchromosomal level, similar progressive genomic changes are observed (i.e., high rates of brain-specific CNVs involving DNA sequences less than 1 Mb) in the developing human brain (McConnell et al. 2013; Rohrback et al. 2018a, b). At the sequence level per se, similar somatic genomic variations are unlikely to exist (Knouse et al. 2014; Muyas et al. 2020). Thus, (sub)chromosomal mosaicism and instability (aneuploidy) are hallmarks of the developing mammalian brain.

Molecular cytogenetic analysis of aneuploidy in the fetal human brain. (a–c). Interphase FISH with chromosome-enumeration DNA probes: (a) two nuclei characterized by additional chromosomes Y and X and a normal nucleus; (b) a nucleus with monosomy of chromosome 15 and a normal nucleus; and (c) a nucleus with monosomy of chromosome 18 and a normal nucleus. (d–g) Interphase chromosome-specific MCB: nuclei with monosomy, disomy, trisomy, and G-banding ideograms with MCB color-code labeling of a chromosome (from left to right),Fig. 4.1 (continued) (d) – chromosome 9, (e) – chromosome 16, and (f) – chromosome 18. (g) Interphase QFISH: (1) a nucleus with two signals for chromosomes 18 (relative intensities: 2058 and 1772 pixels), (2) a nucleus with one-paired signal mimics monosomy of chromosome 18 (relative intensity: 4012 pixels), (3) a nucleus with two signals for chromosomes 15 (relative intensities: 1562 and 1622 pixels), and (4) a nucleus with one signal showing monosomy of chromosome 15 (relative intensity: 1678 pixels). (From Yurov et al. 2007a, an open-access article distributed under the terms of the Creative Commons Attribution License)
Taking into account a correlation between number of aneuploid cells (30–35%) and number of cells cleared by the programmed cell death (30–50%) in the developing brain, aneuploidization (progressive accumulation of aneuploid cells) is suggested as a mechanism for cell number regulation during early brain ontogeny (Iourov et al. 2006c; Muotri and Gage 2006; Yurov et al. 2010a; Fricker et al. 2018). Considering observations evaluating functional effects of aneuploidy either at the single cell level or at the tissular level (Iourov et al. 2008a; Dierssen et al. 2009; Hultén et al. 2013), mitotic catastrophe (a cascade of abnormal mitotic cell divisions producing aneuploidization) has been proposed as a mechanisms for cell number decreases in the developing brain because of aneuploid cell death (Iourov et al. 2006d, 2019d; Yurov et al. 2007a; Fricker et al. 2018). This hypothesis has been supported by studying chromosomal mosaicism in embryonic and extraembryonic tissues, which has shown that this mosaicism type is able to cause prenatal death or spontaneous abortions (Vorsanova et al. 2005, 2010a). Since aneuploidy is likely to have an adverse effect on cellular homeostasis, an alteration to the clearance of aneuploid cells during prenatal period may result in high rates of aneuploidy in the postnatal human brain, mediating neuropsychiatric and neurodegenerative diseases or childhood brain cancer (Iourov et al. 2006c, 2009c, 2019d; Kingsbury et al. 2006; McConnell et al. 2017; Yurov et al. 2018a, b, 2019b). On the other hand , aneuploidy may represent a mechanism for neuronal diversity in the unaffected human brain inasmuch as aneuploid neural cells are functionally active and integrated into brain circuitry (Kingsbury et al. 2005). To gain further insights into the role of chromosomal variation in the human brain in later ontogeny, one has to study interphase chromosome in the childhood and adult human brain.
During the prenatal period , rates of chromosomal and subchromosomal changes or instability decrease to 10% or lower (Yurov et al. 2005, 2018b, 2019b; Iourov et al. 2006a, 2009b; McConnell et al. 2013; Rohrback et al. 2018a). Interestingly, the way of variation in cell numbers mediated by aneuploidization in the developmental brain and programmed cell death is likely to be specific for humans in contrast to other vertebrates studied in this context (Rehen et al. 2001; Yurov et al. 2005, 2007a; Iourov et al. 2006c; Zupanc 2009; Rohrback et al. 2018a). Probably, the functional uniqueness of the human brain is achieved by such a kind of selective pressure at cellular/chromosomal level (Iourov et al. 2012, 2019d). Additionally, intercellular differences between DNA content (~250 Mb) in the adult human brain have been reported (Westra et al. 2008, 2010). The variability of the chromosomal numbers (aneuploidy) allowed to hypothesize that aneuploidy rates may be higher in late ontogeny. In other words, aneuploidization may be a mechanism for brain aging (Iourov et al. 2008a; Yurov et al. 2009b, 2010a, b; Faggioli et al. 2011). However, there is no consensus on the matter. Thus, a number of studies report increased rates of aneuploidy in the aged brain (Fischer et al. 2012; Andriani et al. 2017), whereas other reports do not (Van den Bos et al. 2016; Shepherd et al. 2018). The lack of consensus is more likely to be a result of technological differences between these reports. Single-cell sequencing studies report low rates of genomic changes in moderate cell numbers (~100 cell analyzed with the highest resolution possible) (Knouse et al. 2014; Van den Bos et al. 2016; Rohrback et al. 2018a), whereas molecular cytogenetic studies report high rates of chromosomal variations in large cell populations (reviewed by Iourov et al. 2012; Yurov et al. 2018b, 2019b). One can propose that combination of sequence-based single-cell techniques and molecular cytogenetic (cytogenomic ) methods may solve the problem.
The devastating effect of chromosomal abnormalities (aneuploidy and structural aberrations) suggests that these genomic variations are able to produce functional and structural alterations to the human brain. The confinement of aneuploidy and other types of chromosomal variations (instability ) to the central nervous system has been systematically associated with brain diseases (Yurov et al. 2001, 2018b; Iourov et al. 2006c, d, 2013; Tiganov et al. 2012; McConnell et al. 2017; Leija-Salazar et al. 2018; Iourov 2019; Potter et al. 2019; Heng 2020). It is highly likely that each form of brain pathology is linked to a specific type of brain-specific genomic alterations .
Interphase Chromosomes in the Diseased Brain
Chromosomal variations cause functional brain alterations in a wide spectrum of psychiatric and neurological diseases (DeLisi et al. 1994; Iourov et al. 2008b; Vorsanova et al. 2010d; Graham et al. 2019; Potter et al. 2019). Somatic genome variations at chromosomal and subchromosomal levels are repeatedly associated with neurodevelopmental, neurodegenerative, and/or psychiatric disorders (Iourov et al. 2008b, 2010b, 2019d; Smith et al. 2010; Paquola et al. 2017; Vorsanova et al. 2017; Graham et al. 2019). Chromosomal abnormalities and instability confined to the brain have been reported in schizophrenia and neurodegenerative diseases. Several neuropsychiatric diseases (e.g., autism and epilepsy) are also hypothesized to be associated with neurocytogenetic and neurocytogenomic variations.
The first report on two cases of mosaic aneuploidy (trisomy X and 18) in the schizophrenia brain (Yurov et al. 2001) has formed the basis for further neurocytogenomic studies of the diseased brain. As a result, several schizophrenia cases have been additionally associated with chromosome-1-specific instability and gonosomal instability, which are almost exclusively manifested as aneuploidy (Yurov et al. 2008, 2016, 2018a). Brain-specific structural chromosomal abnormalities (microdeletions) and CNV have been also found in a number of schizophrenia cases (Kim et al. 2014; Sakai et al. 2015). These data allow suggesting that a number of schizophrenia cases are the result of chromosomal abnormalities and/or instability in the diseased brain (Yurov et al. 2018a, b). Further molecular neurocytogenetic (neurocytogenomic ) studies would certainly shed light on the involvement of “neurochromosomal variation” in schizophrenia and would likely to define the exact proportion of schizophrenia cases associated with neural aneuploidy, structural chromosome aberrations and chromosomal/genomic instability.
Somatic mosaic aneuploidy is one of the commonest types of genomic variations in autistic individuals inasmuch as ~10% of autistic males are likely to exhibit low-level 47,XXY/46,XY mosaicism (Yurov et al. 2007b). More importantly, gonosomal mosaicism is common in autistic individuals and their relatives. Several familial cases of behavioral abnormalities co-segregating with X chromosome aneuploidy and chromosomal instability have been reported (Vorsanova et al. 2007, 2010b). These data have been used for theoretical explanation of the male-to-female ratio in autism (Iourov et al. 2008c). Additionally, the neurocytogenetic hypothesis of autism (i.e., a proportion of autism cases may be associated with chromosome abnormalities and instability confined to the brain) has been recently described using systems biology methodology (Vorsanova et al. 2017). Our preliminary studies have demonstrated a possible involvement of brain-specific chromosome instability (chromothripsis) and aneuploidy in pathogenic cascades associated with autistic behavior (Iourov et al. 2017a). In the behavioral context, one has to mention studies suggesting that genome/chromosome instability probably shapes behavior in individuals suffering from neurodevelopmental diseases (Vorsanova et al. 2018) and gulf war illness (Liu et al. 2018). However, direct evaluation of interphase chromosomes in the autistic brain is still in process.
Somatic aneuploidy and other types of chromosome instability have been found to mediate neurodegeneration (Iourov et al. 2009a; Leija-Salazar et al. 2018; Shepherd et al. 2018; Yurov et al. 2019a). The Alzheimer’s disease brain has been systematically shown to exhibit genome/chromosome instability and related phenomena (i.e., abnormal cell cycle entry, endomitosis, replication stress, abnormal DNA damage response, and micronuclei in mitotic tissues) (Herrup and Yang 2007; Mosch et al. 2007; Iourov et al. 2011; Yurov et al. 2011, 2019a; Arendt 2012; Bajic et al. 2015; Coppedè and Migliore 2015; Hou et al. 2017; Lin et al. 2020; Nudelman et al. 2019). Taking into account neurological parallels between Alzheimer’s disease and Down syndrome or trisomy of chromosome 21 (Snyder et al. 2020), Professor Huntington Potter’s group has proposed that brain-specific copy number changes of either whole chromosome 21 or chromosome 21 region containing APP gene are able to mediate neurodegeneration in Alzheimer’s disease (Granic et al. 2010; Potter et al. 2019). Actually, chromosome 21-psecific instability in the diseased brain is one of the most probable mechanisms for Alzheimer’s disease (Iourov et al. 2009b). Additionally, genes mutated in rare familial cases of the diseases are involved in processes granting proper chromosome segregation during the cell division (Boeras et al. 2008; Granic et al. 2010). Similarly, altered chromosome segregation induced by LDL/cholesterol seems to contribute to Alzheimer’s disease as well as to Niemann-Pick C1 and atherosclerosis (Granic and Potter 2013). Moreover, X chromosome aneuploidy (X chromosome loss) — a cytogenetic biomarker of human aging — has been reported to have higher rates in the Alzheimer’s disease brain as to the unaffected brain (Yurov et al. 2014) (Fig. 4.2). Selective cell death of aneuploid neurons (i.e., aneuploidy causes neuron death as it is the case in the developmental brain) has been reported to hallmark the neurodegeneration in the Alzheimer’s disease brain (Arendt et al. 2010). Abnormal DNA damage response resulting in chromosome/genome instability is likely to result in neurodegeneration in the Alzheimer’s disease brain (neural cells with aneuploidy or structurally altered chromosomes produced by DNA damage are susceptible to programmed cell death) (Fielder et al. 2017; Lin et al. 2020). Finally, Alzheimer’s disease has been associated with subchromosomal instability (e.g., nonspecific CNVs) involving the APP gene (Kaeser and Chun 2020). In total, chromosome instability, including aneuploidy, represents an element of the Alzheimer’s disease pathogenic cascade (Iourov et al. 2011; Yurov et al. 2019a). To link observations on aneuploidy/chromosome instability, abortive cell cycle, DNA damage, replication stress , and APP, a hypothesis depicted by Fig. 4.3 has been proposed.

Molecular neurocytogenetic analyses of the AD brain. (a) Multiprobe (two-probe) and quantitative FISH using DNA probes for chromosomes 1 (two red signals/D1Z1) and X (one green signal/DXZ1; relative intensity is 2120 pixels) demonstrating true X chromosome monosomy; (b) multiprobe (two-probe) and quantitative FISH using DNA probes for chromosomes 1 (two red signals/D1Z1) and X (one green signal/DXZ1; relative intensity is 4800 pixels) demonstrating overlapping of two X chromosome signals, but not a chromosome loss; (c) ICS-MCB with a probe set for chromosome X showing one nucleus bearing two chromosomes X and another nucleus bearing single chromosome X. (From Yurov et al. 2014, an open-access article distributed under the terms of the Creative Commons Attribution License)

(a) Simplified schematic presentation of the cell cycle theory of AD. Quiescent neuronal cells (G0 phase) demonstrate the cell cycle reactivation by either endogenous or environmental mitogenic stimuli followed by reentry into the G1 phase. The G0/G1 phase transition is critical for a postmitotic neuron and potentially causes neuronal cell death. During G1 phase, diploid neurons (chromosomal complement: 2 N; number of chromosomes: 46; DNA content: 2C) demonstrate G1-specific cell cycle markers (cyclin D and CDK4/6 complex, cyclin E, and CDK2 complex) which are involved in the regulation of G1 phase progression. Cells successfully passing G1 enter the S phase (phase of DNA replication). During the S phase, CDK2/cyclin E should be silenced to repress additional round of replication of genomic DNA. Protein markers of the S phase are A-type cyclins (cyclin A/CDK2 complex). This complex is essential for proper completion of S phase and transition from S to G2 phase. DNA content of cells during S phase changes from 2C toFig. 4.3 (continued) 4C (chromosome number is still 2 N, but DNA content after replication is tetraploid). During G2 phase, cyclin A is degraded, and cyclin B/CDC2 complex (protein biomarker of late S/early G2 phases) is formed. Cyclin B/CDC2 complex is essential for triggering mitosis. Neuronal cells in G2 phase demonstrate tetraploid (4 N) DNA content or, more precisely, possess a nucleus with 46 replicated chromosomes. Chromosomal complement (genomic content) of cells in G2 consists of one set of 46 duplicated chromosomes (DNA content: 4 N or 4C; diploid nucleus with replicated chromosomes; for more details see, [20]), each having two chromatids—“mitotic” tetraploidy. It is to note that true constitutional polyploidy is a term used to describe cell containing more than two homologous sets of chromosomes (4 N or 92 chromosomes, DNA content: 4C). We suggest that postmitotic neurons are able to replicate DNA but are not able to make a G2/M transition and divide into two daughter cells. (b) The DNA replication stress hypothesis of AD. Interplay between essential elements of the AD-type dementia pathogenetic cascade is proposed. The genetic influences (PSEN or APP mutations, trisomy 21, APOE4 genotype), metabolic changes, and environmental factors affecting neuronal homeostasis in the aging brain lead to activation of neuronal proliferation. Mitogens, which do exist in the human brain (neuronal cells), induce additional stimuli of extensive adult neurogenesis in the hippocampus. In the AD brain, such events would lead to increased hippocampal neurogenesis. A side effect could be that these mitogenic stimuli activate cell cycle reentry in postmitotic neurons. The latter is a pathological activation of neuronal cell cycle, including reentry into G1 and S phases and initiation of DNA replication. Neurons showing protein markers of G2/M phase, probably, contain chromosome set of 23 duplicated chromosome pairs with unseparated chromatids (DNA content, 4C; chromosome complement, 2 N) and become tetraploid in a sense of DNA content (4C). According to the commonly accepted theory of neuronal cell cycle reentry and death, some neuronal populations complete the DNA synthesis but are arrested during the G2/M transition. Therefore, neuronal death occurs in G2 phase. Alternatively, one can propose that a large proportion of activated postmitotic neurons in the AD brain are unable to pass properly the S phase. This would lead to accumulation of genomic and chromosomal instabilities throughout ontogeny (DNA breaks, aneuploidy). In addition, replication-induced DNA damages would lead to fork stalling, incomplete or inefficient DNA replication, together designated as replication stress. Replication stress may be considered the leading cause of neuronal cell death due to processing into S phase or accumulation of genetic instabilities, which together constitute an important element of the AD pathogenetic cascade. According to the present hypothesis, the possibility to link the two main pathways of AD arises from the introduction of accumulation of genomic instabilities associated with DNA replication stress, which is able to produce as neuronal cell death (replicative cell death) as chromosomal aneuploidy due to natural selection in neural cell populations probably causing extra APP in the diseased brain. (From Yurov et al. 2011, an open-access article distributed under the terms of the Creative Commons Attribution License)
Non-Alzheimer’s disease neurodegeneration has been associated with chromosomal variations in the diseased human brain as well. Thus, Lewy body diseases exhibit high rates of neural aneuploidy in the neurodegenerating brain (Yang et al. 2015). MAPT mutations that lead to mitotic defects, neuronal aneuploidy and extensive apoptosis are likely to cause frontotemporal lobar degeneration (Caneus et al. 2018). Subchromosomal instability involving α-synuclein (SNCA) has been associated with Parkinson’s disease and multiple system atrophy (Mokretar et al. 2018). Probably, the most intriguing example of a neurodegenerative disease associated with brain-specific chromosome instability is ataxia-telangiectasia, an autosomal recessive chromosome instability syndrome caused by ATM gene mutations and characterized by cerebellar degeneration (Iourov et al. 2007b; Potter et al. 2019). In fact, neurodegeneration caused by chromosome instability has been firstly demonstrated during the molecular cytogenetic analysis of the ataxia-telangiectasia brain (previously, chromosome instability has been suggested to be almost exclusive mechanism for cancer) (Iourov et al. 2009a, b). The ataxia-telangiectasia brain demonstrates chromosome-14 instability (interphase chromosomal breaks and additional rearranged chromosomes) in ~40% of cells in the degenerating cerebellum (Iourov et al. 2009a). These data have been used as a basis for potential therapeutic strategies for neurodegeneration mediated by chromosome (genome) instability (Yurov et al. 2009a; Iourov et al. 2019b). There are striking differences between cancerous chromosome instability and neurodegenerative chromosome instability. The differences are as follows: Cancer : Cancer-susceptibility mutations interact with environment producing genome and chromosome instabilities. These processes lead to clonal evolution and, thereby, malignancy. Neurodegeneration : Chromosome instability and abnormalities are present in a significant proportion of cells, and genetic-environment interactions trigger progressive neuronal cell loss (neurodegeneration) by natural selection and/or programmed cell death (Iourov et al. 2013; Yurov et al. 2019a). Schematically, this model is shown by Fig. 4.4.

Theoretical model for CIN mediating (a) cancer and (b) neurodegeneration. (a) Genetic defects and genetic-environmental interactions may cause chromosomal/genomic changes, which produce CIN; alternatively, cell populations may adapt to aneuploidy and CIN evolving to a cell population with a fitness advantage. Cells affected by CIN and tolerating deteriorating effects of CIN on cellular homeostasis are able to evolve clonally to produce malignancy. (b) CIN/somatic mosaicism affecting a significant proportion of cells interacting with environmental triggers may result into progressive neuronal cell loss (neurodegeneration) under natural selection pressure and through the programmed cell death (N, normal neurons; CIN, neuronal cell affected by CIN). The model is based on the observations of CIN in the neurodegenerating brain and cancers. (From Yurov et al. 2019a, an open-access article distributed under the terms of the Creative Commons Attribution License)
In the previous version of the book (Yurov et al. 2013), we proposed a hypothesis describing the role of neural aneuploidy and chromosome instability. During the last 7 years, more evidences for supporting the hypothesis have been provided (Iourov et al. 2014, 2019a, b, d; Yurov et al. 2014, 2018a, b, 2019a, b; Bajic et al. 2015; Andriani et al. 2017; McConnell et al. 2017; Vorsanova et al. 2017, 2020; Leija-Salazar et al. 2018; Rohrback et al. 2018b; Shepherd et al. 2018; Graham et al. 2019; Iourov 2019; Potter et al. 2019; Jourdon et al. 2020). Accordingly, we would like to reproduce schematically the hypothesis (Fig. 4.5).

Schematic representation of the hypothesis on the role of aneuploidy in normal CNS development and aging as well as in pathogenesis of brain diseases. During the normal prenatal brain development, developmental chromosome instability is cleared leading to three-time decrease of aneuploidy rates. Brain aging is likely to be associated with slight increase of aneuploidy. Total failure of clearance of developmental chromosome instability would lead to the persistence as observed in chromosome instability syndromes with brain dysfunction (ataxia-telangiectasia) and brain cancers. Clearance may not affect low-level chromosomal mosaicism confined to the developing brain, which is extremely frequent among human fetuses. In such cases, the postnatal brain exhibits low-level chromosome-specific mosaic aneuploidy. The latter is shown to be associated with diseases of neuronal dysfunction and degeneration (mental retardation, autism, schizophrenia, Alzheimer’s disease). (From Yurov et al. 2013 (previous edition of the book — Figure 4.9), reproduced with permission of Springer Nature in the format reuse in a book/textbook via Copyright Clearance Center)
Interphase Chromosomes and Genome Organization in the Human Brain
Nuclear genome organization in interphase is crucial for regulating chromatin remodeling, genome activity (transcription), genome safeguarding (DNA damage response, proper chromosome segregation, mitotic checkpoint, etc.), DNA repair and replication, and programmed cell death (for details, see Chaps. 1, 2, and 9). Previously, we have systematically indicated the importance of neurocytogenetic analysis of chromosome organization in interphase nuclei of the human brain (Iourov et al. 2006c, 2010a, 2012; Yurov et al. 2013, 2018b). Unfortunately, no significant progress has been, as yet, made in this field. Nonetheless, we have attempted to list known properties of interphase chromosome behavior in the human brain along with molecular cytogenetic FISH-based techniques, which are used for the analysis.
To perform a successful study of chromosomal arrangement in interphase, one has to be aware about the spatial preservation of interphase nuclei during tissue/cell suspension preparation for molecular cytogenetic analysis. Although brain cell preparation for molecular neurocytogenetic analysis requires specific procedures, it does provide an opportunity to preserve interphase nuclei of the human brain (Iourov et al. 2006b; Yurov et al. 2017b). Pairing of homologous chromosomes (chromosomal associations/locus associations) is common in the postnatal human brain (Iourov et al. 2005, 2017b; Yurov et al. 2017b). To make accurate scoring of the associations , QFISH may be applied (Iourov et al. 2005; Iourov 2017). Finally, functional complexity and structural variability of neural cell populations lead to requirement of studying integral interphase chromosomes at molecular resolutions in a “band-by-band” manner. This technical opportunity is offered by interphase chromosome-specific multicolor banding (ICS-MCB) (Iourov et al. 2006a, 2007a). An example of ICS-MCB is shown by Fig. 4.6. Nuclear genome organization at the chromosomal level may be a mechanism for brain diseases (Iourov 2012; Yurov et al. 2013). However, there are no, as yet, studies attempting to correlate specific nuclear chromosome organization in neural cells and central nervous system dysfunction .

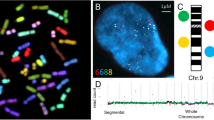
FISH using MCB probes on interphase nuclei of the human brain. (a): FISH with MCB probe for chromosome 1. R110 signals correspond to 1p32.3Yp36.3 and 1q32Yq43. SO (Spectrum Orange) signals Y 1p13Yq21 including constitutive heterochromatin (1qh). TR (Texas Red) signals Y 1p31.1Yp33 and 1q21.3Yq31. Cy5 signals Y 1p13.1Yp22.3 and 1q32Yq43. DEAC signals Y 1q21.3Yq31. Note the upper chromosome 1 is folded around 1qh and bent in the proximal part of the q-arm. (b): FISH with MCB probe for chromosome 9. R110 signals correspond to 9p13Yq13 including constitutive heterochromatin (9qh). SO (Spectrum Orange) signals Y 9p21Yp24 and 9q32Yq34. TR (Texas Red) signals Y 9q22.2Yq34.1. Cy5 signals Y 9p13Yp23. DEAC signals Y 9q13Yq22.2. (c): FISH with MCB probe for chromosome 16. R110 signals correspond to 16p11.1Yp13.1 SO (Spectrum Orange) signals Y 16p13.3Yp21. TR (Texas Red) signals Y 16q11.1Yq21 including constitutive heterochromatin (16qh). Cy5 signals Y 16q21Yq24. Note the single Texas Red signal instead of two; this implies that 16qh regions of two homologous chromosomes 16 are overlapped. Therefore, somatic pairing of two homologous chromosomes 16 by 16qh region should be suspected. (d): FISH with MCB probe for chromosome 18. R110 signalsFig. 4.6 (continued) correspond to 18p11.2Yq12.2. SO (Spectrum Orange) signals Y 18p11.2Yp11.3. TR (Texas Red) signals Y 18q22Yq23. Cy5 signals Y 18q11.2Yq21.3. (e): FISH with MCB probe for chromosome X. R110 signals correspond to Xp21.3Yp22.3 and Xq25Yq28. SO (Spectrum Orange) signals Y Xp11.22Yp22.1 and Xq25Yq28. TR (Texas Red) signals Y Xq12Yq21.1. Cy5 signals Y Xq21.1Yq26. DEAC signals Y Xp11.3Yq13. Note the upper chromosome X appears as a white condensed spot (merged image). Since facultative heterochromatin, a feature of X chromosome inactivation, should appear as a highly condensed structure, the upper X chromosome was assumed to be inactivated one (Xi) in contrast to the active X chromosome (Xa) appearing as a slightly diffused structure. (f): Example of a trisomic nucleus (trisomy of chromosome 9); left side, Y black-and-white picture of DAPI-counterstained nucleus, and right side, Y merged MCB true color picture showing the presence of three chromosomes 9 in this nucleus. (g): Example of a monosomic nucleus (monosomy of chromosome 18); left side, Y black-and-white picture of DAPI-counterstained nucleus, and right side, Y merged MCB true color picture showing the presence of one chromosome 18 in this nucleus. (From Iourov et al. 2006a, reproduced with permission of Springer Nature in the format reuse in a book/textbook via Copyright Clearance Center)
Conclusion
The present chapter is dedicated to behavior and variation of interphase chromosomes in the human brain. Aneuploidy and other types of chromosome instability are mechanisms for neuronal diversity and brain diseases. As repeatedly noted before, brain-oriented interphase chromosome (neurocytogenetic and neurocytogenomic) analysis brings new insights to neuroscience, human genomics, and molecular medicine.
Molecular (neuro)cytogenetic and (neuro)cytogenomic studies seem to benefit from bioinformatics approaches based on network- or pathway-based analysis, i.e., systems biology methodology (Yurov et al. 2017a, b). Actually, pathway-based classification of human diseases is considered the most promising way to unravel complex relationship between molecular/cellular processes and phenotypes (Iourov et al. 2019b). We suggest that systems biology methodology considered in the molecular cytogenomic context is able to provide new information about interphase chromosomes in the human brain (Yurov et al. 2017a, b; Iourov et al. 2019c). These approaches toward the definition of molecular basis of human brain diseases have been already found successful: (i) uncovering molecular mechanisms for somatic mosaicism (Iourov et al. 2015), (ii) genomic instability associated with neurological and psychiatric diseases (McConnell et al. 2017; Vorsanova et al. 2017), and (iii) molecular/cellular alterations causing brain dysfunction (Iourov et al. 2009b, 2019b, c). To this end, one has to conclude that interphase chromosome studies certainly contribute to our knowledge about the human central nervous system.
References
Andriani GA, Vijg J, Montagna C (2017) Mechanisms and consequences of aneuploidy and chromosome instability in the aging brain. Mech Ageing Dev 161:19–36
Arendt T (2012) Cell cycle activation and aneuploid neurons in Alzheimer’s disease. Mol Neurobiol 46(1):125–135
Arendt T, Mosch B, Morawski M (2009) Neuronal aneuploidy in health and disease: a cytomic approach to understand the molecular individuality of neurons. Int J Mol Sci 10(4):1609–1627
Arendt T, Brückner MK, Mosch B et al (2010) Selective cell death of hyperploid neurons in Alzheimer’s disease. Am J Pathol 177:15–20
Bajic V, Spremo-Potparevic B, Zivkovic L et al (2015) Cohesion and the aneuploid phenotype in Alzheimer’s disease: a tale of genome instability. Neurosci Biobehav Rev 55:365–374
Boeras DI, Granic A, Padmanabhan J et al (2008) Alzheimer’s presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol Aging 29:319–328
Caneus J, Granic A, Rademakers R et al (2018) Mitotic defects lead to neuronal aneuploidy and apoptosis in frontotemporal lobar degeneration caused by MAPT mutations. Mol Biol Cell 29:575–586
Coppedè F, Migliore L (2015) DNA damage in neurodegenerative diseases. Mutat Res 776:84–97
DeLisi LE, Friedrich U, Wahlstrom J et al (1994) Schizophrenia and sex chromosome anomalies. Schizophr Bull 20(3):495–505
Dierssen M, Herault Y, Estivill X (2009) Aneuploidy: from a physiological mechanism of variance to down syndrome. Physiol Rev 89:887–920
Faggioli F, Vijg J, Montagna C (2011) Chromosomal aneuploidy in the aging brain. Mech Ageing Dev 132(8–9):429–436
Fielder E, von Zglinicki T, Jurk D (2017) The DNA damage response in neurons: die by apoptosis or survive in a senescence-like state? J Alzheimers Dis 60:S107–S131
Fischer HG, Morawski M, Brückner MK et al (2012) Changes in neuronal DNA content variation in the human brain during aging. Aging Cell 11(4):628–633
Fricker M, Tolkovsky AM, Borutaite V et al (2018) Neuronal cell death. Physiol Rev 98:813–880
Graham EJ, Vermeulen M, Vardarajan B et al (2019) Somatic mosaicism of sex chromosomes in the blood and brain. Brain Res 1721:146345
Granic A, Potter H (2013) Mitotic spindle defects and chromosome mis-segregation induced by LDL/cholesterol-implications for Niemann-Pick C1, Alzheimer’s disease, and atherosclerosis. PLoS One 8:e60718
Granic A, Padmanabhan J, Norden M et al (2010) Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for au and APP. Mol Biol Cell 21(4):511–520
Heng HH (2020) New data collection priority: focusing on genome-based bioinformation. Res Res Biomed 6(1):5–8
Herrup K, Yang Y (2007) Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci 8:368–378
Hou Y, Song H, Croteau DL et al (2017) Genome instability in Alzheimer disease. Mech Ageing Dev 161:83–94
Hu Q, Maurais EG, Ly P (2020) Cellular and genomic approaches for exploring structural chromosomal rearrangements. Chromosom Res 28(1):19–30
Hultén MA, Jonasson J, Iwarsson E et al (2013) Trisomy 21 mosaicism: we may all have a touch of down syndrome. Cytogenet Genome Res 139(3):189–192
Iourov IY (2012) To see an interphase chromosome or: how a disease can be associated with specific nuclear genome organization. BioDiscovery 4:e8932
Iourov IY (2017) Quantitative fluorescence in situ hybridization (QFISH). Methods Mol Biol 1541:143–149
Iourov IY (2019) Cytopostgenomics: what is it and how does it work? Curr Genomics 20(2):77–78
Iourov IY, Soloviev IV, Vorsanova SG et al (2005) An approach for quantitative assessment of fluorescence in situ hybridization (FISH) signals for applied human molecular cytogenetics. J HistochemCytochem 53:401–408
Iourov IY, Liehr T, Vorsanova SG et al (2006a) Visualization of interphase chromosomes in postmitotic cells of the human brain by multicolour banding (MCB). Chromosom Res 14(3):223–229
Iourov IY, Vorsanova SG, Pellestor F et al (2006b) Brain tissue preparations for chromosomal PRINS labeling. Methods Mol Biol 334:123–132
Iourov IY, Vorsanova SG, Yurov YB (2006c) Chromosomal variation in mammalian neuronal cells: known facts and attractive hypotheses. Int Rev Cytol 249:143–191
Iourov IY, Vorsanova SG, Yurov YB (2006d) Intercellular genomic (chromosomal) variations resulting in somatic mosaicism: mechanisms and consequences. Curr Genomics 7:435–446
Iourov IY, Liehr T, Vorsanova SG et al (2007a) Interphase chromosome-specific multicolor banding (ICS-MCB): a new tool for analysis of interphase chromosomes in their integrity. Biomol Eng 24(4):415–417
Iourov IY, Vorsanova SG, Yurov YB (2007b) Ataxia telangiectasia paradox can be explained by chromosome instability at the subtissue level. Med Hypotheses 68:716
Iourov IY, Vorsanova SG, Yurov YB (2008a) Chromosomal mosaicism goes global. Mol Cytogenet 1:26
Iourov IY, Vorsanova SG, Yurov YB (2008b) Molecular cytogenetics and cytogenomics of brain diseases. Curr Genomics 9(7):452–465
Iourov IY, Yurov YB, Vorsanova SG (2008c) Mosaic X chromosome aneuploidy can help to explain the male-to-female ratio in autism. Med Hypotheses 70:456
Iourov IY, Vorsanova SG, Liehr T et al (2009a) Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. Hum Mol Genet 18(14):2656–2669
Iourov IY, Vorsanova SG, Liehr T et al (2009b) Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: differential expression and pathological meaning. Neurobiol Dis 34(2):212–220
Iourov IY, Vorsanova SG, Yurov YB (2009c) Developmental neural chromosome instability as a possible cause of childhood brain cancers. Med Hypotheses 72:615–616
Iourov IY, Vorsanova SG, Solov’ev IV et al (2010a) Methods of molecular cytogenetics for studying interphase chromosome in human brain cells. Russ J Genet 46(9):1039–1041
Iourov IY, Vorsanova SG, Yurov YB (2010b) Somatic genome variations in health and disease. Curr Genomics 11:387–396
Iourov IY, Vorsanova SG, Yurov YB (2011) Genomic landscape of the Alzheimer’s disease brain: chromosome instability – aneuploidy, but not tetraploidy – mediates neurodegeneration. Neurodegener Dis 8:35–37
Iourov IY, Vorsanova SG, Yurov YB (2012) Single cell genomics of the brain: focus on neuronal diversity and neuropsychiatric diseases. Curr Genomics 13(6):477–488
Iourov IY, Vorsanova SG, Yurov YB (2013) Somatic cell genomics of brain disorders: a new opportunity to clarify genetic-environmental interactions. Cytogenet Genome Res 139(3):181–188
Iourov IY, Vorsanova SG, Liehr T et al (2014) Mosaike im Gehirn des Menschen. Diagnostische Relevanz in der Zukunft? Med Genet 26(3):342–345
Iourov IY, Vorsanova SG, Zelenova MA et al (2015) Genomic copy number variation affecting genes involved in the cell cycle pathway: implications for somatic mosaicism. Int J Genomics 2015:757680
Iourov IY, Vorsanova SG, Liehr T et al (2017a) Chromothripsis as a mechanism driving genomic instability mediating brain diseases. Mol Cytogenet 10(1):20(O2)
Iourov IY, Vorsanova SG, Yurov YB (2017b) Interphase FISH for detection of chromosomal mosaicism. In: Liehr T (ed) Fluorescence in situ hybridization (FISH) — application guide (springer protocols handbooks), 2nd edn. Springer-Verlag, Berlin, Heidelberg, pp 361–372
Iourov IY, Liehr T, Vorsanova SG et al (2019a) The applicability of interphase chromosome-specific multicolor banding (ICS-MCB) for studying neurodevelopmental and neurodegenerative disorders. Res Results Biomedicine 5(3):4–9
Iourov IY, Vorsanova SG, Yurov YB (2019b) Pathway-based classification of genetic diseases. Mol Cytogenet 12:4
Iourov IY, Vorsanova SG, Yurov YB (2019c) The variome concept: focus on CNVariome. Mol Cytogenet 12:52
Iourov IY, Vorsanova SG, Yurov YB et al (2019d) Ontogenetic and pathogenetic views on somatic chromosomal mosaicism. Genes (Basel) 10(5):E379
Jourdon A, Fasching L, Scuderi S et al (2020) The role of somatic mosaicism in brain disease. Curr Opin Genet Dev 65:84–90
Kaeser GE, Chun J (2020) Mosaic somatic gene recombination as a potentially unifying hypothesis for Alzheimer’s disease. Front Genet 11:390
Kim J, Shin JY, Kim JI et al (2014) Somatic deletions implicated in functional diversity of brain cells of individuals with schizophrenia and unaffected controls. Sci Rep 4:3807
Kingsbury MA, Friedman B, McConnell MJ et al (2005) Aneuploid neurons are functionally active and integrated into brain circuitry. Proc Natl Acad Sci U S A 102:6143–6147
Kingsbury MA, Yung YC, Peterson SE et al (2006) Aneuploidy in the normal and diseased brain. Cell Mol Life Sci 63:2626–2641
Knouse KA, Wu J, Whittaker CA et al (2014) Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci U S A 111:13409–13414
Leija-Salazar M, Piette C, Proukakis C (2018) Somatic mutations in neurodegeneration. Neuropathol Appl Neurobiol 4:267–285
Lin X, Kapoor A, Gu Y et al (2020) Contributions of DNA damage to Alzheimer’s disease. Int J Mol Sci 21:1666
Liu G, Ye CJ, Chowdhury SK et al (2018) Detecting chromosome condensation defects in gulf war illness patients. Curr Genomics 19:200–206
McConnell MJ, Lindberg MR, Brennand KJ et al (2013) Mosaic copy number variation in human neurons. Science 342:632–637
McConnell MJ, Moran JV, Abyzov A et al (2017) Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science 356:eaal1641
Mokretar K, Pease D, Taanman JW et al (2018) Somatic copy number gains of α-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain 141:2419–2431
Mosch B, Morawski M, Mittag A et al (2007) Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J Neurosci 27:6859–6867
Muotri AR, Gage FH (2006) Generation of neuronal variability and complexity. Nature 441:1087–1093
Muyas F, Zapata L, Guigó R et al (2020) The rate and spectrum of mosaic mutations during embryogenesis revealed by RNA sequencing of 49 tissues. Genome Med 12:49
Nudelman KNH, McDonald BC, Lahiri DK et al (2019) Biological hallmarks of cancer in Alzheimer’s disease. Mol Neurobiol 56:7173–7187
Paquola ACM, Erwin JA, Gage FH (2017) Insights into the role of somatic mosaicism in the brain. Curr Opin Syst Biol 1:90–94
Potter H, Chial HJ, Caneus J et al (2019) Chromosome instability and mosaic aneuploidy in neurodegenerative and neurodevelopmental disorders. Front Genet 10:1092
Rehen SK, McConnell MJ, Kaushal D et al (2001) Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci U S A 98:13361–13366
Rohrback S, April C, Kaper F et al (2018a) Submegabase copy number variations arise during cerebral cortical neurogenesis as revealed by single-cell whole-genome sequencing. Proc Natl Acad Sci U S A 115:10804–10809
Rohrback S, Siddoway B, Liu CS et al (2018b) Genomic mosaicism in the developing and adult brain. Dev Neurobiol 78:1026–1048
Sakai M, Watanabe Y, Someya T et al (2015) Assessment of copy number variations in the brain genome of schizophrenia patients. Mol Cytogenet 8:46
Shepherd CE, Yang Y, Halliday GM (2018) Region- and cell-specific aneuploidy in brain aging and neurodegeneration. Neuroscience 374:326–334
Smith CL, Bolton A, Nguyen G (2010) Genomic and epigenomic instability, fragile sites, schizophrenia and autism. Curr Genomics 11(6):447–469
Snyder HM, Bain LJ, Brickman AM et al (2020) Further understanding the connection between Alzheimer’s disease and down syndrome. Alzheimers Dement 16(7):1065–1077
Soloviev IV, Yurov YB, Vorsanova SG et al (1995) Prenatal diagnosis of trisomy 21 using interphase fluorescence in situ hybridization of post-replicated cells with site-specific cosmid and cosmid contig probes. Prenat Diagn 15:237–248
Tiganov AS, Iurov IB, Vorsanova SG et al (2012) Genomic instability in the brain: etiology, pathogenesis and new biological markers of psychiatric disorders. Vestn Ross Akad Med Nauk 67(9):45–53
Van den Bos H, Spierings DC, Taudt AS et al (2016) Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer’s disease neurons. Genome Biol 17:116
Vorsanova SG, Kolotii AD, Iourov IY et al (2005) Evidence for high frequency of chromosomal mosaicism in spontaneous abortions revealed by interphase FISH analysis. J Histochem Cytochem 53(3):375–380
Vorsanova SG, Yurov IY, Demidova IA et al (2007) Variability in the heterochromatin regions of the chromosomes and chromosomal anomalies in children with autism: identification of genetic markers of autistic spectrum disorders. Neurosci Behav Physiol 37(6):553–558
Vorsanova SG, Iourov IY, Kolotii AD et al (2010a) Chromosomal mosaicism in spontaneous abortions: analysis of 650 cases. Rus J Genet 46:1197–1200
Vorsanova SG, Voinova VY, Yurov IY et al (2010b) Cytogenetic, molecular-cytogenetic, and clinical-genealogical studies of the mothers of children with autism: a search for familial genetic markers for autistic disorders. Neurosci Behav Physiol 40(7):745–756
Vorsanova SG, Yurov YB, Iourov IY (2010c) Human interphase chromosomes: a review of available molecular cytogenetic technologies. Mol Cytogenet 3:1
Vorsanova SG, Yurov YB, Soloviev IV et al (2010d) Molecular cytogenetic diagnosis and somatic genome variations. Curr Genomics 11(6):440–446
Vorsanova SG, Yurov YB, Iourov IY (2017) Neurogenomic pathway of autism spectrum disorders: linking germline and somatic mutations to genetic-environmental interactions. Curr Bioinforma 12(1):19–26
Vorsanova SG, Zelenova MA, Yurov YB et al (2018) Behavioral variability and somatic mosaicism: a cytogenomic hypothesis. Curr Genomics 19(3):158–162
Vorsanova SG, Yurov YB, Iourov IY (2020) Dynamic nature of somatic chromosomal mosaicism, genetic-environmental interactions and therapeutic opportunities in disease and aging. Mol Cytogenet 13:16
Westra JW, Peterson SE, Yung YC et al (2008) Aneuploid mosaicism in the developing and adult cerebellar cortex. J Comp Neurol 507:1944–1951
Westra JW, Rivera RR, Bushman DM et al (2010) Neuronal DNA content variation (DCV) with regional and individual differences in the human brain. J Comp Neurol 518:3981–4000
Yang Y, Shepherd C, Halliday G (2015) Aneuploidy in Lewy body diseases. Neurobiol Aging 36:1253–1260
Yurov YB, Soloviev IV, Vorsanova SG et al (1996) High resolution multicolor fluorescence in situ hybridization using cyanine and fluorescein dyes: rapid chromosome identification by directly fluorescently labeled alphoid DNA probes. Hum Genet 97(3):390–398
Yurov YB, Vostrikov VM, Vorsanova SG et al (2001) Multicolor fluorescent in situ hybridization on post-mortem brain in schizophrenia as an approach for identification of low-level chromosomal aneuploidy in neuropsychiatric diseases. Brain and Development 23(1):S186–S190
Yurov YB, Iourov IY, Monakhov VV et al (2005) The variation of aneuploidy frequency in the developing and adult human brain revealed by an interphase FISH study. J Histochem Cytochem 53(3):385–390
Yurov YB, Iourov IY, Vorsanova SG et al (2007a) Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS One 2(6):e558
Yurov YB, Vorsanova SG, Iourov IY et al (2007b) Unexplained autism is frequently associated with low-level mosaic aneuploidy. J Med Genet 44(8):521–525
Yurov YB, Iourov IY, Vorsanova SG et al (2008) The schizophrenia brain exhibits low-level aneuploidy involving chromosome 1. Schizophr Res 98:139–147
Yurov YB, Iourov IY, Vorsanova SG (2009a) Neurodegeneration mediated by chromosome instability suggests changes in strategy for therapy development in ataxia-telangiectasia. Med Hypotheses 73:1075–1076
Yurov YB, Vorsanova SG, Iourov IY (2009b) GIN‘n’CIN hypothesis of brain aging: deciphering the role of somatic genetic instabilities and neural aneuploidy during ontogeny. MolCytogenet 2:23
Yurov YB, Vorsanova SG, Iourov IY (2010a) Ontogenetic variation of the human genome. Curr Genomics 11(6):420–425
Yurov YB, Vorsanova SG, Solov’ev IV et al (2010b) Instability of chromosomes in human nerve cells (normal and with neuromental diseases). Russ J Genet 46(10):1194–1196
Yurov YB, Vorsanova SG, Iourov IY (2011) The DNA replication stress hypothesis of Alzheimer’s disease. ScientificWorldJournal 11:2602–2612
Yurov YB, Vorsanova SG, Iourov IY (2013) Human interphase chromosomes — biomedical aspects. Springer New York, Heidelberg, Dordrecht, London
Yurov YB, Vorsanova SG, Liehr T et al (2014) X chromosome aneuploidy in the Alzheimer’s disease brain. Mol Cytogenet 7(1):20
Yurov YB, Vorsanova SG, Demidova IA et al (2016) Genomic instability in the brain: chromosomal mosaicism in schizophrenia. Zh Nevrol Psikhiatr Im S Psychiatry Korsakova 116(11):86–91
Yurov YB, Vorsanova SG, Iourov IY (2017a) Network-based classification of molecular cytogenetic data. Curr Bioinforma 12(1):27–33
Yurov YB, Vorsanova SG, Soloviev IV et al (2017b) FISH-based assays for detecting genomic (chromosomal) mosaicism in human brain cells. NeuroMethods 131:27–41
Yurov YB, Vorsanova SG, Demidova IA et al (2018a) Mosaic brain aneuploidy in mental illnesses: an association of low-level post-zygotic aneuploidy with schizophrenia and comorbid psychiatric disorders. Curr Genomics 19(3):163–172
Yurov YB, Vorsanova SG, Iourov IY (2018b) Human molecular neurocytogenetics. Curr Genet Med Rep 6(4):155–164
Yurov YB, Vorsanova SG, Iourov IY (2019a) Chromosome instability in the neurodegenerating brain. Front Genet 10:892
Yurov YB, Vorsanova SG, Iourov IY (2019b) FISHing for unstable cellular genomes in the human brain. OBM Genetics 3(2):11
Zupanc GK (2009) Towards brain repair: insights from teleost fish. Semin Cell Dev Biol 20:683–690
Acknowledgments
We would like to express our gratitude to Dr. OS Kurinnaia and Dr. MA Zelenova for help in chapter preparation. Professors SG Vorsanova and IY Iourov are partially supported by RFBR and CITMA according to the research project No. 18-515-34005. Prof. IY Iourov’s lab is supported by the Government Assignment of the Russian Ministry of Science and Higher Education, Assignment no. AAAA-A19-119040490101-6. Prof. SG Vorsanova’s lab is supported by the Government Assignment of the Russian Ministry of Health, Assignment no. AAAA-A18-118051590122-7.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Yurov, Y.B., Vorsanova, S.G., Iourov, I.Y. (2020). Interphase Chromosomes of the Human Brain. In: Iourov, I., Vorsanova, S., Yurov, Y. (eds) Human Interphase Chromosomes. Springer, Cham. https://doi.org/10.1007/978-3-030-62532-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-62532-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-62531-3
Online ISBN: 978-3-030-62532-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)