
Abstract
Next-generation sequencing (NGS) has revolutionised the way we perform clinical and public health microbiology. This technology combined with the bioinformatics research field has led to a deeper understanding of the mechanisms involved in the pathogenesis of clinical infections, antimicrobial-resistant determination and dissemination of microorganisms inside the hospital, in the community, in animals and the environment. New approaches, like machine-learning methods, will even further potentiate the understanding of the information that is generated through NGS. In this book chapter, we aim to show the possibilities for NGS, including whole-genome sequencing (WGS) and metagenomics, in the fields of clinical and public health microbiology and speculate on its future importance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

2.1 Introduction
For several decades, we have been confined in the clinical microbiology laboratory to techniques that are limited in the amount of information they provide, e.g. limited to species identification or antimicrobial susceptibility; limited with respect to the turnaround time, e.g. culture of slow-growing or obligate intracellular pathogens, and/or limited in the sensitivity of the tests due to, for example, previous antimicrobial therapy administered to the patient before sample collection. These limitations lead to significant consequences for both the patient and the health care system in general, like higher morbidity and mortality due to inappropriate antimicrobial therapy and increased medical costs due to the long turnaround time and limited sensitivity of the diagnostic assays and consequently a longer stay of the patient in the hospital. Next Generation Sequencing (NGS) has the potential to revolutionise the way we perform microbiology as it can become a ‘one test fits all’ [1]. With NGS, pathogen identification, therapeutic resistance, pathogenicity, outbreak transmission, and within-host evolution (in case of chronic infections) can be studied at the same time [1, 2]. NGS is already applied in several medical microbiology laboratories, including our laboratory at the University Medical Center Groningen (UMCG), where it is used for outbreak management and infection prevention within the hospital and within the region, identification of bacteria using the 16S-23S rRNA encoding region, and metagenomics approaches for identification and typing of pathogens. However, numerous limitations need to be supplanted in order to make it feasible and affordable in any Medical Microbiology laboratory, independent of it being a local, regional or academic hospital, or a national reference centre.
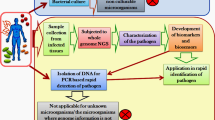
Several decisions have to be made when applying NGS to clinical and public health microbiology. There is no flawless workflow and every step in the process needs constant optimisation. Usually, an NGS workflow comprises the following steps: (1) sample collection; (2) DNA/RNA extraction; (3) library preparation; (4) sequencing; and (5) bioinformatics analysis, as it is shown in Fig. 2.1. The description/optimisation of these steps is not the focus of this book chapter, which will concentrate first on the practical issues of implementing NGS in diagnostic microbiology and second on a series of case studies that show the potential value of NGS for the surveillance and control of microorganisms.

Steps and challenges involved in an NGS workflow implemented in a diagnostic microbiology laboratory
2.2 Implementation of NGS in Clinical Microbiology Laboratories
In most countries, NGS was first introduced to microbiology in academic and/or reference laboratories, due to capital investment, operational costs, and requirements for expertise in the laboratory and bioinformatics processes [3]. The implementation of the NGS competencies at the reference laboratory depends on the type of national health system and may mirror a hierarchal structure that favours a more centralised microbiological surveillance and reference functions [4]. This hierarchal structure reduces the costs per sample at the reference laboratory, by collecting samples from different sources; however, this comes with the cost of prolonged turnaround times [5]. Nevertheless, the decrease in sequencing costs, the introduction of bench-top or portable and low-to-medium throughput devices [6, 7], the growing availability of free, user-friendly bioinformatics tools [8, 9] and the availability of specialised technicians resulted in a broad and rapid introduction of the NGS technology into non-academic laboratories, enabling a transition from a hierarchical to a network-like structure [1]. This significantly reduces the turnaround time, empowers hospital-based microbiology, and positively impacts local efforts such as infection control interventions [10].
To implement NGS in routine diagnostics, several adjustments in the laboratory workflow are required [3]. Both parts of the procedure, i.e. the wet laboratory part (nucleic acid extraction, library preparation, sequencing), and the bioinformatics part (analyses of the sequence data and translating them into easy to understand reports) should be performed by dedicated staff members specialised in NGS. The use of NGS fits best in a batch-wise approach; however, this is typical for high-throughput laboratories or surveillance projects, and it is far from ideal for routine diagnostics [3]. Recent equipment releases, like the MinION from Oxford Nanopore Technologies and the iSeq 100 from Illumina Inc., may overcome such limitations, as such sequencers either are smaller and less expensive (MinION) or provide a low-to-medium output (iSeq 100), which allows them to be more cost-effective. In any way, a balance should be kept between costs, quality (e.g. accuracy), turnaround time and complexity of the laboratory and bioinformatics processes.
Like for any new laboratory method, NGS requires validation. Yet, this process is far from being forthright and is required at both the laboratory and the bioinformatics level [3]. One of the most challenging points is the fact that there are many different kits, platforms and bioinformatics tools that can be used for NGS. The microbiologist should be aware of the stability, shelf life of the reagents and flow cells, and robustness of the bioinformatics tools used in the workflow, to ensure the repeatability and reproducibility of every step.
Additionally, NGS often is superior to other methods currently used within the laboratory; for example, it has higher discriminatory power compared to the current reference standard typing methods [3]. As whole-genome sequencing (WGS) can be used for all microbial species, it is almost impossible, with respect to time and costs, to perform an independent validation for all known species. Therefore, one may consider choosing several indicator species (e.g. one aerobe Gram-positive, one aerobe Gram-negative, one anaerobe Gram-positive, one anaerobe Gram-negative and one slow-growing microorganism) for the validation of the WGS workflow. The guidelines already developed for the validation of NGS in oncology and by the College of American Pathologists may serve as a model for worldwide guidelines of using NGS for pathogen detection [11, 12].
2.3 Whole-Genome Sequencing
Whole-genome sequencing (WGS) is here defined as the process of determining the complete DNA sequence of an organism from a sample that only contains that organism, e.g., a pure culture of bacterial isolate (as opposed to metagenomics which refers to the process of identifying the entire genomic material of a sample containing many organisms). This sample can contain the organism’s genome and other genetic elements (i.e. plasmids or phages) that can be present within the organism’s cell. Whole-genome sequencing has been applied in clinical and public health microbiology for several purposes, but we will focus on the four aspects we consider more important for this book chapter, i.e., outbreak management and infection prevention within the hospital, outbreak management and infection prevention within the region, transmission of zoonotic microorganisms between animals and humans and antimicrobial resistance characterisation.
2.3.1 Outbreak Management and Infection Prevention within the Hospital
Until recently, pathogen surveillance was performed by laborious techniques, that could only discriminate to a certain level (i.e. multi-locus sequence typing [MLST] for bacteria, or gene-specific sequencing for viruses), were limited to one specific pathogen (i.e. spa typing in Staphylococcus aureus) or could not be efficiently shared between laboratories (i.e. pulsed-field gel electrophoresis [PFGE] -based typing results) [13]. WGS, in principle, has the advantage of being applicable to any pathogen and, in fact, one of the most widely used applications of WGS today is for outbreak surveillance and infection prevention within healthcare institutes and foodborne related infections within a defined region. Several studies have proven the usefulness of WGS-based typing for disclosing and tracing the dissemination of microbial pathogens and, to a lesser extent, of mobile genetic elements (MGEs). In Fig. 2.2, we show a simple example of how this can be achieved. At the UMCG, it has been used to characterise both antimicrobial-resistant Gram-positive and Gram-negative bacterial outbreaks within the hospital and also for transmission of MGEs between different bacterial isolates obtained from the same or different patients. Additionally, since in the Netherlands there is a strict policy of “search and destroy”, we have identified and characterised pathogens in the water and the environment that could have potentially resulted in transmission to patients, but that were under control through disinfection measures of the corresponding areas. A few examples are presented below.

Illustration of a possible outbreak episode within the hospital and the role of NGS to understand transmission events
In 2012, a newly emerging blaCTX-M-15 producing Klebsiella pneumoniae clone with sequence type (ST) 1427 was detected in a patient previously hospitalised in Germany, South-Africa and Gambia, who was admitted to the UMCG university hospital [14]. After 2.5 months, regular surveillance screening (once per week) identified two blaCTX-M-15 K. pneumoniae positive roommates of this patient. After whole-genome phylogenetic analysis and patient contact tracing, an epidemiological link between the affected patients was identified. In total, five patients were involved in the outbreak, of which three developed an infection. In addition, environmental contamination with the outbreak clone was found in the patients’ rooms [14]. Interestingly, there was an in-host polymorphism detected among multiple isolates obtained from different body sites of the index patient, which were probably related to antibiotic treatment and/or host adaptation [14]. To prevent further spread, stringent infection control measures consisting of strict patient and staff cohorting were introduced. Contact screening up to 2 weeks after the discharge of all blaCTX-M-15 K. pneumoniae positive patients revealed no further cases and the outbreak was declared to be under control after 3 months [14]. Unfortunately, due to the unavailability of a single room at the time of admission and because initial screening results for highly resistant microorganisms were negative, the index patient was placed in a room shared with multiple patients, which enabled the spread of the resistant K. pneumoniae and so, this study highlighted once more the importance of isolating patients previously hospitalised in countries with high rates of antimicrobial-resistant bacteria.
In 2014, a retrospective analysis of vancomycin-resistant Enterococcus faecium (VREfm) outbreaks that occurred in the UMCG was performed [15]. It included 75 patients, but only 36 VREfm isolates obtained from 34 patients from seven VREfm outbreak investigations were analysed. The core genome MLST (cgMLST) analysis further divided the ST into different cluster types (CTs), however, only four different vanB transposons were found among the isolates. Within VREfm isolates belonging to ST117 CT103, two different vanB transposons were found, while, VREfm isolates belonging to ST80 CT104 and CT106 harboured an identical vanB transposon [15]. The presence of the same vanB transposon in VREfm isolates belonging to distinct lineages combined with the epidemiological data suggested an exchange of genomic material between VREfm and vancomycin-susceptible Enterococcus faecium (VSEfm). Thus, transposon typing resolved this series of outbreaks and demonstrated that an outbreak can be caused by a mobile element rather than a specific strain. Transposons with low DNA sequence homology among them were also found indicating that they probably originated from other species [15]. The presence of insertion sequences originating from anaerobic bacteria suggested transposon acquisition from anaerobic gut bacteria by VSEfm [15]. The occurrence of these two events is an important factor in the emergence of (vanB) VREfm. This study highlighted the importance of analysing additional transposon structures to detect horizontal gene transfer between phylogenetically unrelated strains.
In 2017, we identified four isolates of Legionella anisa in water from dental chair units (DCUs) at the UMCG hospital dental ward [16]. Whole-genome sequencing combined with whole-genome MLST (wgMLST) analysis indicated that all four isolates (two isolates from the same chair) belonged to the same cluster with two to four allele differences. This suggested that a common contamination source was present in the dental unit waterlines, which was resolved by replacing the chairs and the main pipeline of the unit. L. anisa, the most common non-pneumophila Legionella species in the environment, has a role as the causative agent of Legionnaires’ Disease (LD) and Pontiac fever [17] and it may be hospital-acquired [18]. Although a direct link between the dental unit and the patients is rarely shown, the water delivered by the dental unit waterlines has been shown to be one of many possible sources for Legionella infection [19]. This highlights the need to monitor water quality to protect patients and health-workers from acquiring legionella, or other potentially pathogenic bacteria.
2.3.2 Outbreak Management and Infection Prevention within the Region
In collaboration with other regional, national and international reference centres, the UMCG has been characterising the transmission of relevant pathogens between institutions within the region and at the national and international level.
Between May 2012 and September 2013, the transmission of a blaCTX-M-15-producing Klebsiella pneumoniae ST15 occurred between patients treated in a single centre [20]. Additionally, one of these patients was treated in three different institutions located in two cities and was involved in further intra- and inter-institutional spread of this high-risk clone (local expansion, blaCTX-M-15 producing, and containing hypervirulence factors). Environmental contamination and lack of consistent patient screening were identified as the responsible factors for the dissemination of this specific clone. The design of a tailor-made real-time -PCR specific for the outbreak clone based on the whole-genome sequences of the strains allowed the early detection of this K. pneumoniae high-risk-clone with prolonged circulation in the regional patient population [20] and helped prevent further spread. This study raised awareness to the necessity for inter-institutional/regional collaborations for infection/outbreak management of relevant pathogens [20].
In a large cohort study, WGS was used for molecular characterisation of Shiga toxin-producing Escherichia coli (STEC) isolated from faeces of patients obtained from two regions in the Netherlands to reveal the relation between molecular determinants and disease outcome [21]. STEC is a significant public health concern associated with both outbreaks and sporadic cases of human gastrointestinal illness worldwide [22]. A subpopulation of STEC, the enterohaemorrhagic E. coli, can cause bloody diarrhoea in humans, and some can cause haemolytic uremic syndrome (HUS) [23]. This study concluded that there was no clear correlation between serogenotype, stx subtype or ST and disease outcome and the latter was probably influenced by other host factors. Additionally, this study demonstrated that there was substantial genetic diversity and distinct phylogenetic groups observed in the two studied regions, showing that the STEC populations within these two geographically regions were not genetically linked [21].
More recently, a study was conducted to understand the epidemiology of resistant bacteria, including extended-spectrum β-lactamase (ESBL)- and plasmid AmpC (pAmpC)-, and carbapenemase (CP)-producing Enterobacteriaceae and vancomycin-resistant enterococci (VRE) across the Northern Dutch-German border region [24]. The Netherlands and Germany are bordering countries that created a cooperative network to prevent the spread of multidrug-resistant microorganisms (MDRO), such as ESBL and CP-producing Enterobacteriaceae and VRE, and to harmonise guidelines in healthcare settings as patients are regularly transferred between healthcare institutions within the two countries [25]. However, it was concluded that cross-border transmission of ESBL-producing E. coli and VRE was unlikely, based on the cgMLST analysis performed [24]. Yet, the authors reinforced that continuous monitoring is required to control the spread of these pathogens and to stay informed about their epidemiology, in order to implement effective infection prevention measures [24].
2.3.3 Transmission of Zoonotic Microorganisms Between Animals and Humans
Human health is influenced by several factors in the environment, including contact with animals, animal products or contaminated habitats. At the UMCG, we are working in collaboration with other non-hospital institutions to understand the dynamics of transmission of microbial pathogens between humans, animals and the environment. Figure 2.3 shows a model for early outbreak and infection prevention surveillance response that is needed due to detect spilling of new emerging infectious diseases from non-human reservoirs to humans.

Model for outbreak and infection prevention surveillance
Recently, a K. pneumoniae clone (ST348) in a horse was found, which had previously been isolated from humans in Portugal and a few other countries [26,27,28,29,30,31]. The allele differences provided by the cgMLST analysis suggested there was a genetic link, although an epidemiological link could not be found [32]. This indicated that either this particular clone is circulating in humans and horses in Portugal or there was a transfer of this particular isolate from a person to the horse during hospitalisation. In any case, this study demonstrated the importance of identifying and controlling this type of hospital-acquired infections, in both the human and veterinarian hospital settings, in order to avoid antimicrobial resistance dissemination [32].
2.3.4 Antimicrobial Resistance Characterisation Through NGS
Before NGS, finding new mechanisms of antimicrobial resistance was demanding since it involved different laborious techniques (e.g. hybridisation, cloning or primer-walking sequencing) until the gene and/or mutation responsible for resistance could be detected. With the entire genome sequenced through NGS, we can, by homology, identify potential new mechanisms. Further experiments can then be performed to determine if these genes are indeed responsible for the observed antimicrobial resistance pattern [33].
In one study, a possible in vivo selection of a clinical Klebsiella oxytoca isolate showing increased minimum inhibitory concentrations to ceftazidime was described [33]. The patient had been treated with ceftazidime (4 g/day) for a septic episode caused by multiple bacterial species, including K. oxytoca, but after 11 days of treatment, another K. oxytoca was isolated from a pus sample drained from his wound. The wound isolate showed increased resistance to ceftazidime (MIC ≥64 mg/L) compared with the original K. oxytoca isolate. WGS revealed the presence of a novel blaOXY-2 allele, termed blaOXY-2-15, with a two amino acid deletion at Ambler positions 168 and 169 compared to blaOXY-2-2. This report showed the risk of in vivo selection of ceftazidime-resistant K. oxytoca isolates after prolonged ceftazidime treatment and it was the first description of a K. oxytoca isolate conferring resistance to ceftazidime by a two amino acid deletion in the omega loop of blaOXY-2-2 [33].
More recently, a novel nim gene was found in three metronidazole-resistant Prevotella bivia strains, the nimK gene, which was located on a mobile genetic element [34]. For decades, metronidazole has been the antibiotic of choice when dealing with anaerobic infections. However, metronidazole-resistant bacteria have been reported [35]. The nimK gene was associated with an IS1380 family transposase on a mobile genetic element that also contained a gene encoding an efflux small MDR (SMR) transporter associated with a crp/fnr regulator. This was the first description of the presence of a novel nim gene in metronidazole-resistant P. bivia clinical isolates [34]. The detection of MGEs harbouring nim and other relevant genes among anaerobic bacteria is worrying, because these elements may cause a rapid emergence of resistance to the most commonly used antibiotics in anaerobic infections.
2.4 Metagenomics
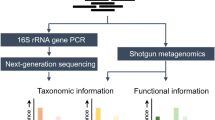
Metagenomics is here defined as the process of determining the complete DNA or RNA sequence(s), either after reverse transcription to cDNA or directly, of microorganisms and/or viruses from a complex sample that contains several microorganisms and/or viruses. This complex sample can contain the microorganisms’ genomes and other genetic elements (i.e. plasmids or phages) that can be present within the organisms’ cell or are freely floating in the sample. Sequencing of DNA, cDNA or RNA within a sample can be based on the amplification of (a) specific sequence(s) (amplicon-based or targeted metagenomics) or on the entire genomes (shotgun metagenomics). We will focus this section on the use of metagenomics for three specific purposes that are currently under optimisation and implementation at the UMCG.
2.4.1 Amplicon-Based Metagenomics of the 16S–23S rRNA Encoding Region
The conventional culturing method has long been considered the gold standard for bacterial identification. However, it can take days to weeks to successfully culture bacteria, as some clinically relevant bacteria are slow-growing, difficult to grow, fastidious or sometimes even non-culturable [36, 37]. The 16S rRNA gene has proven to be a useful molecular target since it is present in all bacteria, either as a single copy or in multiple copies, and it is highly conserved over time [38]. Consequenttly, until recently, most microbiome studies used this amplicon-based metagenomic approach to investigate the microbial communities of different body sites, and vast literature has been published using this technique.
Nonetheless, this method does not always allow the identification of bacteria to the species level due to high sequence similarities between some species [1]. To overcome this problem, Sabat and colleagues [39] developed an innovative approach based on the sequencing of the 16S–23S rRNA encoding region (~4.5 kb). The method proved to be superior to other commonly used identification methods and enabled concurrent identification of several pathogens in clinical samples that were negative by culture and PCR [39]. In order to further improve this method, an in-house database was developed, which, combined with a de novo assembly and BLAST (Basic Local Alignment Search Tool) approach, significantly reduced the time needed for analysis [40].
2.4.2 Shotgun Metagenomics for the Identification and Typing of Microbial Pathogens
Several molecular detection techniques have been implemented in the diagnostic laboratory, but these are generally geared towards specific pathogens (e.g. specific RT-PCR or microarrays) and even when unbiased molecular approaches are used, such as 16S/18S rRNA gene sequencing, these do not provide all the information that can be obtained by culturing, e.g., antimicrobial susceptibility and molecular typing information [41]. For this reason, the use of shotgun metagenomics as a single method that could provide rapid identification and characterisation of clinically relevant pathogens directly from a sample was evaluated [41]. As the complexity of data analysis is a challenge encountered in shotgun metagenomics, a comparison of a diverse set of bioinformatics tools (commercial and non-commercial) was performed to investigate their performance in taxonomic classification, antimicrobial resistance gene detection and typing [41]. Based on the results obtained, the authors concluded that the tools and databases used for taxonomic classification and antimicrobial resistance had a key impact on the results, suggesting that efforts need to be directed towards standardisation of the methods if shotgun metagenomics is to be used routinely in clinical microbiology.
A study was also conducted to optimise a shotgun metagenomics workflow for the identification and typing of Dengue viruses (DENV), a positive-stranded RNA virus, directly from clinical samples [42]. DENV infection continues to be one of the most prevalent arboviral diseases in tropical and subtropical regions [43]. Nevertheless, to date, there is no fully successful vaccine or specific treatment for DENV [44]. It is therefore essential to monitor circulating DENV. Genotyping is mostly based on sequencing parts of the genes coding for the structural proteins through Sanger sequencing of the E region [45] or the CprM region [46, 47]. However, these methods have poor resolution and do not allow for the detection of recombinant events and detection of escape mutants [42]. A shotgun metagenomics approach was used successfully to sequence whole genomes of DENV directly from clinical samples, without the need for prior sequence-specific amplification steps. The method enabled the identification of intra-host DENV diversity (quasi-species), detection of multiple DENV serotypes in a single sample and generation of phylogenetic trees to understand the dynamics of DENV. Results were obtained within 3 days and the associated reagent costs were low enough to be suitable for a clinical setting.
2.4.3 Shotgun Metagenomics to Characterise the Gut Microbiome/Resistome
In Europe, more than 80% of the total antibiotic consumption in the human sector is prescribed in the community, and the rate of prescription increases with age leading to collateral damage and antibiotic pressure. Although the importance of a healthy gut microbiome and the consequences of dysbiosis have been extensively reported, less is known about the resistome present in the healthy population [48]. A study was conducted at the UMCG to describe the gut resistome of healthy middle-aged people in Northern Netherlands, by using samples from the Lifelines cohort [49]. A total of 60 samples were sequenced, in which several resistance genes were identified, and among them the tetracycline resistance genes were predominant. No extended-spectrum β-lactamases (ESBLs) or carbapenemases were found [48]. This study highlighted the importance of monitoring healthy people to identify potential sources of antimicrobial resistance genes and implement effective control measures.
Another study characterised the human intestinal microbiota in faecal samples from STEC-infected patients. The objective was to investigate possible changes in the composition of the intestinal microbiota in samples from STEC-infected patients compared to healthy and healed controls [50]. The stool samples collected from the STEC infected patients had a lower abundance of Bifidobacteriales and Clostridiales members in comparison to controls where these microorganisms predominated. This was the first evidence that changes occur in the intestinal microbiota of patients with STEC infection and it highlighted the importance of metagenomics for the culture-independent diagnosis of infection, as it was able to detect genomic traits associated with STEC in stool samples from infected subjects [50].
2.5 Clinical Impact of NGS
The above-mentioned examples show the power of NGS in the clinical microbiology laboratory. In addition, there is an increasing interest in NGS for clinical microbiology both by laboratories and companies. This becomes clear when looking at the enormous increase in the number of symposia and capacity building workshops related to these topics, and in the available software (commercial and non-commercial) to be used for analyses of shotgun metagenomics and WGS data. We anticipate that the use of NGS in medical microbiology laboratories will further increase over the next years, not only for the characterisation and surveillance of pathogens, the investigation of outbreaks, infection prevention and the detection of novel resistance genes but also for the application of metagenomic approaches in clinical samples for (routine) molecular diagnostics. The latter will have a significant impact on the diagnosis of infectious diseases, on understanding host-pathogen interactions [12], and on the correlation between genotype (provided by NGS) and phenotype [51]. However, the NGS workflow needs further improvement, especially for shotgun metagenomics, to shorten the turnaround time and further reduce costs [1], and to ensure the quality and reproducibilityof the results (including validation and external proficiency testing). Although these and other challenges need to be tackled, we are convinced that NGS will become a powerful tool within the clinical microbiology laboratory and will lead to a personalised approach for diagnosing and monitoring treatment of infectious diseases.
References
Deurenberg R, Bathoorn W, Chlebowicz MA, Couto N, Ferdous M, Garcia-Cobos S et al (2017) Application of next generation sequencing in clinical microbiology and infection prevention. J Biotechnol 243:16–24
Padmanabhan R, Mishra AK, Raoult D, Fournier PE (2013) Genomics and metagenomics in medical microbiology. J Microbiol Methods 95:415–424
Rossen JWA, Friedrich AW, Moran-Gilad J (2018) Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin Microbiol Infect 24:355–360
EFSA (2014) Use of whole genome sequencing (WGS) of food-borne pathogens for public health protection. EFSA Scientific Colloquium Summ Rep 20
Reuter S, Elington MJ, Cartwright EJP, Köser CU, Török ME, Gouliouris T et al (2013) Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA Intern Med 173(15):1397–1404
Gardy JL, Loman NJ (2018) Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Gen 19:9–20
Illumina: iSeq™ 100 Sequencing System (2017) https://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/iseq100-sequencing-system-spec-sheet-770-2017-020.pdf. Accessed 24 Jul 2018
Thomsen MCF, Ahrenfeldt J, Cisneros JLB, Jurtz V, Larsen MV, Hasman H et al (2016) A bacterial analysis platform: an integrated system for analysing bacterial whole genome sequencing data for clinical diagnostics and surveillance. PLoS One 11(6):e0157718
Visconti A, Martin TC, Falchi M (2018) YAMP: a containerized workflow enabling reproducibility in metagenomics research. GigaScience 7(7):giy072
Fricke WF, Rasko DA (2014) Bacterial genome sequencing in the clinic: bioinformatic challenges and solutions. Nat Rev Genet 15:49–55
Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J et al (2017) Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the association for molecular pathology and college of American pathologists. J Mol Diagn 19:341–365
Schlaberg R, Chiu CY, Miller S, Procop GW, Weinstock G, the Professional Practice Committee and Committee on Laboratory Practices of the American Society for Microbiology, and the Microbiology Resource Committee of the College of American Pathologists (2017) Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch Pathol Lab Med 41(6):776–786
Quainoo S, Coolen JPM, van Hijum SAFT, Huynen MA, Melchers WJG, van Schaik W et al (2017) Whole-genome sequencing of bacterial pathogens: the future of nosocomial outbreak analysis. Clin Microbiol Rev 30:1015–1063
Zhou K, Lokate M, Deurenberg RH, Arends J, Lo-Ten Foe J, Grundmann H et al (2015) Characterization of a CTX-M-15 producing Klebsiella pneumoniae outbreak strain assigned to a novel sequence type (1427). Front Microbiol 6:1250
Zhou X, Chlebowicz MA, Bathoorn E, Rosema S, Couto N, Lokate M et al (2018) Elucidating vancomycin-resistant Enterococcus faecium outbreaks: the role of clonal spread and movement of mobile genetic elements. J Antimicrob Chemoth 73(12):3259–3267
Fleres G, Couto N, Lokate M, van der Sluis LWM, Ginevra C, Jarraud S et al (2018) Detection of Legionella anisa in water from hospital dental chair units and molecular characterization by whole-genome sequencing. Microorganisms 6(3):pii:E71
Vaccaro L, Izquierdo F, Magnet A, Hurtado C, Salinas M, Gomes T et al (2016) First case of legionnaire’s disease caused by Legionella anisa in Spain and the limitations on the diagnosis of Legionella non-pneumophila infections. PLoS One 11(7):e0159726
Bornstein N, Mercatello A, Marmet D, Surgot M, Deveaux Y, Fleurette J (1989) Pleural infection caused by Legionella anisa. J Clin Microbiol 27(9):2100–2101
Schönning C, Jernberg C, Klingenberg D, Andersson S, Pääjärvi A, Alm E et al (2017) Legionellosis acquired through a dental unit: a case study. J Hosp Infect 96(1):89–92
Zhou K, Lokate M, Deurenberg RH, Tepper M, Arends JP, Raangs EGC et al (2016) Use of whole-genome sequencing to trace, control and characterize the regional expansion of extended-spectrum β-lactamase producing ST15 Klebsiella pneumoniae. Sci Rep 6:20840
Ferdous M, Friedrich AW, Grundmann H, de Boer RF, Croughs PD, Islam MA et al (2016) Molecular characterization and phylogeny of Shiga toxin-producing Escherichia coli isolates obtained from two Dutch regions using whole genome sequencing. Clin Microbiol Infect 22(7):642.e1–642.e9
Paton JC, Paton AW (1998) Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin Microbiol Rev 11:450–479
Mellmann A, Bielaszewska M, Köck R, Friedrich AW, Fruth A, Middendorf B et al (2008) Analysis of collection of hemolytic uremic syndrome associated enterohemorrhagic Escherichia coli. Emerg Infect Dis 14:1287–1290
Zhou X, García-Cobos S, Ruijs GJHM, Kampinga GA, Arends JP, Borst DM et al (2017) Epidemiology of extended-spectrum β-lactamase-producing E coli and vancomycin-resistant enterococci in the northern Dutch–German cross-border region. Front Microbiol 8:1914
Muller J, Voss A, Köck R, Sinha B, Rossen JW, Kaase M et al (2015) Cross-border comparison of the Dutch and German guidelines on multidrug-resistant Gram-negative microorganisms. Antimicrob Res Infect Control 4:7
Rodrigues C, Machado E, Ramos H, Peixe L, Novais  (2014) Expansion of ESBL-producing Klebsiella pneumoniae in hospitalized patients: a successful story of international clones (ST15, ST147, ST336) and epidemic plasmids (IncR, IncFIIK). Int J Med Microbiol 304:1100–1108
Rodrigues C, Bavlovič J, Machado E, Amorim J, Peixe L, Novais  (2016) KPC-3-producing Klebsiella pneumoniae in Portugal linked to previously circulating non-CG258 lineages and uncommon genetic platforms (Tn4401d-IncFIA and Tn4401d-IncN). Front Microbiol 7:1000
Rodrigues C, Mendes A, Sima F, Bavlovič J, Machado E, Novais Â, Peixe L (2017) Long-term care facility (LTCF) residents colonized with multidrug-resistant (MDR) Klebsiella pneumoniae lineages frequently causing infections in Portuguese clinical institutions. Infect Control Hosp Epidemiol 38:1127–1130
Baraniak A, Izdebski R, Fiett J, Sadowy E, Adler A, Kazma M et al (2013) Comparative population analysis of Klebsiella pneumoniae strains with extended-spectrum β-lactamases colonizing patients in rehabilitation centers in four countries. Antimicrob Agents Chemother 57:1992–1997
Mshana SE, Hain T, Domann E, Lyamuya EF, Chakraborty T, Imirzalioglu C (2013) Predominance of Klebsiella pneumoniae ST14 carrying CTX-M-15 causing neonatal sepsis in Tanzania. BMC Infect Dis 13:466
Vubil D, Figueiredo R, Reis T, Canha C, Boaventura L, da Silva G (2016) Outbreak of KPC-3-producing ST15 and ST348 Klebsiella pneumoniae in a Portuguese hospital. Epidemiol Infect 143:595–599
Trigo da Roza F, Couto N, Carneiro C, Cunha E, Rosa T, Magalhães M et al (2019) Commonality of Multidrug-Resistant Klebsiella pneumoniae ST348 Isolates in Horses and Humans in Portugal. Front Microbiol 10:1657
Nijhuis RH, Oueslati S, Zhou K, Bosboom RW, Rossen JW, Naas T (2015) OXY-2-15, a novel variant showing increased ceftazidime hydrolytic activity. J Antimicrob Chemother 70:1429–1433
Veloo ACM, Chlebowicz M, Winter HLJ, Bathoorn D, Rossen JWA (2018) Three metronidazole-resistant Prevotella bivia strains harbour a mobile element, encoding a novel nim gene, nimK, and an efflux small MDR transporter. J Antimicrob Chemother 73(10):2687–2690
Hartmeyer GN, Sóki J, Nagy E, Justesen US (2012) Multidrug-resistant Bacteroides fragilis group on the rise in Europe? J Med Microbiol 61:1784–1788
Didelot X, Bowden R, Wilson DJ, Peto TEA, Crook DW (2012) Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Gen 13:601–612
Salipante SJ, Sengupta DJ, Rosenthal C, Costa G, Spangler J, Sims EH et al (2013) Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections. PLoS One 8:e65226
Petti CA (2007) Detection and identification of microorganisms by gene amplification and sequencing. Clin Infect Dis 44:1108–1114
Sabat AJ, van Zanten E, Akkerboom V, Wisselink G, van Slochteren K, de Boer RF et al (2017) Targeted next-generation sequencing of the 16S-23S rRNA region for culture-independent bacterial identification – increased discrimination of closely related species. Sci Rep 7(1):3434
Peker N, Garcia-Croes S, Dijkhuizen B, Wiersma HH, van Zanten E, Wisselink G et al (2019) A comparison of three different bioinformatics analyses of the 16S-23S rDNA region for bacterial identification. Front Microbiol 10:620
Couto N, Schuele L, Raangs EC, Machado M, Mendes CI, Jesus TF et al (2018) Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens. Sci Rep 8:13767
Lizarazo E, Couto N, Tami A, Vincenti-Gonzalez M, Raangs EC, Velasco Z, Bethencourt S et al (2019) Applied shotgun metagenomics approach for the genetic characterization of dengue viruses. J Biotechnol X 2:100009
Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL et al (2013) The global distribution and burden of dengue. Nature 496:504–507
Lizarazo E, Couto N, Vincenti-Gonzalez M, Raangs EC, Jaenisch T, Friedrich AW et al (2018) Complete coding sequences of five dengue virus type 2 clinical isolates from Venezuela obtained through shotgun metagenomics. Genome Announc 6:e00545–e00518
Wang E, Ni H, Xu R, Barrett ADT, Watowich SJ, Gubler DJ, Weaver SC (2000) Evolutionary relationships of endemic/epidemic and sylvatic dengue viruses. J Virol 74:3227–3234
Avilés G, Rowe J, Meissner J, Manzur Caffarena JC, Enria D, Jeor S (2002) Phylogenetic relationships of dengue-1 viruses from Argentina and Paraguay. Arch Virol 147:2075–2087
Kukreti H, Chaudhary A, Rautela RS, Anand R, Mittal V, Chhabra M et al (2008) Emergence of an independent lineage of dengue virus type 1 (DENV-1) and its co-circulation with predominant DENV-3 during the 2006 dengue fever outbreak in Delhi. Int J Infect Dis 12(5):542–549
García-Cobos S, Rabbers T, Bossers A, Schmitt H, Harmsen HJM, Sinha B, et al (2018) A snapshot of the gut resistome of a middle-aged healthy population in the northern Netherlands (LifeLines). Oral communication (O0350) presented at the 28th European Congress of Clinical Microbiology and Infectious Diseases, Madrid, Spain (available at https://www.escmid.org/escmid_publications/escmid_elibrary/material/?mid=64330)
Stolk RP, Rosmalen JG, Postma DS, de Boer RA, Navis G, Slaets JP et al (2008) Universal risk factors for multifactorial diseases: LifeLines: a three-generation population-based study. Eur J Epidemiol 23(1):67–74
Gigliucci F, von Meijenfeldt FAB, Knijn A, Michelacci V, Scavia G, Minelli F et al (2018) Metagenomic characterization of the human intestinal microbiota in fecal samples from STEC-infected patients. Front Cell Infect Microbiol 8:25
Motro Y, Moran-Gilad J (2017) Next-generation sequencing applications in clinical bacteriology. Biomol Detect Quantif 14:1–6
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Couto, N., Rossen, J.W. (2021). Overview of Microbial NGS for Clinical and Public Health Microbiology. In: Moran-Gilad, J., Yagel, Y. (eds) Application and Integration of Omics-powered Diagnostics in Clinical and Public Health Microbiology . Springer, Cham. https://doi.org/10.1007/978-3-030-62155-1_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-62155-1_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-62154-4
Online ISBN: 978-3-030-62155-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)