
Abstract
Introduction
Pericyte serve myriad roles in the developing and adult brain, but many of their functions remain poorly understood. In less than a decade, the advent of new tools and approaches has revolutionized brain pericyte research by enabling direct visualization of pericyte structure and function in the living mouse brain. In addition to accessing pericytes in a physiological context, in vivo imaging has revealed fascinating new biology on how distinct pericyte subtypes influence cerebral blood flow in both health and disease. This chapter highlights novel approaches and provides up-to-date guidance on pericyte research using two-photon microscopy and related optical applications.
Methods
We surveyed literature for studies that have imaged brain pericytes in vivo in order to summarize their methodologies and approaches. Keywords included pericyte , mural cell, cerebrovasculature, brain , capillary , blood-brain barrier, cerebral blood-flow, two-photon microscopy, cranial window, endothelium, cre recombinase, in vivo imaging, microvasculature, calcium imaging, cell ablation.
Results
We discuss the identification of true capillary pericytes and set them apart from transitional phenotypes in murine cerebral cortex. An array of available murine Cre drivers and other approaches to target pericytes and brain mural cells are compared and contrasted. Surgical and experimental parameters to perform rigorous quantification of pericyte influence on capillary flow and integrity are also discussed. Lastly, we move beyond correlative observation to cutting-edge approaches for optical ablation and manipulation of pericytes, concomitant with real-time imaging of cerebrovascular function.
Conclusion
The information provided can serve as a roadmap to rigorous in vivo imaging of pericytes in the healthy and diseased brain .
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1.1 Introduction
Deficiency in the function of small brain vessels is a common link between many age-related brain diseases. A recent emergence of new tools and approaches has allowed researchers to observe and modify cerebral blood vessels, as well as their cellular components, in the living brain . In particular, pericytes have taken center stage, and their role in maintaining a functional neurovascular unit is now in a deep phase of exploration. Pericytes find their niche within the basement membrane of all brain capillaries, and are characterized by their long filamentous processes stemming from protruding ovoid cell bodies. Pericytes, along with vascular smooth muscle cells, fall under the broad category of “mural cells,” which exhibit diverse morphologies along cerebrovascular networks. The abundance of pericytes along the walls of small cerebral vessels in the brain compared to the vascular beds of other tissues suggests a critical contribution to cerebrovascular health and stability (Armulik et al. 2005). Indeed, pericytes are integral to the formation of the blood-brain barrier (BBB) (Blanchette and Daneman 2015; Daneman et al. 2010; Armulik et al. 2010) and the stabilization of vascular structure and tone (von Tell et al. 2006; Berthiaume et al. 2018a; Hellstrom et al. 2001), and may even play some roles in immune function (Stark et al. 2018; Rustenhoven et al. 2017) and scar formation following injury (Reeves et al. 2019; Zehendner et al. 2015). The congenital loss of pericytes through genetic knockout is often prenatally lethal (Lindahl et al. 1997; Lindblom et al. 2003), and decreased pericyte numbers has been reportedly involved in aging and Alzheimer’s disease (Sengillo et al. 2013; Montagne et al. 2015; Halliday et al. 2016; Miners et al. 2019). Loss of pericytes under these circumstances has been related to increased BBB permeability (Miners et al. 2019; Dore-Duffy et al. 2000; Berthiaume et al. 2018b; Arango-Lievano et al. 2018).
Much of what we know about cerebrovascular pericytes has come from important studies in primary pericyte cultures and brain slices (Kamouchi et al. 2004; Mishra et al. 2014; Hall et al. 2014; Nakagawa et al. 2007; Peppiatt et al. 2006; Orlidge and D’Amore 1987; Sato et al. 1990). These systems provide the benefit of studying pericytes in relative isolation to determine their fundamental attributes and functions. However, there are also clear limitations to an in vitro approach. Pericytes function within the context of a complex neurovascular network. There are aspects of the intact brain that cannot be replicated with in vitro systems, notably blood flow, blood pressure, and network dynamics of the vascular beds that house pericytes. Furthermore, evidence suggests that cultured pericytes may develop a gene expression profile that differs dramatically from their in vivo counterparts, making it difficult to relate in vitro findings to true pericyte physiology (Guimaraes-Camboa et al. 2017). Accordingly, there has been a recent surge in the number of studies that use in vivo imaging to study brain pericytes, particularly with respect to blood flow regulation (Hall et al. 2014, 2015; Grutzendler and Nedergaard 2019; Hartmann et al. 2020). This move to in vivo preparations was facilitated by the development of several key techniques to target, visualize, and manipulate pericytes within the intact mouse brain . This chapter will focus on methods to study pericyte in vivo, including guidance on genetically engineered mouse lines, gaining optical access to the live brain , and in vivo two-photon microscopy. In addition, we describe approaches to identify different pericyte subtypes, and to properly characterize and quantify aspects of these cells in their native environment.
Early studies that expressed bright fluorescent reporters in mural cells of the brain revealed their remarkable morphological heterogeneity (Grant et al. 2017; Hartmann et al. 2015) (Fig. 1.1a). These range from the circumferential smooth muscle cells on arteries to the filamentous pericytes of the capillary network, with transitional cells between these classic forms. Venules and veins are also covered by a mixture of stellate shaped mural cells that do not completely encircle the vessel. The function of venous mural cells remains poorly defined, though their depletion can lead to disturbances in venous structure (Lai et al. 2015). All these cells occupy the abluminal surface of the vessel wall, but each has distinct identities in respect to morphology, topological organization, genetic expression, and physiology (Berthiaume et al. 2018a; Vanlandewijck et al. 2018). Given this diversity, there has been some confusion as to what mural cell subtypes should be considered true “pericytes,” since mural cell classification has not been consistent across research groups https://pubmed.ncbi.nlm.nih.gov/26661200/. For this reason, care should be taken to properly identify and report exact microvascular location and morphology when studying pericytes in vivo.

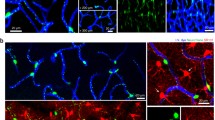
Identifying mural cell subpopulations in the living mouse brain . (a) (Top) Relative expression of contractile protein α-SMA across microvascular zones. (Bottom) Schematic of mural cell morphologies residing on vessels of the mouse cerebral cortex. Colors represent distinct mural cell subpopulations. Adapted from Berthiaume et al. 2018 (Berthiaume et al. 2018a). (b) (Top) Inset from (a), showing a close-up of the vascular network forming the arteriole (red), pre-capillary (yellow), and capillary zones (green). Numbers indicate branch orders from penetrating arteriole, 0, where each bifurcation increases vascular branch order by 1. (Bottom) In vivo two-photon images of the three major pericyte subtypes. From left to right, ensheathing pericytes of the precapillary zone, mesh pericytes of the capillary zone, and thin-strand pericytes of the capillary zone. Left image from PDGFRβ-tdTomato mouse, center and right images from Myh11-tdTomato mice. (c) In vivo i.v. dye image (top) marked with branch order and colors corresponding to colors in (b). Corresponding two-photon image of mural appearance (bottom), with labeled branch orders. These images were from a Myh11-tdTomato mouse. (d) Immunohistological staining for α-SMA using a FITC-conjugated antibody, showing sharp cutoffs in protein abundance. Quantification of average branch order for α-SMA termini per offshoot from penetrating arterioles, showing undetectable expression after fourth branch order. (Adapted from Grant et al. 2017 (Grant et al. 2017))
1.2 Identifying Brain Pericytes
The first challenge of in vivo brain pericyte imaging is knowing how to consistently identify them. Several distinct mural cell morphologies appear as cortical penetrating arterioles branch into pre-capillary arterioles and then capillaries. In our work, we categorize these mural cells based on their morphology, topological location, and expression of contractile protein α-smooth muscle actin (α-SMA) (Fig. 1.1a). We refer to any mural cell type with a protruding ovoid cell body and elongated processes as “pericytes.” However, we have applied nomenclature to three morphological sub-classes of pericytes, which we refer to as ensheathing, mesh, and thin-strand pericytes (Grant et al. 2017; Vanlandewijck et al. 2018; Grubb et al. 2020) (Fig. 1.1b).
Ensheathing pericytes are found primarily on pre-capillary arterioles of the mouse cortex. Though the term “pre-capillary arteriole” is not universally used, we have denoted this zone as the first few branch orders that diverge from the penetrating arteriole. Ensheathing pericytes fully cover the microvessel with tightly packed processes running circumferential to the endothelial tube (Fig. 1.1c), suggesting they are well equipped for vascular contraction and dilation (Grubb et al. 2020). Their circumferential processes are reminiscent of vascular smooth muscle cells (SMCs) on upstream arteries, though they are set apart by their protruding cell bodies and roughly two-fold greater length along the longitudinal capillary axis. Ensheathing pericytes share genetic similarities with SMCs, most notably the high expression of α-SMA (Vanlandewijck and Betsholtz 2018). In contrast, this contractile protein is low or absent in mesh and thin-strand pericytes downstream of the pre-capillary arteriole, reflecting an important functional distinction between ensheathing pericytes and other microvascular pericytes (Grant et al. 2017; Vanlandewijck et al. 2018). While robust α-SMA expression is ideally used to identify ensheathing pericytes, its expression level is not easily ascertained in vivo without ACTA2 reporter mice (Hill et al. 2015). However, they can be generally identified based on their circumferential morphology and topological location (i.e. branch order). Several groups have now shown that, in mouse cortex, α-SMA expression on penetrating arteriole offshoots does not surpass the 4th branch order (Grant et al. 2017; Grubb et al. 2020; Wei et al. 2016)(Fig. 1.1d). Thus, examining microvessel branches that are within the 4th order increases the likelihood of studying a region occupied by ensheathing pericytes, but does not guarantee it. However, examining beyond 4th branch order guarantees that one is within the capillary zone, occupied by mesh or thin-strand pericytes.
As one goes distally from ensheathing pericyte territory, α-SMA expression becomes low or absent and pericyte processes begin to lose their ring-like appearance, instead taking on a complex mesh-like shape (Fig. 1.1c). These mesh pericytes also have characteristic protruding cell bodies, but unlike ensheathing pericytes, their processes do not completely cover the underlying endothelium. Beyond the 4th and 5th order branches, pericyte processes with the classic thin-strand morphology become prominent. Thin-strand pericytes are the most abundant pericyte form, as they cover the majority of the capillary bed. These pericytes are easily recognizable with several long, thin processes emerging from the soma, which protrudes from the capillary wall like a “bump on a log.” We have grouped mesh and thin-strand pericyte types together into the category “capillary pericytes” because their morphologies are difficult to objectively distinguish. Capillary pericytes form an extensive network along the brain microvasculature, where neighboring pericytes occupy non-overlapping territories (Berthiaume et al. 2018b). Each cell therefore acts as a “link” in a chain of capillary pericytes distributed throughout the capillary bed. These morphological features, as well as their topological location within the cerebrovascular network (beyond 4th branch order), make capillary pericytes identifiable with certainty using in vivo optical imaging.
1.3 Distinct Functions of Pericyte Subtypes
The morphological and topological heterogeneity of pericytes translates into functional differences in vivo. Ensheathing pericytes and capillaries pericytes show distinct dynamics during neurovascular coupling, and in response to ischemic pathology and small vessel disease. In the normal brain , ensheathing pericytes are first responders to local neural activity, resulting in rapid dilation of the pre-capillary arteriole and influx of blood to the brain ’s capillary bed (Hall et al. 2014; Cai et al. 2018; Rungta et al. 2018). This dilation outpaces even the dilation speed of upstream SMC-covered arterioles, suggesting that ensheathing pericytes are critical sensors to neural activity and a central locus for blood flow control. In contrast, capillary dilations are absent or small in neurovascular coupling, and appear to be slower if present (Hill et al. 2015; Rungta et al. 2018; Kisler et al. 2017). As expected, intracellular calcium levels are strongly anti-correlated with diameter change in pre-capillary arterioles, suggesting engagement in classic actomyosin contractile machinery, but this relationship is less clear in capillary pericytes (Hill et al. 2015; Rungta et al. 2018). This likely indicates that a separate, slower mechanism of contraction and tone generation exists in capillary pericytes (Hartmann et al. 2020).
The response of these two pericyte subtypes is also strikingly different during brain ischemia. Capillary pericytes are particularly vulnerable to blood flow loss, dying from necrosis within tens of minutes to hours (Hall et al. 2014), undergoing pathological contraction (Yemisci et al. 2009), and producing matrix-metalloproteinase activity, which leaves breaches in the BBB (Underly et al. 2017). In contrast, ensheathing pericytes are more resilient, but still undergo a sustained pathological contraction at the pre-capillary-capillary interface, which impedes flow to the capillary bed even after successful re-cannulation of the occluded artery (Hill et al. 2015). There is also emerging evidence that pericyte subtypes behave differently during chronic pathologies, such as small vessel disease. In a mouse model carrying a mutation in collagen 4 (Col4) relevant to human small vessel disease, spontaneous hemorrhagic stroke occurs in deep brain regions. Interestingly, ensheathing pericytes (referred to as transitional pericytes in this study) proliferate and heighten their expression of contractile proteins in Col4 mutant mice (Ratelade et al. 2020), which does not occur with capillary pericytes or arteriolar SMCs. This is thought to produce hyper-contractility, specifically at the transitional precapillary zone, leading to buildup of intravascular pressure and rupture of upstream arterioles.
Even within the capillary zone, there may be further division of pericyte subtypes to be made, but this remains an open area of research. Under conditions of brain injury , some pericyte populations respond differently than others. For example, NG2-positive capillary pericytes can be either nestin-negative (Type 1) or nestin-positive (Type 2) under basal conditions. Following brain injury , Type 1 pericytes proliferate around the injury site and contribute to scar tissue, unlike Type 2 pericytes (Birbrair et al. 2014a). Type 1 pericytes differ from NG2-positive oligodendrocyte precursor cells, and from PDGFRβ-positive cells that might be perivascular or meningeal fibroblasts, confirming a distinct pericyte type. Further, when Type 1 or Type 2 pericytes are injected into brain tumor masses in vivo, Type 2 pericytes are preferentially recruited into the tumor and promote angiogenesis over a 5-week period (Birbrair et al. 2014b). In contrast, Type 1 pericytes do not have this effect, instead decreasing in number over the same time frame. Thus, there is mounting evidence that pericytes are heterogeneous in both the normal and diseased brain . Referring to all pericytes simply as “pericytes” is now insufficient, as there are subdivisions that require further delineation.
1.4 Targeting Brain Pericytes for In Vivo Imaging
1.4.1 Cre/Lox System for Genetic Targeting of Pericytes
The Cre/lox system is an effective way to achieve a stable fluorescent labeling of brain mural cells, when bred with Cre-dependent fluorescent reporter mice (Madisen et al. 2010). There are several Cre-driver options, each with advantages and disadvantages. The optimal choice of Cre-lox pairing will vary depending on the experimental requirements (Fig. 1.2b).

Targeting brain pericytes for in vivo optical imaging. (a) The NG2-DsRed mouse endogenously expresses DsRed under the NG2 promoter independent of Cre recombination. (b) Outline of commonly used Cre/lox breeding schemes for brain pericyte research. (c) Constitutive labeling of capillary pericytes (arrowhead) in the NG2-DsRed mouse, with off-target labeling of oligodendrocyte precursor cells (OPCs)(arrows). Pericyte processes are difficult to visualize in this line. (d) Constitutive labeling of mural cells (arrowhead) in NG2-Cre::Ai14 mouse. Developmental off-target labeling of OPCs and astrocyte progenitors results in high background fluorescence. (e) Tamoxifen-inducible NG2-CreER::Ai14. OPC labeling persists (arrows), but can be spatially distinguished from mural cells (arrowhead). Pericyte processes are more easily visualized with tdTomato. (f) Sparse labeling of mural cells with tamoxifen-inducible Myh11-CreERT2::Ai14. Notice extensive arterial smooth muscle cell labeling relative to capillary pericyte and venular mural cell labeling. (g) Tamoxifen-inducible sparse labeling of pericytes and other mural cells with Tbx18-CreERT2::Ai14. (h) Constitutive mural cell reporter expression in Pdgfrb-Cre::Ai3 mouse. (i) Membrane-localized tdTomato in all cell membranes and membrane-localized eGFP in capillary pericytes with the Pdgfrb-Cre::mT/mG mouse. No i.v. dye is present in mT/mG images. (J) High resolution image from Pdgfrb-Cre::mT/mG mouse, showing the gap between two adjacent pericyte processes (green), near a faintly visible endothelial cell body (arrowhead). Note how the mGFP label reveals thin sheet-like outcroppings of pericyte processes that are less visible with cytosolic fluorescent proteins (arrows). (k) Images from a Pdgfrb-Cre::Ai14 animal, 5 hours after cortical application of the capillary pericyte-specific dye, NeuroTrace 500/525
1.4.2 PDGFRβ-Cre
For constitutive labeling of all mural cells in brain vasculature, the PDGFRβ-Cre mouse line is a reliable and convenient line (Cuttler et al. 2011) (Fig. 1.2h, i). When these mice are crossed with fluorescent reporters (e.g. Ai14 for tdTomato), mural cell labeling is bright and contiguous throughout the cerebrovasculature, illuminating the entire spectrum of mural cells. Comparing overlap of tdTomato expression with CD13 immunolabeling, an established marker of pericytes, we find that 99% of CD13-positive pericytes are labeled in these mice (Hartmann et al. 2015; Damisah et al. 2017). PDGFRβ-Cre mice are thus very useful for studying the capillary pericyte network as a whole, as well as the interactions between adjacent mural cells under basal conditions and with pericyte manipulation. The PDGFRβ-Cre line is not appropriate for lineage tracing studies, however, because Cre is constitutively active and PDGFRβ expression occurs in non-mural cell types during early development (Guimaraes-Camboa et al. 2017).
PDGFRβ-Cre mice provide highly specific labeling for mural cells, but there is some off-target labeling. Indeed, recent transcriptomic studies have discovered PDGFRβ-expressing fibroblasts that occupy a perivascular location similar to that of mural cells (Vanlandewijck et al. 2018). Perivascular fibroblasts appear mainly on larger vessels, which could allow them to be confused with arteriolar mural cells or ensheathing pericytes. Another consideration in using the PDGFRβ-Cre line for in vivo imaging is the high expression of PDGFRβ in meningeal fibroblasts, which seem to increase in abundance following craniotomy, likely indicating a role of these cells in the fibrotic response to invasive surgery. The dense labeling of the meninges can sometimes make it difficult to obtain clean images of mural cells in pial vessels and pericytes of superficial capillaries. Furthermore, caution should be taken if one wishes to breed PDGFRβ-Cre mice to homozygosity because the location of the gene insertion is unclear.
1.4.3 NG2-Cre, NG2-CreER, and NG2-DsRed
Another widely used promotor to target brain mural cells pericytes is the Cspg4 promoter, which encodes the NG2 protein. This driver has the advantage of not labeling meningeal or perivascular fibroblasts as strongly as PDGFRβ-Cre (Vanlandewijck et al. 2018). However, along with smooth muscle cells and pericytes, several other cells are NG2-positive in development, notably astrocytes and oligodendrocyte precursor cells (OPCs) (Zhu et al. 2008) (Fig. 1.1d). However, it is possible to visually distinguish astrocytes and OPCs in the adult brain . In mice with constitutive Cre expression under the Cspg4 promoter (NG2-Cre), most mural cells are labeled. That being said, the background parenchymal fluorescence from neurons and OPCs may be too dense for some applications, such as those seeking to study pericyte morphology. However, one unexpected advantage found when crossing these mice with mice expressing a genetically-encoded calcium indicator was ectopic transgene expression by neurons, providing a convenient way to examine both neuronal and mural cell activity in unison during neurovascular coupling (Hill et al. 2015).
An NG2-CreER driver is available for tamoxifen-inducible labeling of brain mural cells (Huang et al. 2014). By administering tamoxifen at post-developmental timepoint, the problem of astrocyte progenitor labeling is reduced. OPC expression of NG2 occurs throughout the lifetime, so this off-target labeling remains even with the inducible form of the driver (Fig. 1.1e). Interestingly, a recent study showed strong correlation between OPC calcium transients and neuronal activity, again providing a convenient tool for perform neurovascular coupling, where the timing of local neural activity is crucial to measure (Rungta et al. 2018). As with all tamoxifen-inducible labeling schemes, the sparseness of fluorescent cells is titratable based on tamoxifen dose administered. With minimal tamoxifen (i.e. one i.p. injection of 100 mg/kg), extremely sparse labeling of mural cells can be achieved. This strategy allows for the morphologies of individual pericytes to be discerned (Grant et al. 2017). It also allows single mural cells to be tracked over time in vivo (Berthiaume et al. 2018b).
A related NG2 mouse line, the NG2-DsRed line, has contributed greatly to the in vivo study of brain pericytes (Zhu et al. 2008). They have been used widely for both in vivo and ex vivo imaging studies on blood flow control (Mishra et al. 2014; Hall et al. 2014; Grubb et al. 2020; Cai et al. 2018). In these BAC transgenic mice, DsRed protein expression is driven directly by the Cspg4 promoter, eliminating the need for Cre recombination and increasing the convenience of their use. Mural cells are visible by in vivo two-photon microscopy, and pericyte cell bodies can be easily visualized (Fig. 1.2c). However, the DsRed protein appears insufficiently bright to consistently visualize the fine processes of pericytes, compared to bigenic Cre-reporter mice expressing tdTomato. As with NG2-Cre mice, oligodendrocyte precursors are also labeled in NG2-DsRed mice.
1.4.4 Tbx18-CreERT2
The tamoxifen-inducible Tbx18-CreERT2 mouse line is an excellent tool for sparse-labeling of both smooth muscle cells and pericytes. This mouse line has been successfully utilized for lineage-tracing studies of pericytes in the adult brain . For example, one recent study used the mouse line to show that pericytes of the adult brain do not transdifferentiate into other cell types during brain injury (Guimaraes-Camboa et al. 2017). The tbx18 gene may be highly expressed in certain fibroblast populations, according to the RNA sequencing databank created by the Betsholtz group (http://betsholtzlab.org/VascularSingleCells/database.html), so any cell under study must be positively identified as pericytes through detailed morphological analysis. This also leads to fluorescence expression in some meningeal and perivascular fibroblast populations, though the labeling is not as dense as with the PDGFRβ-Cre mice. Unlike the NG2-CreER inducible labeling system, no parenchymal cells appear to be consistently labeled under this Cre driver. This makes the Tbx18-CreERT2 mouse line a relatively “clean” way to sparsely label pericytes for in vivo imaging (Fig. 1.2g).
1.4.5 Myh11-CreERT2
Another inducible mouse Cre line that has been used for brain pericyte labeling is Myh11-CreERT2 (Wirth et al. 2008). This promoter is not expressed in any non-mural cell type in the brain , including fibroblasts, and therefore leads to a highly specific mural cell label with no meningeal labeling. However, there is considerably greater expression levels of the myh11 promoter in mural cells of arterioles and pre-capillary arterioles compared to capillary pericytes (Betsholtz single cell RNA transcriptome database (Vanlandewijck et al. 2018)). This leads to the strong preferential labeling of mural cells on arterioles with low doses of tamoxifen (Fig. 1.2f). With added doses of tamoxifen, capillary pericytes will also begin expressing the designated reporter. Sparse labeling of capillary pericytes is easily achievable through this method, but the sparse labeling of smooth muscle cells or ensheathing pericytes of pre-capillary arterioles is unlikely due to more complete labeling of these cell types. A disadvantage of the Myh11-CreERT2 mouse line is that the BAC transgene carrying Cre recombinase is inserted within the Y chromosome, which limits reporter expression to only male progeny of crosses. This makes it impossible to run a study with a balanced number of male and female mice if using only this mouse line, which can be a problem when considering sex as a biological variable. Newer versions of the Myh11-CreERT2 line have managed to create a female mouse line to address this limitation, though it has not yet been utilized in brain pericyte research (Liao et al. 2017). In our hands, using the original Y-linked Myh11-CreERT2 mouse line, we have detected some leakiness of Cre activity in the absence of tamoxifen. Constitutive expression is seen specifically in brain smooth muscle cells and ensheathing pericytes when breeding with homozygous Ai14 reporter animals (unpublished observation). This “leaky” Cre is not an issue if the mice are used for cell visualization, but would be problematic if the goal is to modify gene expression in mural cells of the brain in an inducible manner .
1.4.6 Pericyte-CreER
A Cre-driver to specifically target capillary pericytes, while excluding α-SMA positive mural cells, is critical considering the distinct physiological roles of mural cell classes. Recently, a capillary pericyte-specific Cre line was generated by using a combinatorial strategy that requires target cells to express both Cspg4 and Pdgfrb (Nikolakopoulou et al. 2019). While both genes are expressed by smooth muscle cells and ensheathing pericytes, they are expressed at relatively lower levels than in capillary pericytes, reducing probability of Cre expression in α-SMA positive cells. When paired with a fluorescent reporter, this pericyte-CreER line labels capillary pericytes of the brain with remarkable specificity in a tamoxifen-inducible manner. The mural cells of pial/penetrating arterioles, and even pre-capillary arterioles are excluded from expression. As a newly developed mouse line, more characterization will be required from independent groups to fully test the functionality of the line under varying circumstances. In particular, whether pericyte-CreER also excludes mural cells of the veins and venules remains to be seen, as these mural cells express both Cspg4 and Pdgfrb at levels similar to capillary pericytes (Betsholtz single cell RNA transcriptome database (Vanlandewijck et al. 2018)). One application of the pericyte-CreER mouse using a diphtheria toxin strategy to cause mass deletion of brain capillary pericytes has already been implemented, confirming its functionality (Nikolakopoulou et al. 2019; Kisler et al. 2020).
1.4.7 Fluorescent Reporters and Actuators
The most common fluorescent reporters paired with the aforementioned Cre drivers are Ai14 (tdTomato) and Ai3 (eYFP), which are both incorporated into the Rosa26 locus and lead to high expression of their respective fluorescent proteins (Madisen et al. 2010). TdTomato generally performs better than YFP for in vivo two-photon microscopy, partially because red light is less easily scattered than yellow/green. However, the Ai3 YFP reporter can be strategically utilized when mice already have a red fluorescent label. Additional reporters can also be paired with any of the discussed Cre drivers to create useful phenotypes. For instance, the mT/mG reporter (Muzumdar et al. 2007) constitutively labels all cellular membranes with membrane-associated tdTomato. With the action of Cre, however, the targeted cell type will switch to expression of a membrane bound eGFP fluorophore. This phenotype is especially useful in cerebrovascular biology, as eGFP-expressing pericytes and tdTomato-expressing capillary endothelial cells can be visualized concurrently (Fig. 1.2i). The endothelial labeling appears bright relative to other parenchymal cells. Endothelial cell somata can also visualized, though with occasional difficultly due to the diffuse tdTomato expression from the brain parenchyma, (Fig. 1.2j). It is also worth noting that fine sheathes of capillary pericyte processes, beyond thin strands, can be resolved with mGFP labeling compared to cytosolic dyes such as tdTomato. Another useful reporter is Ai95, encoding the GCaMP6f protein (Madisen et al. 2010). This reporter has been successfully utilized to study pericyte fast calcium transients in the living mouse brain (Rungta et al. 2018). The Ai96 reporter (GCaMP6s) is also available to detect slow calcium transients in cells (Madisen et al. 2010), though this reporter line has not yet been directly used in a pericyte study. Looking ahead, an extensive array of next-generation fluorescent reporters have been under development in recent years using the permissive TIGRE locus (Daigle et al. 2018). These reporters generate even higher protein expression than the previous set of Rosa26 locus reporters. Finally, Ai32 mice expressing the original variant of ChR2-YFP have been crossbred with mural cell specific Cre drivers. These reporters have been used to query the contractile dynamics of smooth muscle cells and pericyte in vivo during two-photon imaging (Hill et al. 2015).
1.4.8 Viral Targeting of Brain Pericytes
Viruses are a valuable tool to target cells in vivo. They can be introduced at any time in the lifespan of the mouse, and into any brain region. Genetically engineered viral vectors can lead to high levels of ectopic gene expression while maintaining user safety, as any pathogenic genes of the virus are absent (Wang et al. 2018; Davidson and Breakefield 2003). Several parameters ultimately dictate the level of viral expression, including the volume, titer and penetration of the virus into the brain tissue, the efficiency of its uptake by the targeted cell type, and the activity of its promoter (Packer et al. 2013; Stoica et al. 2013; Lowery and Majewska 2010). The exclusivity of viral uptake to certain cell types is often problematic. Viruses targeting brain pericytes are not exempt from these challenges.
For use with in vivo optical imaging, one virus has been demonstrated to target brain mural cells. This virus is the adeno-associated virus 2/5 (AAV2/5) serotype carrying a Pdgfrb promoter for the expression of the GCaMP6s calcium indicator in vascular mural cells (Khennouf et al. 2018). The virus was microinjected near the somatosensory cortex region of mice. Its specificity was not limited to only mural cells, but also revealed punctate GCaMP6s expression throughout the parenchyma of the injection area with unclear specificity. However, positive expression in pericytes could be confirmed using a pericyte-labeling Cre mouse line in conjunction with the virus. This approach will undoubtedly prove useful as it alleviates a significant bottleneck in the genetic modification of pericytes in vivo for future mechanistic studies. However, specificity for capillary pericytes still needs to be improved. Other studies have also targeted brain pericytes using a viral approach, though none for in vivo optical imaging use. One group selectively manipulated pericytes with a virally expressed shRNA to silence Ephrin-B2 signaling in pericytes, by creating a AAV viral construct with the NG2 promoter (Coucha et al. 2019).
1.4.9 In vivo Dye Labeling of Brain Pericytes
Thus far, the methods discussed for targeting brain pericytes in vivo have relied on genetic approaches. Furthermore, most of these methods are unable to disentangle between capillary pericytes and other populations of mural cells, with the exception of the new pericyte-Cre driver. The use of a specialized dye is an excellent method to temporarily label capillary pericytes for in vivo imaging . Recently, Damisah and colleagues demonstrated that the green fluorescent Nissl dye NeuroTrace 500/525 labeled capillary pericytes with high specificity (Damisah et al. 2017). The preferential uptake of NeuroTrace 500/525 into capillary pericytes requires topical application or intracortical microinjection of the dye into the living mouse brain . Remarkably, only mesh and thin-strand pericytes seem to be labeled by this dye, and mural cells of both pre-capillary arterioles and penetrating arterioles, as well as downstream venules, appear to be excluded. The dye labeling appears several hours after its application, and remains for up to 3 days after. With topical application, dye penetration reaches at most 400 micrometers in cortical depth. Capillary pericytes that internalize the dye exhibit robustly labeled cell bodies and punctate labeling of their processes (Fig. 1.2k). The molecular transporters responsible for selective NeuroTrace 500/525 uptake in capillary pericytes remains unknown.
1.5 Visualization of Brain Pericytes In Vivo
1.5.1 Cranial Windows for in vivo Multi-Photon Imaging
To gain optical access into the living mouse brain , a cranial window must be implanted in the skull (Fig. 1.3). These windows can be created in adult animals, ranging from approximately 2 to 24+ months, with an increase in bone thickness, brittleness, and porosity (and thereby, surgical difficulty) with increased age. The choice of cranial window depends largely on the goals and timeline of the experiment. For example, some studies require long-term access with high imaging quality. Other studies require minimal perturbation of the intracranial environment. With each cranial window there are advantageous and disadvantages that should be carefully weighed during the development of the study.

Cranial windows for optical imaging. (a) Schematic of a mouse with implanted cranial window and metal flange for head fixation. (b) Acute cranial window design, with a layer of agarose separating the coverglass from the cortical surface. (c) Thinned-skull window, with cyanoacrylate glue to stabilize and bind the coverglass to the thin remaining layer of skull. (d) Chronic cranial window schematic, featuring a double coverglass “plug” to impede bone regrowth over time
1.5.2 Acute Cranial Window
For short experiments that require only a single imaging timepoint, an acute cranial window can be generated. This involves the removal of a circular piece of skull (diameter ~ 3–4 mm) over the desired location of mouse cortex. In experiments where cortical location is not critical, windows can most easily be placed over the mouse somatosensory cortex, which lies well within the bounds of the bregma and lambda sutures. The acute cranial window preparation involves the addition of a layer of transparent agarose to provide a buffer between an overlying cover glass and the brain (Shih et al. 2012a) (Fig. 1.3b). The edges of the cover glass are then sealed into place with dental cement. The agarose limits brain swelling and reduces motion artifacts caused by distension of the brain during breathing or heartbeat. Acute windows are best used within ~4 hours after implantation, as the window quality tends to decline over time, likely due to increasing inflammatory responses. An advantage of the acute window is that skull removal allows for the application of compounds directly to the brain before closing the window. This can be done by dissolving compounds into the agarose that sits atop the pial surface, or by leaving an edge of the window unsealed such that dyes or drugs can be injected with a micropipette. In more sophisticated preparations, cannulas for continuous inflow and outflow of fluid can be created as part of the implantation (Nishimura et al. 2010). The full craniotomy also allows optimal imaging depth and resolution since light scattering by the bone is minimized. A disadvantage of the acute cranial window is that the surgical process is invasive and disruption of the brain could affect the physiological processes being studied.
1.5.3 Thinned-Skull Window
A cranial window type which allows for reasonable optical penetration with reduced impact on the brain environment is the thinned-skull window (Xu et al. 2007), or the similar PORTS window (Shih et al. 2012b) that allows for longer term imaging through the thinned-skull. As the name implies, this style of cranial window does not breach the skull, instead leaving a thin layer of bone between the brain and the glass coverslip (Fig. 1.3c). The skull is thinned to a thickness of ~20 μm using a sharp scalpel blade (acute) or very stable hand-held drill (chronic), such as the Osada, EXL-M40. The amount of thinning required to achieve a ~ 20 μm thickness can be fine-tuned over separate surgeries followed by measurement of bone thickness using second harmonic signal generation (blue fluorescence) during two-photon imaging (Drew et al. 2010). In acute uses of this window, application of artificial cerebral spinal fluid to the thinned skull surface will reduce light scattering by the bone (Yang et al. 2010). Acute thinned-skull windows are kept small in part to reduce motion artifacts, which can still occur with distention of the thinned bone. For long-term imaging, the scalp is sutured between imaging sessions and re-opened on subsequent imaging sessions. The skull may regrow during extended intervals and must then be re-thinned for imaging. In a chronic PoRTs version of the thinned-skull window, a layer of cyanoacrylate glue is placed between the bone and cover glass (Shih et al. 2012b). The glue, which has a similar optical index as bone and glass, helps to reduce light scattering for improved light penetration. A second benefit is that the hardened glue preserves optical clarity for longer term imaging, removing the need to re-thin the window. It further adds rigidity to the thinned skull allowing creation of larger windows. In applications seeking to understand microvascular changes across brain regions, entire hemispheres of the skull have been thinned for meso-scale two-photon imaging (Mateo et al. 2017). Thinned skull windows reduce disruption to the underlying brain tissue, though recent studies have shown that brain temperature decreases similarly in thinned-skull and craniotomies for several days post-op (Roche et al. 2019). The maximal depth of imaging achieved through thinned skull windows is approximately 300 μm from the pial surface. Under optimal conditions, thinned-skull windows can be imaged for over a month (Drew et al. 2010), though quality is typically best within the first 7 days after implantation. Loss of imaging quality is usually attributed to new skull growth, which occurs gradually and prevents light penetration. Thinned-skull windows are ideal for experiments where any surgically-induced inflammation or brain swelling would compromise the validity of the study. Thinned skull windows are more difficult to create in older animals (9+ months) due to their more brittle and porous skulls. However, with greater care during surgery, studies can still be performed in aged mice using this approach (Watson et al. 2020; Hyacinth et al. 2019).
1.5.4 Chronic Cranial Window
For long-term experiments over weeks to months requiring optimal imaging resolution, chronic cranial windows are the preparation of choice. Chronic windows are allowed to fully heal for 2–3 weeks between the time of surgery and start of imaging, at which point glial reactivity returns to near baseline levels (Holtmaat et al. 2009; Goldey et al. 2014). While earlier versions of this window used only a single layer of glass, recent studies have implemented a double glass coverslip “plug” that helps to reduce bone regrowth from the edges of the window (Berthiaume et al. 2018b; Goldey et al. 2014) (Fig. 1.3d). These are made by gluing a small circular coverglass (3 mm diameter) onto a larger circular coverglass (4 mm) with optical glue. This plug is then placed into a circular craniotomy that just fits the smaller coverslip, leaving the edges of the larger coverslip to sit atop the skull surface. This plug is then sealed into place with dental cement. For studies requiring a wider visual field, a 4 mm diameter window can also be created by combining a 4 mm and 5 mm round coverglass as the plug. The intracortical injection of a virus pairs well with the creation of a chronic cranial window. The virus can be micro-injected directly into the brain before the glass plug is inserted, and the 2–3 week surgical recovery period can simultaneously provide necessary viral incubation time. This also guarantees that the virally transduced region is within the cranial window. A chronic cranial window with an adjacent access port for the administration of desired compounds can also be created, thereby combining the major benefits of both a chronic and acute cranial window into one preparation (Roome and Kuhn 2014). This allows for longitudinal imaging sessions interleaved with cortical drug applications or intracortical injections to study the long-term effects of various pharmacological interventions. Chronic cranial windows can generally be imaged for many months, though this varies depending on initial quality of surgery (Holtmaat et al. 2009).
1.5.5 Anesthesia during Surgery and Imaging
The method of surgical anesthesia should be chosen based on cranial window type. For chronic cranial windows, fentanyl anesthesia is a good choice. Dexamethasone (4.8 mg/kg, s.c.) is typically administered 4 hours prior to surgery to minimize inflammation and edema (Goldey et al. 2014). Then a cocktail composed of fentanyl citrate (0.05 mg/kg), midazolam (5 mg/kg), dexmedetomidine (0.25 mg/kg) is delivered intraperitoneally. Both dexamethasone and the fentanyl cocktail help to reduce brain swelling upon removal of the skull, which can otherwise lead to compression of the cortex when the cover glass is overlaid. Following surgery, animals require the administration of a “wake-up cocktail” consisting of buprenorphine (0.1 mg/kg), flumazenil (0.5 mg/kg), antisedan (2.5 mg/kg), meloxicam (5 mg/kg), lactated Ringer’s solution (1-2 ml/25 g mouse). After a 2–3 week period of recovery mice can be imaged in the anesthetized state, or awake state if properly habituated to head fixation. For acute cranial windows, imaging immediately follows surgery, and the choice of anesthetic depends upon the goal of the experiment. If stable structural features of the brain will be examined, such as pericyte or microvascular structure, it is be convenient to use isoflurane anesthesia throughout both surgery and subsequent imaging phase. Surgery would require a higher isoflurane concentration (2% MAC), while isoflurane should be minimized to 0.8-1.5% MAC during imaging. However, if vascular dynamics such as blood flow and neurovascular coupling will be measured, isoflurane is not ideal. Instead, surgery can be performed with first with isoflurane, and the animal can then be transitioned to alpha-chloralose anesthesia or chlorprothixene sedation, which better preserve neurovascular responses (Masamoto and Kanno 2012). While isoflurane is more convenient to use for achieving surgical-plane anesthesia, it is a vasodilator that can worsen brain swelling following a craniotomy. However, less invasive procedures such as thinned-skull or PORTS windows can be generated using isoflurane anesthesia without the concern of brain swelling as the skull is not completely breached.
For imaging of pericyte or capillary structure, as well as BBB leakage, the use of light isoflurane anesthesia (0.8–1.5% MAC) is necessary to obtain very stable, high-resolution imaging. Anesthesia may also be appropriate for studying slower aspects of blood flow change that might take place over days, such as those seen after optical ablation of pericytes (Berthiaume et al. 2018b). However, dynamic changes in blood flow or pericyte activity evoked by neurovascular coupling are ideally performed with an awake, habituated or lightly sedated subject. This is because anesthesia significantly dampens the neuronal activity and the neurovascular response (Gao et al. 2017). Awake-imaging involves the habituation of the mouse to head restraint in the days prior to an imaging experiment (Shih et al. 2014). Enclosures, such as hollow tubes, can be utilized to dampen the natural fear response of mice to open spaces. Other strategies include spherical treadmills for the animal to walk on at will during the course of imaging (Dombeck et al. 2007). Once habituated, mice do well under these awake-imaging conditions, and high quality two-photon data can be obtained. Generally speaking, the field is moving towards using awake-imaging as the standard for all two-photon imaging experiments, as this state of consciousness likely gives the most authentic results (Grutzendler and Nedergaard 2019; Gao et al. 2017). However, light sedation using drugs such as chlorprothixene remain a reasonable compromise if study results would be significantly altered by any movement artifact, as is often the case when imaging the characteristics of structures as small as capillaries (Grutzendler and Nedergaard 2019).
1.5.6 Head-Mounting/Stabilization
Stabilization of the mouse’s head is necessary for both the cranial window surgery and for optical imaging. Head fixation can be achieved with a custom-made head-mount setup (Fig. 1.3a). This is usually an adjustable structure that allows the angle of the mouse head to be leveled to the imaging plane, and then fixed in position to prevent further movement. Proper head fixation is critical to minimize motion artifacts caused by breathing or heartbeat during acquisition of high-resolution optical images. The head fixation apparatus is also useful to maintain a similar head positioning for the same mouse on each subsequent imaging day. This facilitates longitudinal imaging studies where exact locations need to be repeatedly imaged and compared over time.
1.5.7 In vivo Two-Photon Microscopy
Once a cranial window has been generated, and the appropriate post-operative recovery period has passed, optical imaging experiments can begin. Two-photon microscopy has been revolutionary for the advancement of neuroscience by allowing visualization of cellular and subcellular processes in vivo, while minimizing photodamage to the delicate brain tissue (Denk et al. 1990). Two-photon imaging has significantly advanced cerebrovascular research, with the ability to visualize and quantify aspects of the dense vascular networks of the brain , their cellular components, and their physiological dynamics (Shih et al. 2012a).
1.5.8 Physiological Variables
It is important to maintain an animal’s health during in vivo imaging. A pulse oximeter can be used to obtain a constant read out of an anesthetized animal’s heart rate and breathing rate while enclosed within the light-tight enclosure of the microscope. Some studies require full control of physiological variables including implantation of femoral artery catheters for continuous blood pressure measurement, and occasional blood gas sampling. Tracheal tubes are also inserted for mechanical ventilation, which helps to ensure a normal breathing rate and proper balance of blood gases (Grubb et al. 2020). However, these implantation procedures are invasive and therefore reserved for acute imaging studies. A feed-back regulated heat pad is necessary to ensure body temperature at normal levels of ~37 °C. Brain temperature is also important to consider, as it can be drastically altered immediately beneath a craniotomy (Kalmbach and Waters 2012). The use of water-immersion microscope objectives poses some additional problems, as room-temperature water has been shown to decrease the temperature of the cortex directly beneath the window, which can affect both blood flow and tissue oxygenation (Roche et al. 2019). The use of objective heaters helps to address this issue.
1.5.9 I.v. Dyes as Vascular Labels
The intricate cerebrovascular networks of the mouse brain can be labeled by a single intravenous (i.v.) injection of fluorescent-dextran dye. Since the i.v. dye labels the blood plasma, any perfused vessel will fluoresce during two-photon imaging. The choice of i.v. dye depends upon which imaging channels are already occupied by genetically expressed fluorophores. The conjugated dextran must also be of sufficient molecular weight so that it is not filtered from the blood stream too quickly. In practice, dextran conjugates at or above 70 kDa are sufficient for hours of imaging. Some common commercially available dye choices are fluorescein isothiocyanate (FITC)-dextran (70 kDa-2 MDa), tetramethylrhodamine (TMR)-dextran (500 kDa), and Texas Red-dextran (70 kDa). Recent studies have also provided protocols to custom conjugate fluorophores to dextran, as was done for Alexa fluor 680 in a deep two-photon imaging study (Li et al. 2020). Ideally, the i.v. dye and fluorophore used to label pericytes in each experiment would have similar two-photon excitation cross-sections, enabling simultaneous imaging of pericytes and vascular structure and flow. For example, an experiment to visualize tdTomato in pericytes and FITC-dextran simultaneously could use a 975 nm excitation wavelength, and capture emissions in the red and green channels, respectively. Alternatively, if pericytes were to express YFP, a Texas Red-dextran could be used and both fluorophores imaged at 900 nm for green and red emission. However, sequential imaging with different excitation wavelengths can also done if the two-photon cross sections of the labels are too dissimilar.
1.5.10 I.v. Dyes as Vascular Permeability Probes
Vascular permeability metrics are often included in pericyte studies seeking to investigate their involvement in blood-brain barrier (BBB) function. For these studies, the molecular weight of the chosen dextran is an important consideration. A dextran weighing ~70,000 Da will be peripherally extravasated from the blood stream within a few hours, but is of an ideal size to test BBB integrity (Armulik et al. 2010). Several blood plasma proteins established as appropriate surrogates for BBB leakage, such as albumin, are almost identical in molecular weight. A dextran of this size may accumulate in the brain parenchyma outside of vessels for minutes to hours after extravasation, and can be readily visualized with two-photon microscopy (Fig. 1.4d). The smallest fluorophores, such as Alexa fluor 488-Cadaverine, are ≤1000 Da and are filtered from the blood stream within minutes. These can only be visualized around the time of bolus injection in vivo (Berthiaume et al. 2018b; Lou et al. 2016) or in tissue slices following fixation (Armulik et al. 2010). This method is useful for detecting small, local breaches in the BBB that involve rapid dye extravasation. When using the smallest molecular weight dyes, it is often necessary to introduce an additional high molecular weight dye to the bloodstream to assess both vessel structure and permeability within one imaging session.

Vascular metrics acquired with in vivo two-photon imaging. (ai) Capillary in maximally-projected image stack with cross-lines used for acquisition of average diameter measurement. (aii) Normalized fluorescence intensity plotted for a single capillary cross-line, used for calculation of full-width at half maximum capillary diameter. (aiii) Intensity plots of full dataset for cross-lines 1–37 from (ai). (aiv) Plot of diameter deviations along the length of a single capillary segment with average and standard deviation of diameter reported. (b) Demonstration of 3D capillary length measured using the ImageJ plugin Simple Neurite Tracer (Longair et al. 2011). (ci) Blood cell velocity is measured using high-speed line-scans during two-photon microscopy. The location of line-scanning is shown for collection of blood cell velocity and flux from two capillaries within the field of view. (cii) Raw image output from the line-scans marked in (ci). Dark bands are generated by passing blood cells that exclude the fluorescent dye, while light bands are i.v. dye within the blood plasma. The number of dark bands over time is used to determine blood cell flux in cells per second. The angle of the streaks is used to calculate the speed of blood cell flow, with streaks closer to the horizontal corresponding to faster flow speed. (ciii) Blood cell velocity in millimeters per second plotted as a function of time for each of the scanned capillaries. (d) Leakage of a 70 kDa i.v. dye immediately following capillary injury . (di) Quantification of fluorescence intensity within a region of interest (ROI) in (d) at pre-injury, 5 minutes post-injury, and 3 days post-injury timepoints
1.5.11 Vascular Characteristics and Dynamics
Pericyte control of cerebral blood flow is a very active area of research. Two-photon imaging is uniquely suited to study this process because it enables the real-time detection of blood flow changes at the level of arterioles, capillaries and venules. This type of imaging also allows for the quantification of vascular structure characteristics such as vessel length and diameter. Vascular length should be measured in three dimensions whenever possible, using a tool such as the ImageJ segmentation analysis tool Simple Neurite Tracer (Longair et al. 2011) (Fig. 1.4b). This allows the full length of the vessel in the z axis to be taken into consideration, rather than only the x and y planes. The diameter of a vessel can be obtained by measuring the full-width at half maximum of a fluorescence intensity profile taken across the width of the capillary (Driscoll et al. 2013) (Fig. 1.4ii). Since brain capillaries are often heterogeneous in diameter even within the same capillary segment, recent studies have also developed strategies to collect diameter at multiple regions along the vessel (Watson et al. 2020; Ivanova et al. 2017) (Fig. 1.4ai). The average of these measurements can then be obtained and reported as an unbiased measure of capillary diameter, while also providing a metric of the variability of diameter along the length of a vessel segment (Watson et al. 2020) (Fig. 1.4aiii, aiv). Blood flow measurements, including blood cell flux and velocity, can be measured at the level of brain capillaries. Since i.v. dyes are excluded by blood cells, they appear as dark shadows within the fluorescent blood plasma. A two-photon line scan through the central axis of the vessel length will reveal moving blood cell shadows as streaks within a resultant line-scan image (Fig 1.4 ci, cii). The number of streaks over time is the flux of blood cells, typically reported as cells per second. The angle of the streaks is used to calculate the velocity of blood cell flow, typically reported as millimeter per second (Shih et al. 2012a; Kleinfeld et al. 1998). Convenient algorithms have been developed to aid extraction of blood cell velocity from line-scan images (Kim et al. 2012; Chhatbar and Kara 2013) (Fig 1.4ciii).
1.6 In Vivo Observation of Pericyte Structure and Function
There exists a spectrum of mural cell morphologies in cortical vasculature, with pericytes being subcategorized into groups based on morphology and α-SMA expression. As capillary pericytes (>4th order from a penetrating vessel) are the most abundant in the mouse cortex, these populations will be the focus of the following section. When targeting the capillary pericyte network for in vivo optical imaging studies, a combination of cell morphology and branching distance from a penetrating arteriole can be used to confirm mural cell identity (Grant et al. 2017).
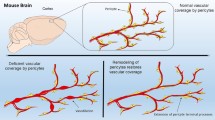
The unique structure of capillary pericytes, and how this structure relates to their in vivo function, has been a question of great interest in past decades. With constitutive genetic labeling, pericytes of the capillary bed form a contiguous network. The discrete domains of individual pericytes may be difficult to discern, but careful inspection reveals occasional small gaps between neighboring pericytes processes, ranging from only a few micrometers in width. The basal growth of individual pericyte processes can be tracked over time, if the boundaries between neighboring cells are sufficiently visible (Berthiaume et al. 2018a). Long-term tracking of these pericyte gaps has shown that pericytes slightly adjust their territories along the capillary bed over time (Fig. 1.5c). This repulsion between neighboring pericytes maintains a non-overlapping chain of pericytes along the capillary bed. The specific mechanisms driving this pericyte-to-pericyte repulsion have yet to be described, but may involve EphrinB2 signaling (Berthiaume et al. 2018a; Foo et al. 2006). In instances where capillary pericytes are lost or damaged, the modulation of this cellular repulsion could play a role in triggering a growth response of the remaining capillary pericytes.

Pericyte structural and functional characteristics. (a) Schematic of methods for quantifying pericytes relative to the capillary endothelium. (Left) Pericyte abundance is quantified as number of pericyte somata over vessel length. (Center) Pericyte contact is the ratio of total pericyte length in a volume over total vessel length. (Right) Pericyte coverage, typically assessed with high resolution imaging of histological sections, is the ratio of total pericyte area over total vessel area. (b) Image of a single capillary pericyte from a Myh11-CreERT2::Ai14 mouse, with measurements for individual and total process lengths, measured in 3D with ImageJ plugin Simple Neurite Tracer (Longair et al. 2011), and soma diameter. (c) Tracking neighboring capillary pericyte territories over time. Inset shows a shift in original boundaries of processes (yellow lines) over 77 days. Image from a Myh11-CreERT2::Ai14 mouse. Adapted from Berthiaume et al. 2018 (Berthiaume et al. 2018a). (d) Super-resolution confocal image of fixed tissue from Pdgfrb-Cre::Ai14 animal. Thin-strand pericyte processes, pseudocolored green, shows serrated-liked edges. Immunostaining for collagen IV, pseudocolored red, labels the basement membrane associated with the underlying capillary . (ei) Mesh pericyte imaged with in vivo two-photon microscopy in a Pdgfrb-Cre::Ai14 mouse. (eii) The pericyte imaged in vivo was re-located post-mortem in fixed tissue and imaged with super-resolution confocal microscopy, revealing the intricate substructure of pericyte processes that is unresolvable with in vivo two-photon imaging. (f) Image of a thin-strand pericyte from a NG2-GCaMP6f mouse. Numbers indicate regions of interest within a single pericyte process analyzed along the line-scan (yellow). (g) Spontaneous spikes in calcium transients plotted over time, separated by ROIs outlined in (f). (Reproduced with permission from Rungta et al. 2018 (Rungta et al. 2018))
Mice with inducible control of Cre activity provide sparse labeling of individual capillary pericytes, allowing their individual structures to be examined. Many structural characteristics of individual pericytes can be quantified from in vivo two-photon imaging data, including: (i) the location of their somata, (ii) the size of the territory (the summated length of capillary contacted by a single pericyte ), and (iii) various aspects of pericyte processes, such as their number, length, complexity and branching pattern. Pericyte somata can be found both at capillary junctions and on capillary segments between junctions. Interestingly, pericyte somata are more common at junctions (~50% of all capillary pericytes in cortex) (Hartmann et al. 2015), suggesting that their occurrence may support physiological roles, such as blood flow control at capillary bifurcations. Abundance of capillary pericytes can be quantified over time or under different experimental conditions by counting the number of pericyte somata within a cortical volume (Fig. 1.5a). When tracked over days to months under basal conditions, pericyte somata are found to be structurally fixed and immobile (Berthiaume et al. 2018b; Cudmore et al. 2017).
The extensive processes of capillary pericytes present another important feature of these cells. More than 90% of the total length of capillaries in cortex is contact by pericyte processes, as opposed to their somata, pointing to the importance this cell compartment in homeostatic function (Underly et al. 2017). Imaging of individual pericytes has revealed that capillary pericytes extend between 2 to 3 primary processes, which are the processes growing directly from a protruding ovoid cell body. These processes can further bifurcate into secondary processes, creating branching patterns that can span hundreds of micrometers of total length per pericyte (Hartmann et al. 2015). Pericyte process length can be assessed in 3D using the ImageJ segmentation plugin “Simple Neurite Tracer,” (Longair et al. 2011) and are measured from the point they emerge from the soma to their terminal tips (Fig. 1.5b). On average, a single capillary pericyte will contact 250 ± 41 μm of total vascular length in the adult mouse somatosensory cortex, though there is a large range in pericyte territory size, with some cells contacting over 300 μm of contiguous capillary length while other nearby pericytes contacting less than 200 μm (Berthiaume et al. 2018b). Super-resolution imaging has revealed detailed sub-structure in pericyte processes (Fig. 1.5d). While there appears to be one or two major strands that run along the capillary length, occasional lamellar sheaths form between these strands to create a more complex footprint on the underlying endothelium (Hartmann et al. 2015). Near the arteriolar or venular poles of the capillary bed, the processes of mesh capillary pericytes are even more extensive, forming intricate networks over the capillary wall. It is important to note that the resolution limits of in vivo two-photon microscopy preclude the examination of this intricate cellular sub-structure (Fig 1.5ei, eii).
Vessel “coverage” by capillary pericytes is often a reported metric in pericyte studies. However, it should be noted that there are discrepancies in the way this characteristic can be measured with optical imaging in vivo versus in fixed tissue. Pericyte “coverage” of the underlying vasculature in fixed tissue is typically quantified as the ratio of pericyte-labeled area over vascular-labeled area, using fluorescence signals from genetic expression or immuno-staining of 2-D images (Fig. 1.5a). This same principle has not yet been applied to 3-D imaging datasets obtain by two-photon imaging, but is in theory possible, especially if using membrane-bound fluorophores that better show the lamellar outcroppings of pericyte processes (i.e. mT/mG reporters). However, it can also present some challenge, as the resolution in the z plane is not as high as x-y planes. As discussed above, the extensive processes form a non-overlapping chain across the capillary network. Thus, an alternative and simple metric to collect is pericyte “contact”, i.e., total capillary length contacted by any portion of a pericyte . This can then be expressed as a proportion of total capillary length to reveal the percentage of capillaries contacted by pericytes. There are many disease conditions and mouse models in which pericyte loss occurs. This would lead to breaches in the capillary pericyte chain and could be captured using the pericyte “contact” metric (Fig. 1.5a). Quantifying length rather than area has the added advantage of supporting measurements in three dimensions, which leverages the 3-D data obtained by two-photon microscopy.
Longitudinal two-photon imaging enables researchers to track features of pericytes in order to gauge structural stability or reactivity to different experimental conditions. Pericytes are not at their maximum size under basal conditions, as they are able to extend their processes to cover a larger territory when challenged with loss of a neighbor (Berthiaume et al. 2018a). This structural plasticity of capillary pericytes can be measured with in vivo two-photon imaging by tracking the same cortical volume over time. Changes to pericyte structure occur on the timescale of days to weeks, requiring cell volume changes (Berthiaume et al. 2018b). This is in contrast to other cell types such as microglial cells that can change shape and migrate on the order of minutes, by re-distributing their existing cell volume. Capillary pericyte remodeling appears to be important for homeostatic functions of brain capillaries, such as blood flow regulation, though the full scope of its physiologic relevance needs to be better delineated. The loss of pericyte coverage or number, as seen in humans and animal models with small vessel disease (Halliday et al. 2016; Park et al. 2014), likely involves a complex interplay between pathological influences leading to decreased pericyte coverage, and reparative pericyte growth to attempt recovery of pericyte coverage. Small vessel disease models suggest that pericytes can also become mal-distributed along the capillary bed. In a mouse model of cerebral amyloid angiopathy, there are larger gaps between adjacent pericytes due to shorter processes (Park et al. 2014). Conversely, in Col4a1 mutant mice that develop spontaneous hemorrhage, mural cells in the transition zone between arterioles and capillaries are packed more tightly leading to hypermuscularization and increased upstream pressure (Ratelade et al. 2020). Both conditions may lead to abnormalities in capillary homeostasis.
Along with pericyte structure, other aspects of pericyte function can also be studied in vivo using two-photon imaging. Pericyte function can be inferred from metrics collected from their adjacent microvessels, such as vessel diameter, blood flow, and BBB integrity. Pericytes control these aspects of vascular function and correlative studies can be performed at the level of single pericytes and capillaries. Recent studies have expanded to the examination of pericyte calcium activity in vivo in relation to neurovascular coupling. With the NG2-CreERT2 mouse line crossed with GCaMP6f reporter mice, it is possible to study moment-to-moment changes in intracellular calcium in pericytes using two-photon imaging (Rungta et al. 2018) (Fig. 1.5f, g). Pericyte neurovascular coupling studies using the NG2-GCaMP6f mouse also benefit from the off-target labeling of OPCs. Because OPC activity has been shown to correlate with neural activity, increased calcium in OPCs can serve as a surrogate for neural activation (Rungta et al. 2018). Using this method, Rungta and colleagues demonstrated that calcium spikes occurred in small, seemingly disjointed compartments of pericyte processes, with no evidence of synchrony or wave-like signal propagation throughout the cell during functional hyperemia (Rungta et al. 2018). In another approach, an adeno-associated virus (AAV2/5) has also been used to transduce pericytes with GCaMP6 transgene for in vivo calcium imaging, with GCaMP6s expression driven under activity of the PDGFRβ promoter (Khennouf et al. 2018). Khennouf and colleagues reported a decrease in cytosolic calcium in pericytes located on dilated vessels during whisker stimulation. Conversely, cortical spreading depression-mediated capillary constriction led to increased intracellular calcium in associated pericytes. As demonstrated by these two studies, cutting-edge calcium imaging approaches can be used to investigate both basal activity of pericytes as well as their response to pathology. For now, the importance of pericyte calcium activity in vivo remains obscure and requires further investigation.
1.7 In Vivo Manipulation of Pericytes
Given that pericytes live within a complex vascular network and continuum of related cells, understanding their functional roles in vivo requires cause-and-effect approaches to experimentally manipulated pericytes in adulthood. While studies with pericyte-specific manipulation are still rare, a few strategies have emerged to either ablate pericytes in the adult brain or to stimulate them. By ablating these cells, we gain a better understanding of the consequence of pericyte loss on the remaining neurovascular unit. Pericyte loss can be achieved on a large scale with partial or global pericyte ablation, or on the much smaller scale of individual pericytes. These two scales of pericyte manipulation can serve complementary roles in investigations of how pericyte loss contributes to cerebrovascular disease. The strategic stimulation of pericytes in vivo, on the other hand, can help our understanding of how capillary pericytes perform their functions in the context of normal brain health, for example in the regulation of capillary blood flow.
1.7.1 Genetic Pericyte Ablation
Ablation of brain pericytes can be accomplished with one of several approaches, depending on the degree and timing of pericyte loss required. There exists an abundance of work using congenital knockouts targeting important signaling components for pericyte recruitment in the developing vasculature. When created as heterozygous mutants, many of these pericyte-deficient mice are able to survive into adulthood. Some of these lines, such as the PdgfrβF7/F7 mice feature an age-related decrease in pericyte numbers, which may mirror some aspects of pericyte loss in age-related human diseases (Nikolakopoulou et al. 2017). One limitation of these congenital ablations is the loss of pericytes from the earliest developmental stages. Pericytes are involved in angiogenesis and formation of a stable vascular network, and vascular abnormalities resulting from reduced pericyte number would create differences in vasculature during adulthood. This is especially true because PDGFRβ mutations do not have effects exclusive to capillary pericytes, but may also affect the function of other cells including smooth muscle cells and neurons (Ishii et al. 2006). With this caveat in mind, pericyte-deficient mice are easily imaged with two-photon microscopy and have been used to show increased BBB disruption, cerebral hypoperfusion, white matter degeneration, and impairment of neurovascular coupling with pericyte loss (Kisler et al. 2017; Montagne et al. 2018; Bell et al. 2010).
A recent study developed an approach to selectively ablate capillary pericytes en masse in the normally developed adult brain . By using the previously described pericyte-CreER mouse line (Nikolakopoulou et al. 2019), non-specific targeting of other cell types is eliminated, including closely related smooth muscle cells of arterioles. When pericyte-CreER::Ai14 are combined with a line encoding an inducible diphtheria toxin receptor (iDTR), DTR expression is exclusive to capillary pericytes (Nikolakopoulou et al. 2019). This method allows for titratable levels of pericyte loss through adjusting the dose of either tamoxifen (to induce DTR expression in Cre-expressing cells) or diphtheria toxin (DT; to induce apoptosis in these cells). At the highest doses administered in this study, a 60% reduction in capillary pericytes was achieved from cortical microvasculature over the following days to weeks. The result of this rapid capillary pericyte loss was brain circulatory failure due to endothelial leakage and edema, which leads to loss of neurovascular coupling and eventually neuronal viability (Nikolakopoulou et al. 2019; Kisler et al. 2020). The administration of lower doses of DT intermittently over weeks to months could reveal a distinct phenotype to model more gradual pericyte drop-out.
1.7.2 Two-Photon Pericyte Ablation
Two-photon optical ablation of individual pericytes is a second approach to ablate pericytes in vivo. The method is tightly coupled with two-photon imaging of the vasculature, and relies on the precise targeting of pericyte somata with two-photon laser light to created localized thermal damage at the pericyte soma, inducing necrotic cell death (Fig. 1.6a). Thermal ablation of single or multiple pericytes is distinct from genetic approaches because it creates localized pericyte loss in an otherwise unperturbed vascular network. This allows researchers to study the effect of pericyte loss on local microvascular responses without disrupting upstream or downstream perfusion. Pericyte loss also occurs with no detectable disruption to the blood-brain barrier (Berthiaume et al. 2018b), which helps to isolate the effects of pericytes on blood flow from complex pathological influences. Further, optical pericyte ablation circumvents the complicating factors of pericyte loss in other tissues and organs that might indirectly influence brain function when using genetic ablation. Two-photon pericyte ablation has been used to study pericyte structural plasticity in reparative responses to maintain coverage of the endothelium in the event of pericyte loss (Berthiaume et al. 2018b) (Fig. 1.6c, d).

Ablating pericyte in vivo with two-photon microscopy. (a) Image of capillary pericytes from NG2-CreER::Ai14 mouse, with pericyte targeted for two-photon thermal ablation encircled in yellow. Thermal ablation line-scan is limited to pericyte soma only. Post-ablation image reveals the immediate disappearance of the targeted pericyte within minutes of ablation. Adapted from Berthiaume et al. 2018 (Berthiaume et al. 2018b). (b) Off-target control for two-photon thermal ablation involves creating a line-scan path in a perivascular location of similar distance to the capillary wall, but devoid of a pericyte cell body. Similar laser powers and time of irradiation must also be used for controls. (c) Longitudinal time-course of response to thermal ablation of two capillary pericytes. Asterisks indicated targeted cells. Both cells are successfully ablated, as determined by their complete disappearance 5 minutes post-ablation. Arrowheads mark the original boundaries of neighboring pericyte processes. Over the course of 7 days, neighboring pericytes extend their processes (arrowheads) into the uncovered capillary area to re-form a contiguous pericyte chain. Arrowheads of the same color denote the process tips that split from an original single process tip of a neighboring pericyte . (d) Change in pericyte process length plotted over time under basal conditions, observed at gaps between neighboring pericytes processes (gray), or following thermal ablation of a pericyte neighbor (red). Adapted from Berthiaume et al. 2018 (Berthiaume et al. 2018b). (e) Image from NG2-CreER::mT/mG mouse demonstrating the 2Phatal method of inducing apoptotic pericyte death over the course of days following laser exposure. (Reproduced with permission from Hill et al. 2017 (Hill et al. 2017))
One drawback of two-photon thermal ablation is that pericytes can only be eliminated in a restricted volume brain volume, typically less than ~200 μm in depth below the pial surface in the cranial window. Additionally, the ablation process can cause off-target damage to the surrounding tissues and extensive controls must be added to understand these effects. Some pericyte cannot be accurately targeted without damaging the blood vessel because they are not oriented in a way that allows for selective ablation. For pericytes genetically labeled with tdTomato, the ideal thermal ablation settings is between 30 and 50 mW of laser power at a wavelength of 725 nm (Berthiaume et al. 2018b). A line-scan is restricted to the pericyte soma and held for 30–90 seconds. In a successful ablation, the tdTomato signal specifically from the targeted pericyte is lost and there is no breach of the underlying capillary , which would indicate endothelial injury . Overt indications of off-target damage, such as photobleaching of mural cells in z-planes above or below the area or the creation of microbleeds from damage to vessel walls, indicate a need to reduce laser power. Ablation settings should be evaluated on a case-by-case basis. Factors such as increased depth of the soma below the pial surface and reduced quality of the cranial window may necessitate higher laser powers. Thermal ablation experiments should always be accompanied with laser-irradiation controls to understand the effect of laser exposure alone. This can be done by administering laser light with settings identical to those used for thermal ablation but to a perivascular area not containing a pericyte cell body (Fig. 1.6b).
To avoid some of the limitations of two-photon thermal ablation (unintended thermal injury to other cells, necrotic cell death), an alternative method of two-photon laser ablation of individual pericytes was developed, named 2Phatal (two-photon chemical apoptotic targeted ablation) (Hill et al. 2017). This technique relies on the administration of the nuclear-binding fluorescent dye Hoechst 33342 (H33342) to living animals, which generates deadly levels of reactive oxygen species (ROS) when selectively activated with 775 nm two-photon laser light. The apoptotic cascade triggered by this reaction causes progressive cell death over the course of several hours to days (Fig. 1.6e). The administration of H33342 via systemic injection or direct cortical application results in the entry of the dye indiscriminately into nuclei of several cell types. Cell specificity in ablation is achieved by controlling the area of two-photon laser irradiation. Capillary pericytes can be identified by pairing any mouse line with labeled pericytes with this dye. This method of induced cell apoptosis has the advantage of requiring shorter laser exposure time to trigger cell death than traditional thermal ablation (775 nm at ~21 to 45 mW of power for 5–20s). Reduced local microglial activation is also reported with 2Phatal (Hill et al. 2017). Further studies will be required to better understand how the presence of the DNA binding agent affects cells during routine two-photon imaging. Since all nuclei contain the DNA dye, any off-target laser light in or out of the plane of focus could create low levels of ROS. Though observational imaging would not be strong enough to directly kill cells, any incidental diffuse damage to the imaging area may affect normal brain and cerebrovascular function.
1.7.3 Two-Photon Optogenetic Manipulation of Pericytes
Optogenetic stimulation provides a means to reversibly depolarize mural cells in vivo. The approach was first applied to pericytes when Hill et al. used channelrhodopsin (ChR2) to depolarize brain pericytes in NG2-CreER::Ai32 mice in vivo (Hill et al. 2015). They found that ChR2 stimulation caused contraction of SMCs, but not of capillary pericytes, supporting a conclusion that capillary pericytes do not regulate cerebral blood flow.
The study by Hill and colleagues used one relatively low ‘dose’ of light stimulation with short stimulation times to test the contractile ability of capillary pericytes. As recent studies have shown that stimulating laser intensity and duration determine the extent of ChR2 activation and mural cell contraction (Wu et al. 2015; Rorsman et al. 2018), we revisited this experiment using different stimulation parameters. In PDGFRβ::Ai32 mice, we carefully counted branch orders (e.g. >4th) to ensure that we examined capillary pericytes (Fig. 1.7a). Though PDGFRβ-Cre mice also target arteriolar SMCs, the activation of ChR2 occurs preferentially at the focal plane, which minimizes optogenetic perturbation of arterioles (Fig. 1.7b). Specialized line-scans can be used to simultaneously stimulate ChR2 and collect vasodynamic data, including diameter, blood cell velocity and flux (Fig. 1.7c). Using higher laser powers and longer illumination times (60 s) than used by Hill et al, we found that optogenetic activation of capillary pericytes can indeed reduce capillary diameter and blood flow in PDGFRβ-Cre:ChR2-YFP mice (Hartmann et al. 2020) (Fig. 1.7d,e). Further, we found that similar excitation protocols caused a much greater contraction of SMCs, suggesting that different mural cell types have a different threshold of ChR2 activation necessary for contraction. Capillary pericytes are contractile but exhibit slower kinetics, thereby requiring longer periods of stimulation than in SMCs.

Optogenetic stimulation of pericytes concurrent with vasodynamic measurements in vivo. (a) Representative image of the cerebrovascular network in a Pdgfrb-Cre::ChR2-YFP mouse. Precapillary and capillary zones are specified, as are vessel branch orders from a penetrating arteriole (0 order). (b) The ideal ChR2 excitation wavelength for mural cells using two-photon microscopy is set at 800 nm. Two-photon activation of ChR2 near the focal plane reduces possibility of stimulating upstream SMCs. (c) A schematic of the multifaceted purposed of the two-photon line-scan pattern utilized for optogenetic stimulation studies. The scan path allows for the two-photon activation of ChR2, the repeated measurement of capillary diameter, and the assessment of blood cell dynamics over time. (d) An example of pericyte stimulation using a multi-segmented two-photon line-scan. (e) (Top) Colored blocks in line correspond to locations along the scan-path in (d). (Middle, and bottom) Raw data acquired from a line-scan demonstrates a reduction on capillary diameter and blood cell flux over a 60 second period of stimulation. FWHM vessel diameter, blood cell flux, and blood cell velocity, can be extracted from the line-scan data. (Adapted from Hartmann et al., 2020 (Hartmann et al. 2020))
A number of important practical considerations arise when using optogenetics to study mural cell contractility in vivo. First, there is a wavelength specificity for activation of ChR2, in this case the H134R variant. While 920–940 nm excitation is the most efficient to drive neuronal firing in cultured neurons (Prakash et al. 2012; Rickgauer and Tank 2009), 800 nm excitation appears most effective for driving contraction of SMCs and pericytes. We and others have found that imaging at 900 nm leads to a considerably lower level of contraction than when imaging at 800 nm, even while using the same laser powers. Although the basis for this discrepancy in optimal wavelengths is unclear, it provides the opportunity to observe and measure hemodynamics at 900 nm, then switch to 800 nm to simultaneously measure hemodynamics and stimulate mural cells. It is also important that laser power be graded based on depth below the pial surface, as deeper pericytes will require more power to stimulate. Second, it is important to recognize that light can alter vasodynamics even in the absence of exogenous opsins like ChR2. In naive animals, blue light (Sikka et al. 2014; Rungta et al. 2017), and two-photon light at 800 nm (Choi et al. 2010; Kimbrough et al. 2015), have been shown to cause vasodilation and vasoconstriction, respectively. To control for these effects, optogenetic stimulation studies should include control animals which express a fluorophore alone (i.e. YFP or mGFP) rather than the ChR2-YFP protein. Critically, the laser powers used between opsin expressing mice and controls must be matched at different cortical depths for an adequate control of the effects of laser light. In our hands, the non-opsin expressing controls have not shown consistent responses to light, allowing us to conclude that ChR2 activation in pericytes mediates capillary constriction.
Using this promising approach, researchers can now test whether capillary pericytes control cerebral blood flow in a cause-and-effect manner. Other questions in pericyte biology, such as how pericytes communicate with other cells in the brain , will undoubtedly benefit from the advancement of this technique. Some limitations remain, however. For one, we need a better understanding of the fluctuations in pericyte membrane potential and intracellular calcium levels in vivo, with and without optogenetic stimulation. This would help us understand how optogenetic stimulation compares to physiologic pericyte function. It will also allow us to design optogenetic stimuli that mimic certain physiologic or pathologic voltage changes in pericytes.
1.8 Concluding Remarks
We have highlighted a variety of new tools and approaches to visualize the structure and function of brain pericytes in their native environment. Imaging provides not only a means to observe fine pericyte structure over time, but also an opportunity for causal studies to delineate pericyte roles in brain physiology. While much has been learned from in vitro and ex vivo studies on pericytes, the field has shifted to discerning which of the diverse functions ascribed to pericytes are fact or fiction in vivo. We have discussed how seemingly basic topics, such as how to name and identify a capillary pericyte , are crucial to the field. This allows for pericytes to be studied consistently among research labs. Careful selection of a pericyte targeting mouse line is similarly crucial, each with their own advantages and disadvantages. Even mice that appear problematic with non-specific targeting can be leveraged for elegant experiments in neurovascular coupling, or comparative analysis across vascular zones. Quality is always preferred over quantity with two-photon imaging, and therefore the utmost care with cranial window construction and attention to animal physiology is important. Optical imaging is not only a means to observe physiological processes, but also a means to delete or stimulate pericytes with single cell precision in vivo. These approaches have already revealed a phenomenon of structural plasticity of pericytes that may be targeted to improve pericyte coverage in ailing cerebrovascular networks. They have also revealed how different pericyte types can have distinct effects on microvascular tone and therefore blood flow regulation. With these tools in hand, the field is now poised for a new era of in vivo studies on pericyte function.
References
Arango-Lievano M et al (2018) Topographic reorganization of cerebrovascular mural cells under seizure conditions. Cell Rep 23(4):1045–1059
Armulik A, Abramsson A, Betsholtz C (2005) Endothelial/pericyte interactions. Circ Res 97(6):512–523
Armulik A et al (2010) Pericytes regulate the blood-brain barrier. Nature 468(7323):557–561
Bell RD et al (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68(3):409–427
Berthiaume AA et al (2018a) Pericyte structural remodeling in cerebrovascular health and homeostasis. Front Aging Neurosci 10:210
Berthiaume AA et al (2018b) Dynamic remodeling of Pericytes in vivo maintains capillary coverage in the adult mouse brain. Cell Rep 22(1):8–16
Birbrair A et al (2014a) Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res Ther 5(6):122
Birbrair A et al (2014b) Type-2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol 307(1):C25–C38
Blanchette M, Daneman R (2015) Formation and maintenance of the BBB. Mech Dev 138(Pt 1):8–16
Cai C et al (2018) Stimulation-induced increases in cerebral blood flow and local capillary vasoconstriction depend on conducted vascular responses. Proc Natl Acad Sci U S A 115(25):E5796–E5804
Chhatbar PY, Kara P (2013) Improved blood velocity measurements with a hybrid image filtering and iterative radon transform algorithm. Front Neurosci 7:106
Choi M, Yoon J, Choi C (2010) Label-free optical control of arterial contraction. J Biomed Opt 15(1):015006
Coucha M et al (2019) Inhibition of Ephrin-B2 in brain pericytes decreases cerebral pathological neovascularization in diabetic rats. PLoS One 14(1):e0210523
Cudmore RH, Dougherty SE, Linden DJ (2017) Cerebral vascular structure in the motor cortex of adult mice is stable and is not altered by voluntary exercise. J Cereb Blood Flow Metab 37(12):3725–3743
Cuttler AS et al (2011) Characterization of Pdgfrb-Cre transgenic mice reveals reduction of ROSA26 reporter activity in remodeling arteries. Genesis 49(8):673–680
Daigle TL et al (2018) A suite of transgenic driver and reporter mouse lines with enhanced brain-cell-type targeting and functionality. Cell 174(2):465–480
Damisah EC et al (2017) A fluoro-Nissl dye identifies pericytes as distinct vascular mural cells during in vivo brain imaging. Nat Neurosci 20(7):1023–1032
Daneman R et al (2010) Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468(7323):562–566
Davidson BL, Breakefield XO (2003) Viral vectors for gene delivery to the nervous system. Nat Rev Neurosci 4(5):353–364
Denk W, Strickler JH, Webb WW (1990) Two-photon laser scanning fluorescence microscopy. Science 248(4951):73–76
Dombeck DA et al (2007) Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron 56(1):43–57
Dore-Duffy P et al (2000) Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res 60(1):55–69
Drew PJ et al (2010) Chronic optical access through a polished and reinforced thinned skull. Nat Methods 7(12):981–984
Driscoll JD et al (2013) Two-photon imaging of blood flow in the rat cortex. Cold Spring Harb Protoc 2013(8):759–767
Foo SS et al (2006) Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell 124(1):161–173
Gao YR et al (2017) Time to wake up: studying neurovascular coupling and brain-wide circuit function in the un-anesthetized animal. NeuroImage 153:382–398
Goldey GJ et al (2014) Removable cranial windows for long-term imaging in awake mice. Nat Protoc 9(11):2515–2538
Grant RI et al (2017) Organizational hierarchy and structural diversity of microvascular pericytes in adult mouse cortex. J Cereb Blood Flow Metab:271678X17732229
Grubb S et al (2020) Precapillary sphincters maintain perfusion in the cerebral cortex. Nat Commun 11(1):395
Grutzendler J, Nedergaard M (2019) Cellular control of brain capillary blood flow: in vivo imaging Veritas. Trends Neurosci 42(8):528–536
Guimaraes-Camboa N et al (2017) Pericytes of multiple organs do not behave as mesenchymal stem cells in vivo. Cell Stem Cell 20(3):345–359
Hall CN et al (2014) Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508(7494):55–60
Halliday MR et al (2016) Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 36(1):216–227
Hartmann DA et al (2015) Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics 2(4):041402
Hartmann DA et al (2020) Brain capillary pericytes exert a substantial but slow influence on blood flow. In: bioRxiv
Hellstrom M et al (2001) Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 153(3):543–553
Hill RA et al (2015) Regional blood flow in the Normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary Pericytes. Neuron 87(1):95–110
Hill RA et al (2017) Targeted two-photon chemical apoptotic ablation of defined cell types in vivo. Nat Commun 8:15837
Holtmaat A et al (2009) Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc 4(8):1128–1144
Huang W et al (2014) Novel NG2-CreERT2 knock-in mice demonstrate heterogeneous differentiation potential of NG2 glia during development. Glia 62(6):896–913
Hyacinth HI et al (2019) Higher prevalence of spontaneous cerebral vasculopathy and cerebral infarcts in a mouse model of sickle cell disease. J Cereb Blood Flow Metab 39(2):342–351
Ishii Y et al (2006) Mouse brains deficient in neuronal PDGF receptor-beta develop normally but are vulnerable to injury. J Neurochem 98(2):588–600
Ivanova E, Kovacs-Oller T, Sagdullaev BT (2017) Vascular Pericyte impairment and Connexin43 gap junction deficit contribute to vasomotor decline in diabetic retinopathy. J Neurosci 37(32):7580–7594
Kalmbach AS, Waters J (2012) Brain surface temperature under a craniotomy. J Neurophysiol 108(11):3138–3146
Kamouchi M et al (2004) Calcium influx pathways in rat CNS pericytes. Brain Res Mol Brain Res 126(2):114–120
Khennouf L et al (2018) Active role of capillary pericytes during stimulation-induced activity and spreading depolarization. Brain 141(7):2032–2046
Kim TN et al (2012) Line-scanning particle image velocimetry: an optical approach for quantifying a wide range of blood flow speeds in live animals. PLoS One 7(6):e38590
Kimbrough IF et al (2015) Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 138(Pt 12):3716–3733
Kisler K et al (2017) Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci 20(3):406–416
Kisler K et al (2020) Acute ablation of cortical pericytes leads to rapid neurovascular uncoupling. Front Cell Neurosci 14:27
Kleinfeld D et al (1998) Fluctuations and stimulus-induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proc Natl Acad Sci U S A 95(26):15741–15746
Lai AY et al (2015) Venular degeneration leads to vascular dysfunction in a transgenic model of Alzheimer’s disease. Brain 138(Pt 4):1046–1058
Li B et al (2020) Two-photon microscopic imaging of capillary red blood cell flux in mouse brain reveals vulnerability of cerebral white matter to hypoperfusion. J Cereb Blood Flow Metab 40(3):501–512
Liao M et al (2017) An X-linked Myh11-CreER(T2) mouse line resulting from Y to X chromosome-translocation of the Cre allele. Genesis 55(9)
Lindahl P et al (1997) Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277(5323):242–245
Lindblom P et al (2003) Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev 17(15):1835–1840
Longair MH, Baker DA, Armstrong JD (2011) Simple neurite tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 27(17):2453–2454
Lou N et al (2016) Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci U S A 113(4):1074–1079
Lowery RL, Majewska AK (2010) Intracranial injection of adeno-associated viral vectors. J Vis Exp (45)
Madisen L et al (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13(1):133–140
Masamoto K, Kanno I (2012) Anesthesia and the quantitative evaluation of neurovascular coupling. J Cereb Blood Flow Metab 32(7):1233–1247
Mateo C et al (2017) Entrainment of arteriole vasomotor fluctuations by neural activity is a basis of blood-oxygenation-level-dependent “Resting-State” connectivity. Neuron 96(4):936–948
Miners JS et al (2019) CSF evidence of pericyte damage in Alzheimer’s disease is associated with markers of blood-brain barrier dysfunction and disease pathology. Alzheimers Res Ther 11(1):81
Mishra A et al (2014) Imaging pericytes and capillary diameter in brain slices and isolated retinae. Nat Protoc 9(2):323–336
Montagne A et al (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85(2):296–302
Montagne A et al (2018) Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat Med 24(3):326–337
Muzumdar MD et al (2007) A global double-fluorescent Cre reporter mouse. Genesis 45(9):593–605
Nakagawa S et al (2007) Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cell Mol Neurobiol 27(6):687–694
Nikolakopoulou AM et al (2017) Regional early and progressive loss of brain pericytes but not vascular smooth muscle cells in adult mice with disrupted platelet-derived growth factor receptor-beta signaling. PLoS One 12(4):e0176225
Nikolakopoulou AM et al (2019) Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci 22(7):1089–1098
Nishimura N et al (2010) Limitations of collateral flow after occlusion of a single cortical penetrating arteriole. J Cereb Blood Flow Metab 30(12):1914–1927
Orlidge A, D’Amore PA (1987) Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J Cell Biol 105(3):1455–1462
Packer AM, Roska B, Hausser M (2013) Targeting neurons and photons for optogenetics. Nat Neurosci 16(7):805–815
Park L et al (2014) Age-dependent neurovascular dysfunction and damage in a mouse model of cerebral amyloid angiopathy. Stroke 45(6):1815–1821
Peppiatt CM et al (2006) Bidirectional control of CNS capillary diameter by pericytes. Nature 443(7112):700–704
Prakash R et al (2012) Two-photon optogenetic toolbox for fast inhibition, excitation and bistable modulation. Nat Methods 9(12):1171–1179
Ratelade J et al (2020) Reducing Hypermuscularization of the transitional segment between arterioles and capillaries protects against spontaneous intracerebral hemorrhage. Circulation
Reeves C et al (2019) Spatiotemporal dynamics of PDGFRbeta expression in pericytes and glial scar formation in penetrating brain injuries in adults. Neuropathol Appl Neurobiol 45(6):609–627
Rickgauer JP, Tank DW (2009) Two-photon excitation of channelrhodopsin-2 at saturation. Proc Natl Acad Sci U S A 106(35):15025–15030
Roche M et al (2019) In vivo imaging with a water immersion objective affects brain temperature, blood flow and oxygenation. Elife 8
Roome CJ, Kuhn B (2014) Chronic cranial window with access port for repeated cellular manipulations, drug application, and electrophysiology. Front Cell Neurosci 8:379
Rorsman NJG et al (2018) Defining the ionic mechanisms of optogenetic control of vascular tone by channelrhodopsin-2. Br J Pharmacol 175(11):2028–2045
Rungta RL et al (2017) Light controls cerebral blood flow in naive animals. Nat Commun 8:14191
Rungta RL et al (2018) Vascular compartmentalization of functional hyperemia from the synapse to the pia. Neuron 99(2):362–375
Rustenhoven J, Jansson D, Smyth LC, Drahunow M (2017) Brain Pericytes as mediators of Neuroinflammation. Trends Pharmacol Sci
Sato Y et al (1990) Characterization of the activation of latent TGF-beta by co-cultures of endothelial cells and pericytes or smooth muscle cells: a self-regulating system. J Cell Biol 111(2):757–763
Sengillo JD et al (2013) Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol 23(3):303–310
Shih AY et al (2012a) Two-photon microscopy as a tool to study blood flow and neurovascular coupling in the rodent brain. J Cereb Blood Flow Metab 32(7):1277–1309
Shih AY et al (2012b) A polished and reinforced thinned-skull window for long-term imaging of the mouse brain. J Vis Exp (61)
Shih AY, Drew PJ, Kleinfeld D (2014) Imaging Vasodynamics in the awake mouse brain with two-photon microscopy. Neurovas Coupling Methods
Sikka G et al (2014) Melanopsin mediates light-dependent relaxation in blood vessels. Proc Natl Acad Sci U S A 111(50):17977–17982
Stark K, Pekayvaz K, Massberg S (2018) Role of pericytes in vascular immunosurveillance. Front Biosci (Landmark Ed) 23:767–781
Stoica L, et al. (2013) Gene transfer to the CNS using recombinant adeno-associated virus. Curr Protoc Microbiol. Chapter 14: p. Unit14D 5
Underly RG et al (2017) Pericytes as inducers of rapid, matrix Metalloproteinase-9-dependent capillary damage during ischemia. J Neurosci 37(1):129–140
Vanlandewijck M, Betsholtz C (2018) Single-cell mRNA sequencing of the mouse brain vasculature. Methods Mol Biol 1846:309–324
Vanlandewijck M et al (2018) A molecular atlas of cell types and zonation in the brain vasculature. Nature 554(7693):475–480
von Tell D, Armulik A, Betsholtz C (2006) Pericytes and vascular stability. Exp Cell Res 312(5):623–629
Wang Y et al (2018) Viral vectors as a novel tool for clinical and neuropsychiatric research applications. Gen Psychiatr 31(2):e000015
Watson AN et al (2020) Mild pericyte deficiency is associated with aberrant brain microvascular flow in aged PDGFRbeta(+/−) mice. J Cereb Blood Flow Metab:271678X19900543
Wei HS et al (2016) Erythrocytes are oxygen-sensing regulators of the cerebral microcirculation. Neuron 91(4):851–862
Wirth A et al (2008) G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14(1):64–68
Wu Y et al (2015) Optogenetic approach for functional assays of the cardiovascular system by light activation of the vascular smooth muscle. Vasc Pharmacol 71:192–200
Xu HT et al (2007) Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci 10(5):549–551
Yang G et al (2010) Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc 5(2):201–208
Yemisci M et al (2009) Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med 15(9):1031–1037
Zehendner CM et al (2015) Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Sci Rep 5:13497
Zhu X, Bergles DE, Nishiyama A (2008) NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development 135(1):145–157
Acknowledgements
Our work is supported by grants to A.Y.S. from the NINDS (NS106138, AG063031, NS097775), the Dana Foundation, the American Heart Association (14GRNT20480366), Alzheimer’s Association NIRG award, and an Institutional Development Award (IDeA) from the NIGMS under grant number P20GM12345. A.A.B. is supported by a scholarship from the American Federation of Aging Research. V.C.S is supported by an American Heart Association Post-doctoral Fellowship (20POST35160001). D.A.H. is supported by awards NIH T32 GM08716, NIH - NCATS (UL1 TR001450 and TL1 TR001451), and NIH-NINDS F30NS096868. We thank Patrick J. Mulholland for use of a Zeiss LSM 880 with Airyscan for super-resolution imaging of pericytes (NIH S10 OD021532). We also thank Tiago Figueiredo for creating artwork used in Figs. 1.3 and 1.5a (www.behance.net/TiagoFigueiredoGD).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Berthiaume, AA., Coelho-Santos, V., Hartmann, D.A., Shih, A.Y. (2021). In Vivo Optical Imaging and Manipulation of Brain Pericytes. In: Birbrair, A. (eds) Biology of Pericytes – Recent Advances. Stem Cell Biology and Regenerative Medicine, vol 68. Humana, Cham. https://doi.org/10.1007/978-3-030-62129-2_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-62129-2_1
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-62128-5
Online ISBN: 978-3-030-62129-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)