
Abstract
Identification of drug-disease association is crucial for drug development and reposition. However, discovering drugs which are associated with diseases from in vitro testing is costly and time-consuming. Accumulating evidence showed that computational approaches can complement biological and clinical experiments for this identification task. In this work, we propose a novel computational method Node2Bio for predicting drug-disease associations using a highly efficient biomolecular network representation model. Specifically, we first construct a large-scale biomolecular association network (BAN) by integrating the associations among drugs, diseases, proteins, miRNAs and lncRNAs. Then, the network embedding model node2vec is used to extract network behavior features of drug and disease nodes. Finally, the feature vectors are taken as inputs for the XGboost classifier to predict potential drug-disease associations. To evaluate the prediction performance of the proposed method, five-fold cross-validation tests are performed on a widely used SCMFDD-S dataset. The experimental results demonstrate that our method achieves competitive performance with a high AUC value of 0.8569, which suggests that our method is a useful tool for identification of drug-disease associations.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Drug-disease association is almost involved in the entire process of drug repositioning, providing a theoretical basis for the discovery of new drug efficacy. Therefore, it is a prospective task to explore as many new drug-disease associations as possible. In recent years, several computational methods of drug-disease association based on drug target information, drug structure information, disease semantic information and other information sources have been proposed. For example, some methods use disease, drug and drug target to predict drug-disease associations (TL-HGBI). Drug - disease association prediction based on drug target information is a popular method [1]. Drug targets are also considered to be one of the sources of information for predicting drug-disease interactions, but the computational conditions for these methods are that the drug can find the corresponding drug target information. In these methods, a three-layer heterogeneous network is typically constructed using drugs, diseases, and drug targets, and the network is constructed based on the distribution of similarity measures [2]. Combining multiple associated sources of information provides more insight into predictive drug-disease association than using only drug targets as sources of information [3]. Therefore, how to effectively integrate more information sources has attracted wide attention [4].
Inspired by graph representation learning, we re-examine some basic relational prediction problems from the perspective of graphs to find better solutions. Graph is a basic and commonly used data structure. Many scenes in the real world can be abstracted into a graph structure, such as social network, traffic network, etc. [5]. The biomolecule in the cell can also be viewed as a graph structure, with the association of different types of biomolecules forming the edges of the graph and the biomolecules serving as the nodes of the graph [6]. Using graph theory to develop reliable bio-association graph technology to solve bio-association prediction problem will have a subversive impact on current bioinformatics research [7]. There is no doubt that the seamless integration of graph with biomacromolecules will drive the development of the post-genomic era [8].
The prediction of nodes and edges is an important task in network analysis [9]. In the node classification task, the most likely node label in the prediction network is the first task [10]. For example, in the drug-target interaction network, the focus is on predicting the functional labeling of drugs [11]. Similarly, in a molecular association network, we want to predict whether a pair of nodes in the network should have an edge that connects them [12, 13]. Predicting nodes and edges can help us discover new interactions between drugs and diseases [14]. Node2vec is an algorithm framework for learning the continuous feature representation of nodes in a network [15]. It defines a flexible concept of node network domain and designs a biased random walk process to effectively explore different network domains [16].
Computational methods used to find new drugs and disease associations can solve the problem of high cost and low efficiency, so it has important practical significance [17]. Based on the similarity of biomolecular association network and graph structure, this paper proposes a biomolecular network representation learning model to predict drug-disease association [18]. The model is based on the biomolecular network representation method Node2Bio and XGboost classifier [19].
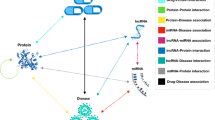
The biomolecular network consists of two parts: nodes (drugs, diseases, proteins, ncRNA (miRNA, lncRNA)) and edges (the relationship of nodes) [20]. Each node can be represented in two ways: attribute information of the node (such as the molecular fingerprint of the drug and the phenotype of the disease) and a vector of relationships with other nodes in the network embedding [21]. Finally, all node features are integrated to form feature descriptors and imported into the XGboost classifier to predict the association of each drug with all diseases [22]. It is worth noting that although the main purpose is to predict drug-disease association, our proposed molecular association network model and iterative update algorithm can be applied to other prediction problems as well [23].
2 Materials and Methods
2.1 Nine Kinds of Molecular Associations
To build a molecular association network, we need to download drugs, diseases, lncRNA, miRNA and protein information from different data sources. Then the feature vectors of drug, disease, lncRNA, miRNA and protein were calculated by different methods. All known interactions are derived from existing databases [24]. Drugs and diseases are downloaded from the CTD database and drugs SMILE is downloaded from DrugBank [25]. Zhang et al. collated 18,416 drug-disease associations from the CTD database and named this data set “SCMFDD-S” [26]. Drug-protein associations were collected from the DrugBank database for a total of 11,107 associations. The Protein-protein association is based on 19,237 associations in the STRING dataset [27]. The Protein-disease association was collected from the DisGeNET [28] database and a total of 25,087 associations were collected. A total of 690 lncRNA-protein associations were collected from the LncRNA2Target [29] database. A total of 1264 lncRNA-disease associations were collected from the LncRNADisease [30] database and the lncRNASNP2 [31] database. 4494 miRNA-protein associations were collected from miRTarBase [32]. The miRNA-disease association was collected from HMDD [33] for a total of 16,427. 8374 miRNA-lncRNA associations were downloaded from lncRNASNP2 [31].
2.2 Disease MeSH Descriptors and Directed Acyclic Graph
In this study, we used the MeSH disease descriptor downloaded from the National Library to calculate the semantic similarity of the disease. This representation is described by a directed acyclic graph (DAG), in which nodes in the DAG represent disease, and the ends of each edge are the parent and child nodes, respectively [34]. If the disease \( p\left( j \right) \) is the parent of the disease \( p\left( i \right) \), the disease \( p\left( i \right) \) can be described as:
where \( N_{p\left( i \right)} \) represents the set of points for all diseases. \( E_{p\left( i \right)} \) contains all the edges in \( DAG_{p\left( i \right)} \).
In \( DAG_{p\left( i \right)} \) of disease \( s \), the contribution of any ancestral disease \( p\left( i \right) \) to disease \( s \) is as the formula:
In addition, disease \( p\left( i \right) \) contributes 1 to its own semantic value. Therefore, the semantic value \( DV\left( {p\left( i \right)} \right) \) of the disease \( p\left( i \right) \) is defined as follows:
We hypothesized that the more DAG Shared between diseases, the higher the semantic similarity score. The DAG similarity value \( SV_{1} \left( {p\left( i \right),p\left( j \right)} \right) \) of the disease \( p\left( i \right) \) and disease \( p\left( j \right) \) is calculated as:
2.3 Stacked Autoencoder
Stacked auto-encoder (SAE) is a multi-layer neural network and is a deep learning model that uses modular units to create deep neural networks [35]. The purpose of Auto-encoder is to make the value of the output as close as possible to the value of the input. Given a drug molecular fingerprint set \( x \), autoencoder input \( x \) through an expression to determine the mapping of hidden:
where \( \sigma \) denotes the logistic sigmoid. Y is the result of the hidden representation, and \( x \) is the reconstructed vector after mapping:
The stack auto-encoder is a combination of multiple autoencoders. The principle is to use the output of the first layer of the autoencoder as the input of the next layer of the autoencoder, and so on, to obtain the output of the last layer of the auto-encoder. In this paper, a drug fingerprint obtains a descriptor representing a structural feature by a stacked autoencoder.
2.4 NcRNA and Protein Sequence
We chose to encode the sequence using a 64 (4 × 4 × 4) dimensional vector encoding ncRNA and analyzed it with K-mer, where k is taken as 3. The 3-mer mode is a sliding window containing 3 nucleotides to analyze each transcription. In the initial state, the number of occurrences of all patterns is set to 0. If the window matches exactly the string in the transcript, the count is incremented by 1 and the slide continues. Finally, divide the number of occurrences by the length of the sequence to get the normalized frequency.
The article by Shen et al. [36] proposes that protein sequences can be encoded into four classes based on the polar side chains of the amino acids. Each protein sequence is characterized by a 3-mer. The ncRNA uses the same normalized frequency calculation method.
2.5 Node Representation
In the molecular association network, many nodes and edges are involved in the prediction task. We chose node2vec to learn the continuous feature representation of nodes in the network [37]. Suppose just traversed go from edge (t, v) to node v. Assume that the transition probability of the next step edge (v, x) is \( \pi_{vx} \). We set the unnormalized transition probability to \( \pi_{vx} = \alpha_{pq} \left( {t,x} \right) \cdot \omega_{vx} \), where \( d_{tx} \) represents the shortest path distance between nodes t and x:
2.6 XGBoost
XGBoost algorithm has been widely applied in the field of bioinformatics. XGBoost is an integration of several weak classifiers, in this case the CART regression tree model. The objective function of XGBoost is defined as:
Here \( l \) is a differentiable convex loss function that measures the difference between the prediction \( \widehat{{y_{m} }} \) and the target \( y_{m} \). The complexity of the \( \Omega \) penalty model. The newly generated tree is to fit the residual error predicted last time. When t trees are generated, the prediction score is:
The target function is updated to:
In general, a second order approximation can be used to quickly optimize the target. The approximate objective function is:
where \( g_{m} \) is the first derivative and \( h_{m} \) is the second derivative.
Since the prediction score of the former \( t - 1 \) tree and the residual of \( y \) do not affect the optimization of the objective function, the objective function can be simplified as:
3 Results and Discussion
3.1 Evaluation Criteria
In order to verify the predictive power of our model. Five-fold cross-validation was performed to verify. All samples were first randomly divided into nearly the same number of five subsets. Each time four subsets are used as a training set and the remaining subsets are used as test sets, the process is repeated five times so that each subset can be used as a test set. Finally, the average of the five groups was taken as the final result. Several evaluation criteria used in our study to estimate the predictive power of our model, including sensitivity (Sen.), specificity (Spec.), precision (Prec.) accuracy (Acc.) and Matthews correlation coefficient (MCC). The calculation method is as follows:
For further evaluation, we also compute the receiver operating characteristic (ROC) curve, sum up the ROC curve in a numerical way, and calculate the area under the ROC curve (AUC). We compute the precision-recall (PR) curve and calculate the area under the PR curve (AUPR).
4 Results and Discussion
4.1 Five-Fold Cross-Validation on SCMFDD-S Dataset
We performed five-fold cross-validation on the SCMFDD-S data set to evaluate the performance of Node2Bio in predicting drug-disease association [38]. The process of cross-validation is to divide the data set into five equal parts, select a different set as the test set each time, and the remaining four sets as the training set, and repeat the experiment five times [39]. Node2Bio yielded an average accuracy of 77.42%, sensitivity of 75.25%, specificity of 79.59%, precision of 78.67%, Matthews correlation coefficient of 54.90% and AUC of 85.69% with standard deviations of 0.24%, 1.01%, 0.74%, 0.41%, 0.46% and 0.12% [40]. To evaluate the performance of Node2Bio, we compare it to some related methods of NTSIM-C. The comparison method uses the same data set for five-fold cross-validation. The experimental results represented by AUC are shown in Table 1. The results from experiments demonstrate that the performance of Node2Bio is significantly better than the related methods of NTSIM-C. Unlike the comparison method, Node2Bio combines nine molecular associations and integrates related information from a cellular perspective to achieve significant predictive effects.
5 Conclusion
In this study, we proposed a computational method for predicting drug-disease associations using a highly efficient biomolecular network representation model. The proposed method leverages multiple types of relational data that are biologically associated and constructs a heterogeneous network on which a graph embedding technique, node2vec, is applied for feature extraction. Using the embedding feature as inputs, we adopted the XGboost algorithm to do classification for drug-disease association. The experimental results are the proposed method to be effective, robust and superior to existing methodologies. It is anticipated that the model we trained can be applied to predict drug effects on different kinds of diseases on a large scale.
References
Chen, Z.-H., et al.: Identification of self-interacting proteins by integrating random projection classifier and finite impulse response filter. BMC Genom. 20(13), 1–10 (2019)
Zheng, K., Wang, L., You, Z.-H.: CGMDA: an approach to predict and validate MicroRNA-disease associations by utilizing chaos game representation and LightGBM. IEEE Access 7, 133314–133323 (2019)
Wang, L., et al.: Identification of potential drug–targets by combining evolutionary information extracted from frequency profiles and molecular topological structures. Chem. Biol. Drug Des. (2019)
Jiang, H.-J., You, Z.-H., Zheng, K., Chen, Z.-H.: Predicting of drug-disease associations via sparse auto-encoder-based rotation forest. In: Huang, D.-S., Huang, Z.-K., Hussain, A. (eds.) ICIC 2019. LNCS (LNAI), vol. 11645, pp. 369–380. Springer, Cham (2019). https://doi.org/10.1007/978-3-030-26766-7_34
Zheng, K., et al.: DBMDA: a unified embedding for sequence-based miRNA similarity measure with applications to predict and validate miRNA-disease associations. Mol. Ther.-Nucleic Acids 19, 602–611 (2020)
Guo, Z.-H., You, Z.-H., Yi, H.-C.: Integrative construction and analysis of molecular association network in human cells by fusing node attribute and behavior information. Mol. Ther.-Nucleic Acids 19, 498–506 (2020)
Wang, M.-N., et al.: LDGRNMF: LncRNA-disease associations prediction based on graph regularized non-negative matrix factorization. Neurocomputing (2020)
Wang, M.-N., et al.: GNMFLMI: graph regularized nonnegative matrix factorization for predicting LncRNA-MiRNA interactions. IEEE Access 8, 37578–37588 (2020)
Wong, L., et al.: LNRLMI: linear neighbour representation for predicting lncRNA-miRNA interactions. J. Cell Mol. Med. 24(1), 79–87 (2020)
Hu, P., et al.: Learning multimodal networks from heterogeneous data for prediction of lncRNA-miRNA interactions. IEEE/ACM Trans. Computat. Biol. Bioinform. (2019)
Huang, Y.-A., et al.: ILNCSIM: improved lncRNA functional similarity calculation model. Oncotarget 7(18), 25902 (2016)
You, Z.-H., et al.: Highly efficient framework for predicting interactions between proteins. IEEE Trans. Cybern. 47(3), 731–743 (2016)
Huang, Y.-A., Chan, K.C., You, Z.-H.: Constructing prediction models from expression profiles for large scale lncRNA–miRNA interaction profiling. Bioinformatics 34(5), 812–819 (2018)
Zheng, K., et al.: iCDA-CGR: Identification of circRNA-disease associations based on Chaos Game Representation. PLoS Comput. Biol. 16(5), e1007872 (2020)
Guo, Z.-H., Yi, H.-C., You, Z.-H.: Construction and comprehensive analysis of a molecular association network via lncRNA–miRNA–disease–drug–protein graph. Cells 8(8), 866 (2019)
Guo, Z.-H., et al.: A learning-based method for lncRNA-disease association identification combing similarity information and rotation forest. iScience 19, 786–795 (2019)
Jiang, H.-J., You, Z.-H., Huang, Y.-A.: Predicting drug – disease associations via sigmoid kernel-based convolutional neural networks. J. Transl. Med. 17(1), 1–11 (2019)
Jiang, H.-J., Huang, Y.-A., You, Z.-H.: SAEROF: an ensemble approach for large-scale drug-disease association prediction by incorporating rotation forest and sparse autoencoder deep neural network. Sci. Rep. 10(1), 1–11 (2020)
Huang, Y.-A., et al.: Graph convolution for predicting associations between miRNA and drug resistance. Bioinformatics 36(3), 851–858 (2020)
Wang, Y., et al.: A high efficient biological language model for predicting protein–protein interactions. Cells 8(2), 122 (2019)
Wang, Y., et al.: Predicting protein interactions using a deep learning method-stacked sparse autoencoder combined with a probabilistic classification vector machine. Complexity 2018 (2018)
Wang, L., et al.: Combining high speed ELM learning with a deep convolutional neural network feature encoding for predicting protein-RNA interactions. IEEE/ACM Trans. Comput. Biol. Bioinform. (2018)
Huang, Y.-A., You, Z.-H., Chen, X.: A systematic prediction of drug-target interactions using molecular fingerprints and protein sequences. Curr. Protein Pept. Sci. 19(5), 468–478 (2018)
Huang, Y.-A., et al.: Sequence-based prediction of protein-protein interactions using weighted sparse representation model combined with global encoding. BMC Bioinform. 17(1), 184 (2016)
Wishart, D.S., et al.: DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46 (2018)
Zhang, W., et al.: Predicting drug-disease associations by using similarity constrained matrix factorization. BMC Bioinform. 19(1), 233 (2018)
Szklarczyk, D., et al.: The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 45 (2017)
Pinero, J., et al.: DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 45 (2017)
Jiang, Q., et al.: LncRNA2Target: a database for differentially expressed genes after lncRNA knockdown or overexpression. Nucleic Acids Res. 43(Database issue), D193 (2015)
Geng, C., et al.: LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 41(Database issue), D983–D986 (2013)
Miao, Y.R., et al.: lncRNASNP2: an updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucleic Acids Res. 46(Database issue), D276–D280 (2018)
Chou, C.H., et al.: miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 46(Database issue) (2017)
Yang, L., et al.: HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 42(Database issue), D1070 (2014)
Wang, D., et al.: Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 26(13), 1644–1650 (2010)
Jiang, H.-J., Huang, Y.-A., You, Z.-H.: Predicting drug-disease associations via using gaussian interaction profile and kernel-based autoencoder. Biomed. Res. Int. 2019, 11 (2019)
Shen, J., et al.: Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. U.S.A. 104(11), 4337–4341 (2007)
Grover, A., Leskovec, J.: node2vec: scalable feature learning for networks. In: ACM SIGKDD International Conference on Knowledge Discovery & Data Mining (2016)
Guo, Z.-H., et al.: MeSHHeading2vec: a new method for representing MeSH headings as vectors based on graph embedding algorithm. Briefings Bioinform. (2020)
Huang, Y.-A., et al.: Prediction of microbe–disease association from the integration of neighbor and graph with collaborative recommendation model. J. Transl. Med. 15(1), 1–11 (2017)
Guo, Z.-H., et al.: A learning based framework for diverse biomolecule relationship prediction in molecular association network. Commun. Biol. 3(1), 1–9 (2020)
Funding
This work is supported by the Xinjiang Natural Science Foundation under Grant 2017D01A78.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
The authors declare that they have no conflict of interest.
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this paper

Cite this paper
Jiang, HJ., You, ZH., Hu, L., Guo, ZH., Ji, BY., Wong, L. (2020). A Highly Efficient Biomolecular Network Representation Model for Predicting Drug-Disease Associations. In: Huang, DS., Premaratne, P. (eds) Intelligent Computing Methodologies. ICIC 2020. Lecture Notes in Computer Science(), vol 12465. Springer, Cham. https://doi.org/10.1007/978-3-030-60796-8_23
Download citation
DOI: https://doi.org/10.1007/978-3-030-60796-8_23
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-60795-1
Online ISBN: 978-3-030-60796-8
eBook Packages: Computer ScienceComputer Science (R0)