
Abstract
The chromosome karyotyping task is vital and indispensable but tedious work for birth defect diagnosis and biomedical research. In this work, we tackle chromosome automatic karyotyping using a multi-stages chromosome segmentation and mixed classification method. Firstly, we apply a global binary threshold-based method to segment the metaphase chromosome microscope grayscale image into several image slices, consisting of chromosome instances and chromosome clusters. Afterward, we propose a mixed chromosome classification method for identifying a given image is a chromosome cluster or corresponding instance label. After that, we use a deep learning-based approach to segment chromosome cluster images into chromosome instances and apply the mixed chromosome classification model to recognize their corresponding labels. Finally, we synthesize a chromosome karyotype from all corresponding instances and labels. In the mixed classification stage, the proposed method yields 99.53 ± 0.23% classification accuracy on the clinical dataset. In segmentation stages, the proposed method achieves 90.81% comprehensive segmentation accuracy and 85.00% instance segmentation accuracy with 90.63% \(AP_{50}\) precision. The experimental results show that our proposed method is promising for solving chromosome segmentation and classification task of the clinical chromosome automatic karyotyping.
This work was supported by Key-Area Research and Development Program of Guangdong Province(No.2019B010137003), NationalKey-Area Research and Development Program of China (2018YFB1404402), Guangdong Science and Technology Fund (No.2016B030305006, No.2018A07071702, No.201804010314), Guangzhou Science & Technology Fund (No.201804010314), VeChain Foundation (No.SCNU-2018-01).
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Human chromosomes contain human genetic information, which is commonly used for analyzing human genetic diseases. In general, there are 23 pairs of chromosomes in a healthy human body, including 22 pairs of autosomes and a pair of sex chromosomes (X and Y chromosome in male cells and double X in female cells) [1]. Karyotype analysis, illustrated by Fig.1, is a fundamental approach for clinical cytogeneticists to diagnose human chromosomes genetic diseases and birth defects, which is generated by arranging these chromosomes after extracting them from the metaphase chromosome images. For cytogeneticists, karyotyping is laborious work, many researchers have dedicated to auto-karyotyping using computation techniques [2,3,4,5,6] for years.

Example of the chromosome karyotype analysis. The Fig. 1(a) is an grayscale image illustrating a G-band metaphase chromosome cell microphotograph. The Fig. 1(a) is a corresponding chromosome karyotype of Fig. 1(a).
In general, researches on chromosome auto-karyotyping follow the sequential procedure of chromosome classification [1, 5] and segmentation [7, 8]. Although the above research has advanced some progress, there are still some limitations in the automatic chromosomes karyotyping.
Challenges: The most significant challenge of automatic karyotyping is the segmentation of overlapping and touching chromosomes. There are three mainstream methods of chromosome segmentation. The threshold-based segmentation method is the most primal method for overlapping and touching chromosome segmentation. The most notable strength of the threshold-based segmentation method is high running efficiency and the most noticeable weakness is poor segmentation effects. The geometric features-based approach is the most commonly used for overlapping and touching chromosomes segmentation in the last decade. The geometric features-based approach makes a tradeoff between the running efficiency and outcome effects. As it is a rule-based approach, it may not work when encountering more complicated overlapping and touching chromosome clusters. The instance segmentation method based on deep learning is the most promising way to solve the task of overlapping and touching chromosomes segmentation currently. These methods can deal with a variety of complicated overlapping and touching chromosome clusters accurately. However, training and running these models require a large scale of the labeled dataset and enormous computational resources.
Motivations: Motivated by the strengths and weaknesses of the above methods, we propose a novel approach to tackle the chromosome auto-karyotyping problem. We utilize a global threshold method to segment the full-size original metaphase image into several image slices at extremely low running costs. After that, we design a mixed chromosome classifier to identify chromosome cluster images that are required to segment by instance segmentation model. And the classifier will give the corresponding labels of the rest images. The most significant advantage of our proposed method is that it compounds the benefit of threshold-based segmentation and instance segmentation methods.
Results: To evaluate the overall performance and clinical application effect of our proposed method, we train and test our method on the clinical dataset which is constructed by skilled cytogeneticists. In the chromosome classification stage, our proposed method achieves mixed classification performance with \((99.53\pm 0.23)\%\) accuracy, which is a state-of-the-art classification result in previously reported literature. In segmentation stages, the proposed method achieves 90.81% comprehensive segmentation accuracy and 85.00% instance segmentation accuracy with 90.63% \(AP_{50}\) precision, which is promising on the clinical dataset.
Contributions: According to the mentioned explorations, we proposed a multiple-stages method, illustrated in Fig.2, for the chromosome auto-karyotyping task. First, to tackle the chromosome auto-karyotyping task, we decompose the task into multiple-stages. After that, we propose a classification model and a segmentation model based on the deep neural network. Second, to evaluate our proposed model in clinical application, we build a clinical chromosome classification dataset and label a chromosome instance segmentation dataset. Finally, we conduct experiments to demonstrate the effectiveness of the proposed method.
2 Related Work
Chromosome Segmentation
As the chromosome segmentation plays a vital role in chromosome automatic karyotyping, so it attracts numerous researches to try it out.
Enea et al. [2] reviewed and implemented a variety of thresholding strategies applied to human chromosome segmentation. According to the conclusion of [2], over-segmentation and under-segmentation are common phenomena in thresholding strategies methods because it is very difficult to set the threshold value for those methods applied in various situations.
Shervin et al. [3] proposed a geometric-based method for separation chromosome clusters when touching and overlapping chromosome clusters were detected automatically. The authors applied their method to a database containing 62 touching and partially overlapping chromosomes and a success rate of 91.9% is achieved. However, their method is a customized method based on the geometric features of the existing chromosome database, which means the doubtful effectiveness of the clinical application.
R. Lily et al. [4] proposed a neural network-based image segmentation method to the problem of distinguishing between partially overlapping chromosomes. Their method achieved intersection over union (IOU) scores of 94.7% for the overlapping region and 88.94% on the non-overlapping chromosome regions. However, their training and testing chromosome images are semi-synthetically generated. The authors did not report the effectiveness of their method in clinical chromosome application.
Tanvi Arora [9] proposed a human metaphase chromosome images segmentation approach of using region based active contours. The author claimed that this method has been tested on Advanced Digital Imaging Research (ADIR) dataset and yielded quite good performance. However, their method is based on the features active contours in ADIR. Therefore, the clinical effectiveness is still uncertain.
Chromosome Classification
In previous studies, there are some works [1, 5] for chromosome classification. In these studies, the best chromosome classification accuracy was 98.9% reported by Yulei [5] in their private dataset. However, the author did not disclose their dataset for others to verify their results and also did not report sensitivity and specificity which are two important metrics for chromosome classification.
Additionally, all the above methods are 24-classification which are designed for identifying each of 22 autosomes, X, and Y sex chromosomes.
Chromosome Automatic Karyotyping
Reem et al. [10] proposed an automatic segmentation method for chromosome cells using difference of gaussian (DoG) as a sharpening filter. However, all chromosome cell images appeared in this paper have no overlapping chromosomes or touching chromosomes, which is impractical in clinical applications.
Yirui et al. [8] proposed an end-to-end chromosome karyotyping method based on generative adversial networks. Their approach can automatically detect, segment and classify chromosomes from original cell images. However, according to their reported paper, the chromosome brands are too nebulous to recognize by geneticists. It is extremely incorrect and irresponsible to make genetic diseases and birth defect diagnoses based on virtually generated karyotypes.
So far, studies that can effectively tackle the problem of chromosome automatic karyotyping are still very scarce. Motivating by the limitations of the above methods for solving the chromosome auto-karyotyping task, we proposed our chromosome auto-karyotyping framework.
3 Proposed Method
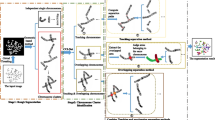
The proposed method, illustrated by Fig. 2, consists of four stages, the global binary threshold segmentation, mixed classification, chromosome cluster instance segmentation, and karyotyping operation stage. In this section, we go through the detail of the above stages.

The plot depicts the operation flow of the proposed method. Forgiven a chromosome cell microphotograph raw image I, the global threshold-based segmentation operation \(\varOmega \) separates I into image slices set \(C_1\) including instances and clusters. Whereafter, the chromosome mixed classifier \(\varPhi \) classifies all instances from \(C_{1}\) into chromosome instance set \(C_{2}\) with their corresponding labels, and chromosome clusters \(C_{3}\). All image slices from C3 are segmented into C1 by the instance segmentation operation \(\varPsi \). When \(C_1\) and \(C_3\) are empty, it means that are clusters have been segmented into instances with corresponding labels. Finally, the chromosome karyotyping operation \(\varUpsilon \) generates the karyotype \(I^{'}\) from \(C_2\).
3.1 Problem Formalization
We use the symbol I to denote a metaphase cell image depicted by Fig.1(a) Similarly, we apply symbol \(I^{'}\) to denote a chromosome karyotype illustrated by Fig. 1(b). We let symbols C1, C2, and C3 represent the images set of chromosome instances & clusters, chromosome instances, and chromosome clusters. We use symbols \(\varOmega \), \(\varPsi \) to denote the operation of the global binary threshold segmentation and chromosome instance segmentation. Furthermore, we use symbols \(\varPhi \) and \(\varUpsilon \) to denote mixed chromosome classification and karyotyping operation.
Global Binary Threshold Segmentation: According to the above symbols, we depict the global binary segmentation procedure of I as \(C1 = \varOmega (I)\).
Mixed Chromosome Classification: The chromosome mixed classification operation \(\varPhi \) identifies the chromosome instance images in C1, and labels these instance images with corresponding chromosome number tags. All labeled chromosome instance images will be moved from C1 to C2, and the rest images in C1 are seen as chromosome clusters, illustrated by \(C2 = \varPhi (C1)\). Therefore, the mixed chromosome classification operation for C1 can be described as \(C3 = C1 - C3\).
Instance Segmentation: The chromosome instance segmentation operation \(\varPsi \) is used for separating a chromosome cluster images into multiple chromosome instance images. The instance segmentation process can be described as \(\forall C \in C3 \rightarrow C1 = C1 \cup \varPsi (C),C3 = C3 - C\).
Chromosome karyotyping: The chromosome karyotyping operation \(\varUpsilon \) refers to the process of generating a karyotype image \(I^{'}\) by arranging chromosome instances of C2 which can be formalized as \(I^{'} = \varUpsilon (C2)\).
3.2 Global Binary Threshold Segmentation
Threshold-based segmentation is a general method in image segmentation applications, which separate images into several parts by the threshold grayscale value. We utilize the global binary threshold method for segmenting the metaphase cell image into chromosome instances and clusters by the following steps.
Firstly, we make statistics on the grayscale distribution of our collected chromosome images. According to the grayscale distribution, we apply the grayscale threshold value 250 to segment a chromosome image into slices. When the total pixels of a slice less than 260, the slice will be seen as non-chromosomal objects and removed. Finally, we save the rest image slices as chromosome instance and cluster images for further processing.
3.3 Mixed Chromosome Classification
We build the mixed chromosome classification model based on the ResNeXt [11] backbone with weakly supervised pre-train weights [12].

This figure describes a standard building block assembled by multiple paths of convolutional neurons. The symbol F denotes the dimension of the input \(x_{i}\) and output \(y_{i}\) while the d represents the total input or output channels number of each inner convolution neuron. The cardinality means the total number of aggregated neuronal paths.
The ResNeXt backbone is an orderly stack of regular neural network modules called building blocks as Fig. 3 depicted. A usual building block is assembled by multiple paths of convolutional neurons. The total quantity of assembled paths is termed by cardinality. The channel amount of the convolutional neurons in the building block is the width of the block, termed by the symbol d. We use F to denote the input and output tensor dimensions of the building block. In our mixed chromosome classifier, we apply ResNeXt 101-32 \(\times \) 16d as the concrete backbone network, which means the cardinality of the building block is 32 and the width is 16. To improve the applicability of the model in the chromosome classification application, we design 9 layers of neurons as the customized header adding to the backbone network as the output of the classifier. Since there are 22 pairs of autosomes and a pair of sex chromosomes (X and Y), we have 24 categories of chromosome instances. Meanwhile, we take all chromosome clusters as an extra category, therefore the output of the classification model is an one-hot vector with the shape of \(25\times 1\). Table 1 shows the detail of the mixed chromosome classification model.
3.4 Chromosome Instance Segmentation
Motivated by the achievements of the PANet [13] in the COCO instance segmentation challenge competition, we transfer this model for solving the task of chromosome instance segmentation, illustrated by Fig. 4. The PANet model consists of five sub-modules: a feature proposal network(FPN), a path augmentation sub-module, an adaptive feature pooling sub-module, a box predict branch and a fully-connected fusion sub-module.

The illustration is the chromosome instance framework transferred from PANet. It has five sub-modules named feature proposal network, path augmentation, adaptive feature pooling, box branch, and fully-connected fusion.
The input of the chromosome instance segmentation framework is an entire chromosome cluster image with 300 \( \times \) 300 pixels. The output of the framework consists of two parts. The output of the framework consists of two parts. The first part of the output is the chromosome instances of proposal boxes and their corresponding probabilities belong to the chromosome. Another output is a pixel mask for each chromosome instance. We separate chromosomes through their respective masks.
3.5 Chromosome Karyotyping
After the preceding processing, we have gathered all chromosome instances in a cell image and their corresponding categories. In the chromosome karyotyping stage, we only need to arrange chromosome instances into a karyotype according to an international system for cytogenetic nomenclature (ISCN) criterion [14].
4 Experiments
4.1 Implementation Details
We implement the global binary threshold segmentation using OpenCV-Python library. According to statistics of the clinical chromosome grayscale, we set up the grayscale segmentation threshold to 225. Meanwhile, we set up the threshold of the minimal pixels filter to 260 which will filter candidate images whose pixels are lower the threshold.
We build the mixed chromosome classification model with PyTorch which is an open source machine learning framework. We adapt the discriminative learning rate [15] to help the model obtain better convergence. First, we load the WSL pre-trained weights [12] as the initial weights of the ResNeXt backbone. Second, we freeze the weights of the backbone and train our customized header with the learning rate at slice(1e-5, 4e-5), 100 maximum epochs. We monitor the training loss and apply early-stopping when the training is no more decrease in the last 5 epochs. Finally, we unfreeze the weights of the backbone and fine-tune the whole model in 100 epochs at slice(1e-6, 4e-6) learning rate. Furthermore, we apply the early-stopping strategy at training loss indicator with 5 epochs patience.
We transfer the chromosome instance segmentation framework and its implementation from PANet [13] where is originally designed for the COCO instance segmentation task. We adapt ResNet50 [16] as the FPN backbone and load the COCO pre-trained wights to the model as initial weights for chromosome instance segmentation task. To alleviate the overfitting of the model, we add a Dropout layer with a 0.25 drop out rate before the last fully-connected fusion layer.
4.2 Dataset Description
This study is motivated by the challenges of the clinical work on genetic disease diagnostic at Guangdong Women and Children Hospital where we obtain 500 privacy-removal clinical metaphase cell images and corresponding karyotypes manually done by skilled geneticists.
We construct a mixed classification dataset with 130 images of each autosome labeled from 0 to 21, and 98 X chromosome images labeled to 22, and 32 Y chromosome images labeled to 23. At the same time, we add 4876 chromosome cluster images with label 24 into this dataset. All chromosome images are padded to 224 \(\times \) 224 pixels.
We annotate 882 chromosome cluster images as the chromosome instance segmented dataset manually. In the chromosome instance segmented dataset, we randomly select 20% images as test image and other as training data. To mitigate the overfitting of the instance segmentation model, we conduct a series of data augmentations in training images.
Firstly, We do a horizontal and vertical flip for individual image and its mask in the training set. Secondary, we rotate all images and their masks in the training dataset every 15\(^\circ \).
4.3 Experimental Evaluation Metrics and Results
As the global binary threshold segmentation stage and chromosome karyotyping stage are engineering tasks, and their operation results are determined. Therefore, the experimental evaluation focus on the chromosome mixed classification and the instances segmentation stages.
Evaluation of Mixed Chromosome Classification: In the mixed classification stage, we apply accuracy, F1, sensitivity, and specificity general metrics to quantitatively evaluate the overall performance of the classification model.
To evaluate the stability of the proposed classification model, we conduct all evaluation experiments utilizing cross-validation by K-Folds. The original chromosome mixed classification dataset is divided into 5 folds by random stratified sampling. One fold data is used as validation data and the other four folds as training data in turn. Table 2 gives the classification evaluation results of the proposed framework on the clinical dataset in five runs.
According to Table 2, the proposed classification model yields \(99.53\pm 0.23\)% accuracy, \(98.85\pm 0.89\)% F1 value, \(98.82\pm 0.86\)% true positive rate (sensitivity), and \(99.98\pm 0.01\)% true negative rate (specificity), which means that our proposed classification method promising for solving the mixed classification problem. Meanwhile, the kappa value with \(99.23\pm 0.47\)% means that the proposed model works well even in the unbalanced dataset.
Evaluation of Chromosome Instance Segmentation. To evaluate the performance of the instance segmentation model, we adopt \(Accuracy_{m}\) as the major evaluation metric which is calculated by the total number of correct segmented instances dividing by the number of total instances in the chromosome cluster set. When the bounding box of the predicted instance has equal or more than 50% intersection over union (IoU) with a ground-truth instance bounding box, we regard this instance as a correct segmented instance. Meanwhile, we follow the metrics of \(AP_{50}\), one of standard evaluation metrics for instance segmentation tasks [13], for quantitatively evaluating the precision of the instance segmentation. Finally, we introduce \(Accuracy_{t}\) as the general segmentation evaluation metric for our proposed multiple stages segmentation method where \(Accuracy_{t}\) is computed by the number of all correct segmented instances dividing by the number of all ground-truth instances in both global binary threshold segmentation stage and instance segmentation stage.

The plots in the first row are original chromosome cluster images while the plots in the second row are their corresponding instance masks predicted by the instance segmentation model.
Table 3 summarizes the quantitative evaluation results. According to the results, we can draw the following conclusions. First, without data augmentation, the instance segmentation model yields a promising result in the verify set while performs poorly in the test set, which demonstrates the overfitting of the model and the necessity of data augmentation. Second, though small performance gaps in verify set and test set still exist, the proposed method obtains promising results with 90.81% \(Accuracy_{t}\), 85% \(Accuracy_{m}\), and 90.63% \(AP_{50}\) precision.
To better depict the effectiveness of the instance segmentation model, we show several examples in Fig. 5. Chromosome instances in various cluster images are separated precisely by the instance segmentation model.
5 Conclusion
In this paper, we proposed a multi-stages chromosome segmentation and mixed classification method for chromosome automatic karyotyping. According to the experimental results on the clinical chromosome dataset, the proposed classification model obtains a promising result (99.53 ± 0.23% accuracy) for solving the mixed chromosome classification problem. Meanwhile, the chromosome segmentation result is quite encouraging (90.81% accuracy). The above results demonstrate the promises for solving the clinical chromosome auto-karyotyping problem currently.
References
Lin, C., et al.: CIR-Net: automatic classification of human chromosome based on inception-ResNet architecture. IEEE/ACM Trans. Comput. Biol. Bioinform. (2020). https://doi.org/10.1109/TCBB.2020.3003445
Poletti, E., Zappelli, F., Ruggeri, A., Grisan, E.: A review of thresholding strategies applied to human chromosome segmentation. Comput. Methods Prog. Biomed. 108(2), 679–688 (2012)
Shervin, M., Mehran, F., Babak, H.K.: A geometric approach to fully automatic chromosome segmentation. In: 2014 IEEE Signal Processing in Medicine and Biology Symposium (SPMB), pp. 1–6. IEEE (2014)
Lily Hu, R., Karnowski, J., Fadely, R., Pommier, J.-P.: Image segmentation to distinguish between overlapping human chromosomes. arXiv preprint arXiv:1712.07639 (2017)
Qin, Y., et al.: Varifocal-net: a chromosome classification approach using deep convolutional networks. IEEE Transactions on Medical Imaging (2019)
Ni, W., Wang, X., Song, W., Li, Y. (eds.): WISA 2019. LNCS, vol. 11817. Springer, Cham (2019). https://doi.org/10.1007/978-3-030-30952-7
Khan, S., DSouza, A., Sanches, J., Ventura, R.: Geometric correction of deformed chromosomes for automatic karyotyping. In: 2012 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, pp. 4438–4441. IEEE (2012)
Wu, Y., Yue, Y., Tan, X., Wang, W., Lu, T.: End-to-end chromosome karyotyping with data augmentation using GAN. In: 2018 25th IEEE International Conference on Image Processing (ICIP), pp. 2456–2460. IEEE (2018)
Arora, T., Dhir, R.: A novel approach for segmentation of human metaphase chromosome images using region based active contours. Int. Arab J. Inf. Technol. 16(1), 132–137 (2019)
Bashmail, R., Elrefaei, L.A., Alhalabi, W.: Automatic segmentation of chromosome cells. In: Hassanien, A.E., Tolba, M.F., Shaalan, K., Azar, A.T. (eds.) AISI 2018. AISC, vol. 845, pp. 654–663. Springer, Cham (2019). https://doi.org/10.1007/978-3-319-99010-1_60
Xie, S., Girshick, R., Dollár, P., Tu, Z., He, K.: Aggregated residual transformations for deep neural networks. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, pp. 1492–1500 (2017)
Dhruv, M., et al.: Exploring the limits of weakly supervised pretraining. In: Proceedings of the European Conference on Computer Vision (ECCV), pp. 181–196 (2018)
Shu, L., Lu, Q., Qin, H., Shi, J., Jia, J.: Path aggregation network for instance segmentation. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, pp. 8759–8768 (2018)
Shaffer, L.G., Jean, M.-J., Schmid, M., et al.: ISCN 2013: an international system for human cytogenetic nomenclature (2013). Karger Medical and Scientific Publishers (2013)
Yang, Y., Igor, G., Boris, G.: Large batch training of convolutional networks. arXiv preprint arXiv:1708.03888 (2017)
He, K., Zhang, X., Ren, S., Sun, J.: Deep residual learning for image recognition. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, pp. 770–778 (2016)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this paper

Cite this paper
Lin, C., Zhao, G., Yin, A., Ding, B., Guo, L., Chen, H. (2020). A Multi-Stages Chromosome Segmentation and Mixed Classification Method for Chromosome Automatic Karyotyping. In: Wang, G., Lin, X., Hendler, J., Song, W., Xu, Z., Liu, G. (eds) Web Information Systems and Applications. WISA 2020. Lecture Notes in Computer Science(), vol 12432. Springer, Cham. https://doi.org/10.1007/978-3-030-60029-7_34
Download citation
DOI: https://doi.org/10.1007/978-3-030-60029-7_34
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-60028-0
Online ISBN: 978-3-030-60029-7
eBook Packages: Computer ScienceComputer Science (R0)