
Abstract
Patients with heart failure commonly develop organ dysfunction as a consequence of impaired blood perfusion or non-hemodynamic indirect injury, and often subsequent progression of renal dysfunction is associated with poor clinical prognosis. While neurohormonal system activation, systemic inflammatory reaction, and hemodynamic derangement are considered the central pathology in acute (or Type 1) cardio-renal syndrome (CRS), there is growing evidence that intricate networks of mediators participate in the process of cardio-renal injury. However, current treatment strategies are unable to effectively modulate this complex interplay of mediators in order to reverse true cardio-renal injury. The presence of conglomerated cardio-renal mediators in advanced CRS often hampers clinician efforts to recognize proper causal relationships in CRS progress. Either imprecise decongestive treatment or imperfect biomarker-based assessment of renal function can contribute to undesirable outcomes. Hence, there is a pressing need to gain insights into vulnerable cardio-renal substrates during subclinical stages of CRS rather than to overcome such mediators during overt CRS.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
7.1 Introduction
Co-existence of renal dysfunction is common in patients with heart failure (HF) and often leads to adverse clinical outcomes [1]. The term “cardio-renal” was introduced as early as 1913 by Dr. Thomas Lewis, who described a unique form of paroxysmal dyspnea in the setting of concomitant cardiac and renal dysfunction [2]. The following year, Dr. Alfred Stengel proposed the classification of cardio-renal diseases into three distinct forms: (1) primary valvular or myocardial disease with secondary renal disease; (2) primary arterial or arteriolar disease with secondary renal and myocardial disease; and (3) primary renal disease with secondary myocardial and vascular disease [3].
After a century of medical progress, our contemporary classification scheme for cardio-renal syndrome (CRS) remains largely descriptive of such temporal bi-directional relationships between cardiac and renal dysfunction without specifying precise mechanistic culprit(s) [4]. Nevertheless, there is general agreement that adverse interactions between the kidneys and circulatory components promote increased circulating volume, exacerbate HF symptoms, and accelerate subsequent disease progression [5]. In contrast, contribution of various non-cardiac factors that have been proposed some half a century ago may still be under-recognized [6]. This chapter will review the classical mediators of cardio-renal injury through which acute HF aggravates renal dysfunction leading to Type 1 CRS, and outline the directions for further investigation beyond our current management strategies.
7.2 Definition of Acute (Type 1) Cardio-Renal Syndrome
Clinicians have largely considered acute (or “Type 1”) CRS as equivalent to the working definition outlined in a National Institute of Health workshop for acute CRS as “an extreme form of cardio-renal dysregulation in which therapy to relieve congestive symptoms of HF is limited by further decline in renal function.” [7] There are several key words in this definition: (1) “dysregulation” refers to the dysfunctional cross-talk between the heart and the kidneys to maintain salt and water homeostasis; (2) “congestive symptoms” refers to the volume overloaded state related to HF; and (3) “limited by further decline in renal function” refers to the refractoriness to standard diuretic regimen (sometimes considered as “diuretic resistance”). In simpler terms, the intention to treat congestive HF by aggressive diuresis was deemed inadequate as a result of ineffective renal responses.
It is important to emphasize here that considerations of “abnormal renal function” still relied on indirect biomarkers that estimate glomerular filtration or function (e.g. clearance of creatinine/cystatin C, and leakage of albumin/protein) rather than biomarkers of tubular function (e.g. clearance of urea or toxins, and handling of electrolyte homeostasis). On the other hand, reliable insights into renal hemodynamics remained limited. Therefore, the precise processes and mechanisms in which the kidneys endure injury remain unclear in the setting of acute CRS [8].
7.3 Factors Contributing to the Development of Acute CRS
Contributing factors to the development of acute (Type 1) CRS include hemodynamic disturbance, neurohormonal activation, and inflammation (Fig. 7.1).

Key Contributors to Cardio-Renal Syndrome. Despite efforts to establish a hierarchy, there seems to be no such hierarchy among cardio-renal connectors. Inflammatory reaction, an activated neurohormonal system and hemodynamic disruption become connected during the subclinical stage of CRS, starting a vicious cycle but staying in a subclinical stage for a period
Impaired Cardiac Output. In the setting of acute HF, reduced cardiac output can lead to impaired renal blood flow and perfusion, which has long been proposed as the primary driver of renal dysfunction and subsequent injury [9]. Indeed, acute kidney injury (AKI) is more prevalent and severe with impaired cardiac output, being reported more than 70% in cardiogenic shock [10]. Improvement in serum creatinine levels shortly after implantation of left ventricular assist devices also highlights the pathophysiological importance of hemodynamic disruption in CRS [11]. However, this once-prevailing concept of “arterial underfilling” as the single perpetrator of CRS cannot be fully explained by clinical observations, since the majority of patients presented with acute HF also have relatively preserved cardiac output [12,13,14,15]. It is likewise important to note that a rise in serum creatinine may not be the primary abnormality to reflect underlying hemodynamic derangements, as hypochloremia may also be triggered by underlying low cardiac output state [16,17,18].
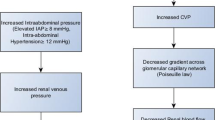
Systemic Venous Congestion . Over the past decade, there is growing understanding of an inverse relationship between central venous pressure (CVP), renal blood flow (RBF), and glomerular filtration rate (GFR) in the setting of HF [19]. Like impaired cardiac output, elevated CVP can lead to increased renal interstitial hydrostatic pressure, resulting in a decreased net filtration pressure, and progressive renal dysfunction [20]. This can be exacerbated in the setting of acute decompensated HF, whereby increased CVP on admission as well as insufficient reduction of CVP during hospitalization can be stronger hemodynamic determinants for the development of worsening renal function compared to diminished cardiac index [21]. Recent mechanistic demonstrations with saline loading experiments have further confirmed the impact of increasing “venous impedance” at the level of the kidney on attenuation of diuresis and natriuresis [22, 23]. These observations may imply that beyond impaired renal perfusion in low cardiac output state, the inability to mobilize venous congestion despite aggressive diuresis can also trigger acute (Type 1) CRS.
Raised Intra-Abdominal Pressure . One of the commonly-overlooked contributors of acute CRS is extra-cardiac hemodynamic alteration in the abdominal cavity [24]. Especially in the setting of overt right-sided HF with significant venous congestion or in post-operative/obstructive settings with ileus or organ swelling, abdominal congestion in the form of splanchnic venous and interstitial congestion can manifest via compromised capacitive function of the splanchnic vasculature and deficient abdominal lymphatic flow resulting in interstitial edema [24]. Increased intra-abdominal pressure detectable via bladder manometry, in extreme cases of abdominal congestion, is correlated with renal dysfunction in advanced refractory congestive heart failure [25].
Pre-existing Renal Insufficiency . The most common scenario whereby acute (Type 1) CRS occurs is due to pre-existing renal dysfunction, which may cause worsening pressure and/or volume overload. Furthermore, chronic uremia can induce left ventricular hypertrophy, promote cardiac fibrosis, and induce systemic oxidant stress [26]. Up to one third of patients hospitalized with acute decompensated HF have concomitant AKI (here referred to rise in biomarkers of glomerular filtration accompanying oligouria), and 60% of patients with acute HF who did not have AKI on admission eventually developed AKI during hospitalization [27]. The co-occurrence of AKI in patients with acute HF worsens survival in those patients [28]. While we do not fully understand the mechanisms leading to increased cardiovascular complications among chronic kidney disease (CKD) patients, worsening renal function in patients with HF is primarily caused by reduced renal perfusion pressure following hemodynamic derangement as the primary culprit. However, when renal dysfunctions become clinically noticeable in the setting of HF, over-activation of neurohormonal systems and systemic inflammation occurs concomitantly with progressive deterioration of cardiac function, making it difficult to single out the culprit among the cardio-renal mediators.
Neurohormonal Mediators . The concept of neurohormonal system activation because of circulatory perturbations plays a large part in our expanded understanding of renal physiology and sodium homeostasis [29]. Activated renin-angiotensin system (RAS) and the sympathetic nervous system (SNS) are prototypical cardio-renal mediators that have diverse influences on hemodynamic components such as right atrial/ventricular compliance, venous capacitance, and returning volume of venous blood [30]. Teleologically, over-activated RAS restores renal perfusion pressure by sustaining intraglomerular pressure and promoting volume expansion [31]. However, while angiotensin restores intraglomerular pressure by constricting efferent arterioles, ensuing vasoconstriction of systemic resistance vessels results in increased afterload and detrimental cardiac function [31]. Excessive urinary sodium and chloride loss caused by aggressive diuresis may induce renin release that increases renal sodium avidity, which is a natural response to dehydration [32]. Avid sodium reabsorption and water retention in the presence of an overactive RAS further aggravates HF and sets up the vicious cycle of CRS [8, 33, 34].
An over-activated RAS can also worsen renal dysfunction through non-hemodynamic mechanism [8]. For example, angiotensin II stimulates production of proinflammatory mediators (e.g. tumor necrosis factor [TNF]-α, interleukin-6, monocyte chemoattractant protein-1, nuclear factor kappa-light-chain-enhancer of activated B cells [NF-κB]) and mobilizes inflammatory cells in the glomeruli. Following cell proliferation, fibrosis and apoptosis eventually progress in the heart and kidneys [35]. Of note, mineralocorticoid in concert with angiotensin II stimulates macrophages in the kidney to secrete galectin-3, a HF biomarker in recent spotlight, which in turn induces proliferation of pericytes, deposition of collagen, and eventual renal fibrosis [36].
Although its deleterious effects in renal injury are less elucidated than in HF [31], the over-activated SNS also contributes to the development of renal dysfunction [37]. First, efferent sympathetic nerves are activated by ischemia/reperfusion injury, a common clinical cause of AKI in various clinical settings [38]. Renal ischemia increases glomerular expression of tyrosine hydroxylase, a rate limiting enzyme of noradrenaline production, suggesting morphological alterations of adrenergic nerve terminals in glomeruli of ischemic AKI [39]. The activated SNS facilitates renal fibrogenesis, tubular vasoconstriction, and reduces GFR in manners dependent on endothelial dysfunction and inflammation, acting jointly with elevated angiotensin II and increased oxidative stress [38]. Adrenergic receptors and endothelin receptors are a superfamily of G protein coupled receptors (GPCR). Transverse aortic constriction elevated renal GPCR signaling and endothelin expression in mice, and then led to deterioration of renal function. In addition, pharmacologic inhibition of GPCR alleviated renal dysfunction [40].
Sympathetic nerve denervation can increase basal renal flow, urine flow rate, fractional sodium excretions, and GFR in rats after renal ischemia/reperfusion injury. The denervated rats had less congestion in the medullary portion, lower level of inflammation, and reduced tubular damage than rats with intact sympathetic activity [41, 42]. In mice with transverse aortic constriction, sympathetic renal denervation did not only blunt the increase in norepinephrine level but also blocked reno-cardiac signaling, which was essential for cardiac hypertrophy in response to pressure overload [43]. Recently, a few small-sized human studies reported renal denervation improved cardiac and renal function [44, 45]. Despite skepticism, observations of renal sympathetic over-activity in patients with CRS support continuing innovative investigational strategies for renal sympathetic denervation [31].
Inflammatory Mediators . Ample evidence has supported the inflammatory process as an important pathology of both cardiovascular disease and CKD. In humans, the circulating level of TNF-α was elevated in severe HF with cachexia and was associated with adverse clinical status as well as RAS system activation [46, 47]. When HF with reduced ejection fraction (HFrEF) patients had acute decompensation, biomarkers for inflammatory response such as high sensitivity C-reactive protein, myeloperoxidase, TNF-α, and galectin-3 continued to increase even after clinical improvement, which implied a unique role of inflammation in the pathophysiology of HF exacerbation [48]. In addition, activation of the complement system occurs in HFrEF, where dysregulated alternative pathways of the complement system can worsen the disease severity [49]. Increase in interleukin-6 may also be mechanistically linked with cardio-renal dysregulation [50].
When we induced chronic HF in mice after coronary artery ligation, the peripheral fraction of pro-inflammatory monocytes/macrophages increased with profound splenic remodeling, representative of augmented antigen processing. In particular, splenectomy resulted in cardiac reverse remodeling and attenuated tissue infiltration of inflammatory cells, while adaptive transfer of splenocytes into naïve mice led to resumption of immune-cell mediated injury, which suggested the central role of the mononuclear cell phagocyte network in chronic inflammation and HF progression [51]. In a similar animal model, activated monocytes and macrophages increased in kidney as well as peripheral blood, mRNA expression of inflammatory cytokines was augmented, and microvascular endothelial permeability and renal tubular cell apoptosis increased through the acute and subclinical phases [52]. Further, depletion of monocytes or macrophages led to alleviation of tubular cell apoptosis and renal fibrosis [52]. Meanwhile, pharmacologic therapy targeting interleukin-1 inhibition [53] or glucocorticoid therapy to promote uricosuria [54] have provided some proof-of-concept demonstrations regarding the inflammatory hypothesis of acute CRS but would require further validation.
Metabolic Contributions . Patients with HF had more permeable intestinal walls than healthy controls, and more pathogenic bacteria were cultured in stool from HF patients. These findings were prominent in patients with severe HF symptoms. Particularly higher serum inflammatory markers in HF patients alluded to bacterial translocation through intestinal walls, which, in turn, is attributable to increased intestinal permeability resulting from intra-abdominal venous congestion [55, 56]. When we incubated renal tubular cells in plasma obtained from CRS septic patients, they had higher levels of apoptosis and caspase-3,-8,-9 expression in plasma with higher endotoxin activity than in plasma with lower endotoxin activity. Plasma inflammatory cytokines were associated with high endotoxin activity and assumed to mediate both extrinsic and intrinsic apoptosis of renal tubular cells, suggesting the presence of detrimental humoral factors in cross-talk between distant organs [57, 58]. Therefore, it is possible that translocation of bacterial endotoxin through intestinal walls worsens renal function in HF patients [59]. Phagocytic systems can generate catecholamine when exposed to bacterial endotoxins, while the disconnection of phagocytes from the autonomic nervous systems leads to reduced inflammatory responses [60]. The autonomic nervous system can influence immunity such as toll-like receptor ligation. During the inflammatory reflex, cytokines locally released from immune cells can transmit signals to the central nervous system through activated vagal afferent nerves [61].
Uremic Toxins . Deterioration of renal function leads to accumulation of protein-bound uremic toxins, such as indoxyl sulfate and p-cresyl sulfate and a tryptophan metabolite produced by gut microbiota, which are excreted by the healthy kidney. Exposure to these uremic toxins can cause, in part, the loss of kidney function [62,63,64]. Uremic toxins originate mainly from protein metabolism, food intake, and can be produced by gut microbiota. In addition to the rise in production, there is an increase in intestinal permeability in CKD allowing a greater absorption of those uremic toxins. [65] The retention of these substances has been associated with an inflammatory state, progression of CKD, cardiovascular disease, and risk of death in CKD patients [66,67,68].
There have been reports that oxidative stress can induce cardiac injury, [69] and urinary indoxyl sulfate excretion was reported to have a positive linear relationship to oxidative stress markers in cardiac tissue [70]. Increased levels of indoxyl sulfate were also associated with chronic inflammation, through indoxyl sulfate-associated pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, leading to left ventricular hypertrophy and cardiac fibrosis [71]. Indoxyl sulfate caused cardiac fibrosis and cardiomyocyte hypertrophy in salt-sensitive hypertensive rats, accompanied by increased oxidative stress marker expression, and decreased anti-oxidative protein expression in cardiac tissue [72]. Reduction in serum indoxyl sulfate levels caused decreased myocardial fibrosis in subtotal-nephrectomized rats [73]. Indoxyl sulfate entered cardiac fibroblasts through OAT1/3, and significantly increased collagen synthesis via activating p38, p42/44 MAPK, and NFκB pathways [71, 74]. Elevated levels of indoxyl sulfate were associated with an increased risk of left ventricular diastolic dysfunction in humans [75]. Thus, emerging evidences from clinical and experimental studies reveal that indoxyl sulfate plays a role in the progression of cardiovascular disease in CKD patients. Although other protein-bound uremic toxins possibly also are involved in the pathogenesis of cardiovascular disease, investigation of the cardiovascular effects of the uremic toxins has been limited to a few toxins. Furthermore, a demonstration that treating indoxyl sulfate leads to improved cardiovascular outcomes is lacking.
7.4 Preventing Type 1 CRS: Identifying Sub-Clinical Cardio-Renal Injury
The key to managing acute (Type 1) CRS is to prevent cardio-renal injury by recognizing the underlying substrates at subclinical stages and preventing the development of cardiac and renal failure (Fig. 7.2). This concept, while logical, has not been fully embraced due to the lack of insights into these potential treatable targets.

Conceptual Framework of Acute (Type 1) Cardio-Renal Syndrome. Once overt cardio-renal syndrome ensues, it seems very difficult to reverse the natural course of disease. Therefore, early detection of patients at risk of cardio-renal syndrome may be a better therapeutic strategy. At the subclinical period of cardio-renal syndrome, there are substrates for renal dysfunction in, particularly, patients with heart failure. Medical resources may be concentrated on these patients to prevent further deterioration of renal function
Biomarkers to Detect Cardio-Renal Injury. The Acute Decompensated Heart Failure National Registry (ADHERE) reported the prevalence of renal insufficiency was about 30% but also likely underestimated [76]. With technological advances, more sensitive and specific novel biomarkers of early organ injuries have been proposed in order to help identify high-risk patients before progression to irreversible stages of CRS [77, 78]. It is therefore postulated that like cardiac troponins for acute coronary syndromes, early detection with AKI biomarker may identify the cohort of patients at higher risk of developing Type 1 CRS and then be triaged to appropriate interventions. Biomarkers of renal tubular damage, such as neutrophil gelatinase-associated lipocalin (NGAL), kidney injury molecule 1 (KIM1), interleukin-18 (IL-18), liver-type fatty acid binding protein (L-FABP), and tissue inhibitor of metalloproteinase 2 plus insulin-like growth factor-binding protein 7 (TIMP2-IGFBP7), have all been investigated for this purpose [79]. However [80], circulating NGAL (a protein of the lipocalin superfamily) was not superior to creatinine for the prediction of worsening renal function (WRF) or adverse in-hospital outcomes [81, 82]. In contrast, few if any acute HF patients who experienced WRF had elevated urinary NGAL levels, and even if levels were high they did not track with poor outcomes despite having pre-existing renal insufficiency [83, 84]. Despite early optimism, few studies have demonstrated the ability of urinary kidney injury biomarkers to provide any prognostic insights or therapeutic directives [84,85,86].
Weight Loss . Obese individuals, even without frank diabetes mellitus, are at risk of CRS development. Obesity per se can induce long-standing glomerular hyperfiltration and obesity-related glomerulopathy, evidenced by focal segmental glomerular sclerosis, foot process effacement, and glomerulomegaly [87,88,89,90]. In an animal model of HFpEF and insulin resistance, glycosuria/proteinuria and microvascular fibrosis were highly analogous to the earliest change of the human cardio-renal syndrome, suggesting the presence of CRS substrate in humans as well [91]. Indeed, phenomapping of HFpEF subtypes has identified a “natriuretic peptide deficient” subtype that likely promotes fluid retention [92]. Intensive lifestyle intervention reduced the incidence of CKD after long-term follow-up, through reductions in bodyweight, HbA1c, and systolic blood pressure [93,94,95]. An enhanced metabolic profile via weight reduction in patients with obesity-associated cardio-renal disease draws attention for a novel therapeutic option [96, 97].
7.5 Managing Type 1 CRS
The latest consensus statement in diuretic use highlighted this goal-targeted strategy (Fig. 7.3), with the introduction of assessing urine output or urine sodium excretion following initial dosing of loop diuretics to assess diuretic efficacy [98]. This is based on observations that urine sodium excretion is diminished in acute HF requiring pharmacologic augmentation, and that insufficient natriuresis either due to abnormal drug delivery at the site of action and/or inadequate urine excretion due to renal sodium avidity may contribute to poor diuretic responses and adverse long-term outcomes [99, 100].

European Society of Cardiology Heart Failure Association Recommendations of the Use of Diuretics in Acute Heart Failure and Cardio-Renal Syndrome. (a) Treatment algorithm for the first 24 hours of admission; (b) Treatment algorithm of second day of admission until discharge
Loop Diuretics. Escalation of intravenous (IV) loop diuretic has been the mainstay of decongestion in HF, and often the key adjustment in Type 1 CRS since most patients remain diuretic responsive. The key determination remains whether loop diuretic dosing is insufficient or whether diuretic resistance is inevitable. Effective diuresis with good urine output despite a rise in serum creatinine or “worsening renal function” should not be classified as CRS. In fact, these patients actually have favorable long-term outcomes [101]. The Diuretic Optimal Strategy Evaluation in Acute Heart Failure (DOSE-AHF) study attempted to address the question of whether higher-dose or continuous administration is superior than standard-dose or bolus administration [102]. While the overall findings were largely neutral except for a statistically significant subjective assessment of well-being in the high-dose arm, a recent post-hoc analysis suggested that when adjusted for total amount of diuretic use, the high-dose strategy may have provided benefits [103].
Part of the challenge has been the inability of the kidneys to excrete loop diuretics to their sites of activity (luminal Na-K-Cl cotransporter at the ascending limb of the Loop of Henle). Indeed, diminished urine sodium per urine furosemide levels in patients with advanced HF receiving IV loop diuretics has been associated with impaired diuresis and natriuresis and poor long-term outcomes [100]. Hence, increasing loop diuretic dosing can be an effective strategy, although doses above the ceiling dose are only moderately effective (despite relatively predictable dose-response curves). Other strategies include increasing frequency of administration (including continuous dosing) or add other types of diuretics for synergistic effects to achieve maximal urinary sodium excretion.
Other Diuretic Drugs . In the stepped pharmacologic uptitration arm of the Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) study [104], patients who experienced worsening renal function were treated with a goal-directed escalation of diuretic drugs including continuous loop diuretic infusion and addition of thiazide diuretics (sequential tubular blockade strategy) [105]. In the majority of cases, urine output goals of 3–5 L negative per day can be achieved.
While there was early enthusiasm on mineralocorticoid receptor antagonist to attenuate distal sodium reabsorption, such strategy was deemed not incremental to standard therapy in a prospective trial [106]. An ongoing multicenter study testing the role of acetazolamide to augment proximal sodium excretion by attenuating tubular renin release is ongoing [107, 108].
Inotropic and Vasoactive Drugs . The typical inotropes used in cardiac intensive care units include dobutamine and milrinone (or to a lesser extend oral digoxin loading), and they are effective in restoring hemodynamics in the “cold and wet” patients under hemodynamic guidance. However, prospective data supporting their use is limited [109, 110]. Vasodilators may improve hemodynamic derangements, although overzealous use can lead to hypotension and worsening renal function [111]. In the setting of vasoplegia, norepinephrine (and to a lesser degree dopamine) may be also be used as it has beta adrenergic activity. Less popular now, is the use of dopamine as an inotrope and pressor especially with no added benefit to the renal vasculature as previously thought [112].
Ultrafiltration/Aquapheresis. Ultrafiltration provides mechanical removal of isotonic fluid independent of the kidneys, thus providing effective and consistent salt and volume removal. Although early studies were promising, subsequent randomized controlled trials have more mixed results [104, 113, 114]. Interestingly, ultrafiltration may even exacerbate hyponatremia as the effluent is relatively more hypertonic [115]. This can exacerbate the cycle of renal vascular constriction and neurohormonal activation if the settings are too aggressive. Peritoneal dialysis has also been employed as an alternative treatment strategy [116].
Hypertonic Saline. Considerations of electrolyte depletion leading to renal sodium avidity has implied potential benefits of hypertonic saline (HSS) infusions during aggressive IV diuretics. This was suggested a decade ago in early Italian series, in which low-volume, intermittent, 1.4–4.6% sodium chloride (depending on serum sodium levels) coupled with high-dose loop diuretics can produce effective diuresis and prevent decline in renal function [117,118,119]. Recent reports using 1.7% salt supplementation (500 mg) with lower doses of IV diuretics also demonstrated improved diuretic efficiencies, especially in those with elevated urinary BUN/creatinine levels [120, 121]. Real-world experience have also supported such a potential strategy in selected patients [122]. However, nephroprotection was not observed in patients with baseline creatinine over >2.2 mg/dL [123]. This was confirmed by preliminary results from a randomized, double-blind study of 50 patients with acute heart failure and renal insufficiency (creatinine >2 mg/dL, BUN >60 mg/dL) that demonstrated a non-significant increase in diuresis with HSS but also BUN elevation from baseline [124]. Hence, further investigations are warranted.
Mechanical Circulatory Assist Support . With the advent of temporary mechanical support such as the Impella® devices, a bridge-to-decision strategy can be instituted as demonstrated in animal models that improve renal blood flow [125]. After a test period to see if there is myocardial recovery, a durable left ventricular assist device (LVAD) may be considered [126]. Renal recovery following LVAD maybe transient [127], and renal function may deteriorate again after early improvement [11]. However if there is right ventricular dysfunction, orthotopic heart transplantation is the only durable solution. Implantable ventricular assist devices are rarely performed in patients reaching end-stage kidney diseases due to their high mortality rates and are not recommended by clinical guidelines [126, 128].
Temporary Renal Support Device Therapies . Recently, a handful of intriguing hemodynamic support devices have emerged targeting venous congestion and/or renal hemodynamics support. Examples include transcatheter intra-aortic pump [129], transcatheter renal venous decongestion system, innovative fluid/diuretic management systems (RenalGuard) [130]. Other examples of volume removal strategies include implantable pump or device designed to continuously remove excess abdominal fluid or direct sodium removal [131, 132], and catheter-based enhancement of lymphatic drainage [133]. The majority are in early clinical development.
7.6 Conclusions
Acute (Type 1) CRS is associated with an acute cardiogenic disturbance leading to acute worsening of renal function. However this cascade also forms a feedback loop further perpetuating cardiac dysfunction, hormonal dysregulation, and treatment resistance. Once ADHF is recognized, treatment must be initiated quickly to break the cycle but despite medical therapy, short term and long term aftereffects can make treatment a challenge. Ultimately changes in traditional renal biomarkers may not accurately reflect the state of the renal system while new insight into electrolyte metabolism may more accurately predict clinical outcomes.
While increases in serum creatinine have been closely tied to renal function, long term predictors of mortality and rehospitalization have not been closely linked. The response of the kidney in light of an acute cardiac insult should be largely viewed as appropriate and natural in the physiologic setting. However, the clinician should note that breaking the renal cycle will ultimately lead to a decongested patient with a chronic illness rather than an acute hospitalization.
Once developed, CRS becomes a serious medical and economic burden. Although hemodynamic derangement, an over-activated neurohormonal system, and systemic inflammation have been recognized as major players in CRS pathophysiology, there are also other cardio-renal mediators contributing to the development of CRS. The intricate network of these mediators makes their pathophysiologic hierarchy opaque. Investigators may need to divert their attention from overt cardio-renal connector in clinical CRS, to more fundamental substrates during a period of subclinical CRS as a part of an early detection and prevention strategy.
References
Liang KV, Williams AW, Greene EL, Redfield MM. Acute decompensated heart failure and the cardiorenal syndrome. Crit Care Med. 2008;36:S75–88.
Lewis T. A clinical lecture on paroxysmal dyspnoea in cardiorenal patients: with special reference to “Cardiac” and “Uraemic” asthma: delivered at University College Hospital, London, November 12th, 1913. Br Med J. 1913;2:1417–20.
Stengel A. The clinical determination of cardiovascular and renal responsibility, respectively, in its disturbances. JAMA. 1914;LXIII:1463–9.
Ronco C, McCullough P, Anker SD, Anand I, Aspromonte N, Bagshaw SM, Bellomo R, Berl T, Bobek I, Cruz DN, Daliento L, Davenport A, Haapio M, Hillege H, House AA, Katz N, Maisel A, Mankad S, Zanco P, Mebazaa A, Palazzuoli A, Ronco F, Shaw A, Sheinfeld G, Soni S, Vescovo G, Zamperetti N. Ponikowski P and acute Dialysis quality initiative consensus g. cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. 2010;31:703–11.
Rangaswami J, Bhalla V, Blair JEA, Chang TI, Costa S, Lentine KL, Lerma EV, Mezue K, Molitch M, Mullens W, Ronco C, Tang WHW, McCullough PA. American Heart Association Council on the kidney in cardiovascular D and council on clinical C. Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American Heart Association. Circulation. 2019;139:e840–78.
Starr I. Our changing viewpoint about congestive failure. Ann Intern Med. 1949;30:1–23.
NHLBI Working Group. Cardio-renal connections in heart failure and cardiovascular disease. 2004;2016.
Braam B, Joles JA, Danishwar AH, Gaillard CA. Cardiorenal syndrome—current understanding and future perspectives. Nat Rev Nephrol. 2014;10:48–55.
Ljungman S, Laragh JH, Cody RJ. Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs. 1990;39(Suppl 4):10–21; discussion 22-4.
Jose P, Skali H, Anavekar N, Tomson C, Krumholz HM, Rouleau JL, Moye L, Pfeffer MA, Solomon SD. Increase in creatinine and cardiovascular risk in patients with systolic dysfunction after myocardial infarction. J Am Soc Nephrol. 2006;17:2886–91.
Brisco MA, Kimmel SE, Coca SG, Putt ME, Jessup M, Tang WW, Parikh CR, Testani JM. Prevalence and prognostic importance of changes in renal function after mechanical circulatory support. Circ Heart Fail. 2014;7:68–75.
Bhatia RS, Tu JV, Lee DS, Austin PC, Fang J, Haouzi A, Gong Y, Liu PP. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med. 2006;355:260–9.
de Sa Correa DD, Hodge DO, Slusser JP, Redfield MM, Simari RD, Burnett JC, Chen HH. Progression of preclinical diastolic dysfunction to the development of symptoms. Heart. 2010;96:528–32.
Hillege HL, Nitsch D, Pfeffer MA, Swedberg K, McMurray JJ, Yusuf S, Granger CB, Michelson EL, Ostergren J, Cornel JH, de Zeeuw D, Pocock S, van Veldhuisen DJ. Candesartan in heart failure: assessment of reduction in M and morbidity I. renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation. 2006;113:671–8.
Yancy CW, Lopatin M, Stevenson LW, De Marco T, Fonarow GC, Committee ASA and Investigators. Clinical presentation, management, and in-hospital outcomes of patients admitted with acute decompensated heart failure with preserved systolic function: a report from the Acute Decompensated Heart Failure National Registry (ADHERE) Database. J Am Coll Cardiol. 2006;47:76–84.
Grodin JL, Mullens W, Dupont M, Taylor DO, McKie PM, Starling RC, Testani JM, Tang WHW. Hemodynamic factors associated with serum chloride in ambulatory patients with advanced heart failure. Int J Cardiol. 2018;252:112–6.
Grodin JL, Sun JL, Anstrom KJ, Chen HH, Starling RC, Testani JM, Tang WH. Implications of serum chloride homeostasis in acute heart failure (from ROSE-AHF). Am J Cardiol. 2017;119:78–83.
Grodin JL, Simon J, Hachamovitch R, Wu Y, Jackson G, Halkar M, Starling RC, Testani JM, Tang WH. Prognostic role of serum chloride levels in acute decompensated heart failure. J Am Coll Cardiol. 2015;66:659–66.
Damman K, van Deursen VM, Navis G, Voors AA, van Veldhuisen DJ, Hillege HL. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009;53:582–8.
Burnett JC Jr, Knox FG. Renal interstitial pressure and sodium excretion during renal vein constriction. Am J Physiol. 1980;238:F279–82.
Mullens W, Abrahams Z, Francis GS, Sokos G, Taylor DO, Starling RC, Young JB, Tang WH. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53:589–96.
Nijst P, Martens P, Dupont M, Tang WHW, Mullens W. Intrarenal flow alterations during transition from Euvolemia to intravascular volume expansion in heart failure patients. JACC Heart Fail. 2017;5:672–81.
Nijst P, Verbrugge FH, Martens P, Dupont M, Tang WHW, Mullens W. Renal response to intravascular volume expansion in euvolemic heart failure patients with reduced ejection fraction: mechanistic insights and clinical implications. Int J Cardiol. 2017;243:318–25.
Verbrugge FH, Dupont M, Steels P, Grieten L, Malbrain M, Tang WH, Mullens W. Abdominal contributions to cardiorenal dysfunction in congestive heart failure. J Am Coll Cardiol. 2013;62:485–95.
Mullens W, Abrahams Z, Skouri HN, Francis GS, Taylor DO, Starling RC, Paganini E, Tang WH. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol. 2008;51:300–6.
Chirakarnjanakorn S, Navaneethan SD, Francis GS, Tang WH. Cardiovascular impact in patients undergoing maintenance hemodialysis: clinical management considerations. Int J Cardiol. 2017;232:12–23.
Hata N, Yokoyama S, Shinada T, Kobayashi N, Shirakabe A, Tomita K, Kitamura M, Kurihara O, Takahashi Y. Acute kidney injury and outcomes in acute decompensated heart failure: evaluation of the RIFLE criteria in an acutely ill heart failure population. Eur J Heart Fail. 2010;12:32–7.
Ismail Y, Kasmikha Z, Green HL, McCullough PA. Cardio-renal syndrome type 1: epidemiology, pathophysiology, and treatment. Semin Nephrol. 2012;32:18–25.
Earley LE, Daugharty TM. Sodium metabolism. N Engl J Med. 1969;281:72–86.
Ronco C, Cicoira M, McCullough PA. Cardiorenal syndrome type 1: pathophysiological crosstalk leading to combined heart and kidney dysfunction in the setting of acutely decompensated heart failure. J Am Coll Cardiol. 2012;60:1031–42.
Hatamizadeh P, Fonarow GC, Budoff MJ, Darabian S, Kovesdy CP, Kalantar-Zadeh K. Cardiorenal syndrome: pathophysiology and potential targets for clinical management. Nat Rev Nephrol. 2013;9:99–111.
Mullens W, Verbrugge FH, Nijst P, Tang WHW. Renal sodium avidity in heart failure: from pathophysiology to treatment strategies. Eur Heart J. 2017;38:1872–82.
Cannon PJ. The kidney in heart failure. N Engl J Med. 1977;296:26–32.
Stanton RC, Brenner BM. Role of the kidney in congestive heart failure. Acta Med Scand Suppl. 1986;707:21–5.
Ruiz-Ortega M, Ruperez M, Lorenzo O, Esteban V, Blanco J, Mezzano S, Egido J. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int Suppl. 2002;62:S12–22.
Schrimpf C, Duffield JS. Mechanisms of fibrosis: the role of the pericyte. Curr Opin Nephrol Hypertens. 2011;20:297–305.
Petras D, Koutroutsos K, Kordalis A, Tsioufis C, Stefanadis C. The role of sympathetic nervous system in the progression of chronic kidney disease in the era of catheter based sympathetic renal denervation. Curr Clin Pharmacol. 2013;8:197–205.
Lambert E, Schlaich M. The role of renal sympathetic nerves in ischemia reperfusion injury. Auton Neurosci. 2017;204:105–11.
Mutoh J, Ohsawa M, Hisa H. Involvement of renal sympathetic nerve activation on the progression of ischemic acute kidney injury in the mouse. J Pharmacol Sci. 2014;125:415–21.
Kamal FA, Travers JG, Schafer AE, Ma Q, Devarajan P, Blaxall BC. G protein-coupled receptor-G-protein betagamma-subunit signaling mediates renal dysfunction and fibrosis in heart failure. J Am Soc Nephrol. 2017;28:197–208.
Salman IM, Ameer OZ, Sattar MA, Abdullah NA, Yam MF, Najim HS, Khan AH, Johns EJ. Role of the renal sympathetic nervous system in mediating renal ischaemic injury-induced reductions in renal haemodynamic and excretory functions. Pathology. 2010;42:259–66.
Salman IM, Sattar MA, Abdullah NA, Ameer OZ, Hussain FB, Hye Khan MA, Yam MF, Rathore KR, Kazi RN, Salman HM, Johns EJ. Renal functional & haemodynamic changes following acute unilateral renal denervation in Sprague Dawley rats. Indian J Med Res. 2010;131:76–82.
Fujiu K, Shibata M, Nakayama Y, Ogata F, Matsumoto S, Noshita K, Iwami S, Nakae S, Komuro I, Nagai R, Manabe I. A heart-brain-kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat Med. 2017;23(5):611–22.
Davies JE, Manisty CH, Petraco R, Barron AJ, Unsworth B, Mayet J, Hamady M, Hughes AD, Sever PS, Sobotka PA, Francis DP. First-in-man safety evaluation of renal denervation for chronic systolic heart failure: primary outcome from REACH-pilot study. Int J Cardiol. 2013;162:189–92.
Dai QM, Fen Y, Lu J, Ma GS. Efficacy of regional renal nerve blockade in patients with chronic refractory heart failure. Chin Med J (Engl). 2013;126:1076–80.
Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–41.
Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. 2001;103:2055–9.
Boulogne M, Sadoune M, Launay JM, Baudet M, Cohen-Solal A, Logeart D. Inflammation versus mechanical stretch biomarkers over time in acutely decompensated heart failure with reduced ejection fraction. Int J Cardiol. 2017;226:53–9.
Shahini N, Michelsen AE, Nilsson PH, Ekholt K, Gullestad L, Broch K, Dahl CP, Aukrust P, Ueland T, Mollnes TE, Yndestad A, Louwe MC. The alternative complement pathway is dysregulated in patients with chronic heart failure. Sci Rep. 2017;7:42532.
Hanberg JS, Rao VS, Ahmad T, Chunara Z, Mahoney D, Jackson K, Jacoby D, Chen M, Wilson FP, Tang WHW, Kakkar R, Testani JM. Inflammation and cardio-renal interactions in heart failure: a potential role for interleukin-6. Eur J Heart Fail. 2018;20:933–4.
Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res. 2014;114:266–82.
Cho E, Kim M, Ko YS, Lee HY, Song M, Kim MG, Kim HK, Cho WY, Jo SK. Role of inflammation in the pathogenesis of cardiorenal syndrome in a rat myocardial infarction model. Nephrol Dial Transplant. 2013;28:2766–78.
Buckley LF, Canada JM, Carbone S, Trankle CR, Kadariya D, Billingsley H, Wohlford GF, Kirkman DL, Abbate A, Van Tassell BW. Potential role for interleukin-1 in the cardio-renal syndrome. Eur J Heart Fail. 2019;21:385–6.
Liu C, Liu K. Effects of glucocorticoids in potentiating diuresis in heart failure patients with diuretic resistance. J Card Fail. 2014;20:625–9.
Pasini E, Aquilani R, Testa C, Baiardi P, Angioletti S, Boschi F, Verri M, Dioguardi F. Pathogenic gut Flora in patients with chronic heart failure. JACC Heart Fail. 2016;4:220–7.
Abraham C, Cho JH. Bugging of the intestinal mucosa. N Engl J Med. 2007;357:708–10.
Virzi GM, Clementi A, Brocca A, de Cal M, Marcante S, Ronco C. Cardiorenal syndrome type 5 in Sepsis: role of endotoxin in cell death pathways and inflammation. Kidney Blood Press Res. 2016;41:1008–15.
Brocca A, Virzi GM, Pasqualin C, Pastori S, Marcante S, de Cal M, Ronco C. Cardiorenal syndrome type 5: in vitro cytotoxicity effects on renal tubular cells and inflammatory profile. Anal Cell Pathol (Amst). 2015;2015:469461.
Sandek A, Rauchhaus M, Anker SD, von Haehling S. The emerging role of the gut in chronic heart failure. Curr Opin Clin Nutr Metab Care. 2008;11:632–9.
Flierl MA, Rittirsch D, Nadeau BA, Chen AJ, Sarma JV, Zetoune FS, McGuire SR, List RP, Day DE, Hoesel LM, Gao H, Van Rooijen N, Huber-Lang MS, Neubig RR, Ward PA. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature. 2007;449:721–5.
Olofsson PS, Rosas-Ballina M, Levine YA, Tracey KJ. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev. 2012;248:188–204.
Adijiang A, Shimizu H, Higuchi Y, Nishijima F, Niwa T. Indoxyl sulfate reduces klotho expression and promotes senescence in the kidneys of hypertensive rats. J Ren Nutr. 2011;21:105–9.
Sun CY, Chang SC, Wu MS. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS One. 2012;7:e34026.
Wu IW, Hsu KH, Lee CC, Sun CY, Hsu HJ, Tsai CJ, Tzen CY, Wang YC, Lin CY, Wu MS. P-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol Dial Transplant. 2011;26:938–47.
Vaziri ND. CKD impairs barrier function and alters microbial flora of the intestine: a major link to inflammation and uremic toxicity. Curr Opin Nephrol Hypertens. 2012;21:587–92.
Lin CJ, Wu CJ, Pan CF, Chen YC, Sun FJ, Chen HH. Serum protein-bound uraemic toxins and clinical outcomes in haemodialysis patients. Nephrol Dial Transplant. 2010;25:3693–700.
Poesen R, Viaene L, Verbeke K, Augustijns P, Bammens B, Claes K, Kuypers D, Evenepoel P, Meijers B. Cardiovascular disease relates to intestinal uptake of p-cresol in patients with chronic kidney disease. BMC Nephrol. 2014;15:87.
Ramezani A, Massy ZA, Meijers B, Evenepoel P, Vanholder R, Raj DS. Role of the gut microbiome in uremia: a potential therapeutic target. Am J Kidney Dis. 2016;67:483–98.
Giam B, Kaye DM, Rajapakse NW. Role of renal oxidative stress in the pathogenesis of the cardiorenal syndrome. Heart Lung Circ. 2016;25:874–80.
Fujii H, Nishijima F, Goto S, Sugano M, Yamato H, Kitazawa R, Kitazawa S, Fukagawa M. Oral charcoal adsorbent (AST-120) prevents progression of cardiac damage in chronic kidney disease through suppression of oxidative stress. Nephrol Dial Transplant. 2009;24:2089–95.
Lekawanvijit S, Adrahtas A, Kelly DJ, Kompa AR, Wang BH, Krum H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur Heart J. 2010;31:1771–9.
Yisireyili M, Shimizu H, Saito S, Enomoto A, Nishijima F, Niwa T. Indoxyl sulfate promotes cardiac fibrosis with enhanced oxidative stress in hypertensive rats. Life Sci. 2013;92:1180–5.
Lekawanvijit S, Kompa AR, Manabe M, Wang BH, Langham RG, Nishijima F, Kelly DJ, Krum H. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin, indoxyl sulfate. PLoS One. 2012;7:e41281.
Liu S, Wang BH, Kompa AR, Lekawanvijit S, Krum H. Antagonists of organic anion transporters 1 and 3 ameliorate adverse cardiac remodelling induced by uremic toxin indoxyl sulfate. Int J Cardiol. 2012;158:457–8.
Gondouin B, Cerini C, Dou L, Sallee M, Duval-Sabatier A, Pletinck A, Calaf R, Lacroix R, Jourde-Chiche N, Poitevin S, Arnaud L, Vanholder R, Brunet P, Dignat-George F, Burtey S. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013;84:733–44.
Heywood JT, Fonarow GC, Costanzo MR, Mathur VS, Wigneswaran JR, Wynne J, Committee ASA and Investigators. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail. 2007;13:422–30.
Devarajan P, Mishra J, Supavekin S, Patterson LT, Steven PS. Gene expression in early ischemic renal injury: clues towards pathogenesis, biomarker discovery, and novel therapeutics. Mol Genet Metab. 2003;80:365–76.
Nguyen MT, Ross GF, Dent CL, Devarajan P. Early prediction of acute renal injury using urinary proteomics. Am J Nephrol. 2005;25:318–26.
McCullough PA, Shaw AD, Haase M, Bouchard J, Waikar SS, Siew ED, Murray PT, Mehta RL, Ronco C. Diagnosis of acute kidney injury using functional and injury biomarkers: workgroup statements from the tenth acute Dialysis quality initiative consensus conference. Contrib Nephrol. 2013;182:13–29.
Aghel A, Shrestha K, Mullens W, Borowski A, Tang WH. Serum neutrophil gelatinase-associated lipocalin (NGAL) in predicting worsening renal function in acute decompensated heart failure. J Card Fail. 2010;16:49–54.
Maisel AS, Mueller C, Fitzgerald R, Brikhan R, Hiestand BC, Iqbal N, Clopton P, van Veldhuisen DJ. Prognostic utility of plasma neutrophil gelatinase-associated lipocalin in patients with acute heart failure: the NGAL EvaLuation along with B-type NaTriuretic peptide in acutely decompensated heart failure (GALLANT) trial. Eur J Heart Fail. 2011;13:846–51.
Maisel AS, Wettersten N, van Veldhuisen DJ, Mueller C, Filippatos G, Nowak R, Hogan C, Kontos MC, Cannon CM, Muller GA, Birkhahn R, Clopton P, Taub P, Vilke GM, McDonald K, Mahon N, Nunez J, Briguori C, Passino C, Murray PT. Neutrophil Gelatinase-associated Lipocalin for acute kidney injury during acute heart failure hospitalizations: the AKINESIS study. J Am Coll Cardiol. 2016;68:1420–31.
Dupont M, Shrestha K, Singh D, Awad A, Kovach C, Scarcipino M, Maroo AP, Tang WH. Lack of significant renal tubular injury despite acute kidney injury in acute decompensated heart failure. Eur J Heart Fail. 2012;14:597–604.
Rao VS, Ahmad T, Brisco-Bacik MA, Bonventre JV, Wilson FP, Siew ED, Felker GM, Anstrom KK, Mahoney DD, Bart BA, Tang WHW, Velazquez EJ, Testani JM. Renal effects of intensive volume removal in heart failure patients with preexisting worsening renal function. Circ Heart Fail. 2019;12:e005552.
Ahmad T, Jackson K, Rao VS, Tang WHW, Brisco-Bacik MA, Chen HH, Felker GM, Hernandez AF, O'Connor CM, Sabbisetti VS, Bonventre JV, Wilson FP, Coca SG, Testani JM. Worsening renal function in patients with acute heart failure undergoing aggressive diuresis is not associated with tubular injury. Circulation. 2018;137:2016–28.
Verbrugge FH, Dupont M, Shao Z, Shrestha K, Singh D, Finucan M, Mullens W, Tang WH. Novel urinary biomarkers in detecting acute kidney injury, persistent renal impairment, and all-cause mortality following decongestive therapy in acute decompensated heart failure. J Card Fail. 2013;19:621–8.
Praga M, Morales E. Obesity, proteinuria and progression of renal failure. Curr Opin Nephrol Hypertens. 2006;15:481–6.
Hunley TE, Ma LJ, Kon V. Scope and mechanisms of obesity-related renal disease. Curr Opin Nephrol Hypertens. 2010;19:227–34.
D'Agati VD, Chagnac A, de Vries AP, Levi M, Porrini E, Herman-Edelstein M, Praga M. Obesity-related glomerulopathy: clinical and pathologic characteristics and pathogenesis. Nat Rev Nephrol. 2016;12:453–71.
Kambham N, Markowitz GS, Valeri AM, Lin J, D'Agati VD. Obesity-related glomerulopathy: an emerging epidemic. Kidney Int. 2001;59:1498–509.
van Dijk CG, Oosterhuis NR, Xu YJ, Brandt M, Paulus WJ, van Heerebeek L, Duncker DJ, Verhaar MC, Fontoura D, Lourenco AP, Leite-Moreira AF, Falcao-Pires I, Joles JA, Cheng C. Distinct endothelial cell responses in the heart and kidney microvasculature characterize the progression of heart failure with preserved ejection fraction in the obese ZSF1 rat with Cardiorenal metabolic syndrome. Circ Heart Fail. 2016;9:e002760.
Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, Gheorghiade M, Bonow RO, Huang CC, Deo RC. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation. 2015;131:269–79.
Saleh F, Kim SJ, Okrainec A, Jackson TD. Bariatric surgery in patients with reduced kidney function: an analysis of short-term outcomes. Surg Obes Relat Dis. 2015;11:828–35.
Look ARG. Effect of a long-term behavioural weight loss intervention on nephropathy in overweight or obese adults with type 2 diabetes: a secondary analysis of the Look AHEAD randomised clinical trial. Lancet Diabetes Endocrinol. 2014;2:801–9.
MacLaughlin HL, Hall WL, Patel AG, Blacklock RM, Swift PA, Phanish MK, Dew T, Chowdhury P, Sanders TA, Macdougall IC. Weight loss, adipokines, and quality of life after sleeve gastrectomy in obese patients with stages 3-4 CKD: a randomized controlled pilot study. Am J Kidney Dis. 2014;64:660–3.
Fenske W, Athanasiou T, Harling L, Drechsler C, Darzi A, Ashrafian H. Obesity-related cardiorenal disease: the benefits of bariatric surgery. Nat Rev Nephrol. 2013;9:539–51.
Brinkworth GD, Buckley JD, Noakes M, Clifton PM. Renal function following long-term weight loss in individuals with abdominal obesity on a very-low-carbohydrate diet vs high-carbohydrate diet. J Am Diet Assoc. 2010;110:633–8.
Mullens W, Damman K, Harjola VP, Mebazaa A, Brunner-La Rocca HP, Martens P, Testani JM, Tang WHW, Orso F, Rossignol P, Metra M, Filippatos G, Seferovic PM, Ruschitzka F, Coats AJ. The use of diuretics in heart failure with congestion - a position statement from the heart failure Association of the European Society of cardiology. Eur J Heart Fail. 2019;21:137–55.
Martens P, Dupont M, Verbrugge FH, Damman K, Degryse N, Nijst P, Reynders C, Penders J, Tang WHW, Testani J, Mullens W. Urinary sodium profiling in chronic heart failure to detect development of acute decompensated heart failure. JACC Heart Fail. 2019;7:404–14.
Singh D, Shrestha K, Testani JM, Verbrugge FH, Dupont M, Mullens W, Tang WH. Insufficient natriuretic response to continuous intravenous furosemide is associated with poor long-term outcomes in acute decompensated heart failure. J Card Fail. 2014;20:392–9.
Testani JM, Chen J, McCauley BD, Kimmel SE, Shannon RP. Potential effects of aggressive decongestion during the treatment of decompensated heart failure on renal function and survival. Circulation. 2010;122:265–72.
Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, LeWinter MM, Deswal A, Rouleau JL, Ofili EO, Anstrom KJ, Hernandez AF, McNulty SE, Velazquez EJ, Kfoury AG, Chen HH, Givertz MM, Semigran MJ, Bart BA, Mascette AM, Braunwald E, O’Connor CM, Network NHFCR. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364:797–805.
Hanberg JS, Tang WHW, Wilson FP, Coca SG, Ahmad T, Brisco MA, Testani JM. An exploratory analysis of the competing effects of aggressive decongestion and high-dose loop diuretic therapy in the DOSE trial. Int J Cardiol. 2017;241:277–82.
Bart BA, Goldsmith SR, Lee KL, Givertz MM, O'Connor CM, Bull DA, Redfield MM, Deswal A, Rouleau JL, LeWinter MM, Ofili EO, Stevenson LW, Semigran MJ, Felker GM, Chen HH, Hernandez AF, Anstrom KJ, McNulty SE, Velazquez EJ, Ibarra JC, Mascette AM, Braunwald E, Heart Failure Clinical Research N. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med. 2012;367:2296–304.
Grodin JL, Stevens SR, de Las FL, Kiernan M, Birati EY, Gupta D, Bart BA, Felker GM, Chen HH, Butler J, Davila-Roman VG, Margulies KB, Hernandez AF, Anstrom KJ, Tang WH. Intensification of medication therapy for Cardiorenal syndrome in acute decompensated heart failure. J Card Fail. 2016;22:26–32.
Butler J, Anstrom KJ, Felker GM, Givertz MM, Kalogeropoulos AP, Konstam MA, Mann DL, Margulies KB, McNulty SE, Mentz RJ, Redfield MM, Tang WHW, Whellan DJ, Shah M, Desvigne-Nickens P, Hernandez AF, Braunwald E, National Heart L and Blood Institute Heart Failure Clinical Research N. Efficacy and safety of spironolactone in acute heart failure: the ATHENA-HF randomized clinical trial. JAMA Cardiol. 2017;2:950–8.
Verbrugge FH, Martens P, Ameloot K, Haemels V, Penders J, Dupont M, Tang WHW, Droogne W, Mullens W. Acetazolamide to increase natriuresis in congestive heart failure at high risk for diuretic resistance. Eur J Heart Fail. 2019;21(11):1415–22.
Mullens W, Verbrugge FH, Nijst P, Martens P, Tartaglia K, Theunissen E, Bruckers L, Droogne W, Troisfontaines P, Damman K, Lassus J, Mebazaa A, Filippatos G, Ruschitzka F, Dupont M. Rationale and design of the ADVOR (acetazolamide in decompensated heart failure with volume overload) trial. Eur J Heart Fail. 2018;20:1591–600.
Klein L, Massie BM, Leimberger JD, O’Connor CM, Pina IL, Adams KF Jr, Califf RM, Gheorghiade M, Investigators O-C. Admission or changes in renal function during hospitalization for worsening heart failure predict postdischarge survival: results from the outcomes of a prospective trial of intravenous Milrinone for exacerbations of chronic heart failure (OPTIME-CHF). Circ Heart Fail. 2008;1:25–33.
Madeira M, Caetano F, Almeida I, Fernandes A, Reis L, Costa M, Goncalves L. Inotropes and cardiorenal syndrome in acute heart failure—a retrospective comparative analysis. Rev Port Cardiol. 2017;36:619–25.
Mullens W, Abrahams Z, Francis GS, Skouri HN, Starling RC, Young JB, Taylor DO, Tang WH. Sodium nitroprusside for advanced low-output heart failure. J Am Coll Cardiol. 2008;52:200–7.
Chen HH, Anstrom KJ, Givertz MM, Stevenson LW, Semigran MJ, Goldsmith SR, Bart BA, Bull DA, Stehlik J, MM LW, Konstam MA, Huggins GS, Rouleau JL, O'Meara E, Tang WH, Starling RC, Butler J, Deswal A, Felker GM, O'Connor CM, Bonita RE, Margulies KB, Cappola TP, Ofili EO, Mann DL, Davila-Roman VG, SE MN, Borlaug BA, Velazquez EJ, Lee KL, Shah MR, Hernandez AF, Braunwald E, Redfield MM, Network NHFCR. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA. 2013;310:2533–43.
Costanzo MR, Negoianu D, Jaski BE, Bart BA, Heywood JT, Anand IS, Smelser JM, Kaneshige AM, Chomsky DB, Adler ED, Haas GJ, Watts JA, Nabut JL, Schollmeyer MP, Fonarow GC. Aquapheresis versus intravenous diuretics and hospitalizations for heart failure. JACC Heart Fail. 2016;4:95–105.
Grodin JL, Carter S, Bart BA, Goldsmith SR, Drazner MH, Tang WHW. Direct comparison of ultrafiltration to pharmacological decongestion in heart failure: a per-protocol analysis of CARRESS-HF. Eur J Heart Fail. 2018;20:1148–56.
Kitai T, Grodin JL, Kim YH, Tang WH. Impact of ultrafiltration on serum sodium homeostasis and its clinical implication in patients with acute heart failure, congestion, and worsening renal function. Circ Heart Fail. 2017;10:e003603.
Ponce D, Goes C, Oliveira M, Balbi A. Peritoneal Dialysis for the treatment of cardiorenal syndrome type 1: a prospective Brazilian study. Perit Dial Int. 2017;37:578–83.
Paterna S, Di Pasquale P, Parrinello G, Amato P, Cardinale A, Follone G, Giubilato A, Licata G. Effects of high-dose furosemide and small-volume hypertonic saline solution infusion in comparison with a high dose of furosemide as a bolus, in refractory congestive heart failure. Eur J Heart Fail. 2000;2:305–13.
Paterna S, Fasullo S, Parrinello G, Cannizzaro S, Basile I, Vitrano G, Terrazzino G, Maringhini G, Ganci F, Scalzo S, Sarullo FM, Cice G, Di Pasquale P. Short-term effects of hypertonic saline solution in acute heart failure and long-term effects of a moderate sodium restriction in patients with compensated heart failure with New York heart association class III (class C) (SMAC-HF study). Am J Med Sci. 2011;342:27–37.
Paterna S, Parrinello G, Amato P, Dominguez L, Pinto A, Maniscalchi T, Cardinale A, Licata A, Amato V, Licata G, Di Pasquale P. Tolerability and efficacy of high-dose furosemide and small-volume hypertonic saline solution in refractory congestive heart failure. Adv Ther. 1999;16:219–28.
Ando T, Okuhara Y, Orihara Y, Nishimura K, Yamamoto K, Masuyama T, Hirotani S. Urinary composition predicts diuretic efficiency of hypertonic saline solution with furosemide therapy and heart failure prognosis. Heart Vessels. 2018;33:1029–36.
Okuhara Y, Hirotani S, Naito Y, Nakabo A, Iwasaku T, Eguchi A, Morisawa D, Ando T, Sawada H, Manabe E, Masuyama T. Intravenous salt supplementation with low-dose furosemide for treatment of acute decompensated heart failure. J Card Fail. 2014;20:295–301.
Goljo E, Soufer A, Colna M, Rao VS, D'Ambrosi J, Riello R, Mahoney D, Tang WH, Testani JM. Real world use of hypertonic saline in refractory acute decompensated heart failure: A U.S. Center’s Experience. J Card Fail. 2018;24:S32.
De Vecchis R, Esposito C, Ariano C, Cantatrione S. Hypertonic saline plus i.v. furosemide improve renal safety profile and clinical outcomes in acute decompensated heart failure: a meta-analysis of the literature. Herz. 2015;40:423–35.
Engelmeier RS, Le TT, Kamalay SE, Utecht KN, Nikstad TP, Kaliebe JW, Olson K, Larrain G. Radomized trial of hyigh dose furosemide-hypertonic saline in acute decompensated heart failure with advanced renal disease. J Am Coll Cardiol. 2019;59:E958.
Moller-Helgestad OK, Poulsen CB, Christiansen EH, Lassen JF, Ravn HB. Support with intra-aortic balloon pump vs. Impella2.5(R) and blood flow to the heart, brain and kidneys - an experimental porcine model of ischaemic heart failure. Int J Cardiol. 2015;178:153–8.
Feldman D, Pamboukian SV, Teuteberg JJ, Birks E, Lietz K, Moore SA, Morgan JA, Arabia F, Bauman ME, Buchholz HW, Deng M, Dickstein ML, El-Banayosy A, Elliot T, Goldstein DJ, Grady KL, Jones K, Hryniewicz K, John R, Kaan A, Kusne S, Loebe M, Massicotte MP, Moazami N, Mohacsi P, Mooney M, Nelson T, Pagani F, Perry W, Potapov EV, Eduardo Rame J, Russell SD, Sorensen EN, Sun B, Strueber M, Mangi AA, Petty MG, Rogers J, International Society for H and Lung T. The 2013 International Society for Heart and Lung Transplantation guidelines for mechanical circulatory support: executive summary. J Heart Lung Transplant. 2013;32:157–87.
Ross DW, Stevens GR, Wanchoo R, Majure DT, Jauhar S, Fernandez HA, Merzkani M, Jhaveri KD. Left ventricular assist devices and the kidney. Clin J Am Soc Nephrol. 2018;13:348–55.
Walther CP, Niu J, Winkelmayer WC, Cheema FH, Nair AP, Morgan JA, Fedson SE, Deswal A, Navaneethan SD. Implantable ventricular assist device use and outcomes in people with end-stage renal disease. J Am Heart Assoc. 2018;7.
Vora AN, Schuyler Jones W, DeVore AD, Ebner A, Clifton W, Patel MR. First-in-human experience with Aortix intraaortic pump. Catheter Cardiovasc Interv. 2019;93:428–33.
Putzu A, Boscolo Berto M, Belletti A, Pasotti E, Cassina T, Moccetti T, Pedrazzini G. Prevention of contrast-induced acute kidney injury by furosemide with matched hydration in patients undergoing interventional procedures: a systematic review and meta-analysis of randomized trials. JACC Cardiovasc Interv. 2017;10:355–63.
Feld Y, Hanani N, Costanzo MR. Hydrostatic pressure gradient ultrafiltration device: a novel approach for extracellular fluid removal. J Heart Lung Transplant. 2018;37:794–6.
Bureau C, Adebayo D, Chalret de Rieu M, Elkrief L, Valla D, Peck-Radosavljevic M, McCune A, Vargas V, Simon-Talero M, Cordoba J, Angeli P, Rosi S, MacDonald S, Malago M, Stepanova M, Younossi ZM, Trepte C, Watson R, Borisenko O, Sun S, Inhaber N, Jalan R. Alfapump(R) system vs. large volume paracentesis for refractory ascites: A multicenter randomized controlled study. J Hepatol. 2017;67:940–9.
Haberman D, Jonas M, Zilberman L, Fungenfirov I, Goland S, George K, Costanzo MR, Abraham WT. Catherter-based enhancement of lymphatic drainage in fluid overloaded acute decompensated heart failure: first in human experience. Eur J Heart Fail. 2018;20:393.
Funding
Dr. Tang is funded by grants from the National Institutes of Health (R01HL103931, R01DK106000, R01HL126827).
Disclosure: All authors have no relationships to disclose.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kim, YH., Xu, W., Kitai, T., Tang, W.H.W. (2021). Type 1 Cardio-Renal Syndrome. In: McCullough, P.A., Ronco, C. (eds) Textbook of Cardiorenal Medicine. Springer, Cham. https://doi.org/10.1007/978-3-030-57460-4_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-57460-4_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-57459-8
Online ISBN: 978-3-030-57460-4
eBook Packages: MedicineMedicine (R0)