
Abstract
The great hopes raised by the discovery of the immunoregulatory cytokine interleukin 12 (IL-12) as an anticancer agent were marred during early clinical experimentation because of severe adverse effects, which prompted a search for alternative formulations and routes of administration. Onco-immunotherapeutic viruses (OIVs) are wild-type or genetically engineered viruses that exert antitumor activity by causing death of the tumor cells they infect and by overcoming a variety of immunosuppressive mechanisms put in place by the tumors. OIVs have renewed the interest in IL-12, as they offer the opportunity to encode the cytokine transgenically from the viral genome and to produce it at high concentrations in the tumor bed. A large body of evidence indicates that IL-12 serves as a potent adjuvant for the immunotherapeutic response elicited by OIVs in murine tumor models. The list of OIVs includes onco-immunotherapeutic herpes simplex, adeno, measles, Newcastle disease, and Maraba viruses, among others. The large increase in IL-12-mediated adjuvanticity was invariably observed for all the OIVs analyzed. Indirect evidence suggests that locally delivered IL-12 may also increase tumor antigenicity. Importantly, the OIV/IL-12 treatment was not accompanied by adverse effects and elicited a long-lasting immune response capable of halting the growth of distant tumors. Thus, OIVs provide an avenue for reducing the clinical toxicity associated with systemic IL-12 therapy, by concentrating the cytokine at the site of disease. The changes to the tumor microenvironment induced by the IL-12-armed OIVs primed the tumors to an improved response to the checkpoint blockade therapy, suggesting that the triple combination is worth pursuing in the future. The highly encouraging results in preclinical models have prompted translation to the clinic. How well the IL-12–OIV–checkpoint inhibitors’ combination will perform in humans remains to be fully investigated.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- IL-12
- Oncolytic virus
- Onco-immunotherapeutic virus
- Oncolytic immunotherapy
- Cancer therapy
- OIV
- HSV
- Adenovirus
- Maraba
- Measles
- NDV
- Immune heating
- TME
- Proinflammatory cytokines
- Adjuvanticity
- Antigenicity
- CP blockade
- CPI combination
4.1 IL-12, Basic Features
Interleukin 12 (IL-12) is a proinflammatory and immunoregulatory cytokine discovered independently by two laboratories 30 years ago [1,2,3]. Structurally, IL-12 (p70) is a disulfide-linked heterodimer of IL-12A (p35) and IL-12B (p40) subunits, shared with two additional members of the IL-12 family, IL-35 and IL-23, respectively [4]. IL-12 is expressed and secreted by activated macrophages, dendritic cells (DCs), microglia, monocytes, neutrophils, and B cells in response to microbial infections and malignancies [5]. IL-12 binds the IL-12 receptor (IL-12R) , a beta1-beta2 heterodimer type I cytokine receptor expressed by a number of cells, including natural killer (NK), activated T, and natural killer T (NKT) cells, DCs, B cells, and macrophages [6,7,8,9]. Each of these cells responds to IL-12 through specific signaling pathways and responses. Shortly after the IL-12 discovery, it was recognized that IL-12 exerts a strong adjuvant effect with antipathogens [10] and anticancer vaccines [11, 12]. This cytokine promotes the secretion of interferon gamma (IFNγ) by NK, T, and B cells [13] and additional proinflammatory cytokines, including tumor necrosis factor alpha (TNFα) and granulocyte-macrophage colony-stimulating factor (GM-CSF). In turn, these molecules target, recruit, and activate effector cells of the innate immune response, and, together, they make IL-12 a master regulator. Importantly, IL-12 provides a link between the activation of innate and adaptive responses by priming Th1 cells for activation. The latter is a key part of the anticancer response, as it promotes the reactivation of memory CD4+ T cells and their repolarization from tumor-permissive Th2 to antitumor Th1 cells [14]. IL-12 also triggers NK and CD8+ T-cell activation, proliferation, and differentiation [15], leading to the generation of cytotoxic T cells (CTLs). Specifically, the cytokine primes macrophages for antigen presentation [8] and their M2 to M1 repolarization [16] promotes DC maturation and activity [17] and induces B-cell proliferation, differentiation, and an IgE to IgG1 shift [6].
IL-12 also affects the nonimmune cells of the tumor microenvironment (TME), including stromal cells and blood vessels that feed the tumor and sustain carcinogenesis [18]. Mechanistically, IL-12 downregulates the proangiogenic cytokines CCL2, CCL6, IL-6, VEGF, and other factors, and upregulates angiostatic and antiangiogenic factors, including TNFα, IFNα, IFNβ, IFNγ, CXCL9, and CXCL10 [19, 20]. Finally, the cytokine facilitates immune cells’ recruitment and lymphocyte localization to the tumor through IFNγ-dependent cascades and upregulation of immune-attractants [21]. Globally, IL-12 reprograms the tumor TME from a protumoral hospitable alcove to an antitumor environment.
The potent anticancer effects elicited by systemically administered IL-12 were well documented in preclinical models (reviewed in [22]). However, early studies in humans were marred by limited efficacy and generalized toxicity. The severe to lethal effects included hematopoietic suppression and gastrointestinal, muscular, pulmonary and liver toxicity, and dysfunction [23,24,25,26]. These side effects prompted the search for novel formulations and for new administration strategies capable of achieving higher local IL-12 concentrations. A promising approach consisted in intratumoral delivery by nonreplicating adenoviruses (AdVs) [27, 28]. Additional approaches include subcutaneous injections of the recombinant protein, fine-tunable expression systems [29], delivery of IL-12-encoding plasmid in the tumor bed, coupling of the cytokine with a tumor-targeting antibody (Ab) [30, 31], and transgenic expression by engineered tumor-specific CAR-T, autologous immune or cancer cells [32, 33]. The Clinicaltrials.gov website lists 84 active clinical trials for the evaluation of IL-12 treatments for a variety of solid tumors, including pancreatic, prostatic, colorectal, ovarian, breast, and liver cancers. IL-12 is administered as a recombinant protein (38), as a fusion protein with cancer-specific antibodies (2), is expressed by a plasmid (23), is vectored by bacteria (1), by viruses (15), or by cells, including CAR-Ts (2), or engineered autologous cells (3).
4.2 From Oncolytic to Onco-immunotherapeutic Viruses, a Paradigm Shift
Oncolytic viruses (OVs) are replication-competent wild-type (wt) or engineered viruses that selectively replicate in tumor cells and/or in cells in the TME. The intrinsic properties of tumors are immune tolerance and immune evasion, which, together with defects in innate immune responses, greatly favor virus susceptibility and replication. Early preclinical studies on human cancer cells implanted in nude mice highlighted the antitumor efficacy exerted by OVs, mainly as a consequence of lysis of the infected cells by immunogenic cell death mechanisms, including necroptosis [34]. When preclinical models were shifted to immunocompetent mouse models, it became apparent that, in addition to tumor cell lysis, tumor infection by oncolytic viruses resulted in the tolerance breakdown, the induction of an innate response to the tumor and, ultimately, to immune control of tumor growth. The current resurgence of interest in OVs is the result of an array of effects, among which are the secretion of type I and II interferons and other proinflammatory cytokines, the infiltration of tumors by NK cells and T-lymphocytes, the activation of these cells, and an overall reprogramming of the TME that enhances the adaptive systemic antitumor response. In brief, OVs convert immunologically cold tumors into immunologically hot ones [35]. Through these modifications, OV-infected tumor cells serve as antigen agnostic antitumor vaccines [36,37,38]. These OVs can be renamed as onco-immunotherapeutic viruses (OIVs) .
The OIV-mediated immunotherapeutic effects observed in preclinical models were documented in humans, in particular with talimogene laherparepvec (T-Vec), a mildly attenuated oHSV that expresses GM-CSF to increase macrophage, DC, and neutrophil responses. In cutaneous melanoma patients, the intratumoral administration of T-Vec in some of the lesions resulted in the shrinkage of distant untreated lesions, even though the reduction was not as large as that observed in the treated lesions [39]. The distant response is attributed to an abscopal immune effect, caused by the adaptive immune response to the tumor.
The immunotherapy of cancer has been recently revolutionized by checkpoint inhibitors (CPIs). Unfortunately, their activity is exerted only toward a subset of cancers and to a fraction of patients, and is limited by severe adverse effects. Making tumors immunologically hot by OIVs confers CPI susceptibility to tumors that are otherwise resistant [40]. Thus, OIVs represent the ideal partners for checkpoint blockade [40,41,42,43]. Today, OIVs are considered as most promising tools to increase the efficacy and broaden the spectrum of CPIs.
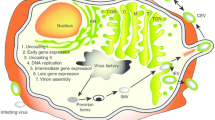
The ability of OIVs to unleash immune suppression and to elicit an innate response to tumors, even in highly immune suppressive tumors, renders OIVs the ideal companions for IL-12. Furthermore, the IL-12 gene can be expressed transgenically from the viral genome in the tumor bed, so as to prevent systemic toxicity of the cytokine. By exerting its adjuvant effect, IL-12 promotes the shift from an innate response to the virus toward an adaptive long-term memory response to the tumor [44, 45] (Fig. 4.1).
The list of IL-12-expressing OIVs and the beneficial effects of OIV-delivered IL-12 in the tumor bed has been documented by numerous studies. Here, we review some select examples.
4.3 oHSVs
In a highly innovative, seminal study, Martuza, Rabkin, and coworkers recognized the ability of oHSVs to confer protection not only in the treated tumor through lysis of the infected cancer cells—the dominant paradigm at that time—but also through elicitation of the host immune response [46]. They employed the oHSV named G207 as a helper virus to generate dvIL12/G207, a defective HSV vector expressing IL-12 [46]. G207 and its derivatives harbor deletions in UL39 (ICP6) and both copies of the γ134.5 gene; hence, they are highly safe yet attenuated. Remarkably, the intralesionally treated tumors responded to the therapy and exhibited reduced growth; 33% of the mice were tumor free (TF). Moreover, the treatment reduced the growth of untreated contralateral tumors; i.e., elicited a long-term in situ vaccination effect [46] dependent on the systemic T-cell response. To improve the antigen presentation, the IL-12 gene was engineered in the G207-derived G47Δ backbone, which additionally harbors the US12 (α47) deletion (G47Δ-mIL12 virus) [47]. This oHSV was used against murine glioblastoma and showed T-cell-dependent reduced tumor growth, reduced intratumoral Treg levels, and inhibition of angiogenesis [47].
Fong and coworkers focused on the development of an IL-12 oHSV for the treatment of squamous cell carcinoma and colorectal and liver cancers [48,49,50]. The NV1042 viral genome carried multiple deletions, namely, US10, US11, and α47 genes and one copy of the γ134.5, α0, α4 genes. Initially, the authors compared the effects of the transgenic expression of IL-12 to that of GM-CSF and found that the IL-12 virus was superior in various models [50,51,52]. The production of the α-promoter-driven IL-12 ranged from 1 to 35 ng/mL at 24–72 h post infection per 5 × 104 cells infected at one plaque-forming unit (PFU) per cell [48]. Although the virus was overall attenuated, a single injection reduced the growth of CT26 murine colorectal cancers; a few mice were completely cured [48]. The local immune stimulation provided by IL-12 resulted in the control of hepatic challenge tumors upon resection of the primary tumor [49]. In a pulmonary metastatic model, the immunotherapeutic effect of IL-12 virus was shown to involve CD4+ and CD8+ T cells [53, 54]. In a model of squamous cell carcinoma, it was also verified that treatment with IL-12-expressing NV1042 resulted in anti-angiogenic effects [53, 54]. The same virus also proved effective upon systemic administration against the pulmonary metastases of squamous cell carcinoma [53, 54], liver metastases of colorectal cancer [55], and of poorly immunogenic prostate adenocarcinoma and metastatic prostatic cancers [51, 52, 56].
Markert and coworkers engineered the IL-12 gene in a less attenuated oHSV, named M002, initially designed for glioblastoma treatment. M002 carries the replacement of both copies of the γ134.5 gene with IL-12 and no other virus genome modification. The two copies of mIL-12 were placed under the murine egr-1 promoter. In vitro, the extent of IL-12 expression was in the range of 0.8–3.2 ng/mL per 5 × 105 cells infected at 1 PFU/cell, at 24 h after infection [57]. Importantly, the M002 treatment resulted in a significant increase in mouse survival [57]. The same virus was also effective in preclinical models of breast cancer metastases to the brain [58], glioma [59], neuroblastoma [60], rhabdomyosarcoma [61], undifferentiated sarcoma [62], and in pediatric high-grade brain tumor and medulloblastoma xenografts [63, 64]. M002 and its cognate M032, an identical recombinant virus expressing hIL-12 in place of mIL-12, have undergone detailed safety analyses in mice and in Aotus primates [65, 66]. A phase 1 clinical trial for glioblastoma multiforme treatment is recruiting participants [67].
Altogether, the three series of studies highlight the superior effects of IL-12-armed oHSVs against murine primary tumors as well as distant T-cell-based immune protection.
A general notion that permeates the OIV field, and particularly the oHSV field, is that the initial safety concerns led to viruses that exhibited a very high safety profile in mice as well as in humans and are effective against murine tumor models but not as effective against human tumors. These considerations led to calls for “safe-and-robust” oHSVs that are more effective than those that are now in clinical practice or trials. To this end, our laboratory engineered tropism-retargeted oHSVs whose safety rests on cancer-specific tropisms, rather than on the cancer-selective replication typical of the oHSVs that are attenuated to varying degrees. The principle of tropism retargeting hinges on two series of modifications, namely, the ablation of HSV tropism for the natural receptors nectin1 and herpesvirus entry mediator (HVEM), and the readdress of the tropism (retargeting) to a cancer-specific receptor of choice. Retargeting was obtained by engineering a single chain Ab (or a ligand) in the receptor-binding glycoprotein, gD [68,69,70]. The cancer receptor we have chosen is the human epithelial growth factor receptor 2 (HER2), expressed in a subset of breast, gastric, gastroesophageal, lung, and other types of cancers [71, 72]. The HER2-retargeted R-LM113 recombinant virus was then armed with IL-12 to generate R-115 [68, 73]. These viruses carry no deletion or mutation in any gene other than the glycoprotein D gene; hence, they are “fully virulent” to their targeted HER2-positive cancer cells. A direct comparison showed that the IL-12-expressing R-115 virus was more efficacious against Lewis lung carcinoma 1 cells expressing human HER2 (LLC-1-HER2) than was the unarmed R-LM113 and conferred distant long-lasting immune protection against challenge tumors [73]. To obtain mechanistic insight into how IL-12 expressed in the tumor bed contributed to the immune therapeutic effect, we compared the tumor-infiltrating lymphocytes and key immune proteins in tumors from mice treated with IL-12-armed or unarmed retargeted oHSVs. In the R-115-treated group, the tumors from responder mice were infiltrated with CD4+ and CD8+ T cells and their activated CD69+ subpopulation, with CD335+ NK cells and their activated CD69+ subpopulation, and with CD141+ DC cells. The tumors were additionally characterized by a decrease in CD11b+ monocytes/macrophages and an increase in the proinflammatory factors IFNγ, IL-2, TNFα, and t-bet [73]. Intriguingly, in the same tumors (from R-115-treated responder mice), there was an increase in anti-inflammatory factors, such as Tregs, tumor PD-L1, and IL-10. The R-LM113-treated mice which underwent tumor reduction recapitulated these responses but to a lesser extent. Altogether, in the LLC-1 model, the immune heating of the tumors and the simultaneous increase in immune-suppressive factors were primed for check point blockade. Thus, the co-administration of R-115 with anti-PD-1 increased the proportion of cured mice from 20 (virus alone) to 60%. The further addition of anti-CTLA-4 cured 100% of the mice (our unpublished results). A notable effect of the treatment with the IL-12-expressing R-115 was an increase in the reactivity of splenocytes and antibodies to tumor cells. The increase was even higher in the mice treated with the combination of R-115 and anti-PD-1. The results suggest that IL-12 boosted not only the adaptive response but also augmented the repertoire of T and B cells that were reactive to tumor-specific antigens. They raise the possibility that IL-12 expression in the tumor bed also increased tumor antigenicity.
These findings were confirmed and extended in a highly immunosuppressive, transplantable glioblastoma model that recapitulated human glioblastoma [74]. A single orthotopic injection of 106 PFU of the IL-12-armed R-115 administered to well-established tumors immediately before the appearance of symptoms caused tumor regression and spared approximately 25% of the mice. The tumor specimens showed CD4+ and CD8+ lymphocytes deeply infiltrating into tumor masses [74]. Thus, HER2-retargeted fully virulent oHSVs emerge as professional igniters of antitumor immunity and IL-12 was their optimal partner.
To summarize, among all the oHSVs analyzed—whether attenuated to varying degrees or not—and in any tumor model tested, IL-12 greatly augmented the OIV-mediated protection against the primary tumor, favored conspicuous immune modifications to the immunosuppressive microenvironment, and contributed to distant abscopal protection; i.e., they had an antigen agnostic vaccination effect. These effects can be interpreted as the result of increased adjuvanticity and, possibly, of increased antigenicity.
4.4 Adenoviruses
First-generation oncolytic adenoviruses (oAdV ) consisted of replication-incompetent viruses, the cancer selectivity of which depended on attenuation provided by knocking out the E1 and E3 genes. E1 is an essential gene; its deletion allows for only one round of replication and prevents uncontrolled virus replication in host tissues. Deletion of the nonessential E3 gene abrogates the major immune-escape mechanisms; the virus becomes unable to counteract the antiviral responses of the infected cells, and its replication is restricted to tumor cells defective in the innate response [75]. The insertion of transgenes in the viral backbone enabled their expression in the infected cancer cells and restricted their accumulation to the tumor bed. Among the engineered cargos were genes encoding cytokines, antigens, tumor suppressors, and suicide proteins [76]. In a comparative study with IL-2 and HSV-1 thymidine kinase (TK), IL-12 emerged clearly as a superior payload [27]. Specifically, a single intratumoral injection of the IL-12-encoding AdV into liver metastatic colon carcinoma and breast cancer cells significantly reduced tumor growth and improved mouse survival [27, 77]. The unarmed virus showed no therapeutic effect. The enhanced efficacy conferred by IL-12 was confirmed in a number of tumor models [78]. In mice bearing CT-26 colon carcinomas, tumor growth was reduced and most of the mice were tumor-free after a single intratumoral treatment. Importantly, a systemic response was elicited, measured as protection from distant untreated tumors and reactive antitumor lymphocytes [79]. When employed against poorly immunogenic tumors, i.e., glioma, prostatic, and thyroid cancers, AdV-IL-12 caused a significant reduction in primary tumor growth; some mice were completely cured; CD4+ and CD8+ T-cell infiltrated tumor masses, and long-lasting protection was established in an IL-12-dependent fashion [80,81,82,83].
Second-generation oAdVs consisted of replication-competent viruses that harbored smaller deletions in the E1 and E3 genes; hence, they were still partially attenuated. [84,85,86]. Because the viral load in the tumor bed increased over time, the level of IL-12 also increased. In vitro, the concentrations were on the order of 4–10 μg/106 cells 48 h after infection, i.e., 80–200-fold higher than those observed with replication-deficient AdV-IL-12 vectors [81, 87]. Replication-competent AdVs proved highly effective as antitumor agents and completely protected 50% of the mice [84]. The reduction in efficiency upon immune cell depletion implied that CD4+/CD8+ T cells [79, 88] and NK cells [81, 87, 89] were the immune populations which contributed more to the anticancer response.
These encouraging results prompted the search for numerous improvements. Thus, for safety and efficacy purposes, a tunable form of IL-12 was obtained by placing the IL-12 gene under a conditionally activated promoter (Ad-RTS-IL-12) [29, 90, 91]. In an alternative approach, IL-12 was engineered as a nonsecreted form [92] or as a p35-p40 fusion protein, named FIL-12 [87, 93]. Both modifications resulted in higher therapeutic activity. Another approach was based on the notion that IL-12 synergizes with a variety of antitumor factors. AdV vectors for combinatorial expression included the proinflammatory factors IL-23, IL-18, GM-CSF, CD80, and 4-1BBL [86, 88, 94,95,96,97,98,99]; anti-immunosuppressive factors, such as shVEGF, decorin, and anti-PD-L1 [100,101,102,103]; and suicidal genes, such as cytosine deaminase (yCD) and HSV-1 thymidine kinase (TK) [87]. An example is Ad5-yCD/mutTKSR39rep-hIL12, which encodes IL-12, yCD, and a mutant form of TK. The enzymes convert systemically administered prodrugs to their active forms, which in turn inhibit DNA synthesis in infected cells. When administered to mice bearing TRAMP-C2 prostate adenocarcinoma as monotherapy, the virus elicited NK and T-cell responses, and cured 40% of the mice. In combination with the yCD- and TK-activated prodrugs the virus cured 70–80% of the mice [87]. In preclinical studies, replication-competent AdV coexpressing IL-12/IL-18, IL-12/IL-23, or IL-12/4-1BBL caused a complete response in mice harboring poorly immunogenic B16-F10 melanoma [86, 94, 95]. In the same model, the combination of IL-12 with GM-CSF or shVEGF led to complete response in 90 and 60% of the mice, respectively [88, 100]. The IL-12/decorin combination proved effective in weakly immunogenic 4 T1 tumors that are refractory to IL-12 as a consequence of high intratumoral TGF-β levels and Treg infiltration [101].
In other developments, oAdVs were administered together with therapies such as radiation, DC infusion, and CAR-T [96, 99, 102, 103]. All approaches resulted in effective therapeutic responses. In head and neck squamous cell carcinoma (HNSCC), local treatment with AdV coexpressing IL-12 and anti-PD-L1 was primed for systemic CAR-T-cell therapy and significantly improved mouse survival [102, 103]. Finally, in a sarcoma model, recombinant AdV was employed to enable IL-12 expression from DCs with the aim of enhancing cross-priming of tumor-specific CD8+ T cells and tumor rejection [104].
A large body of preclinical studies has made AdV the most frequently investigated OIV in clinical trials (#200), about one-half of which are ongoing or recruiting patients. Of these trials, 13 investigated oAdVs armed with IL-12 and included Ad-hIL12 (constitutive IL-12) (#6), Ad-RTS-hIL12 (tunable IL-12) (#6), and Ad5-yCD/mutTKSR39rep-hIL12 (combinatorial) (#1) against pancreatic, breast, prostatic, and pediatric tumors, glioma, glioblastoma, and melanoma. A completed trial with Ad-IL12 against liver, colorectal, and pancreatic human cancers showed a high safety profile, an increase in tumor infiltration by effector immune cells, yet overall mild antitumor effects [105]. Preliminary results of an ongoing trial with Ad-RTS-hIL12/veledimex (the cytokine inducer) against recurrent high-grade glioma showed the safety and tolerability of the treatment and demonstrated that oAdV elicited a sustained intratumoral immune response. The median overall survival (mOS) was higher in patients treated with the armed virus [90].
4.5 oMeV
An interesting example of the benefits provided by targeted IL-12 delivery through OIVs is offered by the oncolytic measles virus (oMeV) . Ungerechts, Engeland, and coworkers engineered a fusion version of murine IL-12 in the virus and named it FmIL-12 MeVac [106]. The backbone was a vaccine strain of MeV (MeVac). In vitro, the infected cells produced large amounts of FmIL-12, up to 2000 ng/mL. In vivo, FmIL-12 MeVac and unarmed MeVac conferred 90 and 40% protection against MC38CEA tumors, respectively. The antitumor efficacy of the IL-12 virus was also superior to that of MeVac expressing anti-PD-L1. The intratumoral administration of FmIL-12 MeVac elicited local and systemic immune responses, documented mainly as intratumoral increases in activated CD8+ T and NK cells, increases in IFNγ and TNFα, splenocyte reactivity to tumor cells, and immune protection from a distant challenge tumor. The unarmed version conferred less protection from a challenge tumor and a very modest or negligible capacity to induce tumor immune-heating. The same authors carried out an interesting comparison of the benefits offered by FmIL-12 MeVac relative to those provided by FmIL-15 MeVac, a virus expressing IL-15 and the sushi-activating portion of its receptor. The former virus was superior in terms of efficacy against primary tumors, even though the two viruses were similar overall with respect to the increase in intratumoral CD8+ T cells and NK cells [107].
4.6 NDV
NDV is an oncolytic virus of bird origin. It replicates in human tumor cells and fails to substantially replicate in noncancerous human cells. An advantage of OIVs of animal origin is the absence of prior immunity in humans, which could neutralize the spread of the OIV, particularly upon systemic OIV administration. NDVs also infect dendritic cells [108]. It was initially recognized that an unarmed version of NDV could overcome the immunosuppressive nature of the TME, at least in part, through the induction of IL-12, IFNγ and additional cytokines, and driving a Th1 response [108, 109].
Various groups have independently investigated the benefits of delivering IL-12 intratumorally with the aid of recombinant NDV in murine tumor models [110,111,112]. In all cases, the IL-12-armed versions were superior to their unarmed NDV counterparts, as assayed in 4 T1 breast cancer, B16 melanoma, and hepatoma models.
Of particular interest is the possibility of encoding both IL-12 and checkpoint inhibitors from the genome of an oncolytic virus to limit the severe adverse effects caused by the systemic administration of immune modulators. An elegant example of this possibility was recently shown with recombinant versions of NVD expressing neutralizing single-chain antibodies (scFvs) against PD-1, PD-L1, or agonistic scFv to the costimulatory CD28 as proteins alone or as fusion proteins with IL-12 (the combination was named checkpoint inhibitor immunocytokines) [112]. The recombinant NDVs were administered to mice bearing B16 melanoma tumors, alone or in combination with systemic anti-CTLA-4. The NDVs expressing the checkpoint inhibitors fused to IL-12 were invariably more potent than their counterparts without IL-12. The IL-12 adjuvant effect converted the highly immunosuppressive and nonresponsive B16 melanoma tumors into immunologically hot tumors, such that the checkpoint inhibitor-immune cytokine synergized with the systemic administration of anti-CTLA-4. Interestingly, these combinations elicited a strong immunotherapeutic effect, highlighted as an abscopal antitumor effect observed on a distant untreated challenge tumor. At present, the strategy of expressing multiple immune-modulatory payloads is being pursued for numerous OIVs [86, 94, 100, 113,114,115,116] and by companies, such as Oncorus, Replimune, Turnstone Biologics, Immvira and others, are developing multiply armed OIVs.
4.7 Maraba Virus
Maraba virus (MRB), an oncolytic rhabdovirus of animal origin, selectively infects human tumor cells. MG1 is an IFN-sensitive mutant selected for safety reasons [117]. The major effect of MRBs consists of their ability to elicit antitumor immunity and exert abscopal protection, which makes them among the most effective oncolytic vectors for antitumor vaccination. MRBs are being employed in a prime-boost modality with AdV [118]. Currently, four first-in-human trials are ongoing or recruiting for patients with advanced/metastatic solid tumors, including melanoma, squamous cell skin carcinoma, non-small-cell lung cancer for testing the effect of MRB as a monotherapy or in combination with CPI or adenovirus vaccination.

Schematic summary of how IL-12 contributes to antitumor immunity and main effects elicited by IL-12-expressing OIVs as compared with those elicited by unarmed OIVs. The IL-12 cassette is engineered in the OIV genome so that IL-12 is transgenically expressed by the OIVs in the infected cells, i.e., the cancerous cells and – in some cases – cells of the TME. Infection provokes cell death. When present, IL-12 induces expression of pro-inflammatory cytokines, IFNγ, TNFα, and GM-CSF, among others. These (1) recruit antitumoral immune cells to the tumor and activate them, (2) reshape the immunosuppressive TME to a proinflammatory and anticancer setting, (3) contrast tumor angiogenesis by reducing proangiogenic and increasing antiangiogenic factors. As a consequent of the combined effects of OIVs and the IL-12-induced immune modifications to the TME, treatment with IL-12-expressing OIVs results in higher inhibition of tumor growth relative to treatment with unarmed versions of OIVs. The administration of IL-12 expressing OIVs also results in higher abscopal protection than the administration of unarmed OVs, and primes for the CP blockade
An IL-12-armed version of MRB MG1 was employed to infect ex vivo autologous tumor cells, which were subsequently administered intraperitoneally as an infected cell vaccine against peritoneal carcinomatosis caused by melanoma B16 or colon carcinoma CT-26 cells in models of metastatic tumors. The treatment promoted the recruitment and activation of NK cells to the peritoneal cavity, causing a reduction in tumor burden and overall improved survival, including complete protection [119].
4.8 Concluding Remarks. If There Were No IL-12, Someone Would Need to Invent it
Compelling evidence indicates that IL-12 serves as a potent adjuvant of the immunotherapeutic response elicited by OIVs in murine models of tumors. The adjuvant effect was invariably observed for all the OIVs analyzed. OIVs offered the opportunity to encode IL-12 as a transgene and thus to express the cytokine—be it wt or as recombinant fusion form—at high concentrations in the tumor bed, without most of the adverse side effects and toxicities that have hampered the systemic application of IL-12 in past human trials. In some studies, it was shown that the changes to the TME induced by the IL-12-armed OIVs primed the tumor for the checkpoint blockade therapy , paving the way for the combined IL-12/OIV/checkpoint blockade treatment. Some features of the IL-12-mediated response support the possibility that IL-12 not only quantitatively boosts the immune response but also increases the antigenic repertoire of B and T cells; this mechanism remains to be analyzed in detail. The highly encouraging results in preclinical models have fueled the translation to the clinic. The extent to which the IL-12/OIV combination holds promise in humans remains to be fully investigated.
References
Gately M, Desai B, Wolitzky A, Quinn P, Dwyer C, Podlaski F, Familletti P, Sinigaglia F, Chizonnite R, Gubler U (1991) Regulation of human lymphocyte proliferation by a heterodimeric cytokine, IL-12 (cytotoxic lymphocyte maturation factor). J Immunol 147(3):874–882
Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G (1989) Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med 170(3):827–845
Stern AS, Podlaski FJ, Hulmes JD, Pan Y, Quinn PM, Wolitzky A, Familletti PC, Stremlo DL, Truitt T, Chizzonite R (1990) Purification to homogeneity and partial characterization of cytotoxic lymphocyte maturation factor from human B-lymphoblastoid cells. Proc Natl Acad Sci 87(17):6808–6812
Vignali DA, Kuchroo VK (2012) IL-12 family cytokines: immunological playmakers. Nat Immunol 13(8):722
Mal X, Trinchieri G (2001) Regulation of interleukin-12 production in antigen-presenting cells. Adv Immunol Actions 79:55–92
Airoldi I, Gri G, Marshall JD, Corcione A, Facchetti P, Guglielmino R, Trinchieri G, Pistoia V (2000) Expression and function of IL-12 and IL-18 receptors on human tonsillar B cells. J Immunol 165(12):6880–6888
Grohmann U, Belladonna ML, Bianchi R, Orabona C, Ayroldi E, Fioretti MC, Puccetti P (1998) IL-12 acts directly on DC to promote nuclear localization of NF-κB and primes DC for IL-12 production. Immunity 9(3):315–323
Grohmann U, Belladonna ML, Vacca C, Bianchi R, Fallarino F, Orabona C, Fioretti MC, Puccetti P (2001) Positive regulatory role of IL-12 in macrophages and modulation by IFN-γ. J Immunol 167(1):221–227
Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu C-Y, Gately MK, Gubler U (1996) A functional interleukin 12 receptor complex is composed of two β-type cytokine receptor subunits. Proc Natl Acad Sci 93(24):14002–14007
Afonso L, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P (1994) The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science 263(5144):235–237
Hall SS (1994) IL-12 holds promise against cancer, glimmer of AIDS hope. Science 263(5154):1685–1687
Heaton KM, Grimm EA (1993) Cytokine combinations in immunotherapy for solid tumors: a review. Cancer Immunol Immunother 37(4):213–219
Trinchieri G (2003) Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 3(2):133
Smits HH, van Rietschoten JG, Hilkens CM, Sayilir R, Stiekema F, Kapsenberg ML, Wierenga EA (2001) IL-12-induced reversal of human Th2 cells is accompanied by full restoration of IL-12 responsiveness and loss of GATA-3 expression. Eur J Immunol 31(4):1055–1065
Perussia B, Chan SH, D’Andrea A, Tsuji K, Santoli D, Pospisil M, Young D, Wolf SF, Trinchieri G (1992) Natural killer (NK) cell stimulatory factor or IL-12 has differential effects on the proliferation of TCR-alpha beta+, TCR-gamma delta+ T lymphocytes, and NK cells. J Immunol 149(11):3495–3502
Watkins SK, Egilmez NK, Suttles J, Stout RD (2007) IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. J Immunol 178(3):1357–1362
Fukao T, Frucht DM, Yap G, Gadina M, O’Shea JJ, Koyasu S (2001) Inducible expression of Stat4 in dendritic cells and macrophages and its critical role in innate and adaptive immune responses. J Immunol 166(7):4446–4455
Birbrair A, Zhang T, Wang Z-M, Messi ML, Olson JD, Mintz A, Delbono O (2014) Type-2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol 307(1):C25–C38
Airoldi I, Di Carlo E, Cocco C, Caci E, Cilli M, Sorrentino C, Sozzi G, Ferrini S, Rosini S, Bertolini G, Truini M, Grossi F, Galietta LJV, Ribatti D, Pistoia V (2009) IL-12 can target human lung adenocarcinoma cells and normal bronchial epithelial cells surrounding tumor lesions. PLos One 4(7):e6119
Strasly M, Cavallo F, Geuna M, Mitola S, Colombo MP, Forni G, Bussolino F (2001) IL-12 inhibition of endothelial cell functions and angiogenesis depends on lymphocyte-endothelial cell cross-talk. J Immunol 166(6):3890–3899
Yago T, Tsukuda M, Fukushima H, Yamaoka H, Kurata-Miura K, Nishi T, Minami M (1998) IL-12 promotes the adhesion of NK cells to endothelial selectins under flow conditions. J Immunol 161(3):1140–1145
Colombo MP, Trinchieri G (2002) Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev 13(2):155–168
Cohen J (1995) IL-12 deaths: explanation and a puzzle. Science 270(5238):908–908
Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, Anichini A (2007) Interleukin-12: biological properties and clinical application. Clin Cancer Res 13(16):4677–4685
Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL (1997) Effects of single-dose interleukin-12 exposure on interleukin-12–associated toxicity and interferon-γ production. Blood 90(7):2541–2548
Motzer RJ, Rakhit A, Schwartz LH, Olencki T, Malone TM, Sandstrom K, Nadeau R, Parmar H, Bukowski R (1998) Phase I trial of subcutaneous recombinant human interleukin-12 in patients with advanced renal cell carcinoma. Clin Cancer Res 4(5):1183–1191
Caruso M, Pham-Nguyen K, Kwong Y-L, Xu B, Kosai K-I, Finegold M, Woo S, Chen S-H (1996) Adenovirus-mediated interleukin-12 gene therapy for metastatic colon carcinoma. Proc Natl Acad Sci 93(21):11302–11306
Qian C, Liu XY, Prieto J (2006) Therapy of cancer by cytokines mediated by gene therapy approach. Cell Res 16(2):182
Barrett JA, Cai H, Miao J, Khare PD, Gonzalez P, Dalsing-Hernandez J, Sharma G, Chan T, Cooper LJ, Lebel F (2018) Regulated intratumoral expression of IL-12 using a RheoSwitch therapeutic system®(RTS®) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther 25(5):106
Rudman SM, Jameson MB, McKeage MJ, Savage P, Jodrell DI, Harries M, Acton G, Erlandsson F, Spicer JF (2011) A phase 1 study of AS1409, a novel antibody-cytokine fusion protein, in patients with malignant melanoma or renal cell carcinoma. Clin Cancer Res 17(7):1998–2005
Strauss J, Heery CR, Kim JW, Jochems C, Donahue RN, Montgomery AS, McMahon S, Lamping E, Marté JL, Madan RA (2019) First-in-human phase I trial of a tumor-targeted cytokine (NHS-IL12) in subjects with metastatic solid tumors. Clin Cancer Res 25(1):99–109
Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE (2018) CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics 8:41–51
Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, Nahvi AV, Ngo LT, Sherry RM, Phan GQ (2015) Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res 21(10):2278–2288
Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM (1991) Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 252(5007):854–856
Vile RG (2018) The immune system in oncolytic immunovirotherapy: gospel, schism and heresy. Mol Ther 26(4):942–946
Andtbacka RH (2016) The role of talimogene laherparepvec (T-VEC) in the age of checkpoint inhibitors. Clin Adv Hematol Oncol 14(8):576
Coffin RS (2015) From virotherapy to oncolytic immunotherapy: where are we now? Curr Opin Virol 13:93–100
Russell SJ, Barber GN (2018) Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell 33(4):599–605
Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S (2010) Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol 17(3):718–730
Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP (2014) Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Trans Med 6(226):226ra232
Biotech J (2019) Checkpoint inhibitors go viral. Nat Biotechnol 37(1):13
Lawler SE, Speranza M-C, Cho C-F, Chiocca EA (2017) Oncolytic viruses in cancer treatment: a review. JAMA Oncol 3(6):841–849
Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RH, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E (2017) Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 170(6):1109–1119.e1110
Gujar S, Pol JG, Kim Y, Lee PW, Kroemer G (2018) Antitumor benefits of antiviral immunity: an underappreciated aspect of oncolytic virotherapies. Trends Immunol 39(3):209–221
Pearl TM, Markert JM, Cassady KA, Ghonime MG (2019) Oncolytic virus-based cytokine expression to improve immune activity in brain and solid tumors. Mol Ther Oncolytics 13:14
Toda M, Martuza RL, Kojima H, Rabkin SD (1998) In situ cancer vaccination: an IL-12 defective vector/replication-competent herpes simplex virus combination induces local and systemic antitumor activity. J Immunol 160(9):4457–4464
Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, Martuza RL, Rabkin SD (2013) Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci 110(29):12006–12011
Bennett JJ, Malhotra S, Wong RJ, Delman K, Zager J, St-Louis M, Johnson P, Fong Y (2001) Interleukin 12 secretion enhances antitumor efficacy of oncolytic herpes simplex viral therapy for colorectal cancer. Ann Surg 233(6):819
Jarnagin W, Zager J, Klimstra D, Delman K, Malhotra S, Ebright M, Little S, DeRubertis B, Stanziale S, Hezel M (2003) Neoadjuvant treatment of hepatic malignancy: an oncolytic herpes simplex virus expressing IL-12 effectively treats the parent tumor and protects against recurrence-after resection. Cancer Gene Ther 10(3):215
Wong RJ, Patel SG, Kim S-H, DeMatteo RP, Malhotra S, Bennett JJ, St-Louis M, Shah JP, Johnson PA, Fong Y (2001) Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Hum Gene Ther 12(3):253–265
Varghese S, Rabkin S, Liu R, Nielsen P, Ipe T, Martuza R (2006b) Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther 13(3):253
Varghese S, Rabkin SD, Nielsen PG, Wang W, Martuza RL (2006a) Systemic oncolytic herpes virus therapy of poorly immunogenic prostate cancer metastatic to lung. Clin Cancer Res 12(9):2919–2927
Wong RJ, Chan M-K, Yu Z, Ghossein RA, Ngai I, Adusumilli PS, Stiles BM, Shah JP, Singh B, Fong Y (2004a) Angiogenesis inhibition by an oncolytic herpes virus expressing interleukin 12. Clin Cancer Res 10(13):4509–4516
Wong RJ, Chan M-K, Yu Z, Kim TH, Bhargava A, Stiles BM, Horsburgh BC, Shah JP, Ghossein RA, Singh B (2004b) Effective intravenous therapy of murine pulmonary metastases with an oncolytic herpes virus expressing interleukin 12. Clin Cancer Res 10(1):251–259
Derubertis B, Stiles B, Bhargava A, Gusani N, Hezel M, D’Angelica M, Fong Y (2007) Cytokine-secreting herpes viral mutants effectively treat tumor in a murine metastatic colorectal liver model by oncolytic and T-cell-dependent mechanisms. Cancer Gene Ther 14(6):590
Varghese S, Rabkin SD, Nielsen GP, MacGarvey U, Liu R, Martuza RL (2007) Systemic therapy of spontaneous prostate cancer in transgenic mice with oncolytic herpes simplex viruses. Cancer Res 67(19):9371–9379
Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM (2000) Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci 97(5):2208–2213
Cody JJ, Scaturro P, Cantor AB, Yancey Gillespie G, Parker JN, Markert JM (2012) Preclinical evaluation of oncolytic Δγ134. 5 herpes simplex virus expressing interleukin-12 for therapy of breast cancer brain metastases. Int J Breast Cancer 2012:628697
Hellums EK, Markert JM, Parker JN, He B, Perbal B, Roizman B, Whitley RJ, Langford CP, Bharara S, Gillespie GY (2005) Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro-Oncology 7(3):213–224
Bauer DF, Pereboeva L, Gillespie GY, Cloud GA, Elzafarany O, Langford C, Markert JM (2016) Effect of HSV-IL12 loaded tumor cell-based vaccination in a mouse model of high-grade neuroblastoma. J Immunol Res 2016:2568125
Waters AM, Stafman LL, Garner EF, Mruthyunjayappa S, Stewart JE, Friedman GK, Coleman JM, Markert JM, Gillespie GY, Beierle EA (2016) Effect of repeat dosing of engineered oncolytic herpes simplex virus on preclinical models of rhabdomyosarcoma. Transl Oncol 9(5):419–430
Ring EK, Li R, Moore BP, Nan L, Kelly VM, Han X, Beierle EA, Markert JM, Leavenworth JW, Gillespie GY (2017) Newly characterized murine undifferentiated sarcoma models sensitive to virotherapy with oncolytic HSV-1 M002. Mol Ther Oncolytics 7:27–36
Friedman GK, Bernstock JD, Chen D, Nan L, Moore BP, Kelly VM, Youngblood SL, Langford CP, Han X, Ring EK (2018) Enhanced sensitivity of patient-derived pediatric high-grade brain tumor xenografts to oncolytic hsv-1 virotherapy correlates with nectin-1 expression. Sci Rep 8(1):13930
Friedman GK, Moore BP, Nan L, Kelly VM, Etminan T, Langford CP, Xu H, Han X, Markert JM, Beierle EA (2015) Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro-Oncology 18(2):227–235
Markert JM, Cody JJ, Parker JN, Coleman JM, Price KH, Kern ER, Quenelle DC, Lakeman AD, Schoeb TR, Palmer CA (2012) Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J Virol 86(9):5304–5313
Roth JC, Cassady KA, Cody JJ, Parker JN, Price KH, Coleman JM, Peggins JO, Noker PE, Powers NW, Grimes SD (2014) Evaluation of the safety and biodistribution of M032, an attenuated herpes simplex virus type 1 expressing hIL-12, after intracerebral administration to aotus nonhuman primates. Hum Gene Ther Clin Dev 25(1):16–27
Patel DM, Foreman PM, Nabors LB, Riley KO, Gillespie GY, Markert JM (2016) Design of a phase I clinical trial to evaluate M032, a genetically engineered HSV-1 expressing IL-12, in patients with recurrent/progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma. Hum Gene Ther Clin Dev 27(2):69–78
Menotti L, Cerretani A, Hengel H, Campadelli-Fiume G (2008) Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J Virol 82(20):10153–10161
Uchida H, Marzulli M, Nakano K, Goins WF, Chan J, Hong C-S, Mazzacurati L, Yoo JY, Haseley A, Nakashima HJMT (2013) Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther 21(3):561–569
Zhou G, Roizman B (2007) Separation of receptor-binding and profusogenic domains of glycoprotein D of herpes simplex virus 1 into distinct interacting proteins. Proc Natl Acad Sci U S A 104(10):4142–4146
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235(4785):177–182
Yan M, Schwaederle M, Arguello D, Millis SZ, Gatalica Z, Kurzrock R (2015) HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev 34(1):157–164
Leoni V, Vannini A, Gatta V, Rambaldi J, Sanapo M, Barboni C, Zaghini A, Nanni P, Lollini P-L, Casiraghi C (2018) A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog 14(8):e1007209
Alessandrini F, Menotti L, Avitabile E, Appolloni I, Ceresa D, Marubbi D, Campadelli-Fiume G, Malatesta P (2019) Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene 38(23):4467
Wold WSM, Toth K (2013) Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr Gene Ther 13(6):421–433
Wu Q, Moyana T, Xiang J (2001) Cancer gene therapy by adenovirus-mediated gene transfer. Curr Gene Ther 1(1):101–122
Bramson J, Hitt M, Addison C, Muller W, Gauldie J, Graham F (1996) Direct intratumoral injection of an adenovirus expressing interleukin-12 induces regression and long-lasting immunity that is associated with highly localized expression of interleukin-12. Hum Gene Ther 7(16):1995–2002
Barajas M, Mazzolini G, Genové G, Bilbao R, Narvaiza I, Schmitz V, Sangro B, Melero I, Qian C, Prieto J (2001) Gene therapy of orthotopic hepatocellular carcinoma in rats using adenovirus coding for interleukin 12. Hepatology 33(1):52–61
Mazzolini G, Qian C, Xie X, Sun Y, Lasarte JJ, Drozdzik M, Prieto J (1999) Regression of colon cancer and induction of antitumor immunity by intratumoral injection of adenovirus expressing interleukin-12. Cancer Gene Ther 6(6):514
Liu Y, Ehtesham M, Samoto K, Wheeler CJ, Thompson RC, Villarreal LP, Black KL, John SY (2002) In situ adenoviral interleukin 12 gene transfer confers potent and long-lasting cytotoxic immunity in glioma. Cancer Gene Ther 9(1):9
Nasu Y, Bangma C, Hull G, Lee H, Hu J, Wang J, McCurdy M, Shimura S, Yang G, Timme T (1999) Adenovirus-mediated interleukin-12 gene therapy for prostate cancer: suppression of orthotopic tumor growth and pre-established lung metastases in an orthotopic model. Gene Ther 6(3):338–349
Zhang R, DeGroot LJ (2000) Genetic immunotherapy of established tumours with adenoviral vectors transducing murine interleukin-12 (mIL12) subunits in a rat medullary thyroid carcinoma model. Clin Endocrinol 52(6):687–694
Zhang R, DeGroot LJ (2003) Gene therapy of a rat follicular thyroid carcinoma model with adenoviral vectors transducing murine interleukin-12. Endocrinology 144(4):1393–1398
Bortolanza S, Bunuales M, Otano I, Gonzalez-Aseguinolaza G, Ortiz-de-Solorzano C, Perez D, Prieto J, Hernandez-Alcoceba R (2009) Treatment of pancreatic cancer with an oncolytic adenovirus expressing interleukin-12 in Syrian hamsters. Mol Ther 17(4):614–622
Endo Y, Sakai R, Ouchi M, Onimatsu H, Hioki M, Kagawa S, Uno F, Watanabe Y, Urata Y, Tanaka N (2008) Virus-mediated oncolysis induces danger signal and stimulates cytotoxic T-lymphocyte activity via proteasome activator upregulation. Oncogene 27(17):2375
Huang J-H, Zhang S-N, Choi K-J, Choi I-K, Kim J-H, Lee M, Kim H, Yun C-O (2010) Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Mol Ther 18(2):264–274
Freytag S, Barton K, Zhang Y (2013) Efficacy of oncolytic adenovirus expressing suicide genes and interleukin-12 in preclinical model of prostate cancer. Gene Ther 20(12):1131
Choi K-J, Zhang S-N, Choi I-K, Kim J-S, Yun C-O (2012) Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther 19(7):711
Divino CM, Chen S-H, Yang W, Thung S, Brower ST, Woo S (2000) Anti-tumor immunity induced by interleukin-12 gene therapy in a metastatic model of breast cancer is mediated by natural killer cells. Breast Cancer Res Treat 60(2):129–134
Chiocca EA, John SY, Lukas RV, Solomon IH, Ligon KL, Nakashima H, Triggs DA, Reardon DA, Wen P, Stopa BM (2019) Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: results of a phase 1 trial. Sci Trans Med 11(505):eaaw5680
Komita H, Zhao X, Katakam AK, Kumar P, Kawabe M, Okada H, Braughler JM, Storkus WJ (2009) Conditional interleukin-12 gene therapy promotes safe and effective antitumor immunity. Cancer Gene Ther 16(12):883
Wang P, Li X, Wang J, Gao D, Li Y, Li H, Chu Y, Zhang Z, Liu H, Jiang G (2017) Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat Commun 8(1):1395
Poutou J, Bunuales M, Gonzalez-Aparicio M, Garcia-Aragoncillo E, Quetglas JI, Casado R, Bravo-Perez C, Alzuguren P, Hernandez-Alcoceba R (2015) Safety and antitumor effect of oncolytic and helper-dependent adenoviruses expressing interleukin-12 variants in a hamster pancreatic cancer model. Gene Ther 22(9):696
Choi I-K, Li Y, Oh E, Kim J, Yun C-O (2013) Oncolytic adenovirus expressing IL-23 and p35 elicits IFN-γ-and TNF-α-co-producing T cell-mediated antitumor immunity. PLoS One 8(7):e67512
Choi I, Lee J, Zhang S, Park J, Lee K-M, Sonn C, Yun CO (2011) Oncolytic adenovirus co-expressing IL-12 and IL-18 improves tumor-specific immunity via differentiation of T cells expressing IL-12Rβ 2 or IL-18Rα. Gene Ther 18(9):898–909
Kim W, Seong J, Oh HJ, Koom WS, Choi K-J, Yun C-O (2011) A novel combination treatment of armed oncolytic adenovirus expressing IL-12 and GM-CSF with radiotherapy in murine hepatocarcinoma. J Radiat Res 52(5):646–654
Lee Y-S, Kim J-H, Choi K-J, Choi I-K, Kim H, Cho S, Cho BC, Yun C-O (2006) Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7-1 in an immunocompetent murine model. Clin Cancer Res 12(19):5859–5868
Pützer BM, Hitt M, Muller WJ, Emtage P, Gauldie J, Graham FL (1997) Interleukin 12 and B7-1 costimulatory molecule expressed by an adenovirus vector act synergistically to facilitate tumor regression. Proc Natl Acad Sci 94(20):10889–10894
Zhang S-N, Choi I-K, Huang J-H, Yoo J-Y, Choi K-J, Yun C-O (2011) Optimizing DC vaccination by combination with oncolytic adenovirus coexpressing IL-12 and GM-CSF. Mol Ther 19(8):1558–1568
Ahn HM, Hong J, Yun C-O (2016) Oncolytic adenovirus coexpressing interleukin-12 and shVEGF restores antitumor immune function and enhances antitumor efficacy. Oncotarget 7(51):84965
Oh E, Choi I-K, Hong J, Yun C-O (2017) Oncolytic adenovirus coexpressing interleukin-12 and decorin overcomes Treg-mediated immunosuppression inducing potent antitumor effects in a weakly immunogenic tumor model. Oncotarget 8(3):4730
Shaw AR, Porter C, Watanabe N, Tanoue K, Sikora A, Gottschalk S, Brenner M, Suzuki M (2017a) Adenovirotherapy delivering cytokine and checkpoint inhibitor augments chimeric antigen receptor T-cell against metastatic head and neck cancer. Mol Ther 25(11):2440–2451
Shaw AR, Porter CE, Watanabe N, Tanoue K, Sikora A, Gottschalk S, Brenner MK, Suzuki M (2017b) Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther 25(11):2440–2451
Tatsumi T, Huang J, Gooding WE, Gambotto A, Robbins PD, Vujanovic NL, Alber SM, Watkins SC, Okada H, Storkus WJ (2003) Intratumoral delivery of dendritic cells engineered to secrete both interleukin (IL)-12 and IL-18 effectively treats local and distant disease in association with broadly reactive Tc1-type immunity. Cancer Res 63(19):6378–6386
Sangro B, Mazzolini G, Ruiz J, Herraiz M, Quiroga J, Herrero I, Benito A, Larrache J, Pueyo J, Subtil JC (2004) Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J Clin Oncol 22(8):1389–1397
Veinalde R, Grossardt C, Hartmann L, Bourgeois-Daigneault M-C, Bell JC, Jäger D, von Kalle C, Ungerechts G, Engeland CE (2017) Oncolytic measles virus encoding interleukin-12 mediates potent antitumor effects through T cell activation. Onco Targets Ther 6(4):e1285992
Backhaus PS, Veinalde R, Hartmann L, Dunder JE, Jeworowski LM, Albert J, Hoyler B, Poth T, Jäger D, Ungerechts G (2019) Immunological effects and viral gene expression determine the efficacy of oncolytic measles vaccines encoding IL-12 or IL-15 agonists. Viruses 11(10):914
Tan L, Zhang Y, Qiao C, Yuan Y, Sun Y, Qiu X, Meng C, Song C, Liao Y, Munir M (2018) NDV entry into dendritic cells through macropinocytosis and suppression of T lymphocyte proliferation. Virology 518:126–135
Lam HY, Yusoff K, Yeap SK, Subramani T, Abd-Aziz S, Omar AR, Alitheen NB (2014) Immunomodulatory effects of Newcastle disease virus AF2240 strain on human peripheral blood mononuclear cells. Int J Med Sci 11(12):1240
Amin ZM, Ani MAC, Tan SW, Yeap SK, Alitheen NB, Najmuddin SUFS, Kalyanasundram J, Chan SC, Veerakumarasivam A, Chia SL (2019) Evaluation of a recombinant newcastle disease virus expressing human IL12 against human breast cancer. Sci Rep 9(1):1–10
Ren G, Tian G, Liu Y, He J, Gao X, Yu Y, Liu X, Zhang X, Sun T, Liu S (2016) Recombinant newcastle disease virus encoding IL-12 and/or IL-2 as potential candidate for hepatoma carcinoma therapy. Technol Cancer Res Treat 15(5):NP83–NP94
Vijayakumar G, McCroskery S, Palese P (2020) Engineering newcastle disease virus as oncolytic vector for intratumoral delivery of immune checkpoint inhibitors and immunocytokines. J Virol 94(3):e01677–e01619
Fukuhara H, Ino Y, Kuroda T, Martuza RL, Todo T (2005) Triple gene-deleted oncolytic herpes simplex virus vector double-armed with interleukin 18 and soluble B7-1 constructed by bacterial artificial chromosome–mediated system. Cancer Res 65(23):10663–10668
Parker JN, Meleth S, Hughes KB, Gillespie GY, Whitley RJ, Markert JM (2005) Enhanced inhibition of syngeneic murine tumors by combinatorial therapy with genetically engineered HSV-1 expressing CCL2 and IL-12. Cancer Gene Ther 12(4):359
Thomas S, Kuncheria L, Roulstone V, Kyula JN, Mansfield D, Bommareddy PK, Smith H, Kaufman HL, Harrington KJ, Coffin RS (2019) Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J Immunother Cancer 7(1):1–17
Yan R, Zhou X, Chen X, Liu X, Tang Y, Ma J, Wang L, Liu Z, Zhan B, Chen H, Wang J, Zou W, Xu H, Lu R, Ni1 D, Roizman B, Zhou GG (2019) Enhancement of Oncolytic Activity of oHSV Expressing IL-12 and Anti PD-1 Antibody by Concurrent Administration of Exosomes Carrying CTLA-4 miRNA. Immunotherapy 5(1). https://www.longdom.org/open-access/enhancement-of-oncolytic-activity-of-ohsv-expressing-il12-and-anti-pd1-antibody-by-concurrent-administration-ofexosomes-carrying--44157.html
Brun J, McManus D, Lefebvre C, Hu K, Falls T, Atkins H, Bell JC, McCart JA, Mahoney D, Stojdl DF (2010) Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol Ther 18(8):1440–1449
Pol JG, Zhang L, Bridle BW, Stephenson KB, Rességuier J, Hanson S, Chen L, Kazdhan N, Bramson JL, Stojdl DF (2014) Maraba virus as a potent oncolytic vaccine vector. Mol Ther 22(2):420–429
Alkayyal AA, Tai L-H, Kennedy MA, de Souza CT, Zhang J, Lefebvre C, Sahi S, Ananth AA, Mahmoud AB, Makrigiannis AP (2017) NK-cell recruitment is necessary for eradication of peritoneal carcinomatosis with an IL12-expressing Maraba virus cellular vaccine. Cancer Immunol Res 5(3):211–221
Acknowledgments
The experimental work carried out in our laboratory reviewed in this chapter was funded by European Research Council (ERC), Advanced Grant number 340060.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Vannini, A., Leoni, V., Campadelli-Fiume, G. (2021). Targeted Delivery of IL-12 Adjuvants Immunotherapy by Oncolytic Viruses. In: Birbrair, A. (eds) Tumor Microenvironment . Advances in Experimental Medicine and Biology, vol 1290. Springer, Cham. https://doi.org/10.1007/978-3-030-55617-4_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-55617-4_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-55616-7
Online ISBN: 978-3-030-55617-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)