
Abstract
Diabetic kidney disease (DKD) is a microvascular complication of progressive renal decline in patients with diabetes, which is the leading cause of end-stage renal disease (ESRD) globally. Early epidemiological and genetic studies demonstrated that DKD is a heritable condition, indicative of an underlying genetic component for its increased susceptibility. Since then, significant work has been conducted to unravel causal genes implicated in DKD development and progression. With the advancement of genomic technologies, genome-wide association studies (GWASs) and next-generation sequencing (NGS) approaches continue to expand our knowledge of the genetic architecture of DKD and uncover novel biological pathways implicated in disease pathogenesis. Additionally, the establishment of large international collaborations has led to significantly increased cohort sizes to improve overall statistical power to detect novel associations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Diabetic kidney disease (DKD)
- End-stage renal disease (ESRD)
- Diabetes
- Genetics
- Genome-wide association studies (GWAS)
- Rare variants
Introduction
The rise in incidence of diabetes and its micro- and macrovascular complications, including diabetic kidney disease (DKD), have become a financial and healthcare burden globally. DKD is a progressive, microvascular complication that affects 30–40% of patients with type 1 (T1D) or type 2 (T2D) diabetes and is the leading cause of end-stage renal disease (ESRD) (Gu, 2019, Alicic et al., 2017, Li and Pezzolesi, 2018, Reutens, 2013). DKD initiation and progression arise through metabolic dysregulation associated with diabetes, e.g., hyperglycemia and hyperlipidemia, and activation of the renin-angiotensin system (Reidy et al., 2014, Alicic et al., 2017). Prescribed medications, such as angiotensin-converting enzyme inhibitors (ACEi) and angiotensin II receptor blockers (ARBs), in conjunction with intense glycemic control interventions, can improve overall clinical outcomes (Brenner et al., 2001). Despite the near universal implementation of renoprotective therapies, there has been little reduction in the rate of DKD and ESRD, and DKD continues to be associated with excess morbidity and premature mortality in patients with diabetes.
Early epidemiological studies demonstrated that DKD is a heritable condition shown to aggregate in families, which thereby provided a foundation for studies aimed at examining the genetic factors underlying its susceptibility (Borch-Johnsen et al., 1992, Quinn et al., 1996, Seaquist et al., 1989, Fioretto et al., 1999, Faronato et al., 1997). Current estimates of the contribution of genetics to the overall risk of DKD (i.e., heritability or h2) range from 0.3 to 0.49 (Freedman et al., 2007, Sandholm et al., 2017), suggesting that genetic variation may contribute up to 49% of the risk of DKD. Completion of efforts to sequence the human genome, through the Human Genome Project (Lander et al., 2001), and characterization of a haplotype map of the genome, through the International HapMap Project (Altshuler et al., 2005), enabled investigators to begin examining sources of genetic variation, such as single nucleotide polymorphisms (SNPs), at a genome-wide scale using genome-wide association studies (GWASs).
Early GWASs of DKD focused on the role of common variation (i.e., SNPs with a minor allele frequency (MAF) > 5%) and identified several SNPs strongly associated with DKD. More recently, large-scale collaborative efforts have led to improved GWASs of DKD, which include larger sample sizes, and facilitated studies that are better powered to detect robust and more reproducible genetic signals. In addition to common variation, which likely explains only a fraction of the associated risk of DKD, next-generation sequencing (NGS) studies involving whole genome sequencing (WGS) and whole exome sequencing (WES) are emerging to help explain some of the ‘missing heritability’ of DKD (Manolio et al. 2009).
Pathophysiology of DKD in T1D and T2D
DKD patients present with numerous histological and clinical phenotypes that vary with the type and duration of diabetes. With regard to renal histology in T1D DKD, glomerulopathy is apparent with an expansion of the mesangium, basement membrane thickening, and podocyte loss, along with tubular atrophy and tubulointerstitial fibrosis (Alicic et al., 2017; Reidy et al., 2014; Fioretto et al. 2008). Furthermore, increasing proteinuria and decreasing estimated glomerular filtration rate (eGFR) occur in T1D DKD patients with advanced glomerulopathy. Contrarily, renal histology in T2D DKD patients is varied and occasionally does not manifest the T1D DKD phenotype. A subset of patients presents similar histological patterns observed in T1D DKD patients; others may, however, present with just some or absent glomerulosclerosis.
Nondiabetic Kidney Deterioration
Early reports of nondiabetic forms of kidney disease (i.e., NDKD), including immunoglobulin A (IgA) nephropathy, focal segmental glomerulosclerosis, and other glomerulopathies, have been reported in < 10% of T2D proteinuric patients (Fioretto et al. 2008) yet, recent estimates from multiple centers report the incidence of NDKD in T2D patients from 33% to 73% (Anders et al., 2018). Unfortunately, the only way to distinguish DKD from NDKD in patients with diabetes is through kidney biopsy and histological analysis. This process is the “gold standard” for diagnosing DKD, albeit physicians rarely perform kidney biopsies on diabetic patients.
Instead, clinical biomarkers are used to assess renal damage and functional decline in patients with diabetes. By measuring urinary albumin excretion and serum or urinary creatinine, physicians can monitor the severity of renal injury and functional impairment to renal insufficiency, respectively. Measurements of progressive renal damage include calculations of urinary albumin-creatinine ratios (ACRs) and renal decline with eGFR, where persistent macroalbuminuria/proteinuria is defined as ACR > 300 mg/g and severe kidney damage is defined by an eGFR < 30 mL/min/1.73m2 (Reutens, 2013).
Disease Trajectory
The natural history of DKD is thought to proceed first with normal glomerular function or hyperfiltration and then with urinary microalbuminuria progressing into macroalbuminuria, which is accompanied by decreasing eGFR and, ultimately, results in ESRD (Alicic et al., 2017, Macisaac et al., 2014, Reutens, 2013) (Fig. 17.1). However, it is difficult to determine the degree of renal decline by the presence of albuminuria alone. Progressive renal impairment can occur without albuminuria in T1D DKD patients who have advanced glomerular lesions (Caramori et al., 2003). In T2D DKD patients, the absence of the transition from microalbuminuria to macroalbuminuria and albuminuria without renal decline can occur (Retnakaran et al., 2006, Gaede et al., 2004). Additionally, individuals can revert to normoalbuminuric status and still have renal insufficiency (Perkins et al., 2010).

Natural History of DKD. Graphical representations demonstrating the progression of DKD using clinical markers of (A) renal injury (albumin-creatinine ratio, ACR) and (B) renal function decline (estimated glomerular filtration rate, eGFR) in patients with diabetes. Macroalbuminuria/proteinuria is defined as ACR > 300 mg/g and severe kidney damage eGFR < 30 mL/min/1.73m2. Regression from microalbuminuria to normoalbuminuria illustrates an example of the phenotypic heterogeneity that can occur with DKD
For extreme phenotypes, overt macroalbuminuria indicates sustained renal damage and a likely progression to ESRD. Because of the dynamic presentation of albuminuria, eGFR and its longitudinal trajectory (i.e., rate of renal function decline) are considered better predictors of progression of DKD (Skupien et al., 2012). Importantly, patients with poor glycemic control do not always progress or ever develop DKD, suggesting that other influences, such as individual genetic risk, may play a role in both susceptibility and progression (Reidy et al., 2014).
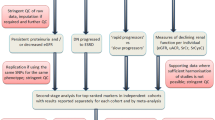
When designing genetic studies of DKD, researchers primarily rely on ACR and eGFR measurements to define cohorts and examine the association of various albuminuria- or eGFR-based phenotypes with genetic variants (i.e., SNPs) (Fig. 17.2). Genetic studies typically dichotomize patients based on these phenotypes and perform case-control association analyses. For example, DKD ‘case’ subjects could be defined as those who have proteinuria or ESRD, and the frequency of SNPs observed in these patients could be compared with frequencies observed in diabetic ‘control’ patients with normoalbuminuria. Studies may also examine associations between SNPs and quantitative traits for ACR or eGFR, instead of dichotomizing cases and controls for association analyses. Unfortunately, no genetic analysis of renal function decline has been conducted to date (Li and Pezzolesi, 2018).

GWASs of DKD. (A) Dichotomous phenotypes examined in case-control-based approaches with presented clinical parameters for each ACR-based, eGFR-based, or combined phenotype in (B). (C) General approaches for conducting a GWAS to identify candidate DKD SNPs. NA, normoalbuminuria; MA, microalbuminuria; PR, proteinuria; CKD, chronic kidney disease; ESRD, end-stage renal disease; eGFR, estimated glomerular filtration rate; DKD, diabetic kidney disease
The Genetic Architecture of DKD through GWASs
While early genetic studies employed positional cloning and linkage analysis to identify potential causal regions for DKD susceptibility, current genetic approaches utilize GWASs to identify susceptibility markers. GWASs detect associations between the frequency of different alleles of SNPs and a disease trait within a given population and have been very powerful in complex, common disease genetics. Generally, GWASs analyze common SNP variation within the genomes of unrelated individuals. Here, we present the major findings from recent GWASs of T1D and T2D DKD patients (Table 17.1).
Early GWASs of DKD were primarily performed on T1D DKD cohorts, as there are less compounding risk factors compared to T2D DKD, such as cardiovascular health and age, which can increase phenotypic heterogeneity. One of the first T1D DKD GWASs identified FRMD3 and CARS in 820 diabetic cases with either persistent macroalbuminuria or ESRD (Pezzolesi et al., 2009b). The original discovery cohort signals did not reach genome-wide significance (defined as a P-value < 5 × 10−8), most likely due to its modest sample size. Associations at these loci were reproducible in a cohort composed of patients from the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study (DCCT/EDIC) (Pezzolesi et al., 2009b).
Another early GWAS analysis and a subsequent replication study identified another reproducible candidate DKD gene, ELMO1, in both T2D and T1D study cohorts (Shimazaki et al., 2005, Pezzolesi et al., 2009a). As genetic associations at both FRMD3 and ELMO1 were independently replicated, the functional roles of both genes in DKD susceptibility have been examined further. In a study by Martini et al., one SNP located in the promoter region of FRMD3 was computationally and functionally demonstrated to show increased binding of transcription factors, which might downregulate FRMD3 expression and lead to increased DKD susceptibility (Martini et al., 2013). Interestingly, for ELMO1, most functional studies have demonstrated that increased renal expression of ELMO1 occurs in DKD mice, overexpression of ELMO1 can promote increased expression of extracellular matrix proteins in vitro, and increased expression can cause pathological features of DKD such as glomerular changes and urinary excretion of albumin (Shimazaki et al., 2005, Hathaway et al., 2016). Contrarily, another study challenged the role of ELMO1 in DKD, demonstrating that introducing additional ELMO1 mRNA in PDX1 morphant diabetic zebrafish is renoprotective, by restoring structural integrity in the pronephros (Sharma et al., 2016).
Since these early genetic studies, realizing that common variants have modest impact on disease risk (e.g., odd ratio (OR) typically 1.10–1.40) and that larger sample sizes can improve detection of these signals, collaborative efforts from investigators across the world have empowered larger GWASs. In 2012, the Genetics of Nephropathy—an International Effort (GENIE) consortium—identified two genome-wide significant loci for associations with ESRD (an intronic SNP rs7583877 in AFF3 and an intergenic SNP rs12437854 located between RGMA and MCTP2), in a meta-analysis of >12,000 individuals (Sandholm et al., 2012). Additionally, although not genome-wide significant, a strong association with diabetic nephropathy (defined as persistent macroalbuminuria or ESRD) was also seen (intronic SNP rs7588550 in the ERBB4 gene).
In addition to increasing sample sizes, to overcome limitations of these early GWASs due to imprecise and variable phenotypic heterogeneity, Sandholm et al. analyzed a range of ACR- and eGFR-based subphenotypes, which span different stages and severities of DKD in a meta-analysis of 4 large cohorts (Sandholm et al., 2017). However, despite being well-powered to detect variants with a MAF ≥ 10% and an OR ≥ 1.55, no SNPs reached genome-wide significance in this study. Suggestive associations for ESRD-based phenotypes (“ESRD vs. no DKD” and “CKD + DKD vs. eGFR >60, normoalbuminuric”) were detected in CNTNAP2 (rs1989248), PTPN13 (rs61277444), and AFF3 (rs7562121). As many of the samples included in this study overlap with those in the previous GWAS from the GENIE consortium, this suggestive association at AFF3 does not provide independent replication of the previously reported association. Interestingly, independent replication was observed in this study for signals reported at ELMO1 (Shimazaki et al., 2005), the MYO16-IRS2 locus on chromosome 13q (Pezzolesi et al., 2009b), and SIK1 (Sambo et al., 2014).
Combined Databases
Following a study design similar to that of Sandholm et al. (Sandholm et al., 2017), the SUrrogate markers for Micro- and Macrovascular hard endpoints for Innovative diabetes Tools (SUMMIT) consortium performed the largest T2D DKD GWAS composed of > 20,000 T2D patients using eight dichotomous and quantitative DKD subphenotypes (van Zuydam et al., 2018). To further increase sample size, a combined T1D and T2D meta-analysis that included samples of European and Asian ancestry was performed. In total, the sample size of this analysis exceeded 40,000 subjects. Despite this, the yield for discovery of novel loci was modest. One SNP, rs2206136 in PLCB4, reached significance with the “CKD” phenotype (P-value = 2.1 × 10−8) in discovery GWAS but not in replication analyses. A novel signal at rs9942471, a position 7 kb upstream of GABRR1, was associated with increased microalbuminuria risk in European subjects. Additionally, the combined meta-analysis for eGFR identified a new genome-wide association mapping near SSB. Interestingly, as the authors note, while this study aimed to overcome some of the limitations of earlier studies, its significantly larger sample size came with increased phenotypic and, likely genetic, heterogeneity that may have impacted its ability to detect additional genetic associations.
Coding vs Noncoding Variants
SNPs included in these studies mostly map to nonprotein coding regions, which limits the interpretation of the candidate SNP’s role in disease susceptibility. Similarly, identified SNPs may be in linkage disequilibrium with causal variants some distance from the identified association, e.g., a causal variant may localize to a gene several hundred kilo-basepair away from the detected association, thereby complicating investigations into the functionality of an associated SNP. Importantly, completion of the National Heart, Lungs, and Blood Institute’s Exome Sequencing Project led to the development of a new generation of commercially available genotyping arrays that, in addition to common noncoding variation, now includes coding or ‘exome’ variants with MAFs as low as 0.5% across the genome (Auer et al., 2016).
Using this technology, the Juvenile Diabetes Research Foundation funded Diabetic Nephropathy Collaborative Research Initiative (JDRF-DNCRI) recently performed the largest T1D DKD GWAS, with 19,406 T1D individuals with and without kidney disease, and identified 16 novel genome-wide significant associations with various dichotomous phenotypes (Salem et al., 2019). The strongest association (rs55703767, a common missense variant in COL4A3) was a protective variant against DN, linked to lower glomerular basement membrane thickness. In contrast to the SUMMIT study, which included T1D and T2D individuals of European and Asian ancestry from 16 different studies genotyped on various platforms, the JDRF-DNCRI study benefited from uniform genotyping, quality control procedures, and standardized phenotype definitions. Adoption of this study design, coupled with an increased sample size, helped foster the discovery of several novel associations with DKD in this study.
Ethnic Patterns
Although the majority of genetic studies of DKD have focused on subjects of European ancestry, several studies have included individuals of different ethnicities and demonstrated potential population-specific signals. The majority of these efforts have, however, been in patients with T2D. In the only trans-ethnic meta-analysis of T2D DKD patients, conducted as part of the Family Investigation of Nephropathy and Diabetes (FIND) study, Iyengar et al. identified SNP rs955333, located between SCAF8 and CNKSR3, among patients of American Indian, Europeans, and Mexican ancestries (Iyengar et al., 2015). No SNP reached genome-wide significance in African American T2D patients, although suggestive associations were seen in APOL1 and MYH9 genes. As APOL1 is a well-known ESRD susceptibility locus in African Americans, this signal may be due to nonDKD influences.
In addition to early GWASs that first reported associations at ELMO1, a more recent GWAS in T2D DKD Japanese patients identified a genome-wide significant association with rs56094641 in FTO as a susceptibility marker for DKD (Taira et al., 2018). The FTO locus has repeatedly been reported to be associated with obesity; however, its association with DKD was not affected by adjustment for BMI in this study. Interestingly, this association was not replicated in European T2D DKD patients. Many studies of T2D attributed ESRD in African Americans have reported additional significant loci, including SLITRK3, ENPP7, GNG7, SFI1, and LIMK2 (Guan et al., 2019, Palmer et al., 2014). With regard to T1D DKD, there have not been any studies to date to address the genetics of T1D DKD in African Americans; intense effort is needed to expand this area of research.
Contributions of Low Frequency and Rare Variations in DKD Patients
Similar to other common, complex diseases, most of the variants identified through GWASs of DKD only explain a small proportion of its overall risk. It has been proposed that some of this ‘missing heritability,’ which is not accounted for by common variation, may be attributed to additional risk conferred by low frequency (0.5% ≤ MAF < 5%) and rare (MAF < 0.5%) variation (Lee et al., 2014; Manolio et al. 2009). Although next-generation GWASs are beginning to explore their role in DKD susceptibility, challenges for investigating low frequency and rare variation persist in all complex disease genetics. Among these, the sample size requirement needed to detect a robust association with low frequency and rare variation remains a major hurdle.
Statistical Challenges
For single variant association tests, the sample sizes to achieve 80% power (at a significance level of 5 × 10−8 with an OR = 1.4) would need to be 6400, 54,000, and 540,000 for variants with MAFs = 0.1, 0.01, and 0.001, respectively (Lee et al., 2014). Unfortunately, current DKD cohorts do not exceed 50,000 patients. Additionally, while newer commercial genotyping arrays do include as many as 200,000–250,000 low frequency and rare variants, these arrays are limited to the variants included in the array’s design, such that novel variants and those not selected for inclusion cannot be assessed. Finally, while NGS-based approaches (e.g., WES and WGS) are ideal for discovering and interrogating low frequency and rare variation, these approaches still remain costly at scale, despite their continuous declining cost and, thereby, have had limited utility in the field.
Despite these challenges, WES and GWASs including low frequency and rare variants have been utilized with some success to explore the contribution of these classes of variants in DKD. In 2017, Sandholm et al. performed WES in 997 T1D DKD subjects (Sandholm et al., 2017). While no variants reached exome-wide significance (P-value < 2.5 × 10−7) for single variant and gene aggregation tests, nominal (P-value < 9 × 10−5) associations with rare missense alleles were identified; the strongest association was observed at an intronic SNP within NVL (rs188427269, MAF = 0.2%, and P-value = 3.3 × 10−7). Importantly, the lack of exome-wide significance in this study is likely due to its modest sample size, as 997 patients may not be enough to detect significant associations with rare variants. In the T1D DKD GWAS from the JDRF-DNCRI, Salem et al. reported several associations with low frequency variants included on a commercial genotyping array associated with ESRD, microalbuminuria, and DN (Salem et al., 2019). The lowest P-value was observed at low frequency intronic variants in PAPLN (rs113554206, MAF = 1.2%, and P-value = 5.39 × 10−7).
Given the excess rate of ESRD in African Americans relative to European Americans, Guan et al. evaluated the impact of low-frequency variants in 47 candidate genes involved in kidney structure (e.g., podocyte, glomerular basement membrane, and renal tubular cell genes) with ESRD in approximately 5000 African American subjects (Guan et al., 2016). After excluding carriers of APOL1 risk alleles, statistically significant associations were observed at two missense variants, located in CLDN8 and COL4A3. A subsequent exome-wide association study by the same group identified suggestive associations at OTUD7B, IFITN3, and DLGAP5 (Guan et al., 2018).
While these studies are the first to begin investigating the role of low frequency and rare variants in DKD, these associations should be interpreted with caution due to an overall lack of genome-wide significance due to their limited sample sizes. These studies may suggest that low frequency or rare variants do not contribute significantly to the underlying genetic component of DKD, as would be seen in monogenic conditions, but may still modestly impact its susceptibility.
From Genetic Causes of DKD to Personalized Therapies
Although progress in personalized therapeutics in DKD has been limited, genetic studies in DKD have shown promise toward translating robust findings to improved patient care. Pharmacogenomics examines how genetic variation can impact beneficial or adverse reactions to therapies and, using genetic data to predict therapeutic response, is beginning to emerge in clinical practice (Roden et al., 2019). This approach has been applied to evaluate reactions to therapies for DKD patients.
One of the earliest studied pharmacogenomic loci in DKD is the insertion-deletion polymorphism of the ACE gene (rs4646994), where the insertion (I) or deletion (D) of a 287 basepair Alu repeat in intron 16 of ACE affects patient responses to angiotensin converting enzyme (ACE) inhibitors, a first-line treatment for DKD. In T1D DKD patients, early reports indicated that individual homozygous for the deletion allele (DD) was more likely to progress to ESRD and lacked a response to renoprotective therapies. However, a study from the Steno Diabetes Center demonstrated that there is no difference in the renoprotective effects in either II or DD individuals (Andersen et al., 2003).
Angiotensin Modulators
These conflicting results in T1D DKD patients could result from the modest sample sizes of these studies or the efficacies of the drugs examined. More recently, Wang et al. investigated the renoprotective efficacy of valsartan, an angiotensin receptor blocker (ARB), in T2D patients carrying the ACE I/D polymorphism and reported an increased risk of developing DKD among carriers of the D allele (Wang et al., 2016). They also observed a beneficial decrease in albuminuria levels for patients with the ID and DD genotypes when treated with valsartan. This observation is likely due to decreased plasma ACE activity in patients carrying this polymorphism.
While no genetic studies of DKD have reported a significant association with CYP2C9, two coding polymorphisms, *2 (Arg144Cys, rs1799853) and *3 (Ile359Leu, rs1057910), can influence CYP2C9 metabolism of ARBs. As ARBs can improve renal function in DKD patients, some studies have examined pharmacological responses with these variants. Lajer et al. described that patients without the CYP2C9*3 polymorphism had a significant change in systolic blood pressure after 4 months compared to *3 carriers. However, both *3 and non*3 carriers had no significant difference in urinary albumin excretion during the study period (Lajer et al., 2007).
Future Directions
Overcoming DKD’s phenotypic heterogeneity has proven to be among the most challenging. As a progressive disease, the lack of consistent findings across studies can, in part, be attributed to the array of clinical definitions established by different studies. Since most genetic approaches are inherently cross-sectional, investigators typically examine multiple phenotypes that span all stages and presentations of renal disease. This approach has drawbacks, as a patient’s renal status could change over time (i.e., a microalbuminuric patient reverts to normoalbuminuria or, alternatively, progresses to proteinuria or ESRD), leading to misclassification. Misclassification bias then could result in a significant number of false-negative associations. Indeed, longitudinal investigations of DKD have shed new light on the natural history of this disease and its heterogeneous nature ( Perkins et al., 2010, Skupien et al., 2012, Krolewski and Bonventre, 2012, Krolewski, 2015). Additionally, the rate of renal function decline varies widely among DKD patients; perhaps, genetic factors influence whether this decline is slow, where ESRD is reached after decades of diabetes, or decline is rapid and progression to ESRD occurs in as little as a few years after the onset of diabetes (Skupien et al., 2012, Krolewski and Bonventre, 2012, Krolewski, 2015). Because of the heterogeneity of the subphenotypes of DKD, considering such disparate patients simultaneously likely poses challenges to uncovering the genetic basis of DKD.
Additionally, it is possible that a subset of DKD patients have nondiabetic kidney disease (NDKD) coincident with diabetes or concurrent DKD and NDKD. Renal biopsies from patients with diabetes and kidney disease suggest that as many as 30–80% of patients diagnosed with DKD actually had kidney disease attributed to nondiabetic causes (Haider et al., 2011, Sharma et al., 2013, Zhuo et al., 2013). Importantly, this has major implications in terms of patient care, monitoring, and family planning, as well as for research studies aimed at understanding the factors that contribute to DKD. To begin investigating this, we have recently shown that a subset of ‘DKD’ patients carry rare pathogenic variants indicative of NDKD, e.g., variants in COL4A3 (associated with focal segmental glomerulosclerosis, thin basement membrane nephropathy, and Alport Syndrome) and REN (associated with autosomal dominant tubulointerstitial kidney disease) (Lazaro-Guevara et al., 2020).
Large research consortiums are now bringing together investigators across the globe to combine resources and enhance the cohort sizes of current genetic studies, such as the Juvenile diabetes research foundation-Diabetic nephropathy collaborative research initiative (JDRF-DNCRI) most recent T1D DKD GWAS. Even though establishing large cohorts increases statistical power, how many samples are sufficient to accurately identify all of the contributing genetic variation underlying DKD? Is there a sample size threshold that diminishes any further efforts of larger GWASs?
Epigenomics
In addition to variation at the DNA sequence level, increasing research shows that epigenetic and epigenomic mechanisms, i.e., heritable changes in gene expression, including DNA methylation and histone modifications, which do not alter the sequence of DNA, are involved in DKD pathogenesis (Kato and Natarajan, 2019). Nucleic acid and chromatin histone complex modifications elicit changes in gene expression by affecting the binding of potential transcriptional regulators. It has been demonstrated that “metabolic memory” of previous metabolic dysregulation exists, where patients with long-standing diabetes can still experience complications even after intense glucose control, highlighting prolonged modification of gene expression patterns in DKD risk (Reidy et al., 2014). Therefore, identifying epigenetic signatures could lead to new treatments and diagnostic biomarkers for DKD initiation and progression.
References
Alicic RZ, Rooney MT, Tuttle KR (2017) Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol 12:2032–2045
Altshuler D, Brooks LD, Chakravarti A, Collins FS, Daly MJ, Donnelly P (2005) A haplotype map of the human genome. Nature 437:1299–1320
Anders HJ, Huber TB, Isermann B, Schiffer M (2018) CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nat Rev Nephrol 14:361–377
Andersen S, Tarnow L, Cambien F, Rossing P, Juhl TR, Deinum J, Parving HH (2003) Long-term renoprotective effects of losartan in diabetic nephropathy: interaction with ACE insertion/deletion genotype? Diabetes Care 26:1501–1506
Auer PL, Reiner AP, Wang G, Kang HM, Abecasis GR et al (2016) Guidelines for large-scale sequence-based complex trait association studies: lessons learned from the NHLBI exome sequencing project. Am J Hum Genet 99:791–801
Borch-Johnsen K, Norgaard K, Hommel E, Mathiesen ER, Jensen JS, Deckert T, Parving HH (1992) Is diabetic nephropathy an inherited complication? Kidney Int 41:719–722
Brenner BM, Cooper ME, De Zeeuw D, Keane WF, Mitch WE et al (2001) Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345:861–869
Caramori ML, Fioretto P, Mauer M (2003) Low glomerular filtration rate in normoalbuminuric type 1 diabetic patients: an indicator of more advanced glomerular lesions. Diabetes 52:1036–1040
Faronato PP, Maioli M, Tonolo G, Brocco E, Noventa F et al (1997) Clustering of albumin excretion rate abnormalities in Caucasian patients with NIDDM. The Italian NIDDM Nephropathy Study Group. Diabetologia 40:816–823
Fioretto P, Steffes MW, Barbosa J, Rich SS, Miller ME, Mauer M (1999) Is diabetic nephropathy inherited? Studies of glomerular structure in type 1 diabetic sibling pairs. Diabetes 48:865–869
Fioretto P, Caramori ML, Mauer M (2008) The kidney in diabetes: dynamic pathways of injury and repair. The Camillo Golgi Lecture 2007. Diabetologia 51:1347
Freedman BI, Bostrom M, Daeihagh P, Bowden DW (2007) Genetic factors in diabetic nephropathy. Clin J Am Soc Nephrol 2:1306–1316
Gaede P, Tarnow L, Vedel P, Parving HH, Pedersen O (2004) Remission to normoalbuminuria during multifactorial treatment preserves kidney function in patients with type 2 diabetes and microalbuminuria. Nephrol Dial Transplant 19:2784–2788
Germain M, Pezzolesi MG, Sandholm N, Mcknight AJ, Susztak K et al (2015) SORBS1 gene, a new candidate for diabetic nephropathy: results from a multi-stage genome-wide association study in patients with type 1 diabetes. Diabetologia 58:543–548
Gu HF (2019) Genetic and epigenetic studies in diabetic kidney disease. Front Genet 10:507
Guan M, Ma J, Keaton JM, Dimitrov L, Mudgal P et al (2016) Association of kidney structure-related gene variants with type 2 diabetes-attributed end-stage kidney disease in African Americans. Hum Genet 135:1251–1262
Guan M, Keaton JM, Dimitrov L, Hicks PJ, Xu J et al (2018) An exome-wide association study for type 2 diabetes-attributed end-stage kidney disease in African Americans. Kidney Int Rep 3:867–878
Guan M, Keaton JM, Dimitrov L, Hicks PJ, Xu J et al (2019) Genome-wide association study identifies novel loci for type 2 diabetes-attributed end-stage kidney disease in African Americans. Hum Genomics 13:21
Haider DG, Peric S, Friedl A, Fuhrmann V, Wolzt M, Horl WH, Soleiman A (2011) Kidney biopsy in patients with diabetes mellitus. Clin Nephrol 76:180–185
Hathaway CK, Chang AS, Grant R, Kim HS, Madden VJ, Bagnell CR Jr, Jennette JC, Smithies O, Kakoki M (2016) High Elmo1 expression aggravates and low Elmo1 expression prevents diabetic nephropathy. Proc Natl Acad Sci U S A 113:2218–2222
Iyengar SK, Sedor JR, Freedman BI, Kao WH, Kretzler M et al (2015) Genome-wide association and trans-ethnic meta-analysis for advanced diabetic kidney disease: family investigation of nephropathy and diabetes (FIND). PLoS Genet 11:e1005352
Kato M, Natarajan R (2019) Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol 15:327–345
Krolewski AS (2015) Progressive renal decline: the new paradigm of diabetic nephropathy in type 1 diabetes. Diabetes Care 38:954–962
Krolewski AS, Bonventre JV (2012) High risk of ESRD in type 1 diabetes: new strategies are needed to retard progressive renal function decline. Semin Nephrol 32:407–414
Lajer M, Tarnow L, Andersen S, Parving HH (2007) CYP2C9 variant modifies blood pressure-lowering response to losartan in Type 1 diabetic patients with nephropathy. Diabet Med 24:323–325
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC et al (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921
Lazaro-Guevara, J., Morales, J. F., Wright, A. H., Gunville, R., Frodsham, S. G., et al. 2020. Targeted next-generation identifies pathogenic variants in kidney-disease related gene in phatients with diabetic kidney disease. https://doi.org/10.1101/2020.05.18.102822.
Lee S, Abecasis GR, Boehnke M, Lin X (2014) Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet 95:5–23
Li M, Pezzolesi MG (2018) Advances in understanding the genetic basis of diabetic kidney disease. Acta Diabetol 55:1093–1104
Macisaac RJ, Ekinci EI, Jerums G (2014) Markers of and risk factors for the development and progression of diabetic kidney disease. Am J Kidney Dis 63:S39–S62
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA et al (2009) Finding the missing heritability of complex diseases. Nature 461:747–753
Martini S, Nair V, Patel SR, Eichinger F, Nelson RG et al (2013) From single nucleotide polymorphism to transcriptional mechanism: a model for FRMD3 in diabetic nephropathy. Diabetes 62:2605–2612
Palmer ND, Ng MC, Hicks PJ, Mudgal P, Langefeld CD, Freedman BI, Bowden DW (2014) Evaluation of candidate nephropathy susceptibility genes in a genome-wide association study of African American diabetic kidney disease. PLoS One 9:e88273
Perkins BA, Ficociello LH, Roshan B, Warram JH, Krolewski AS (2010) In patients with type 1 diabetes and new-onset microalbuminuria the development of advanced chronic kidney disease may not require progression to proteinuria. Kidney Int 77:57–64
Pezzolesi MG, Katavetin P, Kure M, Poznik GD, Skupien J, Mychaleckyj JC, Rich SS, Warram JH, Krolewski AS (2009a) Confirmation of genetic associations at ELMO1 in the GoKinD collection supports its role as a susceptibility gene in diabetic nephropathy. Diabetes 58:2698–2702
Pezzolesi MG, Poznik GD, Mychaleckyj JC, Paterson AD, Barati MT et al (2009b) Genome-wide association scan for diabetic nephropathy susceptibility genes in type 1 diabetes. Diabetes 58:1403–1410
Quinn M, Angelico MC, Warram JH, Krolewski AS (1996) Familial factors determine the development of diabetic nephropathy in patients with IDDM. Diabetologia 39:940–945
Reidy K, Kang HM, Hostetter T, Susztak K (2014) Molecular mechanisms of diabetic kidney disease. J Clin Invest 124:2333–2340
Retnakaran R, Cull CA, Thorne KI, Adler AI, Holman RR (2006) Risk factors for renal dysfunction in type 2 diabetes: U.K. Prospective Diabetes Study 74. Diabetes 55:1832–1839
Reutens AT (2013) Epidemiology of diabetic kidney disease. Med Clin North Am 97:1–18
Roden DM, Mcleod HL, Relling MV, Williams MS, Mensah GA, Peterson JF, Van Driest SL (2019) Pharmacogenomics. Lancet 394:521–532
Salem RM, Todd JN, Sandholm N, Cole JB, Chen WM et al (2019) Genome-wide association study of diabetic kidney disease highlights biology involved in glomerular basement membrane collagen. J Am Soc Nephrol 30:2000–2016
Sambo F, Malovini A, Sandholm N, Stavarachi M, Forsblom C et al (2014) Novel genetic susceptibility loci for diabetic end-stage renal disease identified through robust naive Bayes classification. Diabetologia 57:1611–1622
Sandholm N, Salem RM, Mcknight AJ, Brennan EP, Forsblom C et al (2012) New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet 8:e1002921
Sandholm N, Forsblom C, Makinen VP, Mcknight AJ, Osterholm AM et al (2014) Genome-wide association study of urinary albumin excretion rate in patients with type 1 diabetes. Diabetologia 57:1143–1153
Sandholm N, Van Zuydam N, Ahlqvist E, Juliusdottir T, Deshmukh HA et al (2017) The genetic landscape of renal complications in type 1 diabetes. J Am Soc Nephrol 28:557–574
Seaquist ER, Goetz FC, Rich S, Barbosa J (1989) Familial clustering of diabetic kidney disease. Evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med 320:1161–1165
Sharma SG, Bomback AS, Radhakrishnan J, Herlitz LC, Stokes MB, Markowitz GS, D’agati VD (2013) The modern spectrum of renal biopsy findings in patients with diabetes. Clin J Am Soc Nephrol 8:1718–1724
Sharma KR, Heckler K, Stoll SJ, Hillebrands JL, Kynast K et al (2016) ELMO1 protects renal structure and ultrafiltration in kidney development and under diabetic conditions. Sci Rep 6:37172
Shimazaki A, Kawamura Y, Kanazawa A, Sekine A, Saito S et al (2005) Genetic variations in the gene encoding ELMO1 are associated with susceptibility to diabetic nephropathy. Diabetes 54:1171–1178
Skupien J, Warram JH, Smiles AM, Niewczas MA, Gohda T, Pezzolesi MG, Cantarovich D, Stanton R, Krolewski AS (2012) The early decline in renal function in patients with type 1 diabetes and proteinuria predicts the risk of end-stage renal disease. Kidney Int 82:589–597
Taira M, Imamura M, Takahashi A, Kamatani Y, Yamauchi T et al (2018) A variant within the FTO confers susceptibility to diabetic nephropathy in Japanese patients with type 2 diabetes. PLoS One 13:e0208654
Teumer A, Tin A, Sorice R, Gorski M, Yeo NC et al (2016) Genome-wide association studies identify genetic loci associated with albuminuria in diabetes. Diabetes 65:803–817
Van Zuydam NR, Ahlqvist E, Sandholm N, Deshmukh H, Rayner NW et al (2018) A genome-wide association study of diabetic kidney disease in subjects with type 2 diabetes. Diabetes 67:1414–1427
Wang Y, Peng W, Zhang X, Qiao H, Wang L, Xu Z, Wu C (2016) The association of ACE gene polymorphism with diabetic kidney disease and renoprotective efficacy of valsartan. J Renin-Angiotensin-Aldosterone Syst 17:1470320316666749
Zhuo L, Ren W, Li W, Zou G, Lu J (2013) Evaluation of renal biopsies in type 2 diabetic patients with kidney disease: a clinicopathological study of 216 cases. Int Urol Nephrol 45:173–179
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Simeone, C.A., Lazaro-Guevara, J.M., Pezzolesi, M.G. (2020). The Genetic Basis of Diabetic Kidney Disease. In: Faintuch, J., Faintuch, S. (eds) Obesity and Diabetes. Springer, Cham. https://doi.org/10.1007/978-3-030-53370-0_17
Download citation
DOI: https://doi.org/10.1007/978-3-030-53370-0_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-53369-4
Online ISBN: 978-3-030-53370-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)