
Abstract
Hypoxia (oxygen levels below 1–2%) is a common finding in solid tumors. The development of tumor hypoxia can be viewed as an imbalance between oxygen supply and demand. Tumor hypoxia significantly impacts tumor radiosensitivity and subsequently leads to poor clinical outcomes in patients treated with radiation therapy. The presence of molecular oxygen supports the production of lethal DNA damage in irradiated cells; therefore, the radiation dose required under severely hypoxic conditions to achieve a certain biological effect is generally 2- to 3-fold higher than the dose needed under normoxic conditions (i.e., oxygen enhancement ratio (OER) = 2–3). Several therapeutic approaches have been historically used and are emerging to target tumor hypoxia in order to improve radiation therapy outcomes. These include hyperbaric oxygen, correction of anemia, combination of radiation with carbogen and nicotinamide (ARCON), oxygen mimetics such as nimorazole, hypoxia-activated prodrugs, vascular normalization strategies (reviewed in Chap. 12), and emerging therapies to target tumor oxidative phosphorylation. Even though tumor hypoxia has long been established as a negative factor for radiation therapy outcomes in the clinic, we still lack robust, widely available, and adequately validated biomarkers for assessing tumor hypoxia in patients. This has not only significantly impeded the investigation of the efficacy of hypoxia modifiers, but it has also resulted in an inability to accurately select patients who are likely to benefit from such treatment. It is likely that only when hypoxia biomarkers are widely available will hypoxia modification enter the era of personalized medicine and improve outcomes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Anemia
- ARCON
- Atovaquone
- Hyperbaric oxygen
- Hypoxia, acute
- Hypoxia, chronic
- Hypoxia-inducible factors
- Oxidative phosphorylation
- Oxygen consumption rate
- Oxygen enhancement ratio
- Oxygen mimetics
- Radioresistance
- Tissue oxygenation
- Tumor metabolism
- Tumor microenvironment
- Vascular normalization
1 Introduction
Oxygen is vital for all living cells and plays a primordial role in key cellular activities. Central to our evolution into large and complex multicellular organisms has been the development of an intricate vascular system to ensure the adequate supply of oxygen to support cellular function (Pittman 2011). At the ends of the branching vascular network, capillaries provide the blood–tissue interface for oxygen delivery. In terms of distance, the diffusion limit for oxygen in tissue is in the region of 100–200 μM (Thomlinson and Gray 1955); therefore, cells must be within this proximity of a functional vessel to ensure adequate oxygenation. Given the limit of oxygen diffusion, tissue oxygenation is not binary, but rather exists in continuous gradients. Interestingly, the permission of low and near sub-physiological levels of oxygen are important for certain normal tissue functions, such as fetal development, tissue healing, and liver zonation (Dunwoodie 2009). However, the presence of such low levels of oxygen in healthy tissue is highly restricted and tightly regulated for specific purposes only.
When discussing tissue oxygenation, it is firstly important to define the units used and the relevant nomenclature. Several units are used to report oxygen levels and include the international system pressure unit the Pascal (Pa) beside millimeter of mercury (mmHg) (1 kPa = 7.5 mmHg) and the percentage of oxygen (1 kPa = 1%), both commonly used in medicine. The term “normoxia” refers to the atmospheric oxygen level of 21% (160 mmHg). “Tissue normoxia,” also referred to as “physioxia,” varies depending on tissue type and is significantly lower than atmospheric normoxia (Hammond et al. 2014). For example, in outer layers of skin the oxygen levels are in region of 1% (8 mmHg) (Wang et al. 2003), whilst in lung tissue they are 6% (42 mmHg) (Le et al. 2006) and 10% (72 mmHg) in the kidney (Müller et al. 1998). It is important to note that these values are significantly lower than the majority of in vitro experiments aimed to replicate tissue normoxia, which are conducted using 95% air and 5% carbon dioxide, and thus performed at around 20% oxygen (150 mmHg). The term “hypoxia” should be reserved for oxygen levels below that of the normoxia range for the tissue in question (Carreau et al. 2011), and generally refers to levels below 1–2% (7.5–15 mmHg).
In contrast to normal tissues, hypoxia is a common finding in solid tumors. The development of tumor hypoxia can be viewed as an imbalance between oxygen supply and demand. Rapid proliferation and high metabolic rates result in high oxygen consumption, which quickly surpasses supply. The subsequent hypoxic microenvironment results in the upregulation of angiogenic factors and thus turns on the “angiogenic switch” stimulating tumor neovascularization (Semenza 2012), a phenomenon first described over a century ago (Goldmann 1908). However, despite an abundance of new vessels, the resulting microvasculature is invariably highly dysregulated and “chaotic” (Jain 2014). In particular, the vessels are tortuous, friable, and leaky. The highly abnormal endothelium results in non-laminar flow and further leakiness predisposes to thrombosis and edema, which further impedes perfusion (Hashizume et al. 2000; Dvorak et al. 1999). Overall, the tumor vasculature is sub-functional and unable to match the oxygen demand of tumors and therefore, hypoxic regions arise.
The paradox that increased tumor angiogenesis can result in decreased perfusion is well established (Yang et al. 2017). In fact, frequently used anti-angiogenesis therapies, such as anti-vascular endothelial growth factor (VEGF) agents including bevacizumab, aim to exploit this phenomenon. These drugs improve tumor perfusion by inhibiting uncontrolled angiogenesis, enabling more regulated vessel development and leading to “vascular normalization” (Jain 2014). When combined with chemotherapy, as is often the case, this results in improved drug delivery to tumors.
Traditionally, tumor hypoxia is classified as either chronic or acute. Chronic hypoxia occurs in tumor regions located beyond the diffusion capacity of oxygen and is fairly stable. In contrast, acute hypoxia, also referred to as transient or cycling hypoxia, refers to regions of dynamic changes in oxygenation with the potential for reoxygenation (Brown 1979; Michiels et al. 2016). Transient hypoxia is commonly observed in tumors with in vivo studies demonstrating that up to 20% of the tumor volume may be affected (Bennewith and Durand 2004), with cycle lengths ranging from seconds to days. It is postulated that high frequency hypoxia cycling arises from alterations in vessel perfusion and red cell flux, whereas remodeling of vasculature is responsible for low frequency cycling occurring over a matter of days (Dewhirst 2009). Clinically, in patients with head and neck tumors, for example, significant change in the location of hypoxic regions has been observed using hypoxia imaging a short number of days apart (Nehmeh et al. 2008). Interestingly, chronic and acute hypoxia result in differing cancer cellular signaling responses, thus further highlighting them as distinct pathophysiological entities.
Overall, due to the complex spatiotemporal distribution of tumor hypoxia, accurate characterization of hypoxia within tumors is challenging with results from sample-based measurement potentially not representative of the tumor as a whole. Nevertheless, the presence of measurable hypoxia is an almost universal feature of tumors. Certain tumor types have consistently been shown to contain higher levels of hypoxia than others (Horsman et al. 2012). For example, polarographic oxygen electrode measurements in patients with non-small cell lung carcinoma (NSCLC) have shown median oxygen levels ranging from 14 to 17 mmHg (Le et al. 2006; Falk et al. 1992), whilst in head and neck squamous cell carcinoma (HNSCC) the average level was 9 mmHg (Falk et al. 1992; Nordsmark et al. 2005) and only in the region of 2 mmHg in prostate cancer (Movsas et al. 2000, 2002), therefore highlighting an additional layer of complexity in the study of tumor hypoxia.
2 The Cellular Response to Hypoxia: Hypoxia-Inducible Factors
Central to the cellular response to hypoxia are the hypoxia-inducible factors (HIFs)—the principal oxygen sensing machinery. The HIF family of transcription factors includes the well-studied HIF1 and HIF2, as well as HIF3. Each of these factors consists of an oxygen-sensitive and tightly regulated HIF-α subunit (HIF1-α, HIF2-α, or HIF3-α, respectively), which forms a heterodimer with a constitutively expressed HIF1-β subunit (also known as ARNT) (Wang et al. 1995). The HIF-α subunit contains two highly preserved proline residues (HP402/P564 for HIF1-α and P405/P531 for HIF2-α), which are hydroxylated in the presence of oxygen by prolyl hydroxylase domain-containing proteins (PHDs). This in turn permits binding to the von Hippel-Lindau (VHL) tumor suppressor protein, leading to HIF-α ubiquitination and its targeting for degradation (Ohh et al. 2000; Jaakkola et al. 2001). Under hypoxic conditions, HIF-α is stabilized through decreased hydroxylation, allowing binding with HIF1-β and translocation to the nucleus where it recognizes and binds to hypoxia-responsive elements (HREs), resulting in the transcription of a multitude of target genes (Liu et al. 2012). An additional layer of HIF regulation occurs at the level of recruitment of coactivators. Further oxygen-dependent hydroxylation of HIF1-α, and to a lesser extent HIF2-α, occurs on the C-terminal transactivation domain on aspartate reside 803 by factor-inhibiting HIF1 (FIH) (Lando et al. 2002; McNeill et al. 2002), which results in the inability of HIF1 to transactivate certain target genes (Dayan et al. 2006). The oxygen tension required to inactivate FIH is lower than that of the PHDs (Koivunen et al. 2004), thus enabling a graded transcriptional response dependent upon the severity of hypoxia.
HIF1-α and HIF2-α have differing expression profiles in tissues and tumors, with expression of HIF1-α ubiquitous whereas HIF2-α far more restricted (Wiesener et al. 2003). Together with the difference in their target genes, this provides a differential HIF-responsive transcriptome dependent on tissue and tumor type. Further still, in chronic hypoxia, HIF1-α levels rapidly increase and are stabilized within hours but within days decrease to much lower expression levels (Ginouvès et al. 2008). HIF1 has been shown to upregulate PHDs, which appear to retain enough activity to hydroxylate HIF1-α even under lower oxygen tensions, resulting in its reinstated degradation (Ginouvès et al. 2008; Berra et al. 2003). In contrast, cycling hypoxia results in an enhanced activity and stabilization of HIF1-α, at much greater levels than witnessed in chronic hypoxia (Dewhirst et al. 2008). The sophisticated regulation of HIF signaling points to its importance in regulating wide-ranging aspects of cellular function, with an ever-growing number of HIF target genes identified (Dengler et al. 2014; Choudhry and Harris 2018). Perhaps therefore unsurprisingly, hypoxia-related HIF signaling contributes significantly to tumorigenesis by directly contributing to the majority of the hallmarks of cancer, with its effects ranging from stimulation of invasion and metastasis, to promotion of sustained proliferation and immune system evasion (Petrova et al. 2018; LaGory and Giaccia 2016; Rankin et al. 2016). Overall, through such means, tumor hypoxia results in an aggressive tumor phenotype.
3 Tumor Hypoxia as a Barrier to Radiation Therapy
There is overwhelming evidence that tumor hypoxia significantly impedes tumor radiation sensitivity and subsequently leads to poor clinical outcomes in patients treated with radiation therapy. Nearly three quarters of a century ago the seminal work of Thomlinson and Gray demonstrated that the cellular response to ionizing radiation is highly dependent upon the presence of oxygen (Gray et al. 1953; Wright and Howard-Flanders 1957; Palcic et al. 1982). In the majority of cell types studied, the ratio of radiation dose required under severely hypoxic or anoxic conditions versus normoxic conditions to achieve the same biological effect, termed the oxygen enhancement ratio (OER), is typically 2.5–3.5 for ionizing radiation such as X-rays or γ-rays which are characterized by low linear energy transfer (LET). The most pronounced change in radiosensitivity occurs as oxygen levels increase from 0 to 30 mmHg (0–4%), with further increases in oxygenation having little additional effect. As a significant proportion of tumor oxygen readings fall within this range, with normal tissue readings normally above this range, this highlights the everyday clinical challenge of treating tumors successfully without causing excessive surrounding normal tissue toxicity.
In order to understand how oxygen potentiates cellular radiosensitivity, it is first important to briefly consider how ionizing radiation results in cellular damage. Radiation results in damage through its effects on DNA, with DNA double-strand breaks being the principle cytotoxic lesions. The effects on DNA can be viewed as either direct or indirect. Direct damage results from ionization of the DNA without the involvement of intermediate steps, and is the principal way by which high LET radiation, such as neutrons and α particles, exert their effect (Hall and Giaccia 2011). By contrast, megavoltage (MV) photons do not produce chemical and biological damage by themselves, but rather predominantly as a result of indirect ionization when they are absorbed in the material through which they pass and their energy is transferred to produce fast-moving charged particles that in turn elicit DNA damage (Hall and Giaccia 2011). At the energies used by most clinical linear accelerators, the MV photons predominantly interact with “free” electrons of the absorbing material with some of the photon energy transferred to the electron in the form of kinetic energy and the scattered photon traveling further into the material interacting with more “free” electrons (Compton Effect) (Steel 2002). This ultimately leads to a large number of fast electrons, which can ionize other atoms in the irradiated material. As cells are 80% comprised of water, it is important to consider the radiochemistry of water. Following exposure to ionizing radiation, water becomes ionized as shown:

H2O+ is an ion radical and rapidly interacts with another water molecule to form the highly reactive hydroxyl radical (OH•) as follows:
Such free radicals interact with the DNA molecule and cause damage. The mechanisms responsible for oxygen increasing cellular radiosensitivity can now be explained as outlined by the Oxygen Fixation Hypothesis first described in the 1950s (Alper and Howard-Flanders 1956). Following irradiation, DNA damage can be restored through reactions with free radical scavengers such as sulfydryl-containing compounds (Held et al. 1984; Hutchinson 1961). However, in the presence of molecular oxygen, DNA damage can be “fixed” by the DNA radical reacting with oxygen to form RO2•, which is significantly less amenable to scavenger restoration (Chapman 1979).
4 Tumor Hypoxia and Poor Radiotherapy Clinical Outcomes
Given the multitude of tumor adaptations to hypoxia contributing to many cancer hallmarks, it is not surprising that the presence of tumor hypoxia has been repeatedly shown to be a negative prognostic factor and to be associated with resistance to therapy regardless of treatment modality used (Le et al. 2006; Walsh et al. 2014; Scharping et al. 2017; Doktorova et al. 2015). However, given that oxygen is a direct “facilitator” of radiation-induced DNA damage, it is perhaps unsurprising that the impact of hypoxia on radiation therapy outcomes is most profound. The effect of hypoxia on radiation therapy outcomes has been most studied in patients with HNSCC . In a large analysis of data from numerous HNSCC radiation therapy studies, Nordsmark et al. demonstrated that pre-treatment hypoxia directly measured using oxygen electrodes was a highly significant prognostic factor for overall survival after radiotherapy alone, or in combination with surgery, chemotherapy, or a radiosensitizer (Nordsmark et al. 2005). Multivariate analysis in this study found that pre-treatment proportion of pO2 values ≤2.5 mmHg (HP2.5) was independently prognostic for poor survival using a hypoxia threshold of HP2.5 < 20% (median value). Furthermore, the negative impact of hypoxia on radiation therapy efficacy can be deduced from the large study of 918 patients with HNSCC randomized to receive CHART versus conventional radiation (Dische et al. 1997). Although this study failed to show improved locoregional tumor control (LRC) or overall survival (OS) with CHART, a retrospective subgroup analysis of pre-treatment tissue from nearly 200 patients demonstrated that expression of endogenous markers of hypoxia for the HIF-1 and HIF-2 pathway was strongly associated with post-radiation failure. This led the authors to postulate that hypoxic tumors were resistant to the potential benefit of CHART due to insufficient time for tumor reoxygenation to occur during treatment (Koukourakis et al. 2006).
In other tumor types, hypoxia has also been shown to predict poor radiation therapy outcomes (Rischin et al. 2006; Eschmann et al. 2005; Milosevic et al. 2012). For example, in patients with NSCLC, higher baseline levels of hypoxia, as determined by multiple hypoxia PET imaging parameters, was predictive of lower rates of local control following radical radiotherapy (Eschmann et al. 2005). In another study in patients with localized prostate cancer receiving radical radiotherapy, direct measurement of tumor oxygenation revealed that the presence of hypoxia, using a HP10 cut-off of 63% (median value), was associated with local recurrence within the gland and early biochemical failure (Milosevic et al. 2012).
5 Measuring Tumor Hypoxia in the Clinical Setting
Key to determining the impact of hypoxia on therapy outcomes has been our developing ability to accurately measure tumor hypoxia in patient samples. Over the last few decades, a wide range of methods have been used to study tumor hypoxia in patients, each with specific advantages as well as limitations. The various methods will be summarized here and have been reviewed (Hammond et al. 2014; O'Connor et al. 2019; Lee et al. 2014).
Direct measurements of tumor oxygenation using glass electrodes began in the 1960s with many early studies demonstrating the relationship between tumor oxygen level and radiation response, especially in cervical cancer and HNSCC (Kolstad 1968; Gatenby et al. 1988). However, our understanding of tumor oxygenation increased exponentially in the early 1990s with the introduction of the commercially available Eppendorf pO2 histograph, also referred to as the polarographic electrode (Vaupel et al. 2007). This technique, which is able to measure oxygen tensions as low as 1–2 mmHg, demonstrated the significant intra- and inter-tumor variability in oxygenation and a large number of clinical studies quickly utilized this technique to demonstrate the correlation between hypoxia and poor clinical outcomes, as previously described. There were numerous limitations of electrode-based methods for studying tumor oxygenation. Determining oxygen tensions was limited to only accessible tumors and the invasive nature of the procedure, which requires the insertion of the probe along multiple tracks in order to collect sufficient readings, was understandably viewed as somewhat unattractive by patients and clinicians. Furthermore, due to the complex spatiotemporal heterogeneity of hypoxia within tumors, this method was subject to sampling error. Therefore, although this technique is regarded by many as the gold standard for evaluating hypoxia, it has been universally discontinued.
More recently, the most widely adopted approach for preclinical and clinical assessment of hypoxia has become the use of exogenously administered nitroimidazole-based agents. Such electron-deficient nitroaromatic compounds are selectively reduced by intracellular nitroreductase enzymes under hypoxic conditions to form reactive products, which bind irreversibly to nucleophilic molecules within the cell. Different nitroreductase enzymes in the cytoplasm, mitochondria, and microsomes are capable of the reductive metabolism of nitroheterocycles and include NADPH-cytochrome P450 reductase, cytochrome b5 reductase, xanthine oxidase, aldehyde dehydrogenase, and DT-diaphorase (Kedderis and Miwa 1988). The reduction of nitroimidazoles is accomplished by tissue nitroreductases that are plentiful and which do not represent a rate-limiting factor (Prekeges et al. 1991), thus enabling repeated evaluation of hypoxia by such agents without reaching enzyme saturation. Importantly, bioreduction and intracellular retention of nitroimidazole compounds is inversely proportional to oxygen levels and requires intact nitroreductase enzymes. As a result, it detects only viable hypoxic cells and does not accumulate in regions of tumor necrosis. Typically, such agents have minimal protein binding, permitting efficient transport from blood into tissues (Kizaka-Kondoh and Konse-Nagasawa 2009) and as they are not actively metabolized, their diffusion distance is significantly greater than that of molecular oxygen (Dische 1985), and therefore can still reach poorly perfused areas typical of hypoxic regions. Also of importance is that in vitro experiments have demonstrated that nitroimidazoles are retained over the same oxygen range at which the OER is observed, with sharply increasing retention below oxygen tensions of 10 mmHg (Kizaka-Kondoh and Konse-Nagasawa 2009), therefore highlighting them as clinically relevant markers for the study of tumor hypoxia in radiation oncology. The stability of nitroimidazole adducts in viable hypoxic tissues permits immunohistochemical quantification to be carried out after the administration of nitroimidazole-based agents. Such agents have been widely used to study tumor hypoxia and include EF5 (Lord et al. 1993) and pimonidazole hydrochloride (Raleigh et al. 1999). The degree of immunodetection of such agents has been shown to correlate well with polarographic electrode readings predominantly in preclinical studies (Raleigh et al. 1999). However, as with direct measurements, sampling-specific tumor regions may be unrepresentative of hypoxia throughout the tumor. In addition, obtaining tissue for this purpose is an invasive procedure, which has associated practical limitations.
Nitroimidazoles also provide an exciting and clinically attractive opportunity for the noninvasive evaluation of tumor hypoxia by using PET-CT imaging with radiolabeled nitroimidazole-based tracers (Chapman 1979). [18F]-fluoromisonidazole (FMISO) is the prototypical and most widely used radiotracer for this purpose. In vitro experiments have demonstrated that the under anoxic conditions, [3H]-FMISO binding was approximately 25-fold greater than normoxic controls with binding reduced to 40% at oxygen pressures of 4 mm Hg (Martin et al. 1992), thus demonstrating FMISO as a marker of severe and clinically relevant hypoxia. Relevant to measuring hypoxia in tumors, in vivo data demonstrates no correlation between regional blood flow and FMISO retention with numerous studies concluding that FMISO uptake is independent of blood flow (Martin et al. 1992). In vivo, agreement between FMISO and pimonidazole immunohistochemistry has been observed (Dubois et al. 2004); however, such studies are lacking in patients. In the clinical setting, numerous studies have demonstrated the ability of FMISO to identify heterogeneously distributed hypoxic tissue within human tumors (Rajendran and Krohn 2005).
The partition coefficient describes the lipophilic/hydrophilic nature of molecules and is commonly evaluated as the distribution coefficient between octanol and water. As FMISO partitions nearly equally between octanol and water, once distribution equilibrium is achieved, all tissues should have the same concentration of tracer as blood. Only regions of hypoxia or organs involved in excretion or metabolism will have uptake levels greater than blood. The advantage is that the concentration ratio for normoxic tissue to blood will be very close to one and will be completely independent of blood flow. However, because of such partitioning characteristics of FMISO, whole body clearance is relatively slow, resulting in limited image contrast and requiring imaging following a relatively long period of time after injection of tracer (Rajendran and Krohn 2015). These limitations have led to the development of newer tracers such as [18F]-fluoroazomycin arabinoside (FAZA), which have higher normal tissue clearance kinetics with improved hypoxia-to-normoxia contrast (Fleming et al. 2015).
Clinical imaging protocols for using FMISO PET-CT in the study of tumor hypoxia are fairly well established (McGowan et al. 2019; Koh et al. 1992), with increasing validation and high reproducibility of FMISO demonstrated in NSCLC (Grkovski et al. 2016) and HNSCC (Okamoto et al. 2013). This imaging technique enables the assessment of hypoxia heterogeneity within tumors and for multiple measurements per individual patient. However, the limitations are that it is prohibitively expensive for use in large studies, is associated with logistical challenges in terms of tracer supply and subjects patients to not insignificant radiation exposure. Oxygen-enhanced MRI (OE-MRI) imaging potentially enables the quantification of tumor hypoxia without exposing patients to additional ionizing radiation. Although it has yet to be widely used in clinical studies, OE-MRI might be associated with fewer logistical hurdles than nitroimidazole PET imaging (O'Connor et al. 2019).
As our understanding of the complex tumor response to hypoxia continues to improve, methods used for the assessment of hypoxia have shifted to measuring endogenous markers of this response. The majority of such markers are known to be direct transcription targets of HIF-1 as the detection of HIF-1 itself is challenging due to its instability and rapid degradation.
In terms of tumor tissue hypoxia analysis, immunohistochemistry for carbonic anhydrase IX (CAIX) has been commonly performed. CAIX is a transmembrane metalloenzyme that belongs to the family of α-carbonic anhydrases, which catalyze the reversible hydration of carbon dioxide to bicarbonate ions and protons, thereby regulating the cellular and extracellular pH (Pastorek et al. 1994). Sixteen human CA isoforms have been identified, but it is CAIX that is most strongly induced by hypoxia in a broad range of cell types. Expression of CAIX is tightly regulated by HIF-1-dependent HRE and subsequently, levels are highly responsive to hypoxia (Wykoff et al. 2000). High expression of CAIX has consistently been reported in human tumors and shown to predict poor clinical outcomes (Giatromanolaki et al. 2001; Kaluz et al. 2009). CAIX expression as detected by immunohistochemistry has been shown to grossly co-localize with that of HIF-1 as well as pimonidazole staining (Olive et al. 2001; Gillies et al. 2011) and has also been shown to correlate with direct pO2 measurements (Le et al. 2006).
When considering such endogenous markers of hypoxia, it is important to appreciate that their expression level may be influenced by numerous factors other than oxygen level, such as other cellular signaling pathways (Hughes et al. 2018). In addition, the level of oxygenation and thus severity of hypoxia required to induce HIF signaling (and increase associated response proteins such as CAIX) differs to that required for the retention of exogenous nitroimidazoles. On detailed inspection of tumor tissue staining, binding of pimonidazole is more restricted to severely hypoxic and peri-necrotic regions as compared to HIF-1 expression (Sobhanifar et al. 2005). In addition, HIF1 expression and its response proteins can also be found in regions of cycling hypoxia, which are not necessarily positive for nitroimidazole retention at the time of measurement. In recognition of the complex tumor response to hypoxia, gene expression profiling is becoming more commonly used to evaluate the transcriptional response to hypoxia with numerous hypoxic metagene signatures now identified and increasingly well validated (Buffa et al. 2010; Winter et al. 2007). Importantly, the clinical utility of these gene expression signatures is enhanced by the fact that they can be derived from routine, formalin-fixed tumor samples. For a comprehensive review, see Harris et al. (Harris et al. 2015).
In addition to tumor tissue-based measurements, endogenous markers of hypoxia also provide the opportunity for identifying circulating hypoxic biomarkers, which may ultimately function as a plasma hypoxia signature. One of the most well-studied circulating markers to this effect is VEGF. Numerous studies have demonstrated the correlation between plasma VEGF levels and direct tumor oxygen measurements (Dunst et al. 2001), with higher levels predicting poor treatment response and outcomes in numerous tumor types (Ostheimer et al. 2014).
Another circulating hypoxia marker which has been studied extensively is osteopontin. This multifunctioning glycoprotein is recognized to play an important role in signaling pathways involved in many aspects of cancer progression including promoting proliferation, angiogenesis, and metastasis (Zhao et al. 2018). Osteopontin is upregulated under hypoxic conditions by AKT activation and stimulation of the ras-activated enhancer (RAE) in the OPN promoter (Zhu et al. 2005). Circulating osteopontin levels have been demonstrated to correlate inversely with polarographic electrode oxygen measurements and function as a prognostic and predictive biomarker with regard to radiotherapy outcomes in NSCLC (Ostheimer et al. 2014; Le et al. 2006) and HNSCC (Nordsmark et al. 2007; Petrik et al. 2006; Overgaard et al. 2005; Le et al. 2003). In NSCLC, a recent meta-analysis has demonstrated that osteopontin as measured by both IHC and as a circulating marker, is highly predictive of overall survival (Wang et al. 2015).
A different class of hypoxia-related circulating biomarkers are microRNAs (miRNAs). These approximately 21 nucleotide non-coding RNAs are key post-transcriptional regulators of gene expression (Krol et al. 2010). In the cellular response to hypoxia, and directly targeted by HIF-1α, miR-210 has emerged as the “master hypoxiaMIR” (Dang and Myers 2015). Shown to correlate with other measures of tumor hypoxia, both tissue and circulating miR-210 have been reported to be diagnostic, predictive, and prognostic hypoxia-related biomarkers in a number of tumor types (Chakraborty and Das 2016; Madhavan et al. 2016; Jiang et al. 2018; Świtlik et al. 2019).
6 Historical Hypoxia Modification Strategies with Radiation Therapy
Over the last half-century, numerous strategies have been investigated in an attempt to overcome the detrimental effect of tumor hypoxia on radiation therapy outcomes.
6.1 Increasing Tumor Oxygenation
Early approaches to address tumor hypoxia focused on increasing tumor oxygen delivery. One such method involved patients breathing 100% oxygen under hyperbaric conditions (HBO). A meta-analysis of HBO in patients with HNSCC treated with radiotherapy demonstrated a small but significant improvement in locoregional control (Overgaard 2011). A comprehensive Cochrane review of 19 clinical trials which delivered HBO concluded that HBO improved local control rates in cervix and HNSCC , but these benefits were small and may have only arisen in the context of unconventional fractionation schedules. In addition, HBO was associated with significant adverse effects, including oxygen toxic seizures and severe tissue radiation injury (Bennett et al. 2005). Given the modest benefit observed, practical inconvenience of treatment delivery and safety concerns, HBO was not adopted into clinical practice.
6.2 Correction of Anemia
Additional methods aimed at increasing tumor oxygenation have focused on administering blood transfusions to increase hemoglobin levels. Anemia is a very common finding in patients presenting for radiotherapy with 40 to 64% of patients being anemic prior to treatment with anemia shown to be associated with worse LRC and survival in numerous tumor types (Harrison et al. 2002; Lee et al. 1998). However, although the etiology of anemia in patients can be multifactorial, it often reflects patients with higher disease burden and therefore those who are more likely to have poor clinical outcomes. Numerous studies, mainly in HNSCC and carcinoma of the cervix, have investigated correcting anemia with blood transfusions during radiation treatment with overall mixed results observed (Varlotto and Stevenson 2005). An alternative method explored for correcting anemia during radiotherapy in HNSCC has been erythropoietin injections. However, unfortunately a significant detrimental impact on survival was observed with this approach (Henke et al. 2006), postulated to be attributable to promoting tumor cell proliferation through stimulation of tumor erythropoietin receptors (Henke et al. 2003).
6.3 ARCON
Other studies have aimed to improve oxygen delivery to tumors during radiation therapy by reversing acute hypoxia by combining the vasodilator nicotinamide with inhaled carbogen (95% oxygen and 5% carbon dioxide). This approach has been used as part of the Accelerated Radiation, Carbogen, and Nicotinamide (ARCON ) regime and has demonstrated promising LRC rates and toxicity in large phase II studies in patients with HNSCC (Kaanders et al. 2002) and bladder cancer (Hoskin et al. 2009). Subsequently, in a phase III trial in laryngeal cancer, ARCON was well tolerated and resulted in improved LRC rates, with the poor regional control seen in more hypoxic tumors specifically addressed by this approach (Janssens et al. 2012). Furthermore, in bladder cancer the phase III BCON trial demonstrated improvement in LRC and OS using this treatment (Hoskin et al. 2010). However, due to the not insignificant practical challenges involved in delivering this regime, it has not been widely adopted.
6.4 Oxygen Mimetics
An alternative approach to address hypoxia-mediated radioresistance has been to combine drugs which function as “oxygen mimetics” by promoting the fixation of free radical damage following radiotherapy. These agents belong to the nitroimidazole class of agents, which undergo bioreduction and intracellular trapping under hypoxic conditions, as previously described. Misonidazole was the first of such agents to be shown to result in radiosensitization in vitro (Asquith et al. 1974) and subsequently a number of clinical studies were conducted in many different tumor types combining this agent with radiotherapy (Mäntylä et al. 1982; Bleehen et al. 1981; Overgaard et al. 1989a, b; Papavasiliou et al. 1983). With a few exceptions, generally the results did not demonstrate significant improvements in radiotherapy outcomes and this was associated with high toxicity, in particular with regard to peripheral neuropathy, preventing dose escalation to levels thought to be required to produce tumor radiosensitization (Melgaard et al. 1988). Subsequently, other nitroimidazoles were developed with more favorable toxicity profiles, of which the 5-nitroimidazole nimorazole has been the most widely studied in combination with radiotherapy. In the large randomized phase III Danish Head and Neck Cancer (DAHANCA) study, nimorazole significantly improved LRC in carcinoma of the supraglottic larynx and pharynx (Overgaard et al. 1998). This study, and others in different tumor sites in the head and neck with the addition of cytotoxic chemotherapy, demonstrated acceptable toxicity (Bentzen et al. 2015; Metwally et al. 2014). Despite such promising results, nimorazole is only routinely used in Denmark, with the lack of demonstrated improvement in OS limiting its uptake elsewhere. The results of the randomized nimorazole with radiation versus radiation alone UK phase III study in HNSCC (NIMRAD) are currently awaited (Thomson et al. 2014).
6.5 Hypoxia-Activated Cytotoxic Prodrugs
Hypoxia-activated prodrugs have also been explored in an effort to target hypoxic cells directly and have been reviewed recently (Mistry et al. 2017). Such agents also become preferentially activated and reduced in low oxygen tensions but this time to form highly cytotoxic species (Fig. 11.1). The most well-known molecule in this class is tirapazamine, which has been shown preclinically to be significantly more cytotoxic under hypoxic conditions (Zeman et al. 1986). Unfortunately, randomized phase III studies using tirapazamine in combination with chemoradiation in carcinoma of the cervix (DiSilvestro et al. 2014) and HNSCC (Rischin et al. 2010) have failed to demonstrate improvement in outcomes. More recently, molecularly targeted hypoxia-activated prodrugs have been described which, instead of being reduced to release a cytotoxin, release an inhibitor of a molecular target (reviewed in (Mistry et al. 2017)). It should be noted that patient stratification is likely to be absolutely essential to this approach as mechanism of action for these agents requires hypoxia, i.e., they cannot and should not work in non-hypoxic tumors.

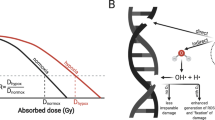
Schematic representation of tumor hypoxia and the mechanism of action of hypoxia-activated prodrugs. As described regions of hypoxia occur at distances of 100–200 μM from functional blood vessels due to the metabolism of oxygen by the tumor cells. Hypoxia-activated prodrugs, which traditionally release a cytotoxin but more recently are molecularly targeted inhibitors, are inactive in oxic conditions but in response to hypoxia become reduced to release the active agent. In some cases, the active agent not only kills the hypoxic cells but also enters the surrounding oxic cells through a bystander mechanism
6.6 Vascular “Normalization”
Intracellular signal transduction pathways are known to play an important role in determining tumor response to radiation with the well-studied EGFR /Ras/PI3K/AKT pathway shown to be of particular importance. Preclinical experiments have demonstrated that inhibitors of EGFR , Ras, PI3K, and AKT produce marked “normalization” of tumor microvasculature with increased perfusion and alleviation of tumor hypoxia (Qayum et al. 2009; Cerniglia et al. 2009). Furthermore, in xenograft models it was observed that PI3K inhibition resulted in significant tumor growth delay after radiation because of vascular remodeling, which was independent and synergistic to increasing intrinsic radiosensitivity (Fokas et al. 2012). Excitingly, recently the specific PI3K inhibitor buparlisib has been shown to result in a reduction in tumor hypoxia as measured by FMISO PET in patients with advanced NSCLC and to be well tolerated in combination with palliative thoracic radiation (McGowan et al. 2019) although no assessment of radiation response was possible in this study.
Novel strategies to normalize tumor angiogenesis and microenvironment to improve hypoxia and radiation outcomes are comprehensively described in Chap. 12.
6.7 Summary of Past Approaches
Over the last three decades, a truly wide range of different approaches has been employed to tackle hypoxia-mediated radioresistance and a meta-analysis of such treatments in HNSCC has demonstrated improvement in LRC and OS, thus providing level 1a evidence for hypoxia modification with radiation. However, due to inconsistency of results observed over the years with any outcome improvements only modest at best, no such treatment is widely used. Concerns regarding toxicity and challenges in practical delivery have further curbed enthusiasm. It is now well recognized that one of the major reasons why hypoxia modification has failed to show significant changes in radiation therapy outcomes is that there has been a complete lack of selecting patients for treatment based on tumor hypoxia and therefore likely to benefit most from such intervention. The reasons for this are largely due to the fact that there is currently no agreement as to which measure of hypoxia should be used for this purpose with many current techniques lacking sufficient validation. We should also consider our selection of in vivo models for testing of hypoxia modifying therapies. The tendency is to choose cell line/tumor models with levels of hypoxia, which are perhaps non-realistic of human tumors. Undeniably, novel and more efficacious hypoxia modification treatments are also required and more recently attention has focused on modulation of tumor metabolism, as will be discussed below.
7 Oxidative Phosphorylation as an Emerging Target to Tackle Tumor Hypoxia
In contrast to previous methods largely aimed at increasing oxygen delivery to tumors, an alternative and exciting novel strategy to tackle tumor hypoxia is to reduce the tumor oxygen consumption rate (OCR), thus addressing the other side of the oxygen supply and demand equation. More specifically, there is growing interest in reducing tumor OCR through inhibition of oxidative phosphorylation (OXPHOS).
7.1 Overview of Cellular Respiration
Cellular respiration describes a series of well-characterized metabolic reactions by which cells convert available nutrients into biochemical energy in the form of adenosine triphosphate (ATP). The most readily available source of cellular energy is glucose and molecular oxygen plays a key role in its conversion to ATP. The first step in cellular respiration in most organisms is glycolysis, also referred to as the Embden-Meyerhof-Parnas pathway. This oxygen-independent pathway constitutes a ten-step multienzyme process occurring in the cytosol and converts one molecule of glucose into two molecules of pyruvate whilst producing two reduced nicotinamide adenine dinuceotide (NADH) and two ATP molecules (net yield). The fate of pyruvate and cellular respiration hereon is highly dependent on the presence of molecular oxygen (Lunt and Vander Heiden 2011). In the absence of oxygen, pyruvate is reduced to lactate by lactate dehydrogenase (LDH). This results in the regeneration of NAD + through NADH oxidation and as NAD+ is required during glycolysis for ATP production, this enables ongoing cellular energy production under anaerobic conditions. Lactate may be converted back to pyruvate by LDH and used for aerobic respiration if oxygenation has increased, or it diffuses into the circulation and can be converted back into glucose by the liver in the Cori cycle.
In contrast, in the presence of oxygen, pyruvate generated during glycolysis enters the mitochondrial matrix, where it undergoes oxidative decarboxylation by a series of enzymes which make up the pyruvate dehydrogenase complex (PDH) and is converted into acetyl coenzyme A (acetyl CoA). Acetyl CoA enters the citric acid cycle, also known as the Krebs or tricarboxylic acid cycle, by combining with oxaloacetate to form citrate, which is systematically converted back to oxaloacetate and then combines with acetyl CoA, and so the cycle repeats. Each full cycle produces one guanosine triphosphate (GTP), one reduced flavin adenine dinucleotide (FADH2) and three NADH molecules. FADH2 and NADH move to the inner mitochondrial membrane and facilitate ATP production during the process of OXPHOS (Lunt and Vander Heiden 2011).
The electron transport chain (ETC) uses NADH and FADH2 produced by the Krebs cycle and NADH from glycolysis to produce ATP. Electrons from NADH and FADH2 are released and transferred systematically through protein complexes located in the inner mitochondrial membrane, thus forming the ETC. These inner membrane protein complexes include four enzyme complexes, complex I, complex II, complex III, and complex IV, and two coenzymes, ubiquinone (also known as co-enzyme Q) and cytochrome c. As electrons are passed from one electron carrier to another, it results in their reduction and then oxidation, which releases enough energy to pump hydrogen ions across the inner membrane into the space between in the inner and outer mitochondrial membranes. The accumulation of protons in this intermembrane space creates a steep electrochemical gradient across the inner membrane. As protons traverse the protein pore complex ATP synthase embedded in the inner membrane back into the mitochondrial matrix, the shaft of the complex rotates and catalyzes the production of ATP by attaching a phosphate to ADP. In total, for every glucose molecule that enters aerobic respiration, a net yield of 36 ATPs is produced. Once the electrons have passed through the ETC, they are combined with molecular oxygen and hydrogen ions to form water molecules. Therefore, oxygen can be viewed as the terminal electron acceptor and electron flow through the ETC ceases without oxygen. The ETC couples the transfer of electrons from donor molecules to oxygen with ATP production, thus representing the process known as OXPHOS (Kühlbrandt 2015).
7.2 Altered Metabolism in Cancer and the Impact of Tumor Hypoxia
A well-established hallmark of cancer is highly abnormal energy metabolism (Jones and Thompson 2009; Hanahan and Weinberg 2011). First observed by Otto Warburg in 1920s (Koppenol et al. 2011), cancer cells reprogram energy metabolism to predominantly rely on glycolysis, even in the presence of normal oxygenation. This phenomenon of “aerobic glycolysis” is characterized by marked increase in glucose uptake by cancer cells, largely by upregulation of glucose transporters such as transporter 1 (GLUT1) (Jones and Thompson 2009). Although relying on glycolysis for energy metabolism may seem counterintuitive due to lower yields of ATP per molecule of glucose molecule compared with OXPHOS , the sheer abundance of glucose due to increased uptake results in far greater ATP production and at a faster rate.
Pro-proliferative signal transduction pathways are known to function as glycolytic drivers with increased PI3K/AKT/mTOR signaling and overexpression of RAS and c-myc known to be of key importance (Jones and Thompson 2009; DeBerardinis et al. 2008). For example, AKT has been shown to be perhaps the most important as it appears to be sufficient to drive glycolysis independently (Elstrom et al. 2004), whilst c-myc increases transcription of GLUT1, as well as multiple key enzymes involved in glycolysis (Osthus et al. 2000; Dang et al. 1997).
Tumor hypoxia also plays an important role in fueling glycolysis, primarily through HIF-1 signaling. For example, HIF-1 upregulates GLUT1 as well as virtually all of the glycolytic enzymes, with key examples being HK, PGI, and LDH (Dang and Semenza 1999; Semenza 2010). In addition, HIF-1 also increases expression of PDH kinase 1 which inhibits PDH complex activity, thus preventing the irreversible conversion of pyruvate into acetyl CoA and entry into the citric acid cycle (Kim et al. 2006), and therefore functions as an important switch between glycolysis and OXPHOS respiration. Within the mitochondria itself, HIF-1 tightly regulates the expression of different isoforms of subunit COX4 and the function of ETC complex IV of the electron transport chain, thus impacting on ATP production and oxygen consumption (Fukuda et al. 2007). More recently, it is increasingly recognized that HIF-1 also regulates the expression of a wide range of miRNAs which fine tune metabolic pathways, as outlined in a comprehensive review (Orang et al. 2019).
In terms of OXPHOS activity in cancer, downregulation has been demonstrated in several tumor types and attributed to mitochondrial DNA (mtDNA) mutations, or reduced mtDNA content, given that mtDNA codes for many of the subunits of OXPHOS protein complexes I to V (Yu 2011). Such downregulation of OXPHOS has been demonstrated to correlate with poor clinical outcomes in many cancers and shown to be associated with invasive and metastatic tumor phenotypes (Gaude and Frezza 2016).
7.3 Emergence of the Importance of OXPHOS in Cancer
Because of the observation that glycolysis is upregulated and OXPHOS is downregulated in tumors, it has become a long-held belief that OXPHOS is largely redundant in cancer and thus has been largely forgotten. However, more recently it has been demonstrated that in a number of tumor types mitochondrial metabolism is not downregulated, including pancreatic and endometrial cancer as well as in lymphoma and high OXPHOS subtype melanoma (Moreno-Sánchez et al. 2007; Weinberg and Chandel 2015). In addition, it has increasingly become recognized that OXPHOS is indeed active in cancer cells and does in fact contribute, although to a varying degree, to cellular energy production (Zu and Guppy 2004). Such is the higher yield of ATP production with OXPHOS compared with glycolysis, that it has been shown that when OXPHOS activity is reduced as low as 6%, it still contributes to 50% of the cell’s energy requirements (Mookerjee et al. 2017). In a meta-analysis of normal cell types and cancer cell lines, the average contribution of OXPHOS to ATP production was 80% in normal cells and 83% in cancer cells (Zu and Guppy 2004). Despite the often severely hypoxic conditions within tumors, the level of oxygen, the key determinant of the oxygen consumption rate (OCR) (Moreno-Sánchez et al. 2009; Vaupel and Mayer 2012), is generally above that which would impair mitochondrial respiration as the KmO2 of COX is as low as 0.1–0.8 μM in different biological systems (Moreno-Sánchez et al. 2007).
The previous explanation that decreased OXPHOS in cancer is attributable to significantly decreased or mutated mitochondrial (mt) DNA is controversial. In fact, many types of cancer have increased mtDNA content relative to normal tissue (Reznik et al. 2016; Yu 2011). With regard to mtDNA mutations, analysis of paired tumor and normal tissue samples has revealed deleterious somatic tumor-specific mtDNA mutations in the majority of rectal and colon adenocarcinomas (Larman et al. 2012). Furthermore, such mutations were identified in all mtDNA genes many of which were predicted to impair OXPHOS . Interestingly, it has been shown that cancer cells with these mutations in OXPHOS-related genes such as complex I subunits, have much higher sensitivity to complex I inhibitors (Birsoy et al. 2014), indicating these mutations play an important role in cancer cell function.
Further still, in vitro experiments using ethidium bromide to deplete mitochondrial DNA have demonstrated that reduction of mitochondrial respiration dramatically changes the behavior of cancer cells and decreases tumorigenic potential (Cavalli et al. 1997). This is supported by a number of different cancer cell in vivo models, which have demonstrated that depletion of mitochondrial respiratory complexes results in delayed tumor formation following transplantation and an inability of cancer cells to metastasize (Tan et al. 2015). Interestingly, when tumors did eventually form and disseminate, they showed marked reactivation of OXPHOS due to acquisition of host mitochondrial DNA. There is also growing evidence that reliance on OXPHOS, and in fact impaired glycolysis, is a key feature of cancer stem cells and quiescent cancer cells resistant to standard therapies and responsible for repopulation of tumors after treatment (Viale et al. 2015).
Perhaps one important role of OXPHOS in carcinogenesis is the production of reactive oxygen species (ROS). High levels of ROS production has long been recognized to be a classical feature of tumors (Szatrowski and Nathan 1991) with many early studies focused on the DNA damaging effects of ROS, which is believed to be tumorigenic by promoting genomic instability (Ames et al. 1993). However, more recently a paradigm shift has occurred in our understanding of ROS with a more nuanced view of the importance of ROS as signaling pathway molecules, promoting tumorigenesis by regulation of cellular proliferation, metabolic alterations, and angiogenesis (Sullivan and Chandel 2014).
Therefore, after decades of disregard, OXPHOS has re-emerged as a functioning and important part of cancer cellular function. As OXPHOS consumes significant quantities of oxygen, inhibition of this process is a novel approach to reduce tumor oxygen consumption and therefore tackle tumor hypoxia.
7.4 OCR Inhibition to Alleviate Tumor Hypoxia
Over the last few years, there has been growing interest in using OXPHOS inhibitors to reduce tumor OCR . Such an approach would permit unconsumed oxygen to diffuse into adjacent low oxygen regions and therefore alleviate tumor hypoxia. Indeed, 3D multicellular spheroid studies have demonstrated that OCR reduction alleviates central hypoxic regions by increasing the availability of free oxygen (Secomb et al. 1995; Grimes et al. 2014). In support of this approach, mathematical modeling suggests that only a 30% decrease in OCR would abolish severe hypoxia (Grimes et al. 2014) and that reduction in OCR is likely to be a more effective strategy to reduce hypoxia than previous methods aimed at increasing oxygen supply (Secomb et al. 1995).
Inhibition of OXPHOS has been shown to reduce OCR in many different cancer cell types, indicating potential wide applicability (Ashton et al. 2016). Also this approach is thought less susceptible to poor tumor perfusion as such inhibitors would initially target OXPHOS in well-perfused and non-hypoxic regions, which would then result in oxygen rapidly diffusing to adjacent regions of chronic hypoxia, thus targeting “hard to reach” tumor regions indirectly (Ashton et al. 2018).
A number of FDA-approved drugs are known to inhibit OXPHOS through off-target effects and have gained attention as potentially clinically useful OCR inhibitors (Ashton et al. 2018). In particular, the anti-diabetic medication metformin has been most studied in this context. This biguanide is the most widely prescribed medication for treatment of type II diabetes mellitus and exerts its glucose-lowering effect through inhibition of hepatic gluconeogenesis (Hunter et al. 2018). The potential of metformin as an anti-cancer agent first came to light when large epidemiological and retrospective studies demonstrated that diabetic patients taking metformin had significantly lower incidence of cancer and better clinical outcomes following treatment, including in the context of radiotherapy, than patients with diabetes taking alternative medication (Cao et al. 2017; Koritzinsky 2015; Pernicova and Korbonits 2014). The observed benefits of metformin have been attributed to a number of its effects, including systemically lowered insulin/insulin-like growth factor-1 (IGF-1) as well as potential inhibition of mammalian target of rapamycin (mTOR), activation of AMP-activated kinase (AMPK), and reduction in ROS (Belfiore et al. 2009; Algire et al. 2012; Dowling et al. 2007). The mechanism by which metformin reduces OCR is through inhibition of complex I of the ETC (Owen et al. 2000). Preclinical data has shown that metformin reduces OCR in numerous cancer cell lines leading to alleviation of hypoxia in spheroids as well as in xenograft models with corresponding improvement in radiation response (Ashton et al. 2016; Zannella et al. 2013; De Bruycker et al. 2019).
However, unfortunately despite such promise, clinically achievable concentrations of metformin are significantly lower than those required in preclinical models to result in meaningful OCR reduction, and thus unlikely to be a good anti-hypoxia agent. Furthermore, in a number of cell types, metformin was shown to be ineffective at inhibiting OCR , suggesting potential limited applicability (Ashton et al. 2016). A recent clinical study in breast cancer has demonstrated that metformin does alter tumor metabolism, switching it to a glycolytic phenotype (Lord et al. 2018), suggesting sufficient tumor metabolism modulatory activity in patients; however, no hypoxia measures were utilized. A recent randomized study of concurrent chemoradiation therapy with or without metformin in locally advanced NSCLC failed to demonstrate improvement in outcomes (Tsakiridis et al. 2019). Recently, Lord et al. reported that treatment of breast cancer patients with metformin, which inhibits OXPHOS through complex I rather than complex III inhibition, resulted in a shift in tumor metabolism from OXPHOS to glycolysis (Lord et al. 2018), therefore supporting that metabolic reprograming is achievable using OXPHOS inhibitors in the clinical setting. As glycolysis has long been associated with tumor progression and metastasis (Heiden and DeBerardinis 2017), some may voice concern that this approach may be detrimental in terms of disease progression. However, retrospective studies have unanimously demonstrated superior outcomes for cancer patients receiving metformin compared to diabetic patients taking alternative medication, with such improvements independent of diabetic control (Cao et al. 2017; Koritzinsky 2015; Pernicova and Korbonits 2014; Belfiore et al. 2009; Algire et al. 2012; Dowling et al. 2007). A number of clinical studies are currently underway to investigate the effect of metformin on tumor hypoxia with no reported results as of yet.
Numerous other FDA-approved drugs, as well as new experimental agents, have been shown to inhibit OXPHOS preclinically but several have significant toxicity and unfavorable pharmacokinetics, and are thus unattractive candidates (Wang et al. 2003; Ashton et al. 2016). In one early phase study of the novel complex I inhibitor BAY87–2243, the trial was terminated early due to unexpected toxicity (Kirkpatrick and Powis 2017), demonstrating the importance of carefully assessing the safety of such agents, especially when combined with chemotherapy or radiation.
Another complex I inhibitor papaverine has also been shown to reduce hypoxia and improve tumor radiation response preclinically (Benej et al. 2018). Parpaverine is an FDA-approved compound conventionally used to treat arterial spasm; this is independent of its ability to inhibit complex I, but instead by inhibiting phosphodiesterase 10A (PDE10A). An attractive property of parpaverine is that pre-treatment of just 45 min resulted in a significant reduction in tumor hypoxia, and subsequent radiosensitization. This relatively short pre-treatment time adds to the potential of parpaverine being translated to a clinical setting. Papaverine is undergoing phase I testing in combination with stereotactic ablative body radiotherapy (SABR) in NSCLC but unfortunately, once again, no hypoxia readouts have been included in the study design.
Overall, preclinical data is highly encouraging for developing OXPHOS inhibitors in order to reduce OCR and thus tumor hypoxia. However, the translation of such agents into the clinical setting has proven challenging with no clinical evidence to date of alleviation of tumor hypoxia. Any agent being developed for this purpose must not only have strong preclinical data to support its effect on OCR and hypoxia, but do so at clinically relevant concentrations and with favorable toxicity profile.
7.5 Atovaquone as a Novel Tumor Hypoxia Modifier and Radiosensitizer
The recent discovery that the commonly prescribed and well-tolerated antimalarial drug atovaquone meets criteria of affecting OCR /hypoxia at clinically relevant concentrations and acceptable toxicity is of great promise. Atovaquone is an FDA-approved drug in widespread clinical use for over 20 years. It is currently used as a single agent for the treatment of the Pneumocystis pneumonia caused by the opportunistic fungus Pneumocystis jirovecii, and in combination with proguanil for the prophylaxis and treatment of malaria caused by the protozoa Plasmodium falciparum.
Atovaquone (2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone, 566C80) is a ubiquinone (co-enzyme Q10) analogue which emerged in 1991 as the leading compound with efficacy against malaria and opportunistic infections such as Pneumocystis pneumonia in immunosuppressed patients with favorable pharmacokinetic properties (Hudson et al. 1991). Following required improvements in synthesis methodology, atovaquone became a commonly used antimicrobial in patients from the mid-1990s. Today, in combination with proguanil, atovaquone accounts for over 70% of malarial prophylaxis prescriptions in the USA (LaRocque et al. 2012).
The mechanism of action of atovaquone against Plasmodium falciparum is mediated through inhibition of mitochondrial complex III (Fry and Pudney 1992; Srivastava et al. 1997), whereas the mechanism responsible for atovaquone’s activity against Pneumocystis species still remains unknown. Complex III, also known as cytochrome bc1 complex or co-enzyme Q cytochrome c reductase, is a multi-subunit transmembrane protein located in the inner mitochondrial membrane which couples the electron transfer from ubiquinol, the reduced form of ubiquinone or co-enzyme Q, to cytochrome c, resulting in the movement of four protons across the lipid bilayer and thereby contributing significantly to ATP production. Atovaquone binds to the catalytic Qo site through hydrogen bonding to the Rieske iron-sulfur proteins which form the most important catalytic subunit of complex III required for transfer of electrons from ubiquinol during the ETC (Birth et al. 2014; Mather et al. 2005). By disrupting mitochondrial respiration in this way, atovaquone significantly prevents energy production in parasites. In addition, atovaquone also acts by blocking essential pyrimidine biosynthesis, which has been shown to be the predominant metabolic function of complex III in Plasmodium falciparum (Painter et al. 2007).
Atovaquone causes very few side effects and has an excellent safety profile. Adverse events which have potentially been attributed to atovaquone are generally very mild and most commonly include rash, fever, vomiting, diarrhea, abdominal pain, and headache (MEPRON (atovaquone) Suspension–GSKSource, Anon. n.d.-a; Wellvone 750 mg/5 ml oral suspension—Summary of Product Characteristics (SmPC)—(emc), Anon. n.d.-b). Encouragingly, an overdose of atovaquone with 31,500 mg resulted in no observed toxicity (Cheung 1999).
Using a Seahorse XF analyzer, Ashton et al. (Ashton et al. 2016) conducted a high-throughput screen of 1697 FDA-approved drugs to assess their effect on OCR in FaDu hypopharyngeal cancer cells. Subsequently, it was demonstrated that 10 μM atovaquone dramatically reduced OCR by approximately 80–90% in A549 NSCLC, H1299 NSCLC, H460 NSCLC, HCT116 colorectal cancer, DLD-1 colorectal cancer, MCF7 breast cancer, and T24 bladder cancer cells. A lesser but still impressive reduction of 58% in OCR was seen in PSN-1 pancreatic cancer cells. Furthermore, the effect on OCR was dose-dependent with smaller but still significant decreases in OCR observed at 2 μM (Ashton et al. 2016). Atovaquone was then demonstrated to completely alleviate hypoxia in 3D spheroids of FaDu, H1299, and HCT116 3D cells, again at clinically achievable concentrations. Combination of atovaquone treatment and radiation in FaDu spheroids resulted in increased growth delay following irradiation. In vivo experiments provided further support for atovaquone as a tumor hypoxia modifier and radiosensitizer . Using FaDu and HT116 mice xenograft models, 7 days of atovaquone treatment was shown to eradicate tumor hypoxia as detected by tumor EF5 staining, with mean mice plasma levels comparable to those seen in patients. There was no effect on tumor growth in FaDu xenografts with atovaquone treatment alone, but marked tumor growth delay was observed when combined with radiation (Ashton et al. 2016).
Mechanistic experiments in FaDu cells demonstrated that, as per its antimicrobial mode of action, atovaquone specifically inhibits mitochondrial complex III. With regard to radiosensitization, colony formation assays performed under different oxygen conditions revealed that atovaquone did not alter radiosensitivity, indicating that atovaquone is not an intrinsic radiosensitizer (Ashton et al. 2016).
Overall, this data provides compelling evidence that atovaquone decreases OCR through mitochondrial complex III inhibition, which results in a reduction in tumor hypoxia and in turn results in tumor radiosensitization. Given that hypoxia is predominantly a tumor-specific phenomenon, it is hypothesized that atovaquone would preferentially sensitize tumors and not normal tissues to radiation and thus improve the therapeutic index for radiation therapy.
A number of recent studies have also demonstrated that atovaquone inhibits cancer cell proliferation. For example, atovaquone has been shown to inhibit proliferation and induce apoptosis in renal cell carcinoma cells through a mechanism dependent upon mitochondrial complex III inhibition and increased ROS production (Chen et al. 2018). This group also showed that in vitro and in vivo atovaquone increased the efficacy of 5-fluorouracil (5-FU) and interferon-α (IFN-α) in this tumor type. Further still, in hepatocellular carcinoma (HCC) cells, atovaquone significantly inhibited proliferation through S phase cell cycle arrest with both intrinsic and extrinsic apoptotic pathway induction associated with upregulation of p53 and p21. Further investigation revealed that atovaquone induced double-stranded DNA breaks, leading to sustained activation of ataxia-telangiectasia mutated (ATM) kinase and its downstream molecules such as cell cycle checkpoint kinase-2 (CHK2) and H2AX. Subsequent in vivo experiments demonstrated that atovaquone-inhibited tumor growth and prolonged survival of tumor-bearing mice (Gao et al. 2018). However, in this study atovaquone did not have any significant effect on tumor growth. Furthermore, as previously described Ashton et al. did not observe any direct antiproliferative effect of atovaquone in numerous cancer cell types or tumor growth delay in xenograft models (Ashton et al. 2016), thus suggesting that perhaps the antiproliferative effects of atovaquone are dependent on cell type, as well as specific experimental conditions.
Lastly, atovaquone has also recently been shown to have direct activity against cancer stem-like cells (CSCs). In MCF7 breast cancer cells, atovaquone inhibits oxygen consumption and induces glycolysis as well as oxidative stress. Given that MCF7-derived CSCs are highly dependent on mitochondrial respiration, atovaquone inhibits CSC proliferation with an IC-50 of just 1 μM. Furthermore, in certain CSC populations, atovaquone induces apoptosis (Fiorillo et al. 2016).
Overall, there is therefore growing evidence that in addition to hypoxia modification and thus its potential as a radiosensitizer, atovaquone may be a multifunctioning anti-cancer agent. Atovaquone is extremely well tolerated, which is of key importance especially for combining it with toxic therapies such as radiation and chemotherapy where the therapeutic window is often already very narrow. The finding that atovaquone is so well tolerated is somewhat surprising given that OXPHOS is an integral part of normal cellular function and the main source of energy production. Perhaps the answer lies in the fact that atovaquone has been shown not to alter the metabolic state of normal cells. For example, in normal human fibroblasts no alteration in metabolic function were observed following atovaquone treatment (Fiorillo et al. 2016). This apparent cancer-specific inhibition of OXPHOS adds further impetus for developing this agent as an anti-cancer treatment.
8 Summary
Tumor hypoxia is arguably the best validated target in oncology due to its multifarious role in tumorigenesis and the profound resistance to radiation therapy it confers. In no other tumor type is the need for improving tumor radiosensitivity more evident than in locally advanced NSCLC where exceptionally poor outcomes following chemoradiation are unfortunately part of everyday clinical practice. Therefore, the development of novel hypoxia modifiers to improve radiation efficacy represents an unmet and urgent clinical need for this patient population. An exciting new strategy to tackle tumor hypoxia is reprogramming cellular metabolism through inhibition of OXPHOS to reduce tumor OCR . The commonly prescribed antimicrobial drug atovaquone has emerged as the most promising agent for this purpose. However, despite decades of clinical research combining promising hypoxia modifiers with radiotherapy, no such agent has been adopted into widespread clinical use. The reasons for this are clear and multifactorial. Although improvement in outcomes has been observed, it has been modest at best and often with concerns regarding toxicity or practical deliverability (Overgaard 2011). However, perhaps of greater importance is that there has been an almost complete absence of proof-of-principle studies to confirm the ability of such agents to reduce tumor hypoxia in patients. The barrier to conducting such studies has undeniably been a lack of robust, widely available and adequately validated biomarkers for the clinical evaluating tumor hypoxia. This has not only significantly impeded the investigation of the efficacy of hypoxia modifiers, but it has also resulted in an inability to accurately select patients who are likely to benefit most from such treatment. It is likely that only when hypoxia biomarkers are widely available will hypoxia modification enter the well-established era of personalized medicine and significant improvements in outcomes be realized.
References
Algire C, Moiseeva O, Deschênes-Simard X, Amrein L, Petruccelli L, Birman E, Viollet B, Ferbeyre G, Pollak MN (2012) Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev Res (Phila) 5(4):536–543. https://doi.org/10.1158/1940-6207.CAPR-11-0536
Alper T, Howard-Flanders P (1956) Role of oxygen in modifying the radiosensitivity of E. coli B. Nature 178(4540):978–979
Ames BN, Shigenaga MK, Hagen TM (1993) Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci U S A 90(17):7915–7922
Anon. (n.d.-a) MEPRON (atovaquone) Suspension-GSKSource
Anon. (n.d.-b) Wellvone 750mg/5ml oral suspension - Summary of Product Characteristics (SmPC)—(emc)
Ashton TM, Fokas E, Kunz-Schughart LA, Folkes LK, Anbalagan S, Huether M, Kelly CJ, Pirovano G, Buffa FM, Hammond EM, Stratford M, Muschel RJ, Higgins GS, McKenna WG (2016) The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat Commun 7. https://doi.org/10.1038/ncomms12308
Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS (2018) Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res 24(11):2482–2490. https://doi.org/10.1158/1078-0432.CCR-17-3070
Asquith JC, Watts ME, Patel K, Smithen CE, Adams GE (1974) Electron affinic sensitization. V. Radiosensitization of hypoxic bacteria and mammalian cells in vitro by some nitroimidazoles and nitropyrazoles. Radiat Res 60(1):108–118
Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R (2009) Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev 30(6):586–623. https://doi.org/10.1210/er.2008-0047
Benej M, Hong X, Vibhute S, Scott S, Wu J, Graves E, Le Q-T, Koong AC, Giaccia AJ, Yu B, Chen C-S, Papandreou I, Denko NC (2018) Papaverine and its derivatives radiosensitize solid tumors by inhibiting mitochondrial metabolism. Proc Natl Acad Sci U S A 115(42):10756–10761. https://doi.org/10.1073/pnas.1808945115
Bennett M, Feldmeier J, Smee R, Milross C (2005) Hyperbaric oxygenation for tumour sensitisation to radiotherapy. Cochrane Database Syst Rev 4:CD005007. https://doi.org/10.1002/14651858.CD005007.pub2
Bennewith KL, Durand RE (2004) Quantifying transient hypoxia in human tumor xenografts by flow cytometry. Cancer Res 64(17):6183–6189. https://doi.org/10.1158/0008-5472.CAN-04-0289
Bentzen J, Toustrup K, Eriksen JG, Primdahl H, Andersen LJ, Overgaard J (2015) Locally advanced head and neck cancer treated with accelerated radiotherapy, the hypoxic modifier nimorazole and weekly cisplatin. Results from the DAHANCA 18 phase II study. Acta Oncol 54(7):1001–1007. https://doi.org/10.3109/0284186X.2014.992547
Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J (2003) HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J 22(16):4082–4090. https://doi.org/10.1093/emboj/cdg392
Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM (2014) Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508(7494):108–112. https://doi.org/10.1038/nature13110
Birth D, Kao W-C, Hunte C (2014) Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat Commun 5:4029. https://doi.org/10.1038/ncomms5029
Bleehen NM, Wiltshire CR, Plowman PN, Watson JV, Gleave JR, Holmes AE, Lewin WS, Treip CS, Hawkins TD (1981) A randomized study of misonidazole and radiotherapy for grade 3 and 4 cerebral astrocytoma. Br J Cancer 43(4):436–442
Brown JM (1979) Evidence for acutely hypoxic cells in mouse tumours, and a possible mechanism of reoxygenation. Br J Radiol 52(620):650–656. https://doi.org/10.1259/0007-1285-52-620-650
Buffa FM, Harris AL, West CM, Miller CJ (2010) Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br J Cancer 102(2):428–435. https://doi.org/10.1038/sj.bjc.6605450
Cao X, Wu Y, Wang J, Liu K, Wang X (2017) The effect of metformin on mortality among diabetic cancer patients: a systematic review and meta-analysis. JNCI Cancer Spectr 1(1). https://doi.org/10.1093/jncics/pkx007
Carreau A, Hafny-Rahbi BE, Matejuk A, Grillon C, Kieda C (2011) Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med 15(6):1239–1253. https://doi.org/10.1111/j.1582-4934.2011.01258.x
Cavalli LR, Varella-Garcia M, Liang BC (1997) Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ 8(11):1189–1198
Cerniglia GJ, Pore N, Tsai JH, Schultz S, Mick R, Choe R, Xing X, Durduran T, Yodh AG, Evans SM, Koch CJ, Hahn SM, Quon H, Sehgal CM, Lee WMF, Maity A (2009) Epidermal growth factor receptor inhibition modulates the microenvironment by vascular normalization to improve chemotherapy and radiotherapy efficacy. PLoS One 4(8):e6539. https://doi.org/10.1371/journal.pone.0006539
Chakraborty C, Das S (2016) Profiling cell-free and circulating miRNA: a clinical diagnostic tool for different cancers. Tumour Biol 37(5):5705–5714. https://doi.org/10.1007/s13277-016-4907-3
Chapman JD (1979) Hypoxic sensitizers--implications for radiation therapy. N Engl J Med 301(26):1429–1432. https://doi.org/10.1056/NEJM197912273012606
Chen D, Sun X, Zhang X, Cao J (2018) Targeting mitochondria by anthelmintic drug atovaquone sensitizes renal cell carcinoma to chemotherapy and immunotherapy. J Biochem Mol Toxicol 32(9):e22195. https://doi.org/10.1002/jbt.22195
Cheung TW (1999) Overdose of atovaquone in a patient with AIDS. AIDS 13(14):1984–1985
Choudhry H, Harris AL (2018) Advances in hypoxia-inducible factor biology. Cell Metab 27(2):281–298. https://doi.org/10.1016/j.cmet.2017.10.005
Dang CV, Lewis BC, Dolde C, Dang G, Shim H (1997) Oncogenes in tumor metabolism, tumorigenesis, and apoptosis. J Bioenerg Biomembr 29(4):345–354
Dang CV, Semenza GL (1999) Oncogenic alterations of metabolism. Trends Biochem Sci 24(2):68–72. https://doi.org/10.1016/S0968-0004(98)01344-9
Dang K, Myers KA (2015) The role of hypoxia-induced miR-210 in cancer progression. Int J Mol Sci 16(3):6353–6372. https://doi.org/10.3390/ijms16036353
Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM (2006) The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res 66(7):3688–3698. https://doi.org/10.1158/0008-5472.CAN-05-4564
De Bruycker S, Vangestel C, Van den Wyngaert T, Pauwels P, Wyffels L, Staelens S, Stroobants S (2019) 18F-Flortanidazole hypoxia PET holds promise as a prognostic and predictive imaging biomarker in a lung cancer xenograft model treated with metformin and radiotherapy. J Nucl Med 60(1):34–40. https://doi.org/10.2967/jnumed.118.212225
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7(1):11–20. https://doi.org/10.1016/j.cmet.2007.10.002
Dengler VL, Galbraith M, Espinosa JM (2014) Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol 49(1):1–15. https://doi.org/10.3109/10409238.2013.838205
Dewhirst MW (2009) Relationships between cycling hypoxia, HIF-1, angiogenesis and oxidative stress. Radiat Res 172(6):653–665. https://doi.org/10.1667/RR1926.1
Dewhirst MW, Cao Y, Moeller B (2008) Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 8(6):425–437. https://doi.org/10.1038/nrc2397
Dische S (1985) Chemical sensitizers for hypoxic cells: a decade of experience in clinical radiotherapy. Radiother Oncol 3(2):97–115
Dische S, Saunders M, Barrett A, Harvey A, Gibson D, Parmar M (1997) A randomised multicentre trial of CHART versus conventional radiotherapy in head and neck cancer. Radiother Oncol 44(2):123–136
DiSilvestro PA, Ali S, Craighead PS, Lucci JA, Lee Y-C, Cohn DE, Spirtos NM, Tewari KS, Muller C, Gajewski WH, Steinhoff MM, Monk BJ (2014) Phase III randomized trial of weekly cisplatin and irradiation versus cisplatin and tirapazamine and irradiation in stages IB2, IIA, IIB, IIIB, and IVA cervical carcinoma limited to the pelvis: a Gynecologic Oncology Group study. J Clin Oncol 32(5):458–464. https://doi.org/10.1200/JCO.2013.51.4265
Doktorova H, Hrabeta J, Khalil MA, Eckschlager T (2015) Hypoxia-induced chemoresistance in cancer cells: the role of not only HIF-1. Biomed Papers 159(2):166–177. https://doi.org/10.5507/bp.2015.025
Dowling RJO, Zakikhani M, Fantus IG, Pollak M, Sonenberg N (2007) Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res 67(22):10804–10812. https://doi.org/10.1158/0008-5472.CAN-07-2310
Dubois L, Landuyt W, Haustermans K, Dupont P, Bormans G, Vermaelen P, Flamen P, Verbeken E, Mortelmans L (2004) Evaluation of hypoxia in an experimental rat tumour model by [(18)F]fluoromisonidazole PET and immunohistochemistry. Br J Cancer 91(11):1947–1954. https://doi.org/10.1038/sj.bjc.6602219
Dunst J, Stadler P, Becker A, Kuhnt T, Lautenschläger C, Molls M, Haensgen G (2001) Tumor hypoxia and systemic levels of vascular endothelial growth factor (VEGF) in head and neck cancers. Strahlenther Onkol 177(9):469–473
Dunwoodie SL (2009) The role of hypoxia in development of the mammalian embryo. Dev Cell 17(6):755–773. https://doi.org/10.1016/j.devcel.2009.11.008
Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM (1999) Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol 237:97–132
Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64(11):3892–3899. https://doi.org/10.1158/0008-5472.CAN-03-2904
Eschmann S-M, Paulsen F, Reimold M, Dittmann H, Welz S, Reischl G, Machulla H-J, Bares R (2005) Prognostic impact of hypoxia imaging with 18F-misonidazole PET in non-small cell lung cancer and head and neck cancer before radiotherapy. J Nucl Med 46(2):253–260
Falk SJ, Ward R, Bleehen NM (1992) The influence of carbogen breathing on tumour tissue oxygenation in man evaluated by computerised p02 histography. Br J Cancer 66(5):919–924
Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, Martinez-Outschoorn UE, Sotgia F, Lisanti MP (2016) Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 7(23):34084–34099. https://doi.org/10.18632/oncotarget.9122
Fleming IN, Manavaki R, Blower PJ, West C, Williams KJ, Harris AL, Domarkas J, Lord S, Baldry C, Gilbert FJ (2015) Imaging tumour hypoxia with positron emission tomography. Br J Cancer 112(2):238–250. https://doi.org/10.1038/bjc.2014.610
Fokas E, Im JH, Hill S, Yameen S, Stratford M, Beech J, Hackl W, Maira S-M, Bernhard EJ, McKenna WG, Muschel RJ (2012) Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res 72(1):239–248. https://doi.org/10.1158/0008-5472.CAN-11-2263
Fry M, Pudney M (1992) Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem Pharmacol 43(7):1545–1553
Fukuda R, Zhang H, Kim J-w, Shimoda L, Dang CV, Semenza GL (2007) HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129(1):111–122. https://doi.org/10.1016/j.cell.2007.01.047
Gao X, Liu X, Shan W, Liu Q, Wang C, Zheng J, Yao H, Tang R, Zheng J (2018) Anti-malarial atovaquone exhibits anti-tumor effects by inducing DNA damage in hepatocellular carcinoma. Am J Cancer Res 8(9):1697–1711
Gatenby RA, Kessler HB, Rosenblum JS, Coia LR, Moldofsky PJ, Hartz WH, Broder GJ (1988) Oxygen distribution in squamous cell carcinoma metastases and its relationship to outcome of radiation therapy. Int J Radiat Oncol Biol Phys 14(5):831–838. https://doi.org/10.1016/0360-3016(88)90002-8
Gaude E, Frezza C (2016) Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun 7:13041. https://doi.org/10.1038/ncomms13041
Giatromanolaki A, Koukourakis MI, Sivridis E, Pastorek J, Wykoff CC, Gatter KC, Harris AL (2001) Expression of hypoxia-inducible carbonic anhydrase-9 relates to angiogenic pathways and independently to poor outcome in non-small cell lung cancer. Cancer Res 61(21):7992–7998
Gillies RM, Robinson SP, McPhail LD, Carter ND, Murray JF (2011) Immunohistochemical assessment of intrinsic and extrinsic markers of hypoxia in reproductive tissue: differential expression of HIF1α and HIF2α in rat oviduct and endometrium. J Mol Hist 42(4):341–354. https://doi.org/10.1007/s10735-011-9338-2
Ginouvès A, Ilc K, Macías N, Pouysségur J, Berra E (2008) PHDs overactivation during chronic hypoxia "desensitizes" HIFalpha and protects cells from necrosis. Proc Natl Acad Sci U S A 105(12):4745–4750. https://doi.org/10.1073/pnas.0705680105
Goldmann E (1908) The growth of malignant disease in man and the lower animals, with special reference to the vascular system. Proc R Soc Med 1(Surg Sect):1–13
Gray LH, Conger AD, Ebert M, Hornsey S, Scott OCA (1953) The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol 26(312):638–648. https://doi.org/10.1259/0007-1285-26-312-638
Grimes DR, Kelly C, Bloch K, Partridge M (2014) A method for estimating the oxygen consumption rate in multicellular tumour spheroids. J R Soc Interface 11(92):20131124. https://doi.org/10.1098/rsif.2013.1124
Grkovski M, Schwartz J, Rimner A, Schöder H, Carlin SD, Zanzonico PB, Humm JL, Nehmeh SA (2016) Reproducibility of 18F-fluoromisonidazole intratumour distribution in non-small cell lung cancer. EJNMMI Res 6. https://doi.org/10.1186/s13550-016-0210-y
Hall E, Giaccia A (2011) Radiobiology for the radiologist, 7th edn. Lippincottt Williams & Wilkins, Philadelphia, PA
Hammond EM, Asselin MC, Forster D, O'Connor JP, Senra JM, Williams KJ (2014) The meaning, measurement and modification of hypoxia in the laboratory and the clinic. Clin Oncol (R Coll Radiol) 26(5):277–288. https://doi.org/10.1016/j.clon.2014.02.002
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
Harris BHL, Barberis A, West CML, Buffa FM (2015) Gene expression signatures as biomarkers of tumour hypoxia. Clin Oncol (R Coll Radiol) 27(10):547–560. https://doi.org/10.1016/j.clon.2015.07.004
Harrison LB, Chadha M, Hill RJ, Hu K, Shasha D (2002) Impact of tumor hypoxia and anemia on radiation therapy outcomes. Oncologist 7(6):492–508
Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM (2000) Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol 156(4):1363–1380
Heiden MGV, DeBerardinis RJ (2017) Understanding the intersections between metabolism and cancer biology. Cell 168(4):657–669. https://doi.org/10.1016/j.cell.2016.12.039
Held KD, Harrop HA, Michael BD (1984) Effects of oxygen and sulphydryl-containing compounds on irradiated transforming DNA. II. Glutathione, cysteine and cysteamine. Int J Radiat Biol Relat Stud Phys Chem Med 45(6):615–626
Henke M, Laszig R, Rübe C, Schäfer U, Haase K-D, Schilcher B, Mose S, Beer KT, Burger U, Dougherty C, Frommhold H (2003) Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 362(9392):1255–1260. https://doi.org/10.1016/S0140-6736(03)14567-9
Henke M, Mattern D, Pepe M, Bézay C, Weissenberger C, Werner M, Pajonk F (2006) Do erythropoietin receptors on cancer cells explain unexpected clinical findings? J Clin Oncol 24(29):4708–4713. https://doi.org/10.1200/JCO.2006.06.2737
Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J (2012) Imaging hypoxia to improve radiotherapy outcome. Nat Rev Clin Oncol 9(12):674–687. https://doi.org/10.1038/nrclinonc.2012.171
Hoskin P, Rojas A, Saunders M (2009) Accelerated radiotherapy, carbogen, and nicotinamide (ARCON) in the treatment of advanced bladder cancer: mature results of a phase II nonrandomized study. Int J Radiat Oncol Biol Phys 73(5):1425–1431. https://doi.org/10.1016/j.ijrobp.2008.06.1950
Hoskin PJ, Rojas AM, Bentzen SM, Saunders MI (2010) Radiotherapy with concurrent carbogen and nicotinamide in bladder carcinoma. J Clin Oncol. https://doi.org/10.1200/JCO.2010.28.4950
Hudson AT, Dickins M, Ginger CD, Gutteridge WE, Holdich T, Hutchinson DB, Pudney M, Randall AW, Latter VS (1991) 566C80: a potent broad spectrum anti-infective agent with activity against malaria and opportunistic infections in AIDS patients. Drugs Exp Clin Res 17(9):427–435
Hughes VS, Wiggins JM, Siemann DW (2018) Tumor oxygenation and cancer therapy—then and now. Br J Radiol 92(1093):20170955. https://doi.org/10.1259/bjr.20170955
Hunter RW, Hughey CC, Lantier L, Sundelin EI, Peggie M, Zeqiraj E, Sicheri F, Jessen N, Wasserman DH, Sakamoto K (2018) Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat Med 24(9):1395. https://doi.org/10.1038/s41591-018-0159-7
Hutchinson F (1961) Sulfhydryl groups and the oxygen effect on irradiated dilute solutions of enzymes and nucleic acids. Radiat Res 14:721–731
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292(5516):468–472. https://doi.org/10.1126/science.1059796
Jain RK (2014) Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26(5):605–622. https://doi.org/10.1016/j.ccell.2014.10.006
Janssens GO, Rademakers SE, Terhaard CH, Doornaert PA, Bijl HP, van den Ende P, Chin A, Marres HA, de Bree R, van der Kogel AJ, Hoogsteen IJ, Bussink J, Span PN, Kaanders JH (2012) Accelerated radiotherapy with carbogen and nicotinamide for laryngeal cancer: results of a phase III randomized trial. J Clin Oncol 30(15):1777–1783. https://doi.org/10.1200/JCO.2011.35.9315
Jiang M, Li X, Quan X, Li X, Zhou B (2018, 2018) Clinically correlated MicroRNAs in the diagnosis of non-small cell lung cancer: a systematic review and meta-analysis. Biomed Res Int. https://doi.org/10.1155/2018/5930951
Jones RG, Thompson CB (2009) Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 23(5):537–548. https://doi.org/10.1101/gad.1756509
Kaanders JHAM, Pop LAM, Marres HAM, Bruaset I, van den Hoogen FJA, Merkx MAW, van der Kogel AJ (2002) ARCON: experience in 215 patients with advanced head-and-neck cancer. Int J Radiat Oncol Biol Phys 52(3):769–778
Kaluz S, Kaluzová M, Liao S-Y, Lerman M, Stanbridge EJ (2009) Transcriptional control of the tumor- and hypoxia-marker carbonic anhydrase 9: a one transcription factor (HIF-1) show? Biochim Biophys Acta 1795(2):162–172. https://doi.org/10.1016/j.bbcan.2009.01.001
Kedderis GL, Miwa GT (1988) The metabolic activation of nitroheterocyclic therapeutic agents. Drug Metab Rev 19(1):33–62. https://doi.org/10.3109/03602538809049618
Kim J-w, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3(3):177–185. https://doi.org/10.1016/j.cmet.2006.02.002
Kirkpatrick DL, Powis G (2017) Clinically evaluated cancer drugs inhibiting redox signaling. Antioxid Redox Signal 26(6):262–273. https://doi.org/10.1089/ars.2016.6633
Kizaka-Kondoh S, Konse-Nagasawa H (2009) Significance of nitroimidazole compounds and hypoxia-inducible factor-1 for imaging tumor hypoxia. Cancer Sci 100(8):1366–1373. https://doi.org/10.1111/j.1349-7006.2009.01195.x
Koh WJ, Rasey JS, Evans ML, Grierson JR, Lewellen TK, Graham MM, Krohn KA, Griffin TW (1992) Imaging of hypoxia in human tumors with [F-18]fluoromisonidazole. Int J Radiat Oncol Biol Phys 22(1):199–212
Koivunen P, Hirsilä M, Günzler V, Kivirikko KI, Myllyharju J (2004) Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem 279(11):9899–9904. https://doi.org/10.1074/jbc.M312254200
Kolstad P (1968) Intercapillary distance, oxygen tension and local recurrence in cervix cancer. Scand J Clin Lab Invest Suppl 106:145–157
Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 11(5):325–337. https://doi.org/10.1038/nrc3038
Koritzinsky M (2015) Metformin: a novel biological modifier of tumor response to radiation therapy. Int J Radiat Oncol Biol Phys 93(2):454–464. https://doi.org/10.1016/j.ijrobp.2015.06.003
Koukourakis MI, Bentzen SM, Giatromanolaki A, Wilson GD, Daley FM, Saunders MI, Dische S, Sivridis E, Harris AL (2006) Endogenous markers of two separate hypoxia response pathways (hypoxia inducible factor 2 alpha and carbonic anhydrase 9) are associated with radiotherapy failure in head and neck cancer patients recruited in the CHART randomized trial. J Clin Oncol 24(5):727–735. https://doi.org/10.1200/JCO.2005.02.7474
Krol J, Loedige I, Filipowicz W (2010) The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11(9):597–610. https://doi.org/10.1038/nrg2843
Kühlbrandt W (2015) Structure and function of mitochondrial membrane protein complexes. BMC Biol 13(1):89. https://doi.org/10.1186/s12915-015-0201-x
LaGory EL, Giaccia AJ (2016) The ever expanding role of HIF in tumour and stromal biology. Nat Cell Biol 18(4):356–365. https://doi.org/10.1038/ncb3330
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK (2002) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16(12):1466–1471. https://doi.org/10.1101/gad.991402
Larman TC, DePalma SR, Hadjipanayis AG, Cancer Genome Atlas Research N, Protopopov A, Zhang J, Gabriel SB, Chin L, Seidman CE, Kucherlapati R, Seidman JG (2012) Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci U S A 109(35):14087–14091. https://doi.org/10.1073/pnas.1211502109
LaRocque RC, Rao SR, Lee J, Ansdell V, Yates JA, Schwartz BS, Knouse M, Cahill J, Hagmann S, Vinetz J, Connor BA, Goad JA, Oladele A, Alvarez S, Stauffer W, Walker P, Kozarsky P, Franco-Paredes C, Dismukes R, Rosen J, Hynes NA, Jacquerioz F, McLellan S, Hale D, Sofarelli T, Schoenfeld D, Marano N, Brunette G, Jentes ES, Yanni E, Sotir MJ, Ryan ET, Global TravEpiNet C (2012) Global TravEpiNet: a national consortium of clinics providing care to international travelers—analysis of demographic characteristics, travel destinations, and pretravel healthcare of high-risk US international travelers, 2009–2011. Clin Infect Dis 54(4):455–462. https://doi.org/10.1093/cid/cir839
Le Q-T, Chen E, Salim A, Cao H, Kong CS, Whyte R, Donington J, Cannon W, Wakelee H, Tibshirani R, Mitchell JD, Richardson D, O'Byrne KJ, Koong AC, Giaccia AJ (2006) An evaluation of tumor oxygenation and gene expression in patients with early stage non–small cell lung cancers. Clin Cancer Res 12(5):1507–1514. https://doi.org/10.1158/1078-0432.CCR-05-2049
Le Q-T, Sutphin PD, Raychaudhuri S, Yu SCT, Terris DJ, Lin HS, Lum B, Pinto HA, Koong AC, Giaccia AJ (2003) Identification of osteopontin as a prognostic plasma marker for head and neck squamous cell carcinomas. Clin Cancer Res 9(1):59–67
Lee CT, Boss MK, Dewhirst MW (2014) Imaging tumor hypoxia to advance radiation oncology. Antioxid Redox Signal 21(2):313–337. https://doi.org/10.1089/ars.2013.5759
Lee WR, Berkey B, Marcial V, Fu KK, Cooper JS, Vikram B, Coia LR, Rotman M, Ortiz H (1998) Anemia is associated with decreased survival and increased locoregional failure in patients with locally advanced head and neck carcinoma: a secondary analysis of RTOG 85-27. Int J Radiat Oncol Biol Phys 42(5):1069–1075
Liu W, Shen S-M, Zhao X-Y, Chen G-Q (2012) Targeted genes and interacting proteins of hypoxia inducible factor-1. Int J Biochem Mol Biol 3(2):165–178
Lord EM, Harwell L, Koch CJ (1993) Detection of hypoxic cells by monoclonal antibody recognizing 2-nitroimidazole adducts. Cancer Res 53(23):5721–5726
Lord SR, Cheng W-C, Liu D, Gaude E, Haider S, Metcalf T, Patel N, Teoh EJ, Gleeson F, Bradley K, Wigfield S, Zois C, McGowan DR, Ah-See M-L, Thompson AM, Sharma A, Bidaut L, Pollak M, Roy PG, Karpe F, James T, English R, Adams RF, Campo L, Ayers L, Snell C, Roxanis I, Frezza C, Fenwick JD, Buffa FM, Harris AL (2018) Integrated pharmacodynamic analysis identifies two metabolic adaption pathways to metformin in breast cancer. Cell Metab 28(5):679–688.e674. https://doi.org/10.1016/j.cmet.2018.08.021
Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27(1):441–464. https://doi.org/10.1146/annurev-cellbio-092910-154237
Madhavan D, Peng C, Wallwiener M, Zucknick M, Nees J, Schott S, Rudolph A, Riethdorf S, Trumpp A, Pantel K, Sohn C, Chang-Claude J, Schneeweiss A, Burwinkel B (2016) Circulating miRNAs with prognostic value in metastatic breast cancer and for early detection of metastasis. Carcinogenesis 37(5):461–470. https://doi.org/10.1093/carcin/bgw008
Mäntylä MJ, Nordman EM, Ruotsalainen PJ, Kylmämaa TT (1982) Misonidazole and radiotherapy in lung cancer: a randomized double-blind trial. Int J Radiat Oncol Biol Phys 8(10):1719–1720. https://doi.org/10.1016/0360-3016(82)90292-9
Martin GV, Caldwell JH, Graham MM, Grierson JR, Kroll K, Cowan MJ, Lewellen TK, Rasey JS, Casciari JJ, Krohn KA (1992) Noninvasive detection of hypoxic myocardium using fluorine-18-fluoromisonidazole and positron emission tomography. J Nucl Med 33(12):2202–2208
Mather MW, Darrouzet E, Valkova-Valchanova M, Cooley JW, McIntosh MT, Daldal F, Vaidya AB (2005) Uncovering the molecular mode of action of the antimalarial drug atovaquone using a bacterial system. J Biol Chem 280(29):27458–27465. https://doi.org/10.1074/jbc.M502319200
McGowan DR, Skwarski M, Bradley KM, Campo L, Fenwick JD, Gleeson FV, Green M, Horne A, Maughan TS, McCole MG, Mohammed S, Muschel RJ, Ng SM, Panakis N, Prevo R, Strauss VY, Stuart R, Tacconi EMC, Vallis KA, McKenna WG, Macpherson RE, Higgins GS (2019) Buparlisib with thoracic radiotherapy and its effect on tumour hypoxia: a phase I study in patients with advanced non-small cell lung carcinoma. Eur J Cancer 113:87–95. https://doi.org/10.1016/j.ejca.2019.03.015
McNeill LA, Hewitson KS, Claridge TD, Seibel JF, Horsfall LE, Schofield CJ (2002) Hypoxia-inducible factor asparaginyl hydroxylase (FIH-1) catalyses hydroxylation at the beta-carbon of asparagine-803. Biochem J 367(Pt 3):571–575. https://doi.org/10.1042/BJ20021162
Melgaard B, Køhler O, Sand Hansen H, Overgaard J, Munck-Hansen J, Paulson OB (1988) Misonidazole neuropathy. A prospective study. J Neuro-Oncol 6(3):227–230
Metwally MAH, Frederiksen KD, Overgaard J (2014) Compliance and toxicity of the hypoxic radiosensitizer nimorazole in the treatment of patients with head and neck squamous cell carcinoma (HNSCC). Acta Oncol 53(5):654–661. https://doi.org/10.3109/0284186X.2013.864050
Michiels C, Tellier C, Feron O (2016) Cycling hypoxia: a key feature of the tumor microenvironment. Biochimica et Biophysica Acta (BBA)—Reviews on Cancer 1866(1):76–86. https://doi.org/10.1016/j.bbcan.2016.06.004
Milosevic M, Warde P, Menard C, Chung P, Toi A, Ishkanian A, McLean M, Pintilie M, Sykes J, Gospodarowicz M, Catton C, Hill RP, Bristow R (2012) Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clin Cancer Res 18(7):2108–2114. https://doi.org/10.1158/1078-0432.CCR-11-2711
Mistry IN, Thomas M, Calder EDD, Conway SJ, Hammond EM (2017) Clinical advances of hypoxia-activated prodrugs in combination with radiation therapy. Int J Radiat Oncol Biol Phys 98(5):1183–1196. https://doi.org/10.1016/j.ijrobp.2017.03.024
Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD (2017) Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem 292(17):7189–7207. https://doi.org/10.1074/jbc.M116.774471
Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E (2007) Energy metabolism in tumor cells. FEBS J 274(6):1393–1418. https://doi.org/10.1111/j.1742-4658.2007.05686.x
Moreno-Sánchez R, Rodríguez-Enríquez S, Saavedra E, Marín-Hernández A, Gallardo-Pérez JC (2009) The bioenergetics of cancer: is glycolysis the main ATP supplier in all tumor cells? Biofactors 35(2):209–225. https://doi.org/10.1002/biof.31
Movsas B, Chapman JD, Greenberg RE, Hanlon AL, Horwitz EM, Pinover WH, Stobbe C, Hanks GE (2000) Increasing levels of hypoxia in prostate carcinoma correlate significantly with increasing clinical stage and patient age: an Eppendorf pO(2) study. Cancer 89(9):2018–2024
Movsas B, Chapman JD, Hanlon AL, Horwitz EM, Greenberg RE, Stobbe C, Hanks GE, Pollack A (2002) Hypoxic prostate/muscle pO2 ratio predicts for biochemical failure in patients with prostate cancer: preliminary findings. Urology 60(4):634–639
Müller M, Padberg W, Schindler E, Sticher J, Osmer C, Friemann S, Hempelmann G (1998) Renocortical tissue oxygen pressure measurements in patients undergoing living donor kidney transplantation. Anesth Analg 87(2):474–476. https://doi.org/10.1097/00000539-199808000-00045
Nehmeh SA, Lee NY, Schröder H, Squire O, Zanzonico PB, Erdi YE, Greco C, Mageras G, Pham HS, Larson SM, Ling CC, Humm JL (2008) Reproducibility of intratumor distribution of 18F-fluoromisonidazole in head and neck cancer. Int J Radiat Oncol Biol Phys 70(1):235–242. https://doi.org/10.1016/j.ijrobp.2007.08.036
Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, Becker A, Adam M, Molls M, Dunst J, Terris DJ, Overgaard J (2005) Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol 77(1):18–24. https://doi.org/10.1016/j.radonc.2005.06.038
Nordsmark M, Eriksen JG, Gebski V, Alsner J, Horsman MR, Overgaard J (2007) Differential risk assessments from five hypoxia specific assays: the basis for biologically adapted individualized radiotherapy in advanced head and neck cancer patients. Radiother Oncol 83(3):389–397. https://doi.org/10.1016/j.radonc.2007.04.021
O'Connor JPB, Robinson SP, Waterton JC (2019) Imaging tumour hypoxia with oxygen-enhanced MRI and BOLD MRI. Br J Radiol 92(1095):20180642. https://doi.org/10.1259/bjr.20180642
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG (2000) Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2(7):423–427. https://doi.org/10.1038/35017054
Okamoto S, Shiga T, Yasuda K, Ito YM, Magota K, Kasai K, Kuge Y, Shirato H, Tamaki N (2013) High reproducibility of tumor hypoxia evaluated by 18F-fluoromisonidazole PET for head and neck cancer. J Nucl Med 54(2):201–207. https://doi.org/10.2967/jnumed.112.109330
Olive PL, Aquino-Parsons C, MacPhail SH, Liao SY, Raleigh JA, Lerman MI, Stanbridge EJ (2001) Carbonic anhydrase 9 as an endogenous marker for hypoxic cells in cervical cancer. Cancer Res 61(24):8924–8929
Orang AV, Petersen J, McKinnon RA, Michael MZ (2019) Micromanaging aerobic respiration and glycolysis in cancer cells. Mol Metab 23:98–126. https://doi.org/10.1016/j.molmet.2019.01.014
Ostheimer C, Bache M, Güttler A, Kotzsch M, Vordermark D (2014) A pilot study on potential plasma hypoxia markers in the radiotherapy of non-small cell lung cancer. Strahlenther Onkol 190(3):276. https://doi.org/10.1007/s00066-013-0484-1
Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV (2000) Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 275(29):21797–21800. https://doi.org/10.1074/jbc.C000023200
Overgaard J (2011) Hypoxic modification of radiotherapy in squamous cell carcinoma of the head and neck—a systematic review and meta-analysis. Radiother Oncol 100(1):22–32. https://doi.org/10.1016/j.radonc.2011.03.004
Overgaard J, Bentzen SM, Kolstad P, Kjoerstad K, Davy M, Bertelsen K, Mäntyla M, Frankendal B, Skryten A, Löftquist I (1989a) Misonidazole combined with radiotherapy in the treatment of carcinoma of the uterine cervix. Int J Radiat Oncol Biol Phys 16(4):1069–1072
Overgaard J, Eriksen JG, Nordsmark M, Alsner J, Horsman MR, Danish H, Neck Cancer Study G (2005) Plasma osteopontin, hypoxia, and response to the hypoxia sensitiser nimorazole in radiotherapy of head and neck cancer: results from the DAHANCA 5 randomised double-blind placebo-controlled trial. Lancet Oncol 6(10):757–764. https://doi.org/10.1016/S1470-2045(05)70292-8
Overgaard J, Hansen HS, Andersen AP, Hjelm-Hansen M, Jørgensen K, Sandberg E, Berthelsen A, Hammer R, Pedersen M (1989b) Misonidazole combined with split-course radiotherapy in the treatment of invasive carcinoma of larynx and pharynx: report from the DAHANCA 2 study. Int J Radiat Oncol Biol Phys 16(4):1065–1068
Overgaard J, Hansen HS, Overgaard M, Bastholt L, Berthelsen A, Specht L, Lindeløv B, Jørgensen K (1998) A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) protocol 5-85. Radiother Oncol 46(2):135–146
Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348(Pt 3):607–614
Painter HJ, Morrisey JM, Mather MW, Vaidya AB (2007) Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446(7131):88–91. https://doi.org/10.1038/nature05572
Palcic B, Brosing JW, Skarsgard LD (1982) Survival measurements at low doses: oxygen enhancement ratio. Br J Cancer 46(6):980–984
Papavasiliou C, Yiogarakis D, Davillas N, Seretakis L, Pappas J, Licourinas M, Theodorou C, Stathopoulos P, Katsoyianni C, Thanos A (1983) Treatment of bladder carcinoma with irradiation combined with misonidazole. Int J Radiat Oncol Biol Phys 9(11):1631–1633
Pastorek J, Pastoreková S, Callebaut I, Mornon JP, Zelník V, Opavský R, Zat'ovicová M, Liao S, Portetelle D, Stanbridge EJ (1994) Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 9(10):2877–2888
Pernicova I, Korbonits M (2014) Metformin—mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol 10(3):143–156. https://doi.org/10.1038/nrendo.2013.256
Petrik D, Lavori PW, Cao H, Zhu Y, Wong P, Christofferson E, Kaplan MJ, Pinto HA, Sutphin P, Koong AC, Giaccia AJ, Le Q-T (2006) Plasma osteopontin is an independent prognostic marker for head and neck cancers. J Clin Oncol 24(33):5291–5297. https://doi.org/10.1200/JCO.2006.06.8627
Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I (2018) The hypoxic tumour microenvironment. Oncogenesis 7(1). https://doi.org/10.1038/s41389-017-0011-9
Pittman RN (2011) Regulation of tissue oxygenation. Integrated systems physiology: from molecule to function to disease. Morgan & Claypool Life Sciences, San Rafael, CA
Prekeges JL, Rasey JS, Grunbaum Z, Krohn KH (1991) Reduction of fluoromisonidazole, a new imaging agent for hypoxia. Biochem Pharmacol 42(12):2387–2395
Qayum N, Muschel RJ, Im JH, Balathasan L, Koch CJ, Patel S, McKenna WG, Bernhard EJ (2009) Tumor vascular changes mediated by inhibition of oncogenic signaling. Cancer Res 69(15):6347–6354. https://doi.org/10.1158/0008-5472.CAN-09-0657
Rajendran JG, Krohn KA (2005) Imaging hypoxia and angiogenesis in tumors. Radiol Clin N Am 43(1):169–187
Rajendran JG, Krohn KA (2015) F18 Fluoromisonidazole for imaging tumor hypoxia: imaging the microenvironment for personalized cancer therapy. Semin Nucl Med 45(2):151–162. https://doi.org/10.1053/j.semnuclmed.2014.10.006
Raleigh JA, Chou SC, Arteel GE, Horsman MR (1999) Comparisons among pimonidazole binding, oxygen electrode measurements, and radiation response in C3H mouse tumors. Radiat Res 151(5):580–589
Rankin EB, Nam J-M, Giaccia AJ (2016) Hypoxia: signaling in the metastatic cascade. Trends Cancer 2(6):295–304. https://doi.org/10.1016/j.trecan.2016.05.006
Reznik E, Miller ML, Şenbabaoğlu Y, Riaz N, Sarungbam J, Tickoo SK, Al-Ahmadie HA, Lee W, Seshan VE, Hakimi AA, Sander C (2016) Mitochondrial DNA copy number variation across human cancers. elife 5. https://doi.org/10.7554/eLife.10769
Rischin D, Hicks RJ, Fisher R, Binns D, Corry J, Porceddu S, Peters LJ (2006) Prognostic significance of [18F]-Misonidazole positron emission tomography–detected tumor hypoxia in patients with advanced head and neck cancer randomly assigned to chemoradiation with or without tirapazamine: a substudy of Trans-Tasman Radiation Oncology Group Study 98.02. J Clin Oncol 24(13):2098–2104. https://doi.org/10.1200/JCO.2005.05.2878
Rischin D, Peters LJ, O'Sullivan B, Giralt J, Fisher R, Yuen K, Trotti A, Bernier J, Bourhis J, Ringash J, Henke M, Kenny L (2010) Tirapazamine, cisplatin, and radiation versus cisplatin and radiation for advanced squamous cell carcinoma of the head and neck (TROG 02.02, HeadSTART): a phase III trial of the Trans-Tasman Radiation Oncology Group. J Clin Oncol 28(18):2989–2995. https://doi.org/10.1200/JCO.2009.27.4449
Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM (2017) Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res 5(1):9–16. https://doi.org/10.1158/2326-6066.CIR-16-0103
Secomb TW, Hsu R, Ong ET, Gross JF, Dewhirst MW (1995) Analysis of the effects of oxygen supply and demand on hypoxic fraction in tumors. Acta Oncol 34(3):313–316
Semenza GL (2010) HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20(1):51–56. https://doi.org/10.1016/j.gde.2009.10.009
Semenza GL (2012) Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci 33(4):207–214. https://doi.org/10.1016/j.tips.2012.01.005
Sobhanifar S, Aquino-Parsons C, Stanbridge EJ, Olive P (2005) Reduced expression of hypoxia-inducible factor-1α in perinecrotic regions of solid Tumors. Cancer Res 65(16):7259–7266. https://doi.org/10.1158/0008-5472.CAN-04-4480
Srivastava IK, Rottenberg H, Vaidya AB (1997) Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J Biol Chem 272(7):3961–3966. https://doi.org/10.1074/jbc.272.7.3961
Steel G (2002) Basic clinical radiobiology, 3rd edn. Oxford University Press, London
Sullivan LB, Chandel NS (2014) Mitochondrial reactive oxygen species and cancer. Cancer Metab 2. https://doi.org/10.1186/2049-3002-2-17
Świtlik WZ, Karbownik MS, Suwalski M, Kozak J, Szemraj J (2019) Serum miR-210-3p as a potential noninvasive biomarker of lung adenocarcinoma: a preliminary study. Genet Test Mol Biomarkers 23(5):353–358. https://doi.org/10.1089/gtmb.2018.0275
Szatrowski TP, Nathan CF (1991) Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res 51(3):794–798
Tan AS, Baty JW, Dong L-F, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, Pesdar EA, Sobol M, Filimonenko A, Stuart S, Vondrusova M, Kluckova K, Sachaphibulkij K, Rohlena J, Hozak P, Truksa J, Eccles D, Haupt LM, Griffiths LR, Neuzil J, Berridge MV (2015) Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 21(1):81–94. https://doi.org/10.1016/j.cmet.2014.12.003
Thomlinson RH, Gray LH (1955) The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer 9(4):539–549
Thomson D, Yang H, Baines H, Miles E, Bolton S, West C, Slevin N (2014) NIMRAD—a phase III trial to investigate the use of nimorazole hypoxia modification with intensity-modulated radiotherapy in head and neck cancer. Clin Oncol (R Coll Radiol) 26(6):344–347. https://doi.org/10.1016/j.clon.2014.03.003
Tsakiridis T, Hu C, Skinner HD, Santana-Davila R, Lu B, Erasmus JJ, Doemer A, Videtic GMM, Coster J, Yang X, Lee R, Werner-Wasik M, Schaner PE, McCormack SE, Esparaz B, McGarry R, Bazan JG, Struve T, Bradley JD (2019) Initial reporting of NRG-LU001 (NCT02186847), randomized phase II trial of concurrent chemoradiotherapy (CRT) +/− metformin in locally advanced non-small cell lung cancer (NSCLC). J Clin Oncol 37(15_suppl):8502–8502. https://doi.org/10.1200/JCO.2019.37.15_suppl.8502
Varlotto J, Stevenson MA (2005) Anemia, tumor hypoxemia, and the cancer patient. Int J Radiat Oncol Biol Phys 63(1):25–36. https://doi.org/10.1016/j.ijrobp.2005.04.049
Vaupel P, Höckel M, Mayer A (2007) Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal 9(8):1221–1236. https://doi.org/10.1089/ars.2007.1628
Vaupel P, Mayer A (2012) Availability, not respiratory capacity governs oxygen consumption of solid tumors. Int J Biochem Cell Biol 44(9):1477–1481. https://doi.org/10.1016/j.biocel.2012.05.019
Viale A, Corti D, Draetta GF (2015) Tumors and mitochondrial respiration: a neglected connection. Cancer Res 75(18):3687–3691. https://doi.org/10.1158/0008-5472.CAN-15-0491
Walsh JC, Lebedev A, Aten E, Madsen K, Marciano L, Kolb HC (2014) The clinical importance of assessing tumor hypoxia: relationship of tumor hypoxia to prognosis and therapeutic opportunities. Antioxid Redox Signal 21(10):1516–1554. https://doi.org/10.1089/ars.2013.5378
Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92(12):5510–5514
Wang W, Winlove CP, Michel CC (2003) Oxygen partial pressure in outer layers of skin of human finger nail folds. J Physiol 549(Pt 3):855–863. https://doi.org/10.1113/jphysiol.2002.037994
Wang Y, Yang J, Liu H, Bi J-R, Liu Y, Chen Y-Y, Cao J-Y, Lu Y-J (2015) The association between osteopontin and survival in non-small-cell lung cancer patients: a meta-analysis of 13 cohorts. Onco Targets Ther 8:3513–3521. https://doi.org/10.2147/OTT.S94082
Weinberg SE, Chandel NS (2015) Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 11(1):9–15. https://doi.org/10.1038/nchembio.1712
Wiesener MS, Jürgensen JS, Rosenberger C, Scholze CK, Hörstrup JH, Warnecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH, Eckardt K-U (2003) Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J 17(2):271–273. https://doi.org/10.1096/fj.02-0445fje
Winter SC, Buffa FM, Silva P, Miller C, Valentine HR, Turley H, Shah KA, Cox GJ, Corbridge RJ, Homer JJ, Musgrove B, Slevin N, Sloan P, Price P, West CML, Harris AL (2007) Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res 67(7):3441–3449. https://doi.org/10.1158/0008-5472.CAN-06-3322
Wright EA, Howard-Flanders P (1957) The influence of oxygen on the radiosensitivity of mammalian tissues. Acta Radiol 48(1):26–32
Wykoff CC, Beasley NJP, Watson PH, Turner KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL (2000) Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res 60(24):7075–7083
Yang W-H, Xu J, Mu J-B, Xie J (2017) Revision of the concept of anti-angiogenesis and its applications in tumor treatment. Chronic Dis Transl Med 3(1):33–40. https://doi.org/10.1016/j.cdtm.2017.01.002
Yu M (2011) Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci 89(3–4):65–71. https://doi.org/10.1016/j.lfs.2011.05.010
Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, Glicksman R, Chaib S, Zamiara P, Milosevic M, Wouters BG, Bristow RG, Koritzinsky M (2013) Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res 19(24):6741–6750. https://doi.org/10.1158/1078-0432.CCR-13-1787
Zeman EM, Brown JM, Lemmon MJ, Hirst VK, Lee WW (1986) SR-4233: a new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int J Radiat Oncol Biol Phys 12(7):1239–1242. https://doi.org/10.1016/0360-3016(86)90267-1
Zhao H, Chen Q, Alam A, Cui J, Suen KC, Soo AP, Eguchi S, Gu J, Ma D (2018) The role of osteopontin in the progression of solid organ tumour. Cell Death Dis 9(3). https://doi.org/10.1038/s41419-018-0391-6
Zhu Y, Denhardt DT, Cao H, Sutphin PD, Koong AC, Giaccia AJ, Le Q-T (2005) Hypoxia upregulates osteopontin expression in NIH-3T3 cells via a Ras-activated enhancer. Oncogene 24(43):6555–6563. https://doi.org/10.1038/sj.onc.1208800
Zu XL, Guppy M (2004) Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun 313(3):459–465. https://doi.org/10.1016/j.bbrc.2003.11.136
Acknowledgments
MS gratefully acknowledges the support of the Howat Foundation. JDW is grateful for support from the Royal College of Radiologists, and Oriel College, Oxford.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Skwarski, M., Bowler, E., Wilson, J.D., Higgins, G.S., Hammond, E.M. (2020). Targeting Tumor Hypoxia. In: Willers, H., Eke, I. (eds) Molecular Targeted Radiosensitizers. Cancer Drug Discovery and Development. Humana, Cham. https://doi.org/10.1007/978-3-030-49701-9_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-49701-9_11
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-49700-2
Online ISBN: 978-3-030-49701-9
eBook Packages: MedicineMedicine (R0)