
Abstract
Biodegradable polymers, especially bacterial synthesized polymers, have gained great attention and interest due to the concerns raised regarding the accumulation of petrochemical plastic wastes in the environment. It is known that biodegradable polymers have advantages such as being biocompatible, biodegradable, natural, renewable and have similar mechanical properties compared to conventional polymers. Poly(hydroxybutyrate) (PHBs) are biodegradable polyesters produced naturally by bacteria. PHBs are highly crystalline polyester, which are brittle and prone to thermal degradation during processing. In the past, attempts have been made to reduce the degree of the brittleness of the PHB by copolymerization with hydroxyhexanoate (HHx) or hydroxyvalerate (HVx) co-monomers. Among PHB copolymers: poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (PHB-co-HHx), poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) and poly(3-hydroxybutyrate-co-4-hydroxybutyrate) (P(3HB-co-4HB)) have improved the flexibility and mechanical properties more than other PHBs. This chapter aims to emphasize the potential on the compatibilization of biodegradable polymer blends with the presence of maleated compatibilizers derived from the host or guest polymers.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
3.1 Introduction
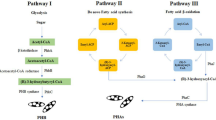
Biodegradable polymers, especially bacterial synthesized polymers, have gained significant attention in recent years, given the growing concerns regarding the accumulation of petrochemical plastics wastes in the environment (Zhang et al. 2012). It is well known that biodegradable polymers have advantages such as biocompatible, biodegradable, natural, renewable, and having comparable mechanical properties compared to conventional polymers. Among the biodegradable polymers, biodegradable polyesters show an important role as biodegradable plastics due to their potentially hydrolysable ester bonds (Environment Australia 2012). As shown in Fig. 3.1, the biodegradable polyester family is divided into two main groups: aromatic (aromatic rings) and aliphatic (linear) polyesters.

Biodegradable polyester family. Reproduced with permission from Environment Australia (2012)
The group of aliphatic thermoplastic polyester (ATP) is the most widely studied given its important diversity, synthetic versatility, variety of monomers and various routes ready for the polyester development (Vroman and Tighzert 2009). During the last decades, several types of ATP, such as poly(butylene succinate) (PBS), poly(lactic acid) (PLA), poly(hydroxybutyrate) (PHB) and its co-polyesters such as poly(hydroxybutyrate-co-hydroxyhexanoate) (PHB-co-HHx) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) have been studied in terms of single polymer, blends and their composite applications. However, some disadvantages of biodegradable polymers include brittleness and their susceptibility to thermal degradation during processing (Chen and Luo 2009), low toughness (T) and melt viscosity in PLA (Pradeep et al. 2017; Zhang et al. 2017), and low glass transition temperature (Tg = -62 °C) and melting temperature (Tm = 57 °C) in poly(ε-caprolactone) (PCL) (Abdul et al. 2013) have limited the processing and application of these polymers. The relatively high production cost of these polymers compared to conventional polymers is also an additional problem. However, to overcome these drawbacks, various strategies such as blending, compositing and copolymerization, are normally used. Among them, blending is the most preferred approach, since it is relatively easy to be carried out with lower costs compared to the copolymerization approach (Si et al. 2016). In addition, most commercially available plastic materials are polymer blends which have been used successfully for numerous applications.
The blending of biodegradable polymer with other biodegradable polymers is favored due to the biodegradability of the blends which could be maintained, while improving the properties of the host polymer. According to Si et al. (2016), a number of studies on the blending of PLA with flexible biodegradable polyesters, such as PBS, PCL, poly(butylene succinate-co-L-lactate) (PBSL), poly(butylenesuccinate-co-ethylene succinate) (PBES) and poly(ethylene succinate) (PES), have been published previously.
The key success of polymer blends is due to their good compatibility and miscibility between the components of the blend. The production of blends with an ideal physical-mechanical performance depends on the interfacial tension between the blends and the size of the dispersed phase droplets. Apart from that, a cohesive interfacial adhesion is also essential to allow the effective stress transfer from the continuous to the dispersed phase (Mani et al. 1999). This ideal condition could be achieved by adopting the compatibilization approach through the introduction of compatibilizer or by adding block or graft copolymers. The production of compatibilizer is relatively easier than the copolymerization process and also has a similar effect on improving the chemical and physical interactions, and phase dispersion between the blend’s components. The compatibilizers can be derived by reacting with the host or via guess polymers with suitable unsaturated polar molecule functional groups such as amines, anhydrides, epoxides, etc. (Pracella et al. 2010).
To the best of our knowledge, very limited compatibilizers commercially available specifically for biodegradable polymers have been produced. In previous studies, many attempts have been made to develop compatibilizer for biodegradable plastics, especially aliphatic thermoplastic polyesters such as glycidyl methacrylate (GMA)- or maleic anhydride (MA)-grafted compatibilizers (Tansiri and Potiyaraj 2015). According to Gardella et al. (2014), moieties such as acrylic acid (AAc), GMA, MA and oxazoline, are suitable to be grafted onto non-reactive polymers. MA is generally preferred, since it is relatively easy to handle, has low toxicity, and is not be likely to be homopolymerized under standard free-radical melt-grafting conditions. In addition, the compatibilizer is expected to have a good miscibility when the components are combined, thus promoting chemical interactions with another, which improves the interfacial adhesion between them (Gardella et al. 2014).
3.2 Maleated Compatibilizer of Biodegradable Polymers
In the last year, the development of biodegradable polymers as an alternative to petroleum-based polymers has motivated researchers to develop MA-based compatibilizer in order to make the polymer more developed for industrial processing and applications. Compatibility by grafting with MA on several conventional polymers such as ethylene vinyl acetate (EVA-g-MA), linear low-density polyethylene (LLDPE-g-MA), polypropylene (PP-g-MA) and styrene-ethylene-butylene-styrene block copolymer (SEBS-g-MA) have been well established, and mainly investigated by previous researchers (Papadopoulou and Kalfoglou 2000).
Normally, a maleated compatibilizer is an additive used to improve interfacial adhesion or interaction between two or more blends and/or composite components. In polymer blend applications, the maleated compatibilizers are added to produce blends with good overall physical-mechanical behavior and can regulate interfacial tension to generate a smaller dispersed phase size and stronger interfacial adhesion, thus allowing the effective stress transfer between the phases of the blends (Mani et al. 1999). The compatibility of the blends is essential since most polymers are commonly immiscible and have little interfacial adhesion. Furthermore, it is known that the miscibility between polymers is determined by a balance of enthalpic and entropic contributions to the free energy of mixing (Fink 2013).
Notwithstanding, the quantity and the effectiveness of the compatibilizer in combination with the ratio of the components has a significant effect on the mechanical, morphological and thermal properties of the blends. According to Markham (1990), other factors may also affect the final properties of the blends such as the cooling rate, the mixing time, the rate of cooling, the shear rate and the temperature during the molding process. The compatibilizers can may also preserve or/and stabilize the morphology of the blends such as agglomeration, delamination, ‘skinning’ and other unwanted phase effects resulting from the blending process (Markham 1990).
3.2.1 Preparation of Maleated Compatibilizer
As is already known, the miscibility between the components of a blend is determined by a balance of enthalpic and entropic of thermodynamic contributions to the free energy of mixing (Fink 2013). However, in reality, thermodynamically compatible blends are difficult to achieve, and in industrial practice, technological compatibility is more than adequate. In addition, technological compatibility can be achieved through chemical or mechanical approaches. As suggested by Fink (2013), technological compatibility of immiscible polymers blends can be achieved by incorporating a compatibilizer in advance or during the blending process.
3.2.1.1 MA Reactive Monomer
MA (furan-2,5-dione) is a well-known monomer for the development of compatibilizer and is commonly used for the modification of polyolefin (Mehrabzadeh 2009; Gao et al. 2012; Paolo et al. 2018). According to Musa (2016), the basic chemical structure of MA is fundamentally versatile where it has a five-member heterocyclic ring, comprising a double bond at the C3-C4 position and two carbonyl groups; one at the C2 position and the other at the C5 position, as shown in Fig. 3.2.

Structure of the MA unit
The C=C of MA is a powerful electron-accepting monomer due to the electron-deficient character of the double bond. The electronic deficiency originates from strong electron-withdrawing forces from the two C=O substituent groups (Musa 2016). Aside from that, the carboxylic acid groups are also known for their high reactivity coupled to two different acid dissociation constants (Musa 2016). The physical properties for MA chemical compounds are summarized in Table 3.1.
In addition, with respect to the economic point of view, MA is an attractive chemical compound, since it can be easily derived from butane gas and benzene that are readily available in world petroleum resources (Musa 2016).
3.2.1.2 Type of Grafting Reaction
Maleated compatibilizer can be produced by a grafting reaction of the polymer with MA functional groups in the presence of a peroxide radical initiator. As reported by Mani et al. (1999), many methods for producing compatibilizer through grafting reaction such as melt, solid-state, solution, solvents redox and suspension grafting in aqueous or organic solvents have been reported by previous studies. Table 3.2 summarizes the advantages, disadvantages and applications of free radical grafting technologies .
The solid-state grafting , also known as mechanochemical grafting, is where the polymer is generally used as a powder and mixed with MA with a high concentration of initiator in the presence of an interfacial wetting agent, e.g. solvent of the polymer. The reaction is carried out in a low-shear mixer or stainless-steel reactor at a temperature ranging from 100–150 °C. Since the grafting reaction does not imply high temperature, degradation of the host polymer is very minimal. However, the homogeneity of the grafted product depends on the solvents used in the particle size of polymer powder and/or other co-monomer that are required to increase the degree of grafting (DG) of the polymer (Qiu and Hirotsu 2005). Solvents or water can further purify the maleated compatibilizer. Nonetheless, this technique only introduces the grafting reaction on the surface of the polymer powders, and the MA could not react with the polymer chain inside the powder particles (Ahmad Thirmizir 2011).
According to Qiu and Hirotsu (2005), the solution grafting process involves the dissolution of polymer in a suitable solvent at an elevated temperature followed by the addition of MA together with a peroxide radical initiator at a predetermined reaction time. The reaction must be carried out in a homogeneous chemical environment to allow a better interaction and reactivity between the polymer and other components. The resulting graft copolymer is further purified via a selective dissolution approach to obtain the compatibilizer with a relatively higher purity. The process is relatively complex and expensive, and it is difficult to eliminate by-products. Aside from that, it is not practical for large-scale production due to the large quantity of solvents recycling involved (Qiu and Hirotsu 2005).
The most practical method to produce compatibilizer is via melt grafting, also known as reactive extrusion (REx) . The process involves grafting of MA onto polymer in the molten state with the presence of a free radical initiator. The functionalized polymer is further purified to remove unreacted MA and a radical initiator. In line with this, Ahmad Thirmizir (2011) obtained maleated PBS.
Normally, the effectiveness of the grafting process depends on the reactive components and the processing parameter applied. Previously, He et al. (2013) conducted a exhaustive study to investigate the effect of the concentration of reactive components and grafting parameters on the DG of maleated LDPE (LDPE-g-MA) via solution grafting in xylene solvent with the presence of dicumyl peroxide (DCP) radical initiator. Table 3.3 shows the studied parameters, and Fig. 3.3 summarizes the findings via an orthogonal experiment.

The changing trend of test index (GD). A: DCP concentration, B: MA concentration, C: reaction time, D: reaction temperature and E: total time for initiator dropping. Reproduced with permission from He et al. (2013)
Following He et al. (2013) the concentration of reactive components (DCP and MA) can affect the DG of the compatibilizer (Fig. 3.3). These authors reported that there is a positive trend between the DG and the concentration of MA and DCP up to an optimal value before dropping with an additional increase in concentration and trends proportional to the reaction temperature. While the reaction time and total time for initiator dropped, thus showing an increasing pattern up to a plateau point where any additional increase does not affect the DG value.
A similar study done by Ahmad Thirmizir et al. (2011) also confirmed that the grafting reaction of MA with biodegradable aliphatic polyester (PBS) is affected by the concentration of MA, and this also affected the DG of the compatibilizer. As shown in Table 3.4, Ahmad Thirmizir et al. (2011) also reported that a maleated PBS at a constant mixing parameter and radical initiator concentration (DCP), as well as an increase of the MA concentration in the compatibilizer significantly increased the DG. In addition, as mentioned by Mani et al. (1999), increasing the MA concentration leads to an increase in the DG due to the better probabilities that free MA will bind to the polymer macro radical sites during the grafting reaction.
Chen et al. (2003) investigated the solution grafting of maleated PHB in chlorobenzene at a temperature of 130 °C with the presence of benzoyl peroxide (BPO) radical initiator. These authors reported the relationship between MA monomer concentrations on the DG. As illustrated in Fig. 3.4, the DG increased with increase in the MA concentration up to a maximum of 0.85%, at an MA concentration of 3% (w/v). Beyond 3% (w/v), the DG then decreased and gradually leveled off. According to Chen et al. (2003), under the solution grafting condition, it is difficult for the MA to homopolymerize given its special molecular structure. However, MA could bind to the PHB backbone in the form of single succinic anhydride rings. The DG is mostly influenced by the number of macroradicals initiated by a radical initiator. Here, as the concentration of MA increases, the chances of PHB macroradicals reacting with MA are higher. When the MA is excessive, unwanted reactions, such as the effect of the cage, easily occur, thus reducing the DG value (Chen et al. 2003).

Grafting degree as a function of monomer concentration. Reaction conditions: BPO: 0.2% (w/v), chlorobenzene: 100 mL, PHB: 5 g, temperature: 130 °C and time: 4 h. Reproduced with permission from Chen et al. (2003)
Chen et al. (2003) also investigated the relationship between DG, MA concentration and Mw. These authors suggested from their results that the increase in the DG is not correlated with the Mw change, which shows that MA grafting is not produced at the ends of the polymer chains, but macro radical sites were formed along the polymer chain. However, in the melt grafting process conducted at relatively high temperature and high shearing rate, chain scission can occur. Meanwhile, our study on the production of maleated PBS and PHB-co-HHx via melt grafting at a temperature of 160 °C and a rotor speed of 50 rpm and a reaction of 5 min, showed that at a constant peroxide concentration, an increase in the MA concentration resulted in an increase in the DG and reduction of Mw (Fig. 3.5) (Own results from the authors). Mani et al. (1999) also reported the occurrence of chain scission accompanying the grafting event showed a reduction of intrinsic viscosity of maleated PBS and poly(butylene succinate-co-adipate) (PBSA) compared to pure polymers with an increasing MA concentration.

Effect of the MA concentration on the grafting degree and Mw of PBS and PHB-co-HHx (PHBHH) (Own results from the authors)
3.2.1.3 Organic Peroxide Radical Initiator
In the grafting process, the reaction begins with the formation of free radicals via homolytic scission of the organic peroxide initiator (Ahmad Thirmizir 2011). The initiator has the ability to dehydrogenation to extract hydrogen atoms from α-carbon atoms in relation to the ester carbonyl group of that particular polyester in order to form a polymeric macro-radical. While, simultaneously the polymeric macro-radical also undergoes a certain degree of degradation via β-scissoring to form an end of the radical chain and an end of the vinylidene chain. The ideal compatibilizer will also achieve an optimum DG and a minimal degree of degradation of the polymer chain (Ahmad Thirmizir et al. 2011). However, the grafting reaction is typically accompanied by a chain scission, which changes the rheological behavior of a polymer. Indeed, determining the optimum conditions of the grafting process is a very complex study. Actually, a great number of variables are involved, such as an additional sequence of the reagents, MA concentration, reaction temperature and time, rotor speed, type and concentration of peroxide, and type of stabilizers if added (Oromiehie et al. 2014).
In general, the grafting of MA onto the polymer chains is carried out via a reactive melt-blending technique in the presence of a peroxide initiator, such as BPO, DCP and t-butyl peroxide (Ahmad Thirmizir et al. 2011). Mani et al. (1999) in the study on the grafting of MA onto PLA via solution grafting comprehensively reported the effect of different types of radical initiators (2,2-azobis(isobutyronitrile) (AIBN), BPO, DCP and di-t-butyl peroxide (DBP)) on the DG and intrinsic viscosity. The condition of this grafting procedure included: toluene, DCP and MA concentration, 1 and 3% (w/w), respectively, temperature: 110 °C and a reaction time 4 hours. The results are shown in Table 3.5.
The maleated PLA obtained the highest DG with the addition of the BPO initiator due to the slow rate of initiator decomposition at that particular reaction temperature (Mani et al. 1999). As reported by Aldrich Chemical Company Inc. (2020), BPO has a 10-hour half-life temperature lower than DBP and DCP, and at 100 °C, the decomposition rate of BPO is considerably high, approx. kd = 5.0 × 10–4. Takamura et al. (2008) reported the lifetime of several peroxides at 190 °C, where DCP peroxide had good stability at that temperature compared to other peroxides. Aside from that, the maleated PLA produced via solution grafting exhibited minimal chain scission/degradation even with different species of radical initiator (AIBN, BPO, DBP and DCP) compared to the one produced by the melt grafting procedure (Mani et al. 1999; Takamura et al. 2008).
In addition, the radical initiator concentration also affects the DG of MA onto a polymer backbone. Phua et al. (2013) in their study on grafting of MA onto PBS via melt grafting procedure, reported that the grafting efficiency increased as the DCP initiator concentration increased from 1 to 1.5 phr due to increased formation of radicals through the initiator decomposition reaction. Moreover, when the concentration of radicals was high, the probability of chain transfer to the polymer backbone was also high, resulting in a greater DG of the compatibilizer. Mani et al. (1999) also studied the effect of initiator (BPO for PLA and DCP for PBS) concentration on MA grafting onto the backbone of the polymer. These authors suggested that melt grafting showed an increase in the DG g with respect to the initiator concentration, which appeared to be quite linear. However, excess of radical initiator can promote termination or a combination reaction between polymer macroradicals due to the available free radical species, thus resulting in a decrease in the DG (Chen et al. 2003). As reported by Chen et al. (2003), the intrinsic viscosity of the compatibilized polymers was reduced by about 30% for PBS, 25% for PBSA and 12% for PLA compared to the unmodified polymer due to the chain scission reaction. The excess radical initiator may cause the polymer to depolymerize, and may also cause an acute reduction of Mw (Chen et al. 2003). Other than that, the presence of peroxide radical initiator could be due to the unwanted crosslinking reaction between the polymer chains, especially for the grafting reaction that was conducted at a high concentration of the initiator.
3.2.1.4 Purification of Compatibilizer
Another critical issue for manufacturing maleated compatibilizer is the purity of the compatibilizer itself. According to Bettini and Agnelli (2000), the MA residue could not be removed by self-evaporation during melt grafting if the reaction was carried out at a temperature below the boiling point of the MA (≈ 202 °C). The purification procedure is essential for producing good quality compatibilizers without the traces of any unreacted residues (Bettini and Agnelli 2000). Ahmad Thirmizir et al. (2011) demonstrated that the removal of ungrafted MA and unreacted DCP residues via solvent extraction improves the fiber-matrix interfacial adhesion. The residuals of the ungrafted MA and the unreacted DCP initiator present in the compatibilizers might interrupt the fiber-matrix interfacial adhesion since it could not form a complete bridging between the composite components (Ahmad Thirmizir 2011).
The high concentration of unreacted MA in the blends causes color fading, degassing, reduces ultraviolet (UV) resistance, release of unpleasant odors and the fogging of proximate surfaces (Martin 2019). In addition, Clasen et al. (2015) informed in its study about the thermoplastic starch (TPS)/PLA blends, that the grafting of MA onto polymer backbone restricts the segmental movement of the polymer chains. However, the presence of ungrafted MA in the blends act as a plasticizer and reduce stiffness of the blends. Here, the MA can act as a plasticizer or compatibilizer in the TPS/PLA blend depending on the concentration of the monomer used and residual of the unreacted monomer. MA as a plasticizer can reduce the rigidity of the blends by reducing the Tg and increasing the mobility of the polymer chain. Furthermore, the grafted MA reduces the crystallinity of the PLA-g-MA, which led to a reduction in the TPS/PLA blends module as a whole. Aside from that, the balance degradation rate and degree of crosslinking are essentials for producing maleated compatibilizer with an optimal Mw distribution, low Mw degradation and a greater number of reactive sites (Rapthel et al. 2018).
3.2.2 Reaction Mechanism of Maleated Compatibilizer
Free radical polymerization is the most commonly used functionalization technique for polyolefins in which organic peroxides are commonly used as initiators. Several formulations of MA-grafted PP (MAPP) are available in the plastics market depending on the processing method and end-use applications. The grafting reaction of MA onto PP by the melt grafting technique involves a reaction between the polymer melt with MA, in the presence of organic peroxides such as BPO, DCP, etc. As reported by Oromiehie et al. (2014), the organic peroxides are thermally unstable and undergo haemolytic scission at the oxygen-oxygen bonds to form primary radicals. The radicals remove hydrogen atoms from the PP chains and form macroradicals to initiate the grafting process. As reviewed by Oromiehie et al. (2014) in other experiments, they found the mechanism of MA grafting onto PP can be divided into three stages: stage 1- initiation, stage 2 - grafting and stage 3 - termination. The schematic reaction mechanism is shown from Eq. 1 to Eq. 21.
-
(i)
Decomposition of a radical initiator:

-
(ii)
Some possible reaction for chain radical:

-
(iii)
Grafting of MA

-
(iv)
Chain transfer

-
(v)
Termination by combination reaction

-
(vi)
Termination by disproportioning

The grafting of MA onto PP was shown in Eqs. 6 and 7. Other than that, the homopolymerization of MA (Eq. 8) is possible to occur mainly at the high content of MA, while the chain transfer reaction was shown in eq. 9, and the different types of termination reactions included combinations and disproportions, which were shown in equations from Eq. 10 to Eq. 15 and Eq. 16 to Eq. 21, respectively.
Mani et al. (1999) investigated the functionalization of PBS and PLA with MA, and suggested the mechanism of grafting reaction of MA onto polyester (Fig. 3.6).

Grafting reaction mechanism for MA grafting onto polyesters. Reproduced with permission from Mani et al. (1999)
According to Ahmad Thirmizir et al. (2013), the organic peroxide initiator formed a homolytic scission as a free radical. The reaction between a free radical and polyester resulted in the hydrogen abstraction of an α-carbon atom to the ester carbonyl group of polyester to form a macroradical. Some of the macroradicals then suffered β-scission to form a radical chain end and a vinylidene chain end.
According to Mani et al. (1999), in solution grafting reactions, in the presence of organic peroxide, the additional reaction of the macroradicals is predominant before β-scission, but in the melt-grafting reaction, β-scission is predominant. Once the MA is grafted onto the polymer macroradical, several reactions such as chain transfer reaction, oligomerization, termination and some side reactions can occur. However, the materials made by Mani et al. (1999), no signs of homopolymerization or oligomerization of poly-MA were observed. Similar findings have been reported in previous studies in which the MA monomer has a poor homopolymerization tendency due to its unique structure consisting of 1,2-disubstituted double bonds and the reaction temperature is higher than the MA ceiling temperature (Mantere 2015; Moghaddam et al. 2012; Muthuraj 2015). However, Mani et al. (1999) suggested based on evidence from Fourier transform infrared (FTIR) spectroscopy and nuclear magnetic resonance (NMR) analysis that the termination reaction of MA onto the polyester could occur via two routes: recombination and radical termination reactions to produce end products I and III as shown in Fig. 3.6. Some examples of biodegradable maleated polymers, mainly aliphatic polyesters, will be presented in Sect. 3.2.3.
3.2.3 Maleated Biodegradable Polymers: Some Examples
Rzayev (2011) used with successfully as a monomer in the grafting of biodegradable polymers such as PBS, PBSA, PCL and PLA. Functionalizing the biodegradable polymers with reactive groups is an effective strategy to produce commercial blends and composites suitable for end-user applications. In this section, some examples of maleated biodegradable will be further developed.
3.2.3.1 Maleated PBS
The grafting of MA onto PBS via the melt grafting technique was initially reported by Mani et al. (1999). In general, the mechanism of MA grafting onto PBS is similar to that of MA grafting onto polyolefins as presented in Fig. 3.6. Many researchers have reported on the production of maleated PBS and its copolymers for the application of polymer blends (Ramaswamy Mani and Bhattacharya 2008; Yin et al. 2015; Muthuraj et al. 2017) and composites (Ahmad Thirmizir et al. 2011; Phua et al. 2013).
In this sense, Mani and Bhattacharya (2008) reported the use of maleated PBS as a compatibilizer for PBS/starch blends. Here, the compatibilizer was produced using the melt grafting method in a twin-screw extruder. The results showed that the tensile strength (σ) values of the PBS/starch blends without PBS-g-MA compatibilizer decreased. For uncompatibilized blends, the increase in starch load decreased the σ values of the blends. At the higher concentration of starch approximately >50 wt.%, a tremendous reduction in σ values of about 60% was observed. Meanwhile, when PBS-g-MA was incorporated into the blends, the σ values increased for all blend’s ratios. In contrast, the σ value decreased slightly at 30 wt.% starch content before increasing with an additional increase in starch content. At 70 wt.% starch content, the σ values were approximately similar to that of the pure PBS. As expected, the strain at break (εb) of the PBS/starch blends was severely reduced with the addition of starch. As reported by Mani and Bhattacharya (2008), at 10 wt.% starch content, only a slight reduction in εb values was observed, while at higher starch load, a tremendous decrease of εb values was reported. At concentrations of 30 wt.% or higher, the εb values of non-compatible and compatibilized blends was comparable (approx. Between 10 and 20% of εb). Starch is brittle material with a modulus of approximately >1 GPa, and the addition of this into blends makes this material rigid.
Another study by Yin et al. (2015), also reported the consumption of maleated PBS as a compatibilizer in thermoplastic starch/PBS (TPS/PBS) blends. The maleated PBS was produced via the melt grafting technique with the presence of a DCP radical initiator. The formulation of maleated PBS was not mentioned in the paper. The TPS/PBS blends were produced in a twin-screw extruder, and then the blends were compression molded at 135 °C into test specimens and were characterized. The FTIR analysis of maleated PBS confirmed the presence of MA attributed to a weak peak at 1633 cm-1, representing the symmetric stretching of the anhydride groups of MA. The 1H-NMR analysis also confirmed the chemical interaction between MA and PBS, as indicated by the existence of new peaks at 2.74 and 3.52 ppm in the maleated PBS spectra. The mechanical properties in terms of σ and εb values for 40TPS/60PBS blends were significantly improved with the incorporation of maleated PBS. The TPS/PBS (40/60) blends showed good flexibility with an εb of about 20% higher compared to the pure blends. Most importantly, the σ values for TPS/PBS (40/60) blends increased almost twice with the same maleated PBS content. However, the tensile properties did not change, obviously, when maleated PBS content was increased even more from 5% to 10%. The result shows that maleated PBS has an excellent compatibilizing effect where it is thermodynamically miscible with PBS and could form a chemical interaction with TPS. With respect to thermal properties, the addition of maleated PBS reduced the crystallization temperature and degree of crystallinity of the TPS/PBS (40/60 wt.%) blends. The higher concentration maleated PBS resulted from the lower crystallization temperature and degree of crystallinity of the blends. It can also be specified that the maleated PBS could make the blends more ductile by promoting the migration of pure PBS molecular chain (Yin et al. 2015).
As reported by Yin et al. (2015) three Tg can be observed by dynamic mechanical analysis (DMA) and be associated with the glycerol-rich phase, the PBS phase and the starch-rich phase. The introduction of maleated PBS resulted in the DMA peaks approaching each other, indicating that PBS and TPS are partially compatible. In addition, the maleated PBS also improved the interfacial compatibilization through a plasticizing effect in the blending system.
Muthuraj (2015) reported the development of maleated PBS as a compatibilizer for poly(butylene adipate-co-terephthalate) (PBAT)/PBS blends. These authors investigated the effect of the concentration of DCP on the maleated PBS properties. The DG of maleated PBS increased with the increase of DCP concentrations, and the highest percentage of 2.56 was achieved at a concentration of 1.0 phr. The MA grafting efficiency of the batch and the continuous process were also compared, and the results showed that the batch-processed samples had a slightly higher grafting yield compared to the continuously processed samples. The grafting yield was slightly higher in the internal batch process, and can be attributed to a longer residence time and air contact of the reaction medium. Muthuraj (2015) reported from the differential scanning calorimetry (DSC) analysis that the crystallization and Tm of the maleated PBS decreased significantly and were lower compared to that of pure PBS. It is believed that the presence of MA groups prevents the nucleation and lamella growth of PBS, thus leading to the formation of the imperfect crystalline structure.
3.2.3.2 Maleated PHB
Among the PHAs, PHB and its copolyesters are the most studied because they are relatively easier to produce and can yield a consistent quality of bioplastics (Avérous 2013). As for physical properties, PHBs are highly crystalline polyester, i.e. they are brittle and prone to thermal degradation during processing. In the past, attempts have been made to reduce the degree of the brittleness of the PHB by copolymerization with hydroxyhexanoate (HHx) or hydroxyvalerate (HVx) co-monomers. Among PHB copolymers, PHB-co-HHx, PHBV, poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (PHB-co-HHx) and poly(3-hydroxybutyrate-co-4-hydroxybutyrate) (P(3HB-co-4HB)) have improved flexibility and mechanical properties over other PHBs. In addition, they have properties comparable to those of conventional polymers such as poly(ethylene terephthalate) (PET), poly(ethylene) (PE) and PP. PHB-co-HHx become soft and flexible, with an increase in the 3-hydroxyhexanoate (3HH) co-monomer fraction. Doi et al. (1995) reported that an increase in the 3HH co-monomer from 0 to 17 mol% greatly increased the εb values from 6 to 850%, but the σ values decreased from 43 to 20 MPa. Another study done by Laycock et al. (2014) reported as the concentration of 3HH comonomer in the melt press P(3HB-co-3HH) copolymers increases from 2.5 to 9.5 mol%, the εb significantly increased from 6.7 to 43% and the σ tremendously reduced from 25.7 to 8.8 MPa.
Beyond that, its mechanical strength and T characteristics can be further improved by blending with ductile biodegradable polymers such as PBS and PCL. The blending approach is widely known for its use in conventional polymers, and is cost effective and relatively easy to be undertaken compared to the biological way. From the study by Thirmizir et al. (2017), it is known that the PBS/PHB-co-HHx blends are immiscible or immiscible limitedly, depending on the ratio of the components where it is expected to observe the morphology associated with phase separation. To overcome this drawback, several methods such as the addition of compatibilizer and/or the introduction of a crosslinking agent at the blending interphase can be employed. According to Chen et al. (2003), very limited studies on the graft copolymerization of PHB have been published. A study on the radiation grafting of 2-hydroxyethyl methacrylate (HEMA), AAc, acrylamide, methyl methacrylate (MMA) and styrene (St) onto PHB and its copolymer, found that grafted polymers are thermally stable and have a faster biodegradability rate (Chen et al. 2003). However, the graft copolymerization only modifies its surface properties and can produce long graft side chains by homopolymerization. These homopolymers can reside in the environment after PHB, which degrades completely and will damage the environment. Therefore, to avoid those drawbacks, Rzayev (2011) chose the MA monomer to be grafted to the PHB chains via free-radical polymerization due to its good reactivity and controllability reaction.
The grafting reaction is depicted in Fig. 3.7. As reported by Chen et al. (2003), grafting reaction of MA onto PHB chains was conducted in chlorobenzene with a solution temperature of about 130 °C using BPO as an initiator.

Grafting of MA onto poly(3-hydroxybutyrate) (P(3HB)). Reproduced with permission from Rzayev (2011)
3.2.3.3 Maleated PLA
Gardella et al. (2014) studied PLA/PCL blends , and reported maleated PLA (PLA-g-MA) which was introduced to promote grafting between PCL and PLA-g-MA backbone and physical compatibility at the PLA-PLA-g-MA interface. The maleated PLA was produced by using the melt grafting technique employing a glass reactor with a mechanical stirrer placed in an aluminium block oven. Before the reaction process, MA, PLA and 2,5-dimethyl-2,5-di-(t-butylperoxy)hexane were purged with helium for 30 min and repeated at least three times, to ensure a moisture free atmosphere. The reaction was conducted at 180 °C under the stirring condition for a reaction time of 10 min. The peroxide content was 0.5 wt.%, and MA was 6 wt.%. The maleated PLA was then purified by dissolving in chloroform and precipitated into methanol. The blends were prepared by mixing different amounts of PCL, PLA and maleated PLA in the same reactor. The suggested reaction mechanism between PCL and maleated PLA is shown in Fig. 3.8.

Reaction scheme between PCL and PLA-g-MA. Reproduced with permission from Gardella et al. (2014)
Ma et al. (2014c) reported the production of maleated PLA via the melt grafting technique using a mini twin screw extruder. Before the grafting process, the PLA pellets were sprayed with a mixture of acetone dissolved in DCP, MA and St and allowed to dry. After that, the pre-treated PLA pellet was fed into the extruder, and the grafting reaction was conducted at 160–190 °C with a screw speed of 35 rpm. In this study, the DCP radical initiator and St co-monomer were used to promote the formation of free radical. St with an electron-donating feature can easily interact/react with the electron-attracting monomers (i.e. MA via a charge transfer complex (CTC)) or through copolymerization (Ma et al. 2014b). St could activate MA to form an asymmetric structure and Π bonds of radical-anion. As a result, the interaction between macroradicals and MA monomers could be bridged by St, and a higher DG could be achieved. The St co-monomer has been widely used for the production of maleated polyolefins, but for maleated aliphatic polyester, it is considerably rare. Keeping this in view, Ma et al. (2014b) reported that the use of the St co-monomer successfully increased the DG of MA onto PLA backbone and achieved an optimum at the St/MA ratio of 2/1. The St co-monomer is necessary in the maleated reaction due to the low MA reactivity towards the macro-radicals attributed to its structural symmetry and low electron density around the –CH=CH– bond. The reaction mechanism of the free radical grafting of MA onto PLA with the presence of DCP radical initiator is shown in Fig. 3.9.

Free radical grafting of MA onto PLA with the presence of DCP radical initiator. Reproduced with permission from Ma et al. (2014b)
The grafting mechanism of MA onto PLA in the presence of St co-monomer is produced in two steps. As proposed by Ma et al. (2014b), first, the reaction begins with the decomposition of DCP to form primary radicals (RO•). This is followed by the initiation of PLA macroradicals (PLA•) by hydrogen abstraction. The PLA macroradicals then react with grafting monomers, and some of them involve inside reactions such as chain scission or recombination. Most of the PLA macroradicals would be used before reacting with MA due to the inert character of the MA towards the macroradicals. St reacts with PLA macroradicals, thus forming stable styryl macroradicals, which then copolymerize with MA. St then reacts with MA to form a CTC which can improve the electric asymmetry on the –CH=CH– bond of MA. The CTC can increase the reactivity and DG of MA, which could be copolymerized with St in the presence of free radicals to form oligomer-radicals (St-co-MA•). The St-co-MA• could also react with macroradicals (PLA•) by a combination reaction. Subsequently, more routes for MA to be grafted onto PLA chains are obtained, thus significantly increases the DG. However, the excess of St may result in the copolymerization of St and MA or the grafting of St rather than MA, which could reduce the DG (Ma et al. 2014b).
The maximum DG of MA was reported by Ma et al. (2014b) at a St/MA ratio of 2/1 in the PLA-g-MA/St system and not at a 1/1 ratio due to polymer chain structures. In the presence of primary radicals (RO•), the residual St may react with PLA macroradicals to produce relatively stable styryl macroradicals (PLA-g-St•) or copolymerize with MA to form short oligomer-radicals (St-co-MA•). The PLA-g-St• could then react with short St-co-MA•, CTC or MA to form branched structures. Ma et al. (2014b) suggested that the grafting of short St-co-MA• onto PLA chains causes the main reaction to occur at the St/MA ratio of around 2/1, as shown in Fig. 3.10.

The main grafting reactions proposed at low St/MA ratios (R = C9H11). Some possible side reactions are not present here. Reproduced with permission from Ma et al. (2014b)
As stated by Ma et al. (2014b), St-co-MA is a random co-oligomer rather than a block co-oligomer, due to the strong free radical reactions. Ma et al. (2014b) also reported the effect of MA concentration on the DG at a constant St/MA/DCP ratio where the DG remained unchanged with the increase in the MA concentration. The DG also depends on the number of reactive species associated with the MA monomers and the DCP concentration. Another factor that affects the DG is the reaction temperature. In this sense, Ma et al. (2014b) reported that at a fix MA concentration of 4.5 phr and constant St/MA/DCP ratio of 2/1/0.1, the DG increased with temperature and achieved optimum at 180 °C. Beyond that, the DG was reduced.
3.2.3.4 Maleated PCL
PCL is a ductile biodegradable polyester commonly used to improve the brittleness of other biodegradable polymers, such as PBS (Can et al. 2014; Gumede et al. 2018), PHB (Barghini et al. 2010) and PLA (Gardella et al. 2014) through the blending approach. However, Gardella et al. (2014) indicated that PCL and PLA are thermodynamically incompatible and form blends with a multiphase structure and an inadequate interfacial bonding, which deteriorates their mechanical performance. Many studies have been carried out to improve the compatibility between them via compatibilization techniques, such as the incorporation of polymeric compatibilizers and reactive compatibilization approaches (Gutiérrez and Alvarez 2017 a,b,c; Herniou--Julien et al. 2019). Wu and Liao (2012) studied PCL/rice straw fiber blends and used maleated PCL as a compatibilizer. The maleated PCL was produced by the solution grafting technique using tetrahydrofuran (THF) as the solvent and BPO as the free radical initiator. The grafting reaction of MA onto PCL was carried out at a temperature of 40 ± 2 °C and a rotor speed of 60 rpm for 10 hours. The optimal DG was 1.02 wt.% with BPO and MA contents of 0.3 and 10 wt.%, respectively. The schematic reaction of MA onto PCL is shown in Fig. 3.11.

The grafting reaction of MA onto PCL. Reproduced with permission from Wu and Liao (2012)
Wu and Liao (2012) assessed the maleated PCL by FTIR and 13C-NMR to confirm the grafting reaction. The FTIR analysis exhibited the presence of two additional bands at 1786 and 1857 cm−1, representing anhydride carboxyl groups in the modified PCL-g-MA. The presence of the bands represents free acid in the PCL-g-MA showing an effective MA grafting onto PCL (Wu and Liao 2012). Meanwhile, solid state 13C-NMR analysis was conducted to confirm this finding. These authors observed six peaks, corresponding to carbon atoms in the unmodified PCL (1, δ = 64.3 ppm; 2, δ = 28.9 ppm; 3, δ = 25.8 ppm; 4, δ = 25.1 ppm; 5, δ = 34.4 ppm; 6, δ = 172.9 ppm). For PCL-g-MA, the additional peaks at (7, δ = 42.3 ppm, 8, δ = 36.2 ppm; 9, −C=O δ = 174.3 ppm) confirmed that MA was covalently grafted onto PCL (Wu and Liao 2012).
An overview on the REx of PCL/starch blends made by Kalambur and Rizvi (2006) was reported using maleated PCL as a compatibilizer in PCL/starch blend systems suggested by John et al. (1997). The maleated PCL was prepared by melt grafting of MA onto PCL in a batch mixer with a roller blades type rotor in a twin-screw extruder using DCP as an initiator. The results indicated that no crosslinking reaction occurred during the grafting process.
According to John et al. (1997), the reaction mechanism begins with the homolytical scission of peroxide, which produces radicals followed by hydrogen extraction of α- carbon atom relative to the carbonyl group. The second step involves the formation of radical on the PCL chain and some degree of β-scission due to the existence of organic peroxide. The third step involves the addition of a double bond to the radical from β-scission. In this case, the termination reaction can occur in three possibilities: homopolymerization, radical termination and recombination. Based on the FTIR and NMR analysis, the termination reaction and recombination reaction are favored and no traces of homopolymerization reaction products were detected in the compatibilizer. The reaction mechanism between the maleated PCL and the starch was further developed by Kalambur and Rizvi (2006) and the schematic reaction is shown in Fig. 3.12.

Reaction mechanism of MA grafting onto PCL and reaction between maleated PCL and starch. Reproduced with permission from Kalambur and Rizvi (2006)
3.3 Free Radical Crosslinking of Biodegradable Polymer
During the last decade, stabilization of polymer blends by the crosslinking reaction has been widely practiced in many industries. Initially, the concept of a system of crosslinked blends was practiced in the production of thermoplastic vulcanizates for replacing pure elastomeric block copolymer which was relatively expensive and complicated to be produced. The blends were dynamically crosslinked using various crosslinking agents such as organic peroxides (e.g. 2,5-dimethyl-2,5 bis(t-butylperoxy) and DCP), phenolic curative (e.g. dimethyl alkyl phenol), sulphur and zinc oxide (Harrats and Groeninckx 2007). Various compatibilizers such as GMA-grafted ethylene propylene diene monomer (EPDM), MA-grafted EPDM and MA-grafted PP, have been to improve the interfacial adhesion between the rubber phase and thermoplastic matrix. As for the mechanical properties, thermoplastic vulcanizates show a full strain recovery compared to pure thermoplastics. They also have higher σ values compared to pure rubbers and are controlled by the size of the vulcanized rubber particles in the blends. As shown in the EPDM-PP blends in Fig. 3.13, the smaller the particle size, the higher the tensile stress of thermoplastic vulcanizate.

Effect of vulcanized-rubber particle size on the mechanical properties of EPDM-PP thermoplastic vulcanizates. Reproduced with permission from Harrats and Groeninckx (2007)
3.3.1 Type of Peroxide Radical Crosslinking
Peroxide initiated crosslinking commonly produced by adding small amounts of peroxide during melt processing. Peroxides can be classified into seven types according to the chemical structures: diacyl peroxides, dialkyl peroxides, diperoxyketals, hydroperoxides, ketoneperoxides, peroxydicarbonates and peroxyesters (Takamura et al. 2008). In the context biodegradable polymers, various types of peroxides have been used in the crosslink or partial crosslink polymer blends, such as BPO, DCP, dilauroyl peroxide (LPO), n-butyl 4,4-di-(t-butyl peroxy) valerate (BTBV), OO-(t-butyl) O-(2-ethylhexyl) peroxycarbonate (TBEC), t-butyl peroxy benzoate (TBPB), t-butyl peroxy-2-ethylhexanoate (TBEH) and t-butyl peroxy-3,5,5-trimethylhexanoate (TBTH) (Takamura et al. 2008). Among these peroxides, DCP has been the most widely used peroxide crosslinking agent in the biodegradable polymer blends system (Semba et al. 2006; Mishra et al. 2007; Takamura et al. 2008; Deng and Thomas 2015).
Takamura et al. (2008) reported the use of various types of peroxides as crosslinking agents of PLA under REx conditions. The peroxides can be divided into three main groups according to their decomposition rates: group I: fast, group II: moderate and group III: slow, as shown in Table 3.6.
3.3.2 Crosslinking Reaction Mechanism
According to Takamura et al. (2008), crosslinking of polymers initiated with peroxides occurs through three key steps:
-
(i)
The generation of primary radicals derived from thermal decomposition of peroxides.
-
RO—OR• → 2RO• (primary radicals)
-
(ii)
Hydrogen abstraction from polymer chains by primary radicals to generate polymer radicals.
-
RO• + P(Polymer) → P•(Polymer radical) + ROH
-
(iii)
The bimolecular recombination of polymer radicals to form carbon-carbon cross-links.
-
2P → P—P (crosslinking).
In contrast, Mishra et al. (2007) proposed the mechanism of crosslinking reaction from PCL/epoxidized natural rubber (ENR) (50/50) blends that occurred via two schemes: (1) formation of PCL macroradical and chain scission of its polymer chain (Fig. 3.14) and (2) inter-chain crosslinking between PCL and ENR at the interface (Fig. 3.15).

Chain scission of PCL during crosslinking with DCP. Reproduced with permission from Mishra et al. (2007)

Inter-chain crosslinking between PCL and ENR at the interfacial region with DCP. Reproduced with permission from Mishra et al. (2007)
On the other hand, Hu et al. (2018) produced crosslinked PBS/PLA blends by using BPO as a peroxide radical initiator in hot chloroform at a temperature of 65 °C as a blending solution. The reactive solution was blended for 120 min, and the blends were them dried for 48 h at 50 °C before compression molding at a temperature of 160 °C. The possible BPO-initiated crosslinking reaction mechanism of PLA/PBS is shown in Fig. 3.16.

Reaction mechanism of BPO-initiated PLA/PBS crosslinking. Reproduced with permission from Hu et al. (2018)
3.3.3 Peroxides Concentration
Fei et al. (2004) and Takamura et al. (2008) reported the influence of the concentration peroxides on the degree of crosslinking of the partially crosslinked copolymer poly(L-lactic acid) (PLLA) and PHBV via the gel content, Mw analysis and thickness swelling. According to Fei et al. (2004), by varying the concentration of peroxide during processing, the degree of crosslinking of PHBV chains can be tailored to a suitable degree. By tailoring the Mw of PHBV via the grafting approach, the undesirable effect of heat processing on the melt viscosity and Mw of PHBV could be compensated, and the resulting material even shows better mechanical properties.
Generally, the crosslinking time can be estimated based on the half-life of the peroxide used at that particular grafting temperature. The grafting period is normally five times than the half-life of the peroxide (Fei et al. 2004). In addition, the crosslinking density can be controlled by the initial amount of peroxide at constant crosslinking parameters.
It can be seen in Fig. 3.17 that the gel fraction is almost zero at 0.17% DCP. Fei et al. (2004) suggested that mainly branched PHBV was produced under these conditions. When the DCP content was increased to 0.5%, the gel fraction of about 2% was obtained, and since the DCP content is increased to 1%, the gel fraction is about 55%. However, additional increases in the DCP content did not result in a significant increase in the gel fraction. Fei et al. (2004) also compared the gel fraction analysis between PHBV with LDPE, where the crosslinking efficiency of PHBV was much lower than that of LDPE when the peroxide concentration used reached between 3 and 5%. While at the lower peroxide concentration (1% of DCP), the crosslinking efficiency in PHBV is comparable to that in LDPE, thus suggesting that the ideal DCP content should be in a range between 0.5 and 1% in order to obtain an optimal degree of crosslinking of the PHBV. On the contrary, the gel swelling ratio decreased as the content of DCP increased, which explains why the crosslink density of the copolymer increased as the content of DCP increased (Fig. 3.18).

Gel fraction based on DCP content for crosslinked PHBV. Reproduced with permission from Fei et al. (2004)

Effect of DCP content on the swelling ratio of crosslinked PHBV obtained via chloroform extraction. Reproduced with permission from Fei et al. (2004)
Peroxide radical crosslinking has also been used to improve the compatibility of biodegradable polymer blends via partial crosslinking of the components of the blend. Previous studies have reported the positive impact of partial crosslinking on the improvement of σ, εb and T of the blends (Dong et al. 2013; Ji et al. 2014; Ma et al. 2014a; Signori et al. 2015). In line with this, the incorporation of peroxides tends to promote the formation of chain branching, as well as crosslinking, thus increasing the degree of crystallinity, T and thermal behavior of the polymer blends. Indeed, the crosslinking reaction is also an effective approach to improve interfacial adhesion of the immiscible polymer blends by initiating the emerging of mixed chains (copolymers), which act as compatibilizers at the interphases (Signori et al. 2015). The formation of branching or crosslinking of the polymer chains by heterogeneous and/or homogeneous radical coupling reactions are controlled by the concentration of peroxides used in the blending system.
Dong et al. (2013) reported the effect of peroxide (i.e. DCP concentration on the properties of PHB/poly(D,L-lactic acid) (PDLLA) blends). It can be seen from the result of the gel fraction (Fig. 3.19) that the gel fraction of the PHB/PDLLA (70/30) blends increased up to 2 wt.% before being leveled with an additional increase in DCP concentration. A similar observation was also reported by Signori et al. (2015) for PLA/PBAT/DCP crosslinked blends where the gel fraction of the blends increased with the peroxide content from 0 to 0.2 wt.%. Ji et al. (2014) also observed similar trends in which the gel fraction of partial crosslinked PLA/PBS blends increased when the DCP peroxide concentration increased from 0.1 to 0.5 phr. In addition, Dong et al. (2013) reported, at a low concentration of DCP (1%) the gel fraction obtained was considerably low, while with a 2% DCP content, the gel fraction was almost doubled. However, a slight drop in the gel fraction was observed at the high DCP concentration (4%). This was related to the domination of the chain scission reaction of the components of the blend’s main chains instead of crosslink/branching reactions.

Gel fraction of the crosslinked PHB/PDLLA (70/30) blends as a function of DCP content. Reproduced with permission from Dong et al. (2013)
According to Dong et al. (2013), PHB/PDLLA blends with a DCP concentration greater than 1% were relatively more difficult to be processed due to the presence of a highly density portion from crosslinking which tended to increase the melt viscosity of the blends. Dong et al. (2013) reported that the incorporation of DCP into PHB/PDLLA (70/30) blends resulted in a large increase in the mixing torque: the higher the DCP content, the higher the mixing torque. This phenomenon was due to the formation of the crosslinking/branching structure by carbon-carbon crosslinks as a result of the recombination of polymer radicals (Takamura et al. 2008).
Regarding the rheology point of view, Dong et al. (2013) reported the effect of the DCP addition on the storage module (G’) and the complex viscosity (ɲ*) of the PHB/PDLLA (70/30) blends (Figs. 3.20 and 3.21). The G’ of the blends increased significantly after adding DCP, while the plots of G’ vs frequency show a flatter curve in the low-frequency zone. The trends denote the presence of branched structures and/or partial crosslinks in the blends. It can be seen from the ɲ* result that the partial crosslinking improved the melt strength of the blends. However, the ɲ* for the partially crosslinked PHB/PDLLA blends at the high frequency zone was not so high, thus indicating that the blends maintain good melt processability even after partial crosslinking. Signori et al. (2015) also found good processability for partial crosslinked PLA/PBAT blends up to the peroxide content of 0.2 wt.%.

Storage module (G’) of the PHB/PDLLA (70/30) blends as a function of DCP content. Reproduced with permission from Dong et al. (2013)

Viscosity of the PHB/PDLLA (70/30) blends as a function of DCP content. Reproduced with permission from Dong et al. (2013)
3.4 Compatibilization of Blends
The polymer blend is a relatively easy and cost-effective approach to produce polymer products with beneficial combinations of valuable properties with respect to the single polymer properties (Rapthel et al. 2018). Many of the polymer blends are immiscible and incompatible, and therefore, a compatibilization process either non-reactive or reactive is essential to ensure that the properties of desired blends can be achieved (Muthuraj 2015). Non-reactive compatibilization usually involves a process in which a prefabricated graft or block copolymers are used. On the other hand, reactive compatibilization is carried out via melt blending with the presence of compatibilizer can effectively form chemical interactions at the interfacial region of the components of the blend (Muthuraj 2015). This procedure is conducted by the addition of pre-produced polymers (block copolymer, graft copolymer, homopolymer, etc.) or by forming reactive compatibilizers in-situ to improve the interfacial adhesion, decrease the interfacial tension and the dispersed phase size and suppress the coalescence of the dispersed phase (Rapthel et al. 2018).
3.4.1 Reactive Compatibilization of Biodegradable Blends
Reactive compatibilization is also known as REx if it is conducted in a continuous mixing system and reactive melt blending if it is conducted in a batch mixing system. Although REx has been well known for conventional polymer processing in the last decade, its application for processing of biodegradable polymer blends is a somewhat a new direction of scientific research. The reactive compatibilization of polymer blends can be produced via one step (in-situ) or two-step REx. In one-step REx, all components are introduced simultaneously during blending. Both the functionalization and reactive blending steps are carried out in the same extrusion process (Sun et al. 1996). The functionalization can be conducted in the first section of the extruder, followed by interfacial reaction compatibilizer and polymer blends. The process is also known as in-situ compatibilization. While in the multi-steps, usually two-step REx, it includes functionalization of the polymer with reactive agents normally with the presence of the free radical initiator in the first step and blend of functionalized polymers with other components through an extrusion process in the second step (Gutiérrez et al. 2017).
3.4.1.1 Single Step or in-situ Reactive Compatibilization
Finding the components of polymer blends that are thermodynamically correct with good miscibility is realistically challenging, since the incompatibility between the components tends to occur. The improvement of compatibility and adhesion between phases can be carried out by incorporating suitable interfacial agents, either block or graft copolymers, which is a relatively complicated and less economical process. Alternatively, these copolymers can be produced in-situ by a blending process through polymer-polymer graft reactions using functionalized polymers (Rzayev 2011). The incorporation of block copolymers or functionalized homopolymers, which can react to form copolymers in-situ, is an effective method for compatibilizing immiscible polymer blends and preventing coalescence (Fink 2013).
Sun et al. (1996) reported in-situ compatibilization based on the REx technological point of view, where the compatibilization and reactive melt blending are carried out in the same barrel. The authors also suggested two types of extruder and screw configuration, specifically for in-situ reactive compatibilization purposes, as shown in Fig. 3.22. For an extruder with type (a) screw configuration, e.g. the PP pellets, the monomers (GMA and St) and the peroxide are fed by the first hopper, while the poly(p-phenylene-2, 6-benzobisthiazolediyl) (PBT) pellets are fed by the second hopper. In this configuration, the functionalization of the PP is produced almost completely in the first zone between the first and second hoppers, after which (in the second zone, from the second hopper to the matrix) the interfacial reaction between the functionalized PP and the PBT occurs. While in type (b) screw configuration, the devolatilization zone can be moved to the end of the first zone before the second hopper. In addition, the devolatilization zone in the screw configuration (b) can also be extended more than in the configuration (a) in order to efficiently remove unreacted monomers and avoid any unwanted reaction between the unreacted monomer and the PBT.

The two screw configurations used for the in situ compatibilization of PP/PBT blends by one-step REx. Reproduced with permission from Sun et al. (1996)
Ma et al. (2014c) reported the preparation of PHB/EVA/starch ternary blends via in-situ grafting between EVA-g-starch and PHB. The EVA-g-starch was prepared separately in a twin-screw extruder at 135 °C and with a rotor speed of 100 rpm. The reactive compatibilization was conducted with the presence of MA, using BPO and glycerol as a radical initiator and plasticizer, respectively. The possible MA-induced chemical interaction in the EVA-g-starch copolymer is shown in Fig. 3.23. The extruded PHB/EVA/starch blends were pelletized and compression-molded at 180 °C into the test specimens. In the grafting process between EVA and starch, the starch gelatinization occurs due to a combination of plasticizer elements, heat and shear during mixing, which resulted in the elimination of crystalline structure of starch (Ma et al. 2014c). However, the phase morphology of starch is usually thick due to its high Mw, hydrophilic nature and strong hydrogen bonds. In this grafting process, MA was introduced to alter the blends in a fine morphology, in order to produce better mechanical properties. The EVA-g-starch copolymers were generated in-situ at the interfaces to prevent the agglomeration of fine starch particles. As a result, a reduction in starch particle size in the blends by a factor of about 100 times was achieved. In addition, the PHB/EVA/starch ternary blends had a better affinity between starch and PHB due to the use of MA, thus reducing their particle size.

Grafting reaction between the EVA and starch via using MA to produce EVA-g-starch . Reproduced with permission from Ma et al. (2014c)
3.4.1.2 Two Steps or more Reactive Compatibilization
For example, Thirmizir et al. (2017) reported the use of maleated PHB-co-HHx (PHB-co-HHx-g-MA) as a compatibilizer for the PBS/PHB-co-HHx blend systems. The process was conducted via two steps REx. In the first step, the compatibilizer was produced by reactive melt grafting of MA onto PHB-co-HHx at 160 °C, using DCP as an initiator in a double-wing co-rotating internal mixer at 50 rpm rotor speed. The compatibilizer was also purified before use. For manufacturing blends, the 5 wt.% of PHB-co-HHx-g-MA was added to the PBS/PHB-co-HHx blends, and the reactive melt blending was conducted at 160 °C for 5 min. The blends were then compression molded at the same temperature and assessed using tensile and morphological analysis. The compatibilized blends had higher σ values in all the blend ratios compared to the uncompatibilized blends. The tensile modulus of the compatibilized blends was also higher than uncompatibilized blends. However, the εb values of the blends only experienced a significant increase effect at 20/80, 30/70 and 40/60 ratio from blends before remaining unchanged at 50/50 blend ratio. It is known that the incorporation of the PHB-co-HHx-g-MA compatibilizer helps promote a good interfacial interaction between both phases. As expected, the compatibilization between brittle and ductile polymers increased the εb values due to the improved stress transfer from the brittle phase to the ductile phase. The increase in the σ and T values denotes the improved mechanical properties. Therefore, these findings demonstrate that the addition of PHB-co-HHx-g-MA had a synergistic effect in improving the properties and morphologies of the blends towards a more cohesive and continuous structure (Thirmizir et al. 2017).
Persenaire et al. (2014) also investigated the reactive compatibilization of PLLA/PBS blends with the addition of maleated PBS and PLLA. These authors used reactive compatibilization of 2 steps, where the grafting process of the compatibilizers was prepared by REx using a twin-screw extruder at 120 °C and a low screw speed of 30 rpm for developing PLLA-g-MA and PBS-g-MA. The compatibility of the blends was carried out separately in a batch mixer bench scale kneader at 190 °C with a reaction time sequence of 3 min at 30 rpm and 6 min at 60 rpm. The MA and the radical initiator Luperox® 101 concentrations were fixed at 3 wt.% and 0.5 wt.%, respectively, for both compatibilizers. The maleated compatibilizer was further purified to remove un-grafted MA and initiator. The grafted MA content was approximated to be 0.65 wt.% for PLLA-g-MA and 0.55 wt.% for PBS-g-MA. In addition, Persenaire et al. (2014) observed that the incorporation of 4 wt.% of PLLA-g-MA into PLLA/PBS blends 80/20 (w/w) exhibited the improvement of σ and εb values by 18% and 61%, respectively. With respect to the morphology of the blends, PLLA/PBS blends 80/20 (w/w) showed a dispersed phase morphology where the PBS drops were dispersed into the PLLA matrix, as shown in Fig. 3.24a. In contrast, the addition of PLLA-g-MA (4 wt.%) resulted in a drastic reduction in the size of the PBS drops, thus indicating a better dispersion of guest polymer. In fact, most of the drops were less than 1 μm in diameter, as shown in Fig. 3.24b. Furthermore, the polymer interface was improved as a result of the chemical interaction between both blend components.

SEM microphotographs of cryo-fractured surfaces of 80/20 (wt/wt) PLLA/PBS blends: (a) uncompatibilized and (b) PLLA-g-MA compatibilized. Reproduced with permission from Persenaire et al. (2014)
Gardella et al. (2014) investigated the potential of PLA-g-MA as a compatibilizing agent for PLA/PCL blends. In this study, the maleated compatibilizer was prepared to be used in 2-step reactive compatibilization. The first step involved free radical grafting of MA onto PLA chains in the presence of 2,5-dimethyl-2,5-di-(t-butylperoxy)hexane peroxide initiator. The second step involved the compatibilization of the PLA/PCL blends with maleated PLA. As expected, the mechanical properties of the uncompatibilized blends were an average between the two polymers, depending on the ratio of the two components of blends. The module of the PLA/PCL blends was intermediate between those of PLA and PCL, while the εb did not significantly improve compared to that of pure PLA, indicating that there is no toughening effect of PCL. However, by incorporating maleated PLA, a direct relationship between the concentration of maleated PLA and the tensile modulus was observed. In addition, εb values were significantly increased (650%) with the use of maleated PLA up to 7 wt.%, since at higher maleated PLA concentrations, εb values declined. According to Gardella et al. (2014) the increase in the tensile modulus and εb values of the PLA/PCL blends was related to the improved compatibility between the two phases. Nonetheless, at the high PLA-g-MA content, the loss of ductility obtained was observed.
3.4.2 Non-reactive Compatibilization of Biodegradable Blends
The method to improve the properties of the polymer with blending with other polymers has been well established in conventional polymers such as acrylonitrile butadiene styrene (ABS), poly(carbonate) (PC), polyolefins, etc. With respect to biodegradable polymers, modification as the toughening approach by adding elastomeric biodegradable polymer such as PCL has been reported in previous studies (Kalambur and Rizvi 2006; Barghini et al. 2010; Gardella et al. 2014). This type of non-reactive compatibilization is also effective for improving the T values by reducing the stiff characteristic of blends. However, it is generally unable to compensate for the reduction in strength of the blends due to two main factors, namely: (1) poor interfacial adhesion and (2) immiscibility between the components of the blend. To overcome these drawbacks and achieve more valuable properties, several methods have been introduced, e.g. the addition of block copolymers such as PCL-poly(ethylene glycol) (PEG) copolymer, PCL-PLA diblock, triblock and random copolymers and poly(ethylene oxide) (PEO)-poly(phenylene oxide) (PPO)-PEO triblock copolymer (Imre and Pukánszky 2013). In the immiscible blends, block-copolymer plays a role as a compatibilizer between the components of the blend, which reduces the interfacial tension between them (Muthuraj 2015). While, the incorporation of diblock-copolymer can improve the stability of the blend as it tends to segregate at the interface between the two components. In addition, the graft or block copolymer is designed to reduce the interfacial tension and create strong interfacial adhesion, which leads to reduced particle size of the dispersed phase and makes it more stable against coalescence during the melt processing (Karami et al. 2019).
According to Kim and Park (1999), a random or block copolymer of two or more biodegradable polymers can be produced by transesterification reaction using an appropriate catalyst (di-n-butyltin-dilaurate at 0.5 wt.%) at high temperature to accelerate the reaction. These authors demonstrated that the degree of transesterification between PBS and PTB increased linearly with increasing reaction time and the use of catalyst. The possible copolymer structure derived from the transesterification reaction between PBS and PBT is listed in Fig. 3.25. Kim and Park (1999) observed from the DSC and the dynamic mechanical thermal analysis (DMTA) that although the PBS/PBT blends were immiscible, with the introduction of the PBS-PBT copolyester, the degree of miscibility between both components was improved.

Possible copolymer structures of the transesterification reaction between PBS and PBT. Reproduced with permission from Kim and Park (1999)
3.5 Crosslinking of Biodegradable Polymer Blends
Polymer blends are materials commonly used in the plastics industry for various applications, such as adhesives, coatings, composites, foams, molded products and many more. Many techniques can improve the blend properties. One of the simplest techniques is the introduction of organic peroxide crosslinking between blend components. The crosslinked blends are more compatible blends compared to simple blends due to improved interfacial adhesion between the components of the blend which can improve the mechanical properties of the blends as a whole (Mishra and Wonho 2011).
Keeping this in view, Mishra et al. (2007) reported on the development of PCL/ENR blends crosslinked with DCP as the peroxide crosslinking agent produced by the melt blending technique in an internal mixer at 160 °C for 8 min. The crosslinking effect was improved the σ and εb values from the PCL/ENR (50/50) blends. It was believed that peroxide (DCP) introduced inter-chain crosslinking between PCL and ENR as presented in Fig. 3.15. Mishra et al. (2007) suggested that the compatibility of the interchain reaction via crosslinking of peroxide radical is similar to that of blends with the addition of block copolymers, which leads to an increased σ and εb values. While in a homopolymer system, the introduction of crosslinking normally increases the stiffness and causes a decrease in the εb values (Fei et al. 2004). Mishra et al. (2007) also reported that the crosslinked blends have a slightly lower modulus compared to uncrosslinked blends, possibly due to the alteration of at least one or more of the following factors: degree of crosslinking, entanglement network, number of binding molecules and crystallinity (Mishra et al. 2007).
Aside from that, many other studies have reported the use of peroxides as a crosslinking agent in the blend systems such as BPO (Hu et al. 2018) and DCP (Fei et al. 2004; Semba et al. 2006; Mishra et al. 2007; Dong et al. 2013; Ji et al. 2014). Dong et al. (2013) reported the introduction of partial crosslinking in PHB/PDLLA blends by using DCP as a free radical initiator via melt blending at 170 °C and a rotation speed of 40 rpm. The DCP was added after 4 min. of mixing, and the blends were processed for another 2 min. The formation of free radicals in the PHB and PDLLA chains was initiated by peroxide via a hydrogen absorption mechanism. The grafting of PHB/PDLLA blends was produced at the interface through a combination of free radicals on both components and also occurred in the PHB and PDLLA rich phases, separately (Dong et al. 2013). As a result, complex products such as branched/crosslinked PHB branched/crosslinked PDLLA, PHB-crosslinked-PDLLA network and PHB-g-PDLLA copolymers could be produced.
Following Dong et al. (2013) the melt blending technique also caused chain scissions due to the low thermal stability of the free radicals and PHB polymer. The crosslinking and chain scissions occurred through a six-member ring transition, which is difficult to avoid. The crosslinking and chain scission are two competitive reactions. These phenomena could be seen during the mixing process, where the torque value increases when the DCP is introduced until it reaches a maximum value and then dropping with a prolonged mixing time. This observation was also recorded by Dong et al. (2013) as indicated by the curves in the crosslinking zone (Fig. 3.26). Since the peroxide crosslinking only occurred somewhere of the blends. This is known as partial crosslinking systems.

Effect of DCP on the torque vs processing time during blending. Reproduced with permission from Dong et al. (2013)
Table 3.7 summarizes the properties of biodegradable blends crosslinked by peroxides that have been studied by previous researchers.
3.6 Crosslinked-Compatibilized of Biodegradable Blends
Compatibilization is a chemical process that was introduced to improve the adhesion between the phases of the blends, facilitate chain dispersion, reduce interfacial tension and stabilize the morphology of the blends (James et al. 2009). On the other hands, crosslinking in the context of polymer blends refers to in-situ free radical crosslinking which introduces partial crosslinking inter- and intra-components from blends. Crosslinking leads to a more cohesive interfacial interaction and phase distribution. In this chapter, the effect of phase compatibilization-crosslinking synergism is further discussed. Many studies on compatibilization of polymer blends via crosslinking have been focused on non-biodegradable polymers, as will be cited in the literatures below. In this section, we will demonstrate that such a technique can also be used to improve the properties of biodegradable blends.
In a review made by Koning et al. (1998) revealed the compatibilization reaction of polymer blends through a combination of peroxides and multifunctional chemicals. The peroxide is used to activate the reaction between a polymer and the functional groups of the chemical. The multifunctional chemical then bound to the polymer chains in graft or branch copolymers, which is the real compatibilizer. The combination of a peroxide and unsaturated monomers such as hydroxypropylmethacrylate, low Mw unsaturated rubber, MA, St, triallyl isocyanurate, and undefined silane is actually similar to the peroxide/co-agent systems that are commonly used for crosslinking of conventional polymers (e.g. EPDM, PE and PP). The main advantage of the crosslinked-compatibilized reaction is to increase the efficiency of the peroxide and the reaction rate. While for a single compatibilization reaction system, the crosslinking or compatibilization will experience a lack of chemical selectivity and the improvement of properties will not be pronounced.
Xu et al. (1993), reported on the synergistic effect of crosslinking-compatibilization in poly(vinyl chloride) (PVC)/LDPE blends in the presence of butadiene-acrylonitrile rubber (NBR) unsaturated monomer as a compatibilizing agent and DCP organic peroxides as a crosslinking agent. A two-roll mixer produced the blends at 155 °C. In compatible PVC/LDPE blends, the NBR was added to shear forces during blending by increasing the melt viscosity of the LDPE and reducing the lubrication effect on PVC phase. As a result, the dispersion of the blends was improved as indicated by the reduction domain size of the dispersed phase. On the other hand, in the DCP-crosslinked PVC/LDPE blends, an increase in mechanical properties was recorded due to the formation of the co-crosslinked product. Without the NBR compatibilizer, the interphase area between two components was quite small, which made the co-crosslinking reaction at the interface region difficult, thus resulting in minimal improvements in the mechanical properties. In PVC/LDPE blends, crosslinked-compatibilized with DCP and NBR, provided a significant improvement in the σ and εb values. In this system, the possibility of a crosslinking agent residing in the interfacial area is greater due to the better phase dispersion which is promoted by the NBR compatibilizer. In addition, with the presence of DCP, a greater number of co-crosslinked products could be formed to promote a more cohesive interfacial interaction. The phase morphology of the crosslinked-compatibilized blends shows an unclear interface and smaller domains, which indicates the successful synergism of crosslinked-compatibilized reaction between the blending system.
James et al. (2009) reported the crosslinked-compatibilized reaction between ultra-high Mw polyethylene (UHMEPE) and natural hyaluronan (HA) polysaccharide for biomaterial applications. These authors used MA-grafted high-density polyethylene (HDPE-g-MA) as a compatibilizer and DCP organic peroxide as a crosslinking agent. In this case, the anhydride group of MA reacted with the hydroxyl groups onto the HA to form ester bonds, while the hydrophobic chain portion of the compatibilizer diffused and entangles with the UHMWPE chains. Aside from that, HDPE-g-MA also helps improve the processability since the HDPE portion of the compatibilizer has a lower Tm compared to pure UHMWPE , which makes the molding process easier and spends less thermal energy compared to the molding of pure blends. To crosslink the compatibilized UHMWPE/HA, DCP was introduced as a crosslinking agent via dipping the compatibilized UHMWPE/HA preform in a DCP/antioxidant solution. This novel crosslinked-compatibilized process is simplified in Fig. 3.27.

Crosslinked compatibilized UHMWPE/HA process. Reproduced with permission from James et al. (2009)
James et al. (2009) also subjected the blends to a wear resistance test conducted in 90% bovine calf serum at 37 ± 1 °C, applying a load cycle with a maximum force of 330 N at a frequency of 1.6 Hz. The duration of the test was almost two million cycles. The authors found that the wear rates of crosslinked and crosslinked compatibilized UHMWPE/HA were non-significant compared to the control (50 kGy irradiated and stabilized PE GUR 1050) at 1.98 million cycles. Within the same cycles, the total wear of crosslinked compatibilized UHMWPE/HA was 5.54 mg, while the total wear of the control sample was 3.56 mg. The lower wear (mg) and wear rates (mg/million cycles) of crosslinked and crosslinked compatibilized blends, compared to pure blends, demonstrated the effectiveness of crosslinking-compatibilization reaction on the wear resistance properties of UHMWPE/HA blends.
3.7 Performance Evaluation
In this section, the evaluation of the performance of biodegradable polymer blends will be further discussed in terms of water performance or exposed to moisture, natural weathering, and biodegradation in the natural environment. The discussion will also cover the effect of the type of polymer, the blending system and type of additives used to alter or improve the properties of the blends.
3.7.1 Water Absorption
Some applications of biodegradable polymers involve a greater understanding of the durability and the possibility of the macroscopic properties. Functional groups, such as carboxylic acid, hydroxyl and lactide, as well as other hydrophilic molecules, can increase the level of moisture, which leads to higher degradation rates. Water absorption causes a severe alteration of the properties of biodegradable polymer blends, including chemical and physical modifications such as hydrolysis, interface debonding and plasticization (Hiljanen-Vainio et al. 1996).
In addition, water absorption is highly dependent on the morphology of polymer blends (Choi et al. 2002). In this case, the morphology cannot only affect water absorption based on crystallinity but also via the existence of microvoids or porosity (Drumright et al. 2000). The degree of crystallinity plays an important role in determining barrier properties, as well as the water absorption of the polymers. Berthé et al. (2010) revealed that crystallinity seemed to be the most influential factor rather than Mw to determine the water absorption properties of two different PLLA resins. These authors also found that PLLA resin with greater crystallinity induced less water absorption. According to Berthé et al. (2010), high crystallinity possibly reduced the mobility of the chain that could hinder the water diffusion into the polymer. Zhang et al. (1995) also investigated water absorption and hydrolytic degradation of PCL, PLA and PLA/PCL blends, and reported that highly crystalline PCL was more hydrophobic compared to amorphous PLA, which could explain the greater water absorption of PLA compared to PCL. Meanwhile, the water absorption of PLA/PCL (25/75 and 50/50) blends was between that of simple PCL and PLA. Interestingly, it was found that the PLA/PCL (75/25) blend exhibited greater water absorption compared to the PLA component. Zhang et al. (1995) also stated that this was contributed by a higher content of the amorphous region and its two-phase nature of the blends. Realizing the importance of having a crystalline phase, some studies have focused on increasing the crystallinity of biodegradable blends (Bucci et al. 2007; Zhang and Thomas 2011). The incorporation of PHB, a highly crystalline biopolymer into the PLA matrix by melt blending, is a relatively easy approach to increase crystallinity and control the properties of the blends. Similar Tm values also allowed both polymers to be blended in the melt state. In the previous work of Arrieta et al. (2014), the PLA was melt blended with 25 w.% of PHB and showed an improved water resistance, the oxygen barrier and the reduction of the high transparency typical of PLA.
The presence of microvoids has also allowed water molecules to migrate into the blends and increase the rate of hydrolysis. Therefore, in some case, water absorption from biodegradable polymer blends is followed by hydrolysis that can also affect the absorption rate. Pitt et al. (1992) reported that the water absorption of the blends from poly(glycolic acid-co-lactic acid) (PGLA) and poly(vinyl alcohol) (PVA) increased rapidly at an early stage and then gradually decreased due to hydrolysis of PGLA in the blends. In contrast, Tsuji and Muramatsu (2001) not observed this behavior for PLLA/PVA blends. Rather, the water adsorption increased with immersion time and then reached saturation in 10 h, regardless of the blend ratio. This was due to the low hydrolysis rate of PLLA compared to that of PGLA.
It has also been reported that the hydrolysis rate constant (k) for PLLA and PGLA is 2.59 × 10–3 day-1 (Tsuji et al. 2000) and 7.44 × 10–2 (Cha and Pitt 1990), respectively. Hydrolysis of polymers is influenced by chemical structure (crosslinking, co-monomers) (Li and McCarthy 1999), crystallinity (Li and McCarthy 1999), Mw (Bastioli 2005), pH (Bastioli 2005), polydispersity (Lépine and Gilbert 2002), shape of the sample (Grizzi et al. 1995), temperature (Dell’Erba et al. 2001) and traces of catalysts (Sodergard and Stolt 2002).
On the other hand, blending biodegradable polymer with starch is a good way to balance cost-effective problems and obtain new material that has outstanding properties (Ren et al. 2009). However, the starch is hydrophilic in nature due to the hydroxyl groups present in its structure. Tianyi and Xiuzhi (2000) studied the effects of the blend ratio of starch and PLA on the physical properties of the blends, including water absorption. Figure 3.28 shows the water absorption as a function of soaking time of PLA/starch blends at different ratios. Water absorption was increased sharply on day 1 and then stabilized on day 3 regardless of the blend ratio. According to Tianyi and Xiuzhi (2000), starch was mainly responsible for the water absorption of the blends. Tianyi and Xiuzhi (2000) also reported that the PLA formed a good continuous phase that covers the starch component at a lower starch content (< 60%), resulting in less water absorption (Fig. 3.29). As the starch content increased to >60%, the degree of the discontinuous phase of PLA increased considerably (Fig. 3.29). The water penetration into the blends could occur via voids and be absorbed by starch, resulting in greater water absorption values. A similar observation was reported in several studies of various biodegradable aliphatic polyester/starch blends (Lai et al. 2006; Yu et al. 2006; Liao and Wu 2009).

Water absorption of starch/PLA blends at different ratios depending on water soaking time. Reproduced with permission from Tianyi and Xiuzhi (2000)

Scanning electron microscopy of corn starch/PLA blends: (a) 20:80 and (b) 80:20. Reproduced with permission from Tianyi and Xiuzhi (2000)
In the literature, a great effort has been made to modify the properties of starch-based blends in order to obtain blends with better dispersion, miscibility and morphology, as well as less water absorption. The increase in water resistivity is important because starch disintegrates in water and loses its properties when exposed to environmental humidity, since both amylose and amylopectin are swollen in the presence of water (Liao and Wu 2009). Zeng et al. (2011) also found that the incorporation of hydrophobic PBS significantly reduces the water sensitivity in TPS/PBS blends compared to TPS alone. This is understandable, since PBS is hydrophobic. Interestingly, Yin et al. (2015) obtained a further improvement in water resistance in maleated PBS-containing blends due to good dispersion, homogeneous microstructure of starch and well dispersion of starch particles hindering the diffusion of water molecules into TPS/PBS blends. Similarly, Wu (2003) found that at the same starch content, maleated PCL (PCL-g-MA/starch) blends showed moderately good water resistance compared to PCL/starch blends. The author deduced that this was due to a good miscibility between the components of the blend because of the presence of the carbonyl ester group in the blends.
3.7.2 Weathering
The weathering test is conducted within a specific time to measure the durability of the material towards the elements of environmental exposure. These elements are relative humidity/water, solar radiation, temperature and/or other environmental variables such as pollutants (dirt, dust, nitrogen oxides, sand and sulphur oxides) that can deteriorate the properties of the polymer, and polymer blends and composites (Li 2000; Falk et al. 2001; Matuana et al. 2002; Ariawan et al. 2017). Several studies have also reported that color fading, loss of mechanical properties, surface erosion and weight loss are the most common harmful effects on the polymeric materials that have been exposed to the weathering test (Lopez et al. 2006). Among all the weathering elements, it is important to analyze the solar radiation that induces photo-degradation of polymers, since it greatly influences the properties of polymers during their in-life service, especially in outdoor applications.
Solar radiation induces photo-degradation due to the absorption of UV radiation, which promotes chemical and physical alterations. The complex process of photodegradation involves a sequence of reactions which produce radicals that progress as a sequence of chain mechanisms where peroxides are the oxidation initiators. The presence of a stabilizer and the material structure influences the properties of the final and stable end products. Fully formed products appear as an oxygen-containing structure caused by the collateral reactions (chain branching) or by the self-decaying of peroxyl radicals (chain termination) as shown in Fig. 3.30.

Photodegradation of most polymers by exposure to UV rays. Reproduced with permission from Rosu and Visakh (2016)
On the other hand, few studies have reported on the degradation mechanism of biodegradable polymer blends. In most studies, comparisons between the blends and the original homopolymers in terms of stability have been made (Kaczmarek and Podgoorski 2004; Therias et al. 2010). In some cases, polymer blends with two or more components have less stable combinations (Waldman and De Paoli, 2008). The formation of free radicals in the less stable polymer degrades the more stable polymer. In most blends, the degradation rate of the blends was among that of the components of the blend. However, such a relationship is not obvious or direct due to several factors involved, and the mechanism of radical transport from one polymer to another is not simple. In addition, the photodegradation behavior of polymer blends is greatly influenced by the components of the blend which may deviate from the single polymer system due to chemical interactions between the different species in the blends during degradation and interactions between the degradation products. These chemical reactions can lead to an acceleration of the degradation rate (Kowalonek 2016) or a stabilizing effect (Botta et al. 2009; Fernandes et al. 2010). For example, Rivaton et al. (1998) studied the photolysis rate on PP/PBT blends, and found that photo yellowing was more prevalent for the PP/PBT blend than PBT alone. These authors clarified that the appearance of a more extensive yellowing was due to the macroradicals formed by the photochemical hemolysis of the ester bonds in PBT that cannot extract hydrogen atoms from the PP chains. Figure 3.31 illustrates the mechanism of photolysis, as suggested by the authors. The mechanism includes cross-degradation of the polymer resulting in acceleration degradation of the blends.

Photolysis of PP/PBT blends. Reproduced with permission from Rivaton et al. (1998)
However, in some polymer blends a stabilizing effect could occur. For example, Rivaton et al. (1998) studied polymer blends made from PP containing between 5 and 80% of PE exposed to UV radiation. A blend containing 5% PE endured the maximum degradation, and the degradation became less severe as the concentration of PE increased. This is in line with the performance of the individual polymers where PE had a better resistance since it does not contain susceptible tertiary hydrogens. In this case, the properties of the blends are the sum of the properties of their components. Meanwhile, Kaczmarek et al. (2011) suggested that the PEC/PVA blend is an example in which the blends have superior properties than the sum of the components. As confirmed by the authors, through spectroscopic analysis, they stated that intermolecular interactions between PEC and PVA explained the effect of mutual stabilization.
Among the various research works conducted on polymer blends, studies on biodegradable polymer blends under the weathering test are limited. With this in mind, Persenaire et al. (2014) investigated the weathering behavior of biodegradable polymers in a varied blending ratio of PLLA/PBS with and without the use of MA as a compatibilizer. These authors found that the addition of PBS to PLLA accelerated the degradation of the blend. In addition, PBS demonstrates a higher degradation rate than PLLA. As a consequence, its dispersion in the PLLA matrix improved the PLLA degradability. When considering the PLLA/PBS blend at a 80/20 (w/w) composition as the optimal ratio of polymer blends, the authors deduced that PBS tends to promote the degradation of PLLA in the presence of compatibilizer. Meanwhile, uncompatibilized blends seem less degradable compared to pure PLLA. This phenomenon was attributed to the existence of homogeneously dispersed carboxylic acids produced by PBS degradation, which preferred the hydrolysis of the PLLA matrix.
It can thus be concluded that the prediction of the degradation of polymer blends is not directly related to the behavior of the pure component. The composition of the blend and the possible presence of a compatibilizer can strongly affect the degradation behavior of a polymer blend. The degradation means of the pure components may differ from the blends since interactions can occur between the different components in the blends during degradation or between the degradation products. Therefore, the additive rule cannot be used for the degradation of polymer blends. Apparently, the degradation rate of a polymer blend may be greater, intermediate or lower than that of the pure components. The chemical and physical changes of degraded blends are comparable to those that occur in the single polymer. The possible changes in carbonyl index, the hydroxyl/hydroperoxide index, the optical density of the material can be determined by deterioration of the mechanical properties, IR, UV-Vis, gel content and cloud point determination (Wypych 2008).
3.7.3 Biodegradability
Most conventional polymers are petroleum based, have good resistance against biological degradation (biodegradation), and therefore, exist in the environment for many years (Muniyasamy et al. 2016). This problem has motivated researchers to develop biobased and biodegradable polymers (Gutiérrez 2018a; Toro-Márquez et al. 2018; Gutiérrez et al. 2019; Herniou--Julien et al. 2019). In addition, these polymers have mechanical properties comparable to conventional polymers and have been used as eco-friendly materials in various industries such as automotive, medical, packaging, pharmaceutical and consumer products (Gutiérrez 2018b; Gutiérrez and Alvarez 2017d). They are also widely used for single-use applications and short-term applications, such as food packaging, trash bags, mulch films, personal products and some home care products (Muniyasamy et al. 2016; Merino et al. 2018a, b; 2019a, b).
However, these biodegradable polymers have several drawbacks, such as poor mechanical properties and resistance to environmental degradation, as well as low thermal resistance. For that reason, several approaches have been imposed to modify biodegradable polymers using blending, compatibilization, crosslinking and a combination of them to improve the chemical, thermal and physical properties. Other than that, the lifetime of a biodegradable polymer can be tailored by incorporating additives (e.g. antioxidants, antioxidants, plasticizers and other chemicals) (Gutiérrez 2018c, d; Gutiérrez and Alvarez 2018; Gutiérrez, Herniou-Julien et al. 2018).
Consequently, further studies should be carried out to understand the influence of the modification of chemical, thermal, physical and mechanical properties on the biodegradability and degradation performance of biodegradable polymers in natural environments. Therefore, it is necessary to carry out an exhaustive study on the degradation and biodegradation mechanisms of these polymers to ensure their safe use in various end-use applications. Several international norms and standards can be adopted and used to test the biodegradability properties of biodegradable polymers in a natural environment. The biodegradation rate can also be measured through loss of mechanical properties, morphology variations and weight reduction under different environmental conditions (Muniyasamy et al. 2016). Several standards can be also used as guidelines for biodegradation analysis, such as ASTM D5338–15, biodegradation under composting conditions and ASTM D5988–12, biodegradation under soil burial condition.
Biodegradation can be described as the decomposition of materials by the action of microorganisms, which involve the recycling of carbon, the mineralization of organic compounds and the production of biomass (Lucas et al. 2008). Biodegradation occurs in three steps: biodeterioration, biofragmentation and assimilation, without neglecting the participation of abiotic factors (Fig. 3.32). The biodegradation of polymeric materials involves several steps, so the process can be stopped at each stage, as shown in Fig. 3.34.

Polymer biodegradation process. Reproduced with permission from Lucas et al. (2008)
According to Lucas et al. (2008), the biodegradation process involves several stages. Starting with biodeterioration via the combined reaction of microbes, other decomposing organisms or/and abiotic factors, biodegradable plastics become small pieces. This is followed by depolymerization by microorganism-secreted catalytic agents (e.g. free radicals and enzymes) that can reduce the polymer molecules into oligomers, dimers and monomers. Certain molecules are identified by microbial cell receptors and can across the plasmic membrane. The other molecules remain in the extracellular environment and may be object to different changes. Next is assimilation , where in the cytoplasm, the transported molecules are integrated by microbial metabolism to produce storage vesicles, energy and new biomass. The last stage is mineralization , where some simple and complex metabolites can be excreted and reach the extracellular environment (e.g. aldehydes, antibiotics, organic acids, terpenes, etc.). Simple molecules such as CH4, CO2, H2O, N2 and various salts from intracellular metabolites are completely oxidized and released into the environment.
In addition, polymeric products exposed to outdoor conditions (i.e. aging, burying and weathering) may experience abiotic degradation that will deteriorate the chemical, light, mechanical and thermal properties of polymers.
Keeping this in view, Muniyasamy et al. (2016) reported the biodegradation of PHBV, PLA and PLA/PHBV blends under composting and soil burial conditions according to ASTM D5338–15 and ASTM D5988–12, respectively. The degree of biodegradation was calculated as a percentage of the overall theoretical CO2. For biodegradation under control compositing condition, the test was conducted for 200 days. According to the results, no significant amount of CO2 emission was observed for both pure PHBV and PLA during the first 32 days of compost incubation. After the initial phase, the blends reached 70% biodegradation rate in 120 days and a slight static phase that achieved a 92% biodegradation in 200 days. This slow biodegradation could be due to the crystalline and brittle nature of the PLA. While the delayed degradation of PHBV could be due to an approx. 12% valeric acid content present in the degradation of PHBV polymer.
On the other hand, Muniyasamy et al. (2016) reported that the PLA/PHBV blends showed about 20% biodegradability in the first 32 days because the blending process could break the crystalline structure of the PLA, as well as the surface-enriched heterogeneous phase in one constituent. In terms of physical degradation, all samples (PHBV, PLA and PLA/PHBV blends) began to experience fragmentation after 32 days. The size of fragmented samples became smaller over time and apparently could not be detected in the composting medium after 45 days, thus indicating that the composting environment conditions were adequate for the primary degradation (i.e. fragmentation/disintegration) to occur.
Regarding biodegradation under the soil burial environment, the pure PHBV and PLA/PHBV blends exhibited about 30% biodegradability after 120 days while pure PLA showed no significant biodegradability even after 200 days of exposition. Following Muniyasamy et al. (2016) results indicated the pure PHBV and PLA/PHBV blends have a better degree of biodegradability under the soil burial environment compared to pure PLA. The PLA had a better biodegradation capability under industrial composting conditions than in soil burial because the PLA requires environment temperature in order to trigger hydrolytic degradation of its Mw before allowing the action of microbial degradation (thermophilic bacteria and fungi).
Lucas et al. (2008) mentioned that another way in which the polymer can undergo chemical degradation during biodegradation in the compost or in the soil environment is by hydrolysis. Polymers containing hydrolysable functional groups, such as amide, anhydride, carbamide, ester and ether can be hydrolyzed. However, the degradation reaction depends on some parameters such as pH, temperature, time and water activity. The crystalline fraction could prevent the diffusion of oxygen and water, thus limiting hydrolytic degradation. While amorphous domains which disorganize molecular regions are prone to oxidative and hydrolytic degradations. Lucas et al. (2008) proposed the schematic reaction of the PLA hydrolysis under acidic and alkaline condition as examples of abiotic degradation via chemical degradation, as shown in Figs. 3.33 and 3.34, respectively.

PLA hydrolysis under acidic conditions. Reproduced with permission from Lucas et al. (2008)

PLA hydrolysis under alkaline conditions. Reproduced with permission from Lucas et al. (2008)
These biodegradation conditions require adequate controls of moisture content, pH and temperature, and a specific thermophilic microorganism, which is only available in an industrial composting facility to adequately test the biodegradation of plastic materials. In addition, the biodegradation of polymer blends depends largely on the ratio of the components and the type of polymer blend. For example, Zhang and Thomas (2011) observed that the PHB/PLA blends and the components of the blends experienced a different degradation mechanism. In this sense, the PHB is mainly degraded by the reaction of several enzymes on the surface, while the degradation of the PLA begins with non-enzymatic hydrolysis, which depends largely on the ambient temperature.
In another study by Liu et al. (2019) reported the species of soil bacteria responsible for the biodegradation of PHB, PLA and their blends. In this accelerated biodegradation test, the soil bacteria were collected and cultured several times, approximately three times in the generation of bacterial culture (Gen III) until a stable bacterial community was achieved before the biodegradation test. The bacteria were then inoculated into a vial filled with several nonwoven samples and grown for 15 days on a shaker at 150 rpm at room temperature. As expected, the morphological observation revealed that the incubation of nonwoven blends for 15 days with the soil bacterial resulted in severe degradation with different changes, such as the presence of pores and microcracks on the nonwoven samples. In line with this, the CO2 evolved during the 15 day of incubation period also exhibited an increasing percentage of biodegradation over time.
Liu et al. (2019) in order to identify bacterial species that may be responsible in the biodegradation process, extracted the Gen III bacteria DNA and then subjected to pyrosequencing analysis. From the sequencing analysis, the relative abundance of marker gene sequences in the samples was associated with members of the genera Citrobacter, Lysinibacillus (phylum Firmicutes) and Pseudomonas (phylum Proteobacteria), which demonstrates that these newly identified bacterial species can play an important role in the biodegradation of PHB/PLA-blended nonwovens (Liu et al. 2019). Several species belong to the genus Citrobacter , such as C. amalo-naticus, C. freundii and C. koseri, which have been reported to use polyhydroxyl compounds as the sole carbon source (Liu et al. 2019).
According to Shah et al. (2008), the biodegradation rate of polymer blends is mainly controlled by the degradation of the most susceptible biodegradation components with respect to the type of presence of bacteria and the environmental condition. Table 3.8 shows a list of microorganisms that could degrade biodegradable polyesters.
To date, limited studies have reported on the biodegradability of biodegradable polymer blends and the effect of the compatibilizer and crosslinking agent on the biodegradation rate. With this in mind, a slower biodegradation for compatibilized blends in another mixing systems (biodegradable/non-biodegradable polymer blends) such as PC/PLA (Lee et al. 2011), PHB/PP (Sadi et al. 2013) and PBS/PP (Bionelle/CPP) blends (Zainuddin et al. 1999) have been informed.
As reported by Zainuddin et al. (1999), the addition of Modic compatibilizer into PBS/PP blends improved the dispersion of the dispersed phase into finer droplets, thus allowing the microbes to attack randomly and penetrate the blends as a whole matrix. However, the biodegradability of the compatibilized blends seems to depend on the ratio of the blends. At the 25/75 blend ratio, the biodegradability was considerably lower is due to the PBS droplets since they were protected from microbial attacks and degradation by the surrounding PP layer as the interfacial interaction between the components of blend. On the contrary, the degradation rates appear to increase exponentially up to 75% PBS content due to the changes in the morphology of the blends and the presence of higher PBS-rich area.
Hu et al. (2018) investigated the effect of BPO as a crosslinking agent on the biodegradable properties of PBS/PLA blends. In this study, the biodegradation rate was determined by the enzymatic degradation of proteinase K from the Tritirachium album. The hydrophobic interaction of proteinase K is well known to be the main cause of degradation of PLA. Aside from that, it could reside on the surface of PLA and be able to change its conformation to create the catalytic site to hydrolyze PLA around the enzyme molecules. Hu et al. (2018) reported that the BPO-crosslinked PBS/PLA blends showed a better resistance to enzymatic degradation of proteinase K than pure PLA. It is known that PBS is relatively less susceptible to degradation of proteinase K than PLA and that the incorporation of PBS in the blends reduces the degradation rate of the blends. In addition, the introduction of peroxide crosslinking can change the structure of the blends and limit degradation by proteinase K (Hu et al. 2018). Along with the degradation, the surface morphology of PBS/PLA/BPO blends changed from the filament to the segments and finally to the particles as structures. It can also be concluded from the work of Hu et al. (2018) that the uncrosslinked PLA fraction was degraded first and left the crosslinked PLA/PBS filaments in the blends. Then, the fragments of crosslinked PLA began to degrade, causing the filaments to change to segments and eventually to particles. The remaining particles or fragments could be associated with the PBS and a portion of non-degraded crosslinked PLA. The results obtained in this study were useful to tailor the biodegradation characteristic of the new biodegradable polymers and can also be used to understand the biodegradation mechanisms in new polymers or mixing systems.
3.8 Conclusions
In recent decades, biodegradable polymers have been developed, along with many published studies on biodegradable polymers. The research in this field is focused on strategies to improve the mechanical, thermal and biodegradable properties of polymers through many approaches, such as compatibilization, copolymerization, crosslinking and addition of reinforcement or functional additives.
In terms of compatibilizer, maleated compatibilizers have been the most studied and are well established in the polymer industry. It has also been demonstrated that the incorporation of maleated compatibilizers improves the mechanical properties of binary and ternary biodegradable polymer blends. The maleated compatibilizer also reduces the interfacial energy, which results in a reduction of the size of the dispersed phase. It is known that the presence of active chemical sites on the MA (i.e. the double bond and the carboxylic acid group) is a powerful electron-accepting monomer and reactive. The most extensive method to produce compatibilizer is by the melt grafting technique, which involves the grafting of MA onto polymer in a molten state with the presence of a free radical initiator. MA has been used successfully as a monomer to graft onto biodegradable polymers such as PBS, PBSA, PCL and PLA to produce maleated compatibilizer.
Other than that, crosslinking by means of radical peroxides is an interesting strategy that has been widely used to stabilize and improve the properties of biodegradable polymer blends due to its simplicity and efficiency. Among these peroxides, DCP is the most widely used peroxide crosslinking agent in the biodegradable polymer blend system. The use of peroxides promotes the formation of chain branches, as well as crosslinking, which increases the Tg, the modulated crystalline content and the thermal behavior of polymer blends. The crosslinking reaction is also an effective means to improve interfacial adhesion in immiscible polymer blends by initiating the appearance of mixed chains (copolymers), which act as compatibilizers at the interfaces. The formation of chain branching or crosslinking by heterogeneous and/or homogeneous radical coupling reactions also depends on the concentration of peroxides and the reaction temperature used.
In addition, reactive compatibilization, either by incorporating a maleated compatibilizer or peroxide-induced crosslinking, can be performed in one step (in-situ) or two-step extrusion, depending on the available processing equipment and the system of blends and additives used. In addition to the compatibilization and crosslinking approaches, the combination of the crosslinked-compatibilization approach seems to be more effective for improving the mechanical properties such as σ, εb and T of the biodegradable polymer blends. The synergistic effect of crosslinking and compatibilization is produced due to the presence of a compatibilizer, which reduces the interfacial energy and forms a better interfacial adhesion which in turn helps the guest polymer to be well dispersed, thus providing larger interface area between the components of the blend. When the peroxide radical crosslinking agent is added, the higher number of crosslinking will be formed at the interface region, thus resulting in a significant increase in the properties of the blends.
Finally, the performance of biodegradable blends depends on the moisture or water exposure. Likewise, the biodegradation rate of polymers in the natural environment depends largely on the characteristics of the components of the blend and how they interact with environmental factors. Additions of compatibilizer or crosslinking agent have improved the resistance of the blends to environmental degradation. However, due to limited studies on the effect of compatibilizer and crosslinking agent on the performance of biodegradable polymer blends and natural degradation, a comprehensive study is necessary to understand the degradation mechanism and its safe use in many applications. International norms and standards should be used to test the degradation properties of biodegradable polymers in the natural environment in order to allow these materials to be used safely in consumer products.
Abbreviations
- 3HH:
-
3-hydroxyhexanoate
- AAc:
-
Acrylic acid
- ABS:
-
Acrylonitrile butadiene styrene
- AIBN:
-
2,2-azobis(isobutyronitrile)
- ATP:
-
Aliphatic thermoplastic polyester
- BPO:
-
Benzoyl peroxide
- BTBV:
-
n-butyl 4,4-di-(t-butyl peroxy) valerate
- CTC:
-
Charge transfer complex
- DBP:
-
di-t-butyl peroxide
- DCP:
-
Dicumyl peroxide
- DG:
-
Degree of grafting
- DMA:
-
Dynamic mechanical analysis
- DMTA:
-
Dynamic mechanical thermal analysis
- DSC:
-
Differential scanning calorimetry
- ENR:
-
Epoxidized natural rubber
- EPDM:
-
Ethylene propylene diene monomer
- EVA:
-
Ethylene vinyl acetate
- FTIR:
-
Fourier transform infrared
- G’:
-
Storage module
- GMA:
-
Glycidyl methacrylate
- HA:
-
Hyaluronan
- HDPE:
-
High-density polyethylene
- HEMA:
-
2-hydroxyethyl methacrylate
- HHx:
-
Hydroxyhexanoate
- HVx:
-
Hydroxyvalerate
- LLDPE:
-
Linear low-density polyethylene
- LPO:
-
Dilauroyl peroxide
- MA:
-
Maleic anhydride
- MAPP:
-
MA-grafted PP
- MMA:
-
Methyl methacrylate
- Mw:
-
Molecular weight
- ɲ*:
-
Complex viscosity
- NBR:
-
Butadiene-acrylonitrile rubber
- NMR:
-
Nuclear magnetic resonance
- P(3HB):
-
Poly(3-hydroxybutyrate)
- P(3HB-co-4HB):
-
Poly(3-hydroxybutyrate-co-4-hydroxybutyrate)
- PBAT:
-
Poly(butylene adipate-co-terephthalate)
- PBES:
-
Poly(butylenesuccinate-co-ethylene succinate)
- PBS:
-
Poly(butylene succinate)
- PBSA:
-
Poly(butylene succinate-co-adipate)
- PBSL:
-
Poly(butylene succinate-co-L-lactate)
- PC:
-
Poly(carbonate)
- PCL:
-
Poly(ε-caprolactone)
- PDLLA:
-
Poly(D,L-lactic acid)
- PE:
-
Poly(ethylene)
- PEG:
-
Poly(ethylene glycol)
- PEO:
-
Poly(ethylene oxide)
- PES:
-
Poly(ethylene succinate)
- PET:
-
Poly(ethylene terephthalate)
- PGLA:
-
Poly(glycolic acid-co-lactic acid)
- PHB:
-
Poly(hydroxybutyrate)
- PHB-co-HHx:
-
Poly(3-hydroxybutyrate-co-3-hydroxyhexanoate)
- PHBHH:
-
PHB-co-HHx
- PHBV:
-
Poly(3-hydroxybutyrate-co-3-hydroxyvalerate)
- PLA:
-
Poly(lactic acid)
- PLA•:
-
PLA macroradicals
- PLLA:
-
Poly(L-lactic acid)
- PP:
-
Poly(propylene)
- PPO:
-
Poly(phenylene oxide)
- PVA:
-
Poly(vinyl alcohol)
- PVC:
-
Poly(vinyl chloride)
- REx:
-
Reactive extrusion
- RO•:
-
Primary radicals
- SEBS:
-
Styrene-ethylene-butylene-styrene
- St:
-
Styrene
- St•:
-
Styryl macroradicals
- T:
-
Toughness
- TBEC:
-
OO-(t-butyl) O-(2-ethylhexyl) peroxycarbonate
- TBEH:
-
t-butyl peroxy-2-ethylhexanoate
- TBPB:
-
t-butyl peroxy benzoate
- TBTH:
-
t-butyl peroxy-3,5,5-trimethylhexanoate
- Tg:
-
Glass transition temperature
- THF:
-
Tetrahydrofuran
- Tm:
-
Melting temperature
- TPS:
-
Thermoplastic starch
- UHMEPE:
-
Ultra-high Mw polyethylene
- UV:
-
Ultraviolet
- εb:
-
Strain at break
- σ:
-
Tensile strength
References
Abdul, R. H., Wahab, M. S., & Wahit, M. U. (2013). Improvement of mechanical properties of polycaprolactone (PCL) by Addition of nano-montmorillonite (MMT) and hydroxyapatite (HA). Applied Mechanics and Materials, 315(2013), 815–819. https://doi.org/10.4028/www.scientific.net/amm.315.815.
Ahmad Thirmizir, M. Z. (2011). Characterization of kenaf bast fibre filled poly(butylene succinate) composites : Mechanical, water absorption and weathering. Universiti Sains Malaysia.
Thirmizir, M. A., Ishak, Z. M., Taib, R. M., Rahim, S., & Jani, S. M. (2011). Kenaf-bast-fiber-filled biodegradable poly(butylene succinate) composites: Effects of fiber loading, fiber length, and maleated poly(butylene succinate) on the flexural and impact properties. Journal of Applied Polymer Science, 122(5), 3055–3063. https://doi.org/10.1002/app.34046.
Sigmaaldrich.com. (n.d.). Applications: free radical initiators. Retrieved June 26, 2020, from https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Aldrich/General_Information/thermal_initiators.pdf?utm_source=redirect&utm_medium=promotional&utm_campaign=insite_thermal_initiators
Arrieta, M. P., Samper, M. D., López, J., & Jiménez, A. (2014). Combined effect of poly(hydroxybutyrate) and plasticizers on polylactic acid properties for film intended for food packaging. Journal of Polymers and the Environment, 22(4), 460–470. https://doi.org/10.1007/s10924-014-0654-y.
Barghini, A., Ivanova, V. I., Imam, S. H., & Chiellini, E. (2010). Poly-(ε-caprolactone) (PCL) and poly(hydroxy-butyrate) (PHB) blends containing seaweed fibers: Morphology and thermal-mechanical properties. Journal of Polymer Science, Part A: Polymer Chemistry, 48(23), 5282–5288. https://doi.org/10.1002/pola.24327.
Bastioli, C. (2005). Handbook of biodegradable polymers. Rapra Technology Limited. Available in: http://appliedchem.unideb.hu/biodegradabilis/bastioli%20c.%20(ed)%20-%20handbook%20of%20biodegradable%20polymers%20-%20(rapra%202005).pdf
Berthé, V., Ferry, L., Bénézet, J. C., & Bergeret, A. (2010). Ageing of different biodegradable polyesters blends mechanical and hygrothermal behavior. Polymer Degradation and Stability, 95(3), 262–269. https://doi.org/10.1016/j.polymdegradstab.2009.11.008.
Botta, L., Dintcheva, N. T., & La Mantia, F. P. (2009). The role of organoclay and matrix type in photo-oxidation of polyolefin/clay nanocomposite films. Polymer Degradation and Stability, 94(4), 712–718. https://doi.org/10.1016/j.polymdegradstab.2008.12.017.
Bucci, D. Z., Tavares, L. B. B., & Sell, I. (2007). Biodegradation and physical evaluation of PHB packaging. Polymer Testing, 26(7), 908–915. https://doi.org/10.1016/j.polymertesting.2007.06.013.
Can, E., Bucak, S., Kinaci, E., Çalikoǧlu, A. C., & Köse, G. T. (2014). Polybutylene succinate (PBS)-polycaprolactone (PCL) blends compatibilized with poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) (PEO-PPO-PEO) copolymer for biomaterial applications. Polymer - Plastics Technology and Engineering, 53(11), 1178–1193. https://doi.org/10.1080/03602559.2014.886119.
Cha, Y., & Pitt, C. G. (1990). The biodegradability of polyester blends. Biomaterials, 11(2), 108–112. https://doi.org/10.1016/0142-9612(90)90124-9.
Chen, C., Peng, S., Fei, B., Zhuang, Y., Dong, L., Feng, Z., Chen, S., & Xia, H. (2003). Synthesis and characterization of maleated poly(3-hydroxybutyrate). Journal of Applied Polymer Science, 88(3), 659–668. https://doi.org/10.1002/app.11771.
Chen, G.-Q., & Luo, R.-C. (2009). Polyhydroxyalkanoate blends and composites polyhydroxyalkanoate blends and composites. In L. Yu (Ed.), Biodegradable polymer blends and composites from renewable resources (pp. 239–286). New Jersey: John Wiley & Sons. https://doi.org/10.1002/9780470391501.ch11.
Choi, N. S., Kim, C. H., Cho, K. Y., & Park, J. K. (2002). Morphology and hydrolysis of PCL/PLLA blends compatibilized with P(LLA-co-εCL) or P(LLA-b-εCL). Journal of Applied Polymer Science, 86(8), 1892–1898. https://doi.org/10.1002/app.11134.
Dell’Erba, R., Groeninckx, G., Maglio, G., Malinconico, M., & Migliozzi, A. (2001). Immiscible polymer blends of semicrystalline biocompatible components: Thermal properties and phase morphology analysis of PLLA/PCL blends. Polymer, 42(18), 7831–7840. https://doi.org/10.1016/s0032-3861(01)00269-5.
Deng, Y., & Thomas, N. L. (2015). Blending poly(butylene succinate) with poly(lactic acid): Ductility and phase inversion effects. European Polymer Journal, 71, 534–546. https://doi.org/10.1016/j.eurpolymj.2015.08.029.
Doi, Y., Kitamura, S., & Abe, H. (1995). Microbial synthesis and characterization of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate). Macromolecules, 28(14), 4822–4828. https://doi.org/10.1021/ma00118a007.
Dong, W., Ma, P., Wang, S., Chen, M., Cai, X., & Zhang, Y. (2013). Effect of partial crosslinking on morphology and properties of the poly(β-hydroxybutyrate)/poly(d,l-lactic acid) blends. Polymer Degradation and Stability, 98(9), 1549–1555. https://doi.org/10.1016/j.polymdegradstab.2013.06.033.
Drumright, R. E., Gruber, P. R., & Henton, D. E. (2000). Polylactic acid technology. Advanced Materials, 12(23), 1841–1846. https://doi.org/10.1002/1521-4095(200012)12:23<1841::aid-adma1841>3.0.co;2-e.
Environment Australia. (2012). Biodegradable plastics – Developments and environmental impacts. Melbourne. Available in: http://www.europeanplasticfilms.eu/docs/australianreportonbiodegradableplastics.pdf
Fei, B., Chen, C., Chen, S., Peng, S., Zhuang, Y., An, Y., & Dong, L. (2004). Crosslinking of poly[(3-hydroxybutyrate)-co-(3-hydroxyvalerate)] using dicumyl peroxide as initiator. Polymer International, 53(7), 937–943. https://doi.org/10.1002/pi.1477.
Fernandes, L. L., Freitas, C. A., Demarquette, N. R., & Fechine, G. J. M. (2010). Photodegradation of thermodegraded polypropylene/high-impact polystyrene blends: Mechanical properties. Journal of Applied Polymer Science, 120(2), 770–779. https://doi.org/10.1002/app.
Fink, J. K. (2013). Reactive polymers fundamentals and applications: A concise guide to industrial polymers (Second ed., p. 576). William Andrew. https://doi.org/10.1016/c2012-0-02516-1.
Gao, H., Xie, Y., Ou, R., & Wang, Q. (2012). Grafting effects of polypropylene/polyethylene blends with maleic anhydride on the properties of the resulting wood-plastic composites. Composites Part A: Applied Science and Manufacturing, 43(1), 150–157. https://doi.org/10.1016/j.compositesa.2011.10.001.
Gardella, L., Calabrese, M., & Monticelli, O. (2014). PLA maleation: An easy and effective method to modify the properties of PLA/PCL immiscible blends. Colloid and Polymer Science, 292(9), 2391–2398. https://doi.org/10.1007/s00396-014-3328-3.
Grizzi, I., Garreau, H., Li, S., & Vert, M. (1995). Hydrolytic degradation of devices based on poly(dl-lactic acid) size-dependence. Biomaterials, 16(4), 305–311. https://doi.org/10.1016/0142-9612(95)93258-f.
Gumede, T. P., Luyt, A. S., & Müller, A. J. (2018). Review on PCL, PBS, and PCL/PBS blends containing carbon nanotubes. Express Polymer Letters, 12(6), 505–529. https://doi.org/10.3144/expresspolymlett.2018.43.
Gutiérrez, T. J., & Alvarez, V. A. (2017a). Data on physicochemical properties of active films derived from plantain flour/PCL blends developed under reactive extrusion conditions. Data in Brief, 15, 445–448. https://doi.org/10.1016/j.dib.2017.09.071.
Gutiérrez, T. J., & Alvarez, V. A. (2017b). Eco-friendly films prepared from plantain flour/PCL blends under reactive extrusion conditions using zirconium octanoate as a catalyst. Carbohydrate Polymers, 178, 260–269. https://doi.org/10.1016/j.carbpol.2017.09.026.
Gutiérrez, T. J., & Alvarez, V. A. (2017c). Properties of native and oxidized corn starch/polystyrene blends under conditions of reactive extrusion using zinc octanoate as a catalyst. Reactive and Functional Polymers, 112, 33–44. https://doi.org/10.1016/j.reactfunctpolym.2017.01.002.
Gutiérrez, T. J., & Alvarez, V. A. (2017d). Films made by blending poly(ε-caprolactone) with starch and flour from sagu rhizome grown at the Venezuelan Amazons. Journal Polymers and the Environment, 25(3), 701–716. https://doi.org/10.1007/s10924-016-0861-9.
Gutiérrez, T. J., & Alvarez, V. A. (2018). Bionanocomposite films developed from corn starch and natural and modified nano-clays with or without added blueberry extract. Food Hydrocolloids, 77, 407–420. https://doi.org/10.1016/j.foodhyd.2017.10.017.
Gutiérrez, T. J. (2018a). Chapter 9. Biodegradability and compostability of food nanopackaging materials. In G. Cirillo, M. A. Kozlowski, & U. G. Spizzirri (Eds.), Composite materials for food packaging (pp. 269–296). EE.UU.. ISBN: 978-1-119-16020-5: WILEY-Scrivener Publisher. https://doi.org/10.1002/9781119160243.ch9.
Gutiérrez, T. J. (2018b). Biological macromolecule composite films made from sagu starch and flour/poly(ε-caprolactone) blends processed by blending/thermo molding. Journal Polymers and the Environment, 26(9), 3902–3912. https://doi.org/10.1007/s10924-018-1268-6.
Gutiérrez, T. J. (2018c). Active and intelligent films made from starchy sources/blackberry pulp. Journal Polymers and the Environment, 26(6), 2374–2391. https://doi.org/10.1007/s10924-017-1134-y.
Gutiérrez, T. J. (2018d). Are modified pumpkin flour/plum flour nanocomposite films biodegradable and compostable? Food Hydrocolloids, 83, 397–410. https://doi.org/10.1016/j.foodhyd.2018.05.035.
Gutiérrez, T. J., Guarás, M. P., & Alvarez, V. A. (2017). Chapter 9. Reactive extrusion for the production of starch-based biopackaging. In M. A. Masuelli (Ed.), Biopackaging (pp. 287–315). Miami, EE.UU.. ISBN: 978-1-4987-4968-8: Editorial CRC Press Taylor & Francis Group. https://doi.org/10.1201/9781315152349-9.
Gutiérrez, T. J., Herniou-Julien, C., Álvarez, K., & Alvarez, V. A. (2018). Structural properties and in vitro digestibility of edible and pH-sensitive films made from guinea arrowroot starch and wastes from wine manufacture. Carbohydrate Polymers, 184, 135–143. https://doi.org/10.1016/j.carbpol.2017.12.039.
Gutiérrez, T. J., Toro-Márquez, L. A., Merino, D., & Mendieta, J. R. (2019). Hydrogen-bonding interactions and compostability of bionanocomposite films prepared from corn starch and nano-fillers with and without added Jamaica flower extract. Food Hydrocolloids, 89, 283–293. https://doi.org/10.1016/j.foodhyd.2018.10.058.
Harrats, C., & Groeninckx, G. (2007). Reactive processing of polymer blend using reactive compatibilization and dynamic crosslinking: Phase morphology control and microstructure – Property relations. In F. Ciardelli & S. Penczek (Eds.), Modification and blending of synthetic and natural macromolecules (pp. 155–199). Dordrecht: Kluwer Academic Publishers. https://doi.org/10.1007/978-1-4020-2735-2_9.
He, X., Zheng, S., Huang, G., & Rong, Y. (2013). Solution grafting of maleic anhydride on low-density polyethylene: Effect on crystallization behavior. Journal of Macromolecular Science, Part B: Physics, 52(9), 1265–1282. https://doi.org/10.1080/00222348.2013.764217.
Hiljanen-Vainio, M., Varpomaa, P., Seppälä, J., & Törmälä, P. (1996). Modification of poly(L-lactides) by blending: mechanical and hydrolytic behavior. Macromolecular Chemistry and Physics, 197(4), 1503–1523. https://doi.org/10.1002/macp.1996.021970427.
Herniou--Julien, C., Mendieta, J. R., & Gutiérrez, T. J. (2019). Characterization of biodegradable/non-compostable films made from cellulose acetate/corn starch blends processed under reactive extrusion conditions. Food Hydrocolloids, 89, 67–79. https://doi.org/10.1016/j.foodhyd.2018.10.024.
Hu, X., Su, T., Li, P., & Wang, Z. (2018). Blending modification of PBS/PLA and its enzymatic degradation. Polymer Bulletin, 75(2), 533–546. https://doi.org/10.1007/s00289-017-2054-7.
Imre, B., & Pukánszky, B. (2013). Compatibilization in bio-based and biodegradable polymer blends. European Polymer Journal, 49(6), 1215–1233. https://doi.org/10.1016/j.eurpolymj.2013.01.019.
James, S. P., Kurkowski Rachael, O., Zhang, M., & Schwartz, H. (2009). UHMWPE/hyaluronan microcomposite biomaterials. In S. M. Kurtz (Ed.), UHMWPE biomaterials handbook: Ultra High Molecular Weight Polyethylene in Total joint replacement and medical devices (2nd ed., pp. 259–276). London: Academic. https://doi.org/10.1016/b978-0-12-374721-1.00018-3.
Ji, D., Liu, Z., Lan, X., Wu, F., Xie, B., & Yang, M. (2014). Morphology, rheology, crystallization behavior, and mechanical properties of poly(lactic acid)/poly(butylene succinate)/dicumyl peroxide reactive blends. Journal of Applied Polymer Science, 131(3), 1–8. https://doi.org/10.1002/app.39580.
John, J., Tang, J., Yang, Z., & Bhattacharya, M. (1997). Synthesis and characterization of anhydride-functional polycaprolactone. Journal of Polymer Science, Part A: Polymer Chemistry, 35(6), 1139–1148. https://doi.org/10.1002/(sici)1099-0518(19970430)35:6<1139::aid-pola17>3.0.co;2-7.
Kaczmarek, H., Dabrowska, A., & Vukovic-Kwiatkowska, I. (2011). Accelerated weathering of pectin/poly(vinyl alcohol) blends studied by spectroscopic methods halina. Journal of Applied Polymer Science, 122(3), 1936–1945. https://doi.org/10.1002/app.34298.
Kaczmarek, H., & Podgoorski, A. (2004). Photostability of poly(vinyl chloride)/poly(vinyl alcohol) blends. Molecular Crystals and Liquid Crystals, 416(3), 479–484. https://doi.org/10.1080/15421400490482916.
Kalambur, S., & Rizvi, S. S. H. (2006). An overview of starch-based plastic blends from reactive extrusion. Journal of Plastic Film and Sheeting, 22(1), 39–58. https://doi.org/10.1177/8756087906062729.
Karami, S., Nazockdast, H., Ahmadi, Z., Rabolt, J. F., Noda, I., & Chase, D. B. (2019). Microstructure effects on the rheology of nanoclay-filled PHB/LDPE blends. Polymer Composites, 40(10), 4125–4134. https://doi.org/10.1002/pc.25273.
Kim, Y. J., & Park, O. O. (1999). Miscibility and biodegradability of poly(butylene succinate)/poly(butylene terephthalate) blends. Journal of Environmental Polymer Degradation, 7(1), 53–66. https://doi.org/10.1023/a:1021846219775.
Koning, D., Pagnoulle, & Jerome. (1998). Strategies for compatibilization of polymer blends. Progress in Polymer Science, 23(97), 707–757. https://doi.org/10.1016/s0079-6700(97)00054-3.
Kowalonek, J. (2016). Surface studies of UV-irradiated poly(vinyl chloride)/poly(methyl methacrylate) blends. Polymer Degradation and Stability, 133, 367–377. https://doi.org/10.1016/j.polymdegradstab.2016.09.016.
Lai, S. M., Don, T. M., & Huang, Y. C. (2006). Preparation and properties of biodegradable thermoplastic starch/poly(hydroxy butyrate) blends. Journal of Applied Polymer Science, 100(3), 2371–2379. https://doi.org/10.1002/app.23085.
Laycock, B., Halley, P., Pratt, S., Werker, A., & Lant, P. (2014). The chemomechanical properties of microbial polyhydroxyalkanoates. Progress in Polymer Science, 39(2), 397–442. https://doi.org/10.1016/j.progpolymsci.2013.06.008.
Lee, J. B., Lee, Y. K., Choi, G. D., Na, S. W., Park, T. S., & Kim, W. N. (2011). Compatibilizing effects for improving mechanical properties of biodegradable poly(lactic acid) and polycarbonate blends. Polymer Degradation and Stability, 96(4), 553–560. https://doi.org/10.1016/j.polymdegradstab.2010.12.019.
Lépine, L., & Gilbert, R. (2002). Thermal degradation of polyacrylic acid in dilute aqueous solution. Polymer Degradation and Stability, 75(2), 337–345. https://doi.org/10.1016/s0141-3910(01)00236-1.
Li, S., & McCarthy, S. (1999). Influence of crystallinity and stereochemistry on the enzymatic degradation of poly(lactide)s. Macromolecules, 32(13), 4454–4456. https://doi.org/10.1021/ma990117b.
Liao, H. T., & Wu, C. S. (2009). Preparation and characterization of ternary blends composed of polylactide, poly(ε-caprolactone) and starch. Materials Science and Engineering A, 515(1–2), 207–214. https://doi.org/10.1016/j.msea.2009.03.003.
Liu, Y., Zhan, Z., Ye, H., Lin, X., Yan, Y., & Zhang, Y. (2019). Accelerated biodegradation of PLA/PHB-blended nonwovens by a microbial community. RSC Advances, 9(18), 10386–10394. https://doi.org/10.1039/c8ra10591j.
Martin, L. W. (2019). Overview of Maleic-Anhydride-Grafted Polyolefin Coupling Agents. Retrieved April 14, 2019, from http://www.temp.speautomotive.com/SPEA_CD/SPEA2012/pdf/TP/TP9.pdf
Lucas, N., Bienaime, C., Belloy, C., Queneudec, M., Silvestre, F., & Nava-Saucedo, J.-E. (2008). Polymer biodegradation: Mechanisms and estimation techniques – A review. Chemosphere, 73(4), 429–442. https://doi.org/10.1016/j.chemosphere.2008.06.064.
Ma, P., Xu, P., Chen, M., Dong, W., Cai, X., Schmit, P., Spoelstra, A. B., & Lemstra, P. J. (2014c). Structure-property relationships of reactively compatibilized PHB/EVA/starch blends. Carbohydrate Polymers, 108(1), 299–306. https://doi.org/10.1016/j.carbpol.2014.02.058.
Ma, P., Cai, X., Wang, W., Duan, F., Shi, D., & Lemstra, P. J. (2014a). Crystallization behavior of partially crosslinked poly(β-hydroxyalkonates)/poly(butylene succinate) blends. Journal of Applied Polymer Science, 131(21), 1–8. https://doi.org/10.1002/app.41020.
Ma, P., Hristova-Bogaerds, D. G., Lemstra, P. J., Zhang, Y., & Wang, S. (2012). Toughening of PHBV/PBS and PHB/PBS blends via in situ compatibilization using dicumyl peroxide as a free-radical grafting initiator. Macromolecular Materials and Engineering, 297(5), 402–410. https://doi.org/10.1002/mame.201100224.
Ma, P., Jiang, L., Ye, T., Dong, W., & Chen, M. (2014b). Melt free-radical grafting of maleic anhydride onto biodegradable poly(lactic acid) by using styrene as a comonomer. Polymers, 6(5), 1528–1543. https://doi.org/10.3390/polym6051528.
Mani, R., Bhattacharya, M., & Tang, J. (1999). Functionalization of polyesters with maleic anhydride by reactive extrusion. Journal of Polymer Science: Part A: Polymer Chemistry, 37(11), 1693–1702. https://doi.org/10.1002/(sici)1099-0518(19990601)37:11<1693::aid-pola15>3.0.co;2-y.
Mani, R., & Bhattacharya, M. (2008). Properties of injection moulded blends of starch and modified biodegradable polyesters. European Polymer Journal, 37(3), 515–526. https://doi.org/10.1016/s0014-3057(00)00155-5.
Mantere, L. (2015). Functional polyethylene as a compatibilizer in blends of recycled polyethylenes and polyamides. Tampere University of technology.
Markham, R. L. (1990). Introduction to compatibilization of polymer blends. Advances in Polymer Technology, 10(3), 231–236. https://doi.org/10.1002/adv.1990.060100307.
Mehrabzadeh, M. (2009). Maleic anhydride grafting onto HDPE by in situ reactive extrusion and its effect on intercalation and mechanical properties of HDPE/clay nanocomposites. Iranian Polymer Journal, 18(10), 833–842. Available in: http://194.225.115.91/manuscripts/ipj-2009-10-4941.pdf
Merino, D., Gutiérrez, T. J., & Alvarez, V. A. (2019a). Potential agricultural mulch films based on native and phosphorylated corn starch with and without surface functionalization with chitosan. Journal Polymers and the Environment, 27(1), 97–105. https://doi.org/10.1007/s10924-018-1325-1.
Merino, D., Gutiérrez, T. J., & Alvarez, V. A. (2019b). Structural and thermal properties of agricultural mulch films based on native and oxidized corn starch nanocomposites. Starch-Stärke, 71(7–8), 1800341. https://doi.org/10.1002/star.201800341.
Merino, D., Gutiérrez, T. J., Mansilla, A. Y., Casalongué, C. A., & Alvarez, V. A. (2018a). Critical evaluation of starch-based antibacterial nanocomposites as agricultural mulch films: Study on their interactions with water and light. ACS Sustainable Chemistry & Engineering, 6(11), 15662–15672. https://doi.org/10.1021/acssuschemeng.8b04162.
Merino, D., Mansilla, A. Y., Gutiérrez, T. J., Casalongué, C. A., & Alvarez, V. A. (2018b). Chitosan coated-phosphorylated starch films: Water interaction, transparency and antibacterial properties. Reactive and Functional Polymers, 131, 445–453. https://doi.org/10.1016/j.reactfunctpolym.2018.08.012.
Mishra, J. K., Chang, Y. W., & Kim, D. K. (2007). Green thermoplastic elastomer based on polycaprolactone/epoxidized natural rubber blend as a heat shrinkable material. Materials Letters, 61(17), 3551–3554. https://doi.org/10.1016/j.matlet.2006.11.119.
Mishra, J. K., & Wonho, Y. C. (2011). The effect of peroxide crosslinking on thermal, mechanical, and rheological properties of polycaprolactone/epoxidized natural rubber blends. Polymer Bulletin, 66(5), 673–681. https://doi.org/10.1007/s00289-010-0376-9.
Moghaddam, L., Rintoul, L., Halley, P. J., George, G. A., & Fredericks, P. M. (2012). In-situ monitoring by fibre-optic NIR spectroscopy and rheometry of maleic anhydride grafting to polypropylene in a laboratory scale reactive extruder. Polymer Testing, 31(1), 155–163. https://doi.org/10.1016/j.polymertesting.2011.10.002.
Muniyasamy, S., Ofosu, O., John, M. J., & Anandjiwala, R. D. (2016). Mineralization of poly(lactic acid) (PLA), poly(3-hydroxybutyrate-co-valerate) (PHBV) and PLA/PHBV blend in compost and soil environments. Journal of Renewable Materials, 4(2), 133–145. https://doi.org/10.7569/jrm.2016.634104.
Musa, O. M. (Ed.). (2016). Handbook of maleic anhydride based materials - synthesis, properties and applications (Pp) (Vol. 638). New Jersey: Springer International Publishing. https://doi.org/10.1007/978-3-319-29454-4.
Muthuraj, R. (2015). Biodegradable polymer blends and their biocomposites: Compatibilization and performance evaluation. Retrieved from chrome-extension://ngpampappnmepgilojfohadhhmbhlaek/captured.html?back=1
Muthuraj, R., Misra, M., & Mohanty, A. K. (2017). Biocomposite consisting of miscanthus fiber and biodegradable binary blend matrix: Compatibilization and performance evaluation. RSC Advances, 7(44), 27538–27548. https://doi.org/10.1039/c6ra27987b.
Oromiehie, A., Ebadi-Dehaghani, H., & Mirbagheri, S. (2014). Chemical modification of polypropylene by maleic anhydride: Melt grafting, characterization and mechanism. International Journal of Chemical Engineering and Applications, 5(2), 117–122. https://doi.org/10.7763/ijcea.2014.v5.363.
Paolo, F., Mantia, L., Ceraulo, M., Mistretta, M. C., Botta, L., & Morreale, M. (2018). Compatibilization of polypropylene/polyamide 6 blend fibers using photo-oxidized polypropylene. Materials, 12(1), 81. https://doi.org/10.3390/ma12010081.
Papadopoulou, C. P., & Kalfoglou, N. K. (2000). Comparison of compatibilizer effectiveness for PET/PP blends: Their mechanical, thermal and morphology characterization. Polymer, 41(7), 2543–2555. https://doi.org/10.1016/s0032-3861(99)00442-5.
Persenaire, O., Quintana, R., Lemmouchi, Y., Sampson, J., Martin, S., Bonnaud, L., & Dubois, P. (2014). Reactive compatibilization of poly(l-lactide)/poly(butylene succinate) blends through polyester maleation: From materials to properties. Polymer International, 63(9), 1724–1731. https://doi.org/10.1002/pi.4700.
Phua, Y. J., Chow, W. S., & Mohd Ishak, Z. A. (2013). Reactive processing of maleic anhydride-grafted poly(butylene succinate) and the compatibilizing effect on poly(butylene succinate) nanocomposites. Express Polymer Letters, 7(4), 340–354. https://doi.org/10.3144/expresspolymlett.2013.31.
Pitt, G. G., Cha, Y., Shah, S. S., & Zhu, K. J. (1992). Blends of PVA and PGLA: Control of the permeability and degradability of hydrogels by blending. Journal of Controlled Release, 19(1–3), 189–199. https://doi.org/10.1016/0168-3659(92)90076-4.
Pracella, M., Haque, M., & Alvarez, V. (2010). Functionalization, compatibilization and properties of polyolefin composites with natural fibers. Polymers, 2, 554–574. https://doi.org/10.3390/polym2040554.
Pradeep, S. A., Kharbas, H., Turng, L. S., Avalos, A., Lawrence, J. G., & Pilla, S. (2017). Investigation of thermal and thermomechanical properties of biodegradable PLA/PBSA composites processed via supercritical fluid-assisted foam injection molding. Polymers, 9(1), 22. https://doi.org/10.3390/polym9010022.
Qiu, W., & Hirotsu, T. (2005). A new method to prepare maleic anhydride grafted poly(propylene). Macromolecular Chemistry and Physics, 206(24), 2470–2482. https://doi.org/10.1002/macp.200500375.
Rapthel, I., Wilms, J., & Piestert, F. (2018). Industrial production and use of grafted polyolefin. In G. Beyer & C. Hopmann (Eds.), Reactive extrusion: Principles and applications (pp. 449–496). Weinhiem: WILEY-VCH. https://doi.org/10.1016/b978-0-12-814509-8.00015-4.
Ren, J., Fu, H., Ren, T., & Yuan, W. (2009). Preparation, characterization and properties of binary and ternary blends with thermoplastic starch, poly(lactic acid) and poly(butylene adipate-co-terephthalate). Carbohydrate Polymers, 77(3), 576–582. https://doi.org/10.1016/j.carbpol.2009.01.024.
Rivaton, A., Serre, F., & Gardette, J. L. (1998). Oxidative and photooxidative degradations of PP/PBT blends. Polymer Degradation and Stability, 62(1), 127–143. https://doi.org/10.1016/s0141-3910(97)00271-1.
Rosu, D., & Visakh, P. M. (Eds.). (2016). Photochemical behavior of multicomponent polymeric-based materials (p. 405). Springer International: Publishing. https://doi.org/10.1007/978-3-319-25196-7.
Rzayev, Z. M. O. (2011). Graft copolymers of maleic anhydride and its isostructural analogues: High performance engineering materials. International Review of Chemical Engineering, 3(2), 153–215. https://doi.org/10.15866/ireamt.v2i5.7001.
Sadi, R. K., Fechine, G. J. M., & Demarquette, N. R. (2013). Effect of prior photodegradation on the biodegradation of polypropylene/poly(3-hydroxybutyrate) blends. Polymer Engineering & Science, 53(10), 2109–2013. https://doi.org/10.1002/pen.23471.
Semba, T., Kitagawa, K., Ishiaku, U. S., & Hamada, H. (2006). The effect of crosslinking on the mechanical properties of polylactic acid/polycaprolactone blends. Journal of Applied Polymer Science, 101(3), 1816–1825. https://doi.org/10.1002/app.23589.
Shah, A. A., Hasan, F., Hameed, A., & Ahmed, S. (2008). Biological degradation of plastics: A comprehensive review. Biotechnology Advances, 26(3), 246–265. https://doi.org/10.1016/j.biotechadv.2007.12.005.
Si, P., Luo, F., & Luo, F. (2016). Miscibility, morphology and crystallization behavior of poly(butylene succinate-co-butylene adipate)/poly(vinyl phenol)/poly(L-lactic acid) blends. Polymers, 8(12), 421. https://doi.org/10.3390/polym8120421.
Signori, F., Boggioni, A., Righetti, M. C., Rondán, C. E., Bronco, S., & Ciardelli, F. (2015). Evidences of transesterification, chain branching and cross-linking in a biopolyester commercial blend upon reaction with dicumyl peroxide in the melt. Macromolecular Materials and Engineering, 300(2), 153–160. https://doi.org/10.1002/mame.201400187.
Sodergard, A., & Stolt, M. (2002). Properties of lactic acid based polymers and their correlation with composition. Progress in Polymer Science, 27(6), 1123–1163. https://doi.org/10.1016/s0079-6700(02)00012-6.
Sun, Y. J., Hu, G. H., Lambla, M., & Kotlar, H. K. (1996). In situ compatibilization of polypropylene and poly(butylene terephthalate) polymer blends by one-step reactive extrusion. Polymer, 37(18), 4119–4127. https://doi.org/10.1016/0032-3861(96)00229-7.
Takamura, M., Nakamura, T., Takahashi, T., & Koyama, K. (2008). Effect of type of peroxide on cross-linking of poly(L-lactide). Polymer Degradation and Stability, 93(10), 1909–1916. https://doi.org/10.1016/j.polymdegradstab.2008.07.001.
Tansiri, V., & Potiyaraj, P. (2015). Compatibilization efficiency of reactively modified poly(butylene succinate) as a compatibilizer for poly(butylene succinate) composites. Advanced Materials Research, 1119, 288–291. https://doi.org/10.4028/www.scientific.net/amr.1119.288.
Therias, S., Tzankova Dintcheva, N., Gardette, J. L., & La Mantia, F. P. (2010). Photooxidative behaviour of polyethylene/polyamide-6 blends. Polymer Degradation and Stability, 95(4), 522–526. https://doi.org/10.1016/j.polymdegradstab.2009.12.017.
Thirmizir, M. Z. A., Hazahar, M. D., & Mohd Ishak, Z. A. (2017). Mechanical and morphological properties of poly(butylene succinate)/poly(hydroxybutyrate-co-hydroxyhexanoate) polymer blends: Effect of blend ratio and maleated compatibiliser. Key Engineering Materials, 737(2003), 313–319. https://doi.org/10.4028/www.scientific.net/kem.737.313.
Tianyi, K., & Xiuzhi, S. (2000). Physical properties of poly(lactic acid) and starch composites with various blending ratios. Cereal Chemistry, 77(6), 761–768. https://doi.org/10.1094/cchem.2000.77.6.761.
Toro-Márquez, L. A., Merino, D., & Gutiérrez, T. J. (2018). Bionanocomposite films prepared from corn starch with and without nanopackaged Jamaica (Hibiscus sabdariffa) flower extract. Food and Bioprocess Technology, 11(11), 1955–1973. https://doi.org/10.1007/s11947-018-2160-z.
Tsuji, H., Mizuno, A., & Ikada, Y. (2000). Properties and morphology of poly(L-lactide). III. Effects of initial crystallinity on long-term in vitro hydrolysis of high molecular weight poly(L-lactide) film in phosphate-buffered solution. Journal of Applied Polymer Science, 77(7), 1452–1464. https://doi.org/10.1002/1097-4628(20000815)77:7%3c1452::aid-app7%3e3.0.co;2-s.
Tsuji, H., & Muramatsu, H. (2001). Blends of aliphatic polyesters. IV. Morphology, swelling behavior, and surface and bulk properties of blends from hydrophobic poly(L-lactide) and hydrophilic poly(vinyl alcohol). Journal of Applied Polymer Science, 81(9), 2151–2160. https://doi.org/10.1002/app.1651.
Vroman, I., & Tighzert, L. (2009). Biodegradable polymers. Materials, 2(2), 307–344. https://doi.org/10.3390/ma2020307.
Waldman, W. R., & De Paoli, M. A. (2008). Photodegradation of polypropylene/polystyrene blends: Styrene-butadiene-styrene compatibilisation effect. Polymer Degradation and Stability, 93(1), 273–280. https://doi.org/10.1016/j.polymdegradstab.2007.09.003.
Wu, C. S. (2003). Physical properties and biodegradability of maleated-polycaprolactone/starch composite. Polymer Degradation and Stability, 80(1), 127–134. https://doi.org/10.1016/s0141-3910(02)00393-2.
Wu, C. S., & Liao, H. T. (2012). Polycaprolactone-based green renewable ecocomposites made from rice straw fiber: Characterization and assessment of mechanical and thermal properties. Industrial and Engineering Chemistry Research, 51(8), 3329–3337. https://doi.org/10.1021/ie202002p.
Wypych, G. (2008). Handbook of material weathering (4th ed.). Toronto: ChemTec Publishing. https://doi.org/10.1016/1352-2310(96)90058-8.
Xu, C., Fang, Z., & Zhong, J. (1993). Study on compatibilization-crosslinking synergism in PVC/LDPE blends. Die Angewandte Makromolekulare Chemie, 212(1), 45–52. https://doi.org/10.1002/apmc.1993.052120105.
Yin, Q., Chen, F., Zhang, H., & Liu, C. (2015). Fabrication and characterisation of thermoplastic starch/poly(butylene succinate) blends with maleated poly(butylene succinate) as compatibiliser. Plastics, Rubber and Composites, 44(9), 362–367. https://doi.org/10.1179/1743289815y.0000000031.
Yu, L., Dean, K., & Li, L. (2006). Polymer blends and composites from renewable resources. Progress in Polymer Science, 31(6), 576–602. https://doi.org/10.1016/j.progpolymsci.2006.03.002.
Zainuddin, Sudradjat, A., Razzak, M. T., Yoshii, F., & Makuuchi, K. (1999). Polyblend CPP and Bionolle with PP-g-MAH as compatibilizer: I. compatibility. Journal of Applied Polymer Science, 72(10), 1277–1282. https://doi.org/10.1002/(sici)1097-4628(19990606)72:10<1277::aid-app6>3.0.co;2-%23.
Zeng, J. B., Jiao, L., Li, Y. D., Srinivasan, M., Li, T., & Wang, Y. Z. (2011). Bio-based blends of starch and poly(butylene succinate) with improved miscibility, mechanical properties, and reduced water absorption. Carbohydrate Polymers, 83(2), 762–768. https://doi.org/10.1016/j.carbpol.2010.08.051.
Zhang, B., Sun, B., Bian, X., Li, G., & Chen, X. (2017). High melt strength and high toughness PLLA/PBS blends by copolymerization and in situ reactive compatibilization. Industrial and Engineering Chemistry Research, 56(1), 52–62. https://doi.org/10.1021/acs.iecr.6b03151.
Zhang, K., Mohanty, A. K., & Misra, M. (2012). Fully biodegradable and biorenewable ternary blends from polylactide, poly(3-hydroxybutyrate-co-hydroxyvalerate) and poly(butylene succinate) with balanced properties. ACS Applied Materials and Interfaces, 4(6), 3091–3101. https://doi.org/10.1021/am3004522.
Zhang, L., Xiong, C., & Deng, X. (1995). Biodegradable polyester blends for biomedical application. Journal of Applied Polymer Science, 56(1), 103–112. https://doi.org/10.1002/app.1995.070560114.
Zhang, M., & Thomas, N. L. (2011). Blending polylactic acid with polyhydroxybutyrate: The effect on thermal, mechanical, and biodegradation properties. Advances in Polymer Technology, 30(2), 67–79. https://doi.org/10.1002/adv.20235.
Acknowledgement
The authors gratefully acknowledge the financial support by the Long Term Research Grant Scheme (Account No: 1001/PKT/6725002) from the Ministry of Education Malaysia and Short Term Research Grant (Account No: 304.PBAHAN.60312042) from the Universiti Sains Malaysia (USM). Thanks for the research facilities by the Cluster for Polymer Composites, Science and Engineering Research Centre, Universiti Sains Malaysia. The authors would also like to express their gratitude to all researchers that make this book chapter possible.
Conflicts of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Thirmizir, M.Z.A., Ishak, Z.A.M., Salim, M.S. (2020). Compatibilization and Crosslinking in Biodegradable Thermoplastic Polyester Blends. In: Gutiérrez, T.J. (eds) Reactive and Functional Polymers Volume Two. Springer, Cham. https://doi.org/10.1007/978-3-030-45135-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-45135-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-45134-9
Online ISBN: 978-3-030-45135-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)