
Abstract
One of the main processes involved in genetic expression is the so-called splicing in which the introns present in the precursor messenger RNA are removed, and exons are joined together, forming the mature form of mRNA. Such a process gives quite a versatility and plasticity to the genome since it can generate different mRNA isoforms from a single pre-mRNA through a process known as alternative splicing (AS). AS is a mechanism through which a higher level of complexity can be achieved, despite the relatively limited capacity of the coding transcriptome. Humans (Homo sapiens), compared to other model organisms, have a higher percentage of AS events. Aberrant splicing events have been correlated with several diseases including those correlated to age, since most of them lead to aberrant protein formation and, therefore, to misfolded protein accumulation, a process very common in these types of diseases. In this context, AS seems to be a critical process not only for therapeutic purposes but also to deeply understand the pathophysiology of diseases. Therefore, in the present chapter, we resume the main characteristics of AS and review its relationship with some of the primary age-related diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
Introduction
One of the most striking characteristics of eukaryotic genomes is the discontinuous arrangement of genes, in which protein-coding segments, known as exons, are interrupted by non-coding sequences, known as introns. Through a highly regulated process known as splicing, the introns present in the precursor messenger RNA (pre-mRNA) are removed, and exons are joined together, forming the mature form of mRNA. While some genes are constitutively spliced, meaning that they will produce the same mature mRNA from a given pre-mRNA, some can generate different mRNA isoforms from a single pre-mRNA through a process known as alternative splicing (AS). AS is a process by which a single gene can give rise to different transcripts through the differential assembly and organization of the sequences present within the exons/introns of a gene. This process can give rise to a wide variety of transcript isoforms from a small number of genes, increasing both transcriptome and proteome [1, 2].
With the recent development of faster and accurate sequencing technologies and the ever-decreasing costs of sequencing whole transcriptomes, it has become apparent that AS has a significant role in organism development. It is estimated that around 95% of all human protein-coding genes undergo a process of AS [3, 4]. After the sequencing of the human genome and that of other model organisms, it became clear that although humans are significantly more complex both morphologically and behaviorally than mice, fruit flies (Drosophila melanogaster), or tapeworms (Caenorhabditis elegans), the difference in the number of genes between these organisms was not the reason behind these differences. This meant that something else besides the number of protein-coding genes was responsible for the increasing level of complexity among different organisms. Evidence from the characterization of expressed cDNA clone sequence tags [5, 6], as well as bioinformatic analysis, indicates that AS events seem to increase with organism complexity. These findings suggest that AS has been used throughout evolutionary history as a mechanism through which higher level of molecular complexity can be achieved. In that regard, humans (Homo sapiens), compared to other organisms, are the ones that have the highest percentage of AS events for multiexon protein-coding genes (around 88%) with at least 2+ mRNA isoforms per gene (comparatively mouse sits at around 63%, fruit fly 45%, and tapeworm 25%). This information is based upon current genome annotations that do not consider recent RNA-seq data. Recent studies based on RNA-seq of the human transcriptome indicate that >95–100% of human genes generate at least two alternative pre-mRNA isoforms (with an average of seven mRNA isoforms per gene) [3, 4]. These observations support that an increase in the occurrence of AS events is directly correlated with the increase in organism complexity [7].
Mechanisms of Pre-mRNA Splicing
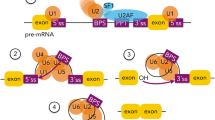
The underlying molecular mechanisms by which introns are removed from the pre-mRNA seem to be very well conserved among all eukaryotic groups [8]. Intron excision from the pre-mRNA and subsequent exon ligation is mediated by a complex and highly regulated molecular machinery known as the spliceosome. The spliceosome is a ribonucleoprotein (RNA-protein complex) which acts in a dynamic cycle of stepwise reactions that is assembled and disassembled for every single splicing reaction [9]. Several efforts have been made to characterize and determine the structure and function of the different components of the spliceosome throughout the different steps of the cycle. Biochemical and structural analyses have demonstrated that the catalytic center of the spliceosome is composed of RNA [10]. The actual biochemical reactions that take place during a splicing event are two consecutive SN2-type transesterification reactions involving functional groups from three reactive regions present in the pre-mRNA. Two of these regions are present at the 5′ and 3′ end of introns and are known as either 5′ or 3′ splice sites (SS). The other region involved is known as the branch point site (BPS) located near the 3′ end of the intron, around 15–50 nucleotides upstream of the 3′ SS [11]. The sequences that define the 5′ and 3′ SS, as well as the BPS, are very small consensus elements [9].
The primary assembly process of the spliceosome requires the recruitment of several small nuclear ribonucleoprotein (snRNP) subunits, namely, snRNPs U1, U2, U4, U5, and U6, as well as other non-snRNP splicing factors. Each of these snRNP subunits is composed of a specific small nuclear RNA (snRNA), as well as other proteins. These snRNAs are the ones that through a base pairing-based mechanism recognize the specific sequences within the DNA (5′ and 3′ SS, BPS) to differentiate exons from introns. Firstly, the U1 snRNP recognizes the 5′ SS of the mRNA forming the early complex (complex E), meanwhile, the 3′ SS is recognized by the U2 snRNP as well as other associated factors, such as splicing factor 1 (SF1) and U2 auxiliary factors (U2AFs), which are also components of complex E. U2 then recognizes sequences at the BPS and interacts with U1 to form the pre-spliceosome complex (complex A). After the assembly of the complex A, the U4, U5, and U6 snRNPs are recruited as a pre-formed tri-snRNP, forming complex B. This resulting complex B is still catalytically inactive and goes through several changes to form the catalytically active complex B∗. The activation of complex B results in the release of U1 and U4. Complex B∗ can complete the first catalytic step of splicing, generating complex C, which contains exon 1 and the intron-exon 2 lariat intermediate. Complex C undergoes a series of rearrangements to carry out the second catalytic step that results in a post-spliceosomal complex that contains the lariat intron and the spliced exons. In the last step of the cycle, the U2, U5, and U6 snRNPs are released alongside the lariat intron and recycled for additional rounds of splicing. As mentioned before, the splicing process is largely RNA-based, mainly through the U2-U5-U6 snRNP complex [12], which seems to be the active structure that catalyzes both steps of the splicing reaction, but it seems that some proteins are also necessary for the formation of the catalytic site [13].
Mechanisms and Regulation of Alternative Splicing
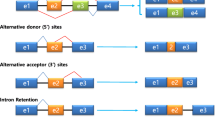
The occurrence of different types of AS events is influenced by a wide variety of factors, including splicing regulatory sequences present in the pre-mRNA, differential activities of splicing factors that either activate or repress AS, chromatin density and structure, as well as transcription elongation rates [14]. The different types of AS events include exon skipping, in which an exon is spliced out of the mature mRNA, along with its flanking introns. Intron retention occurs when an intron is included in the mature transcript; alternative 3′ SS and 5′ SS selection events occur when multiple SS are recognized at the same time at either end of an exon. A mutually exclusive AS event can occur when two exons are included in the mature transcript in a mutually exclusive way (either one of the two exons is included); another type of AS event is known as alternative polyadenylation in which the region where the poly(A) tract is localized can vary. In vertebrates, and specifically in humans, the most prevalent AS event is exon skipping [6, 15]. All these events can be combined into even more complex types of AS, giving rise to a wide variety of isoforms from a single transcript.
The SS selection that eventually gives rise to different transcript isoforms from a single gene is determined by a wide variety of elements which can be both cis- and trans-acting. As mentioned earlier, because most SS consensus sequences are poorly conserved, SS by themselves cannot efficiently direct the spliceosome assembly process. For instance, the cis-acting elements include regulatory sequences which can be found both within exons and introns and act as splicing enhancers or silencers, depending on their position as well as their effect on the usage of a SS [14], meaning that within a specific region of the genome, both intronic splicing enhancers (ISE) and silencers (ISS) can be found, as well as exonic splicing enhancers (ESE) and silencers (ESS). These regulatory sequences function mainly as binding sites for trans-acting factors that in turn recruit the snRNP subunits of the spliceosome to a specific site.
The trans-acting elements function through the binding to splicing enhancer or silencer sequences and include both serine-arginine (SR)-rich and heterogeneous nuclear ribonucleoprotein (hnRNP) families of proteins. It is generally considered that SR proteins act as splicing promoters while hnRNP as repressors, although there is evidence indicating that the activities of some of these splicing regulators are determined by the region of the pre-mRNA in which they bind. A recent study has shown that SR proteins can act both as enhancers and silencers of splicing [16]. Interestingly, these proteins can recognize short RNA sequence motifs and can function as splicing enhancers when bound to exons and as repressors of splicing when bound to introns [17]. hnRNPs can also recognize specific binding sequences in RNA, and depending on its binding position, they can also act as both enhancers and silencers of splicing [18].
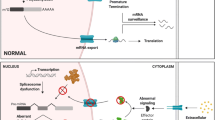
Alterations of pre-mRNA splicing can give rise to several physiological abnormalities that often lead to disease [19]. Even though the process of splicing itself consists of relatively simple steps, the recognition of correct SS is a daunting task for the splicing machinery given the highly complex organization of the genome. Mutations of these (and other) regulatory regions of splicing can sometimes generate faulty transcripts harboring premature termination codons (PTC) that if translated could lead to defective proteins. The nonsense-mediated mRNA decay (NMD) pathway was first described solely as a post-transcriptional surveillance and quality control mechanism responsible for the degradation of transcripts harboring a PTC that if translated could lead to the production of truncated proteins with deleterious effects for the organism. Evidence has also shown the importance of the NMD pathway as a regulatory mechanism that controls the expression of several naturally occurring transcripts [20]. Recently, it has been shown that not all PTC-containing transcripts trigger the activation of the NMD pathway. Moreover, other transcripts that do not contain a PTC are also targets of NMD [20], which indicates that further studies are needed to understand all the factors involved in the activation of the NMD pathway.
AS can generate transcripts harboring PTCs, and canonically these transcripts should be subject to degradation by NMD; however, the NMD pathway does not degrade these transcripts. Instead, it seems that some of these transcripts were able to “hijack” the NMD pathway to serve as a regulatory mechanism for their transcription [21]. This phenomenon, known as AS-NMD regulation, was first reported as a widely spread mechanism of regulation of a variety of transcripts, both naturally occurring and disease-related [22]. More recent evidence seems to suggest that AS-NMD regulation is not as widespread as initially thought [23], but there is evidence supporting an important role of AS-NMD regulation on specific gene families, including well-known regulators of splicing and AS [24,25,26]. It would seem that AS-NMD acts mainly as a repressive regulatory mechanism, but there is evidence that it can also be used as a developmental switch [27].
Coupling of Splicing with Transcription
There has been accumulating evidence showing the coupling of transcription and splicing [14]. The fact that splicing occurs in a cotranscriptional manner is the basis for inferring that transcription elongation rate, chromatin structure, and modifications are coupled with splicing.
One of the key elements that link transcription and splicing is the RNA polymerase II (RNAPII). Transcription elongation rate can affect SS selection and thus the products of AS. A faster elongation rate of transcription encourages skipping of exons with “weak” 3′ SS, while slower elongation rates encourage the inclusion of exons with weak 3′ SS sites [28]. One possible mechanism is that the occupancy of nucleosomes can alter the elongation rate of the RNAPII to facilitate inclusion of weak SS [29]. Nucleosomes can act as barriers for elongating RNAPII altering its elongation rate, as exons flanked by weak SS are more enriched with nucleosomes compared with those containing strong SS [30].
One of the main characteristics of RNAPII is the presence of a C-terminal domain (CTD). Post-translational modifications of this CTD have been shown to play a crucial role in the regulation of its transcriptional activity, particularly phosphorylation. The CTD of RNAPII serves as a scaffold for the recruitment of a wide variety of splicing factors [31], as mutations of the CTD lead to splicing alterations [32]. These post-transcriptional modifications may regulate the physical interactions between the CTD and splicing components creating a binding platform for splicing factors. Apart from nucleosome occupancy, there is evidence that chromatin modifications also have a role in coupling transcription and splicing [33]. Transcriptional elongation rate is regulated by a dynamic cycle of histone acetylation/deacetylation, which is very important for nucleosome dynamics during transcription and is coordinated by the CTD of RNAPII [34]. A wide variety of histone acetyltransferase (HAT) and deacetylase (HDAC) proteins mediate the addition and removal of acetyl groups that modify the interactions between nucleosomes and DNA. During the transcription process, it is necessary that HATs acetylate the nucleosome downstream of the elongation complex in order to destabilize the interactions between histones and DNA. RNAPII elongation causes a displacement of histones, which are subsequently placed onto the DNA behind RNAPII. These deposited nucleosomes are hyperacetylated momentarily. HDAC complexes remove acetylation marks from the chromatin to maintain a stable configuration. This acetylation/deacetylation dynamic can influence the selection of SS in many genes [33].
Chromatin-modifying proteins that recognize and bind specific histone marks can also affect splicing patterns by recruiting several splicing factors to sites of active transcription to modulate the inclusion or exclusion of alternative exons. Specific patterns of histone marks can correlate with particular splicing patterns in many genes [35]. Another evidence is the fact that modulation of histone marks by inhibitors, overexpression of histone modifiers, or knockdown experiment induces changes in the splicing patterns [36].
Connections Between Alternative Splicing and Human Disease
Recently, a vast amount of evidence supporting pre-mRNA splicing, both constitutive and alternative, as an important regulatory mechanism of organismic complexity in humans gave rise to further research the relevance that pre-mRNA splicing alterations could have on disease [37, 38].
The complex genomic arrangement of eukaryotic genomes comes with the implication that every intron-containing gene requires to undergo the process of splicing, meaning that the proper processing of pre-mRNA into mature transcripts needs to be tightly regulated, and the fact that splicing occurs in a cotranscriptional manner adds another layer of complexity to this process. The downside of the functional versatility that comes with pre-mRNA splicing is that this process can be disrupted through a wide variety of ways and these alterations can end up being the cause of several pathological conditions [37].
According to data from the Human Gene Mutation Database (HGMD), of all the single-nucleotide polymorphisms (SNPs) that are the cause of a disease, around 15% are located within SS sequences and 22% of disease alleles are located within splicing elements, meaning that more than one third of all disease-causing SNPs can alter splicing [39, 40]. Also, evidence from the HGMD suggests that around 10% of human inherited diseases are due to single base-pair substitution mutations located in SS [41]. However, these data only take into account mutations located at the relatively well-conserved SS sequences, but not at other cis-acting splicing elements nor mutations located at loci of trans-acting splicing elements. Alterations in pre-mRNA splicing can come from a wide variety of alterations, but they are grouped into these categories: mutations of canonical 5′ and 3′ SS, mutations of the BPS, mutations of cis-acting regulatory elements, mutations of trans-acting splicing elements, and mutations of the splicing machinery.
Mutations in Splice Sites and Regulatory Sequences
The most common types of splicing-related mutations that occur are those of cis-acting elements, such as the core consensus sequences (both 5′ and 3′ SS, as well as the BPS) as well as other splicing regulatory sequences [19]. Among these, familial dysautonomia is a rare recessive disorder in the Ashkenazi Jewish population that affects both the autonomous nervous system and the somatic sensory neurons [42]. Caused by a point mutation in intron 20 (T→C) in the IKBKAP gene, that results in the alteration of a 5′ SS weakening the binding of the spliceosome subunit U1, leading to the skipping of exon 20 which results in the introduction of a PTC in exon 21, making the mRNA susceptible to degradation by the NMD pathway [43].
One of the best studied cases of splicing alterations of cis-acting elements that end up being the cause of a pathological condition is spinal muscular atrophy (SMA). SMA is a prevalent recessive disorder associated with infant mortality; more than 90% of all cases of SMA are the result of mutations of the Survival Motor Neuron 1 (SMN1) gene. Humans carry two copies of the SMN gene: SMN1 and SMN2. SMN is required for proper snRNP synthesis, and its absence leads to degeneration of motor neurons, particularly those of the spinal cord. The main difference between the two copies is the fact that in SMN2 exon 7 is predominantly skipped in most tissues. SMN2 codes for SMNΔ 7, a partially functional and unstable protein. Loss of SMN1 leads to a deficit of the SMN protein and the consequent death of motor neurons. SMN2 presents a C→T change at position 6 of exon 7. This single mutation causes two different outcomes: first, the deletion of an ESE, and second, it creates an ESS, which in turn promotes the skipping of exon 7 [44].
Another well-documented case is Duchenne muscular dystrophy (DMD). While genomic deletions of the dystrophin gene cause the most severe forms of DMD, some mild forms of DMD are caused by point mutations that affect the splicing patterns [37]. Particularly, a T→A substitution in exon 31 simultaneously generates an ESS resulting in exon skipping and introduces a PTC [45]. These splicing alterations produce a partially functioning form of the protein, which explains the mild phenotype of the disease.
Mutations of Splicing Trans-acting and Core Spliceosome Factors
Mutations in trans-acting splicing factors, as well as core spliceosome components, can simultaneously affect a significant number of genes. Unlike mutations in cis-acting elements, there are relatively few examples of genetic disorders caused by alterations of trans-acting factors, including spliceosomal factors, maybe because many of these mutations result in lethality during embryonic development [46].
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease characterized by the degeneration of upper and lower motor neurons. It is a fatal disorder that ultimately causes death within 2–5 years following diagnosis. Around 10% of cases seem to follow a pattern of Mendelian inheritance and high penetrance [47]. Studies demonstrated that RNA processing plays an essential role in the onset of the disease. Specifically, one of the most important genes for ALS, TDP-43 (TARDBP), an RNA-binding protein, seems to be a major component of cytoplasmic inclusions in motor neurons of ALS patients [48]. TDP-43 is normally localized in the nucleus and it is involved in RNA processing as well as AS and neurons with cytoplasmic aggregations show a depletion of nuclear TDP-43. Recent studies have demonstrated that TDP-43 seems to be a direct regulator of several AS events in the brain [49, 50]. This evidence suggests that the loss of function of TDP-43 seems to be a major determinant factor in ALS.
Retinitis pigmentosa is a disorder characterized by the progressive degeneration of the retina. Mutations in the core spliceosomal factors PRPF3, PRPF8, PRPF31, and SNRNP200 are associated with the onset of the disease [51]. These factors are important for the assembly of the tri-snRNP complex of U4/U6/U5 from the spliceosome [11]. Even though the specific pathological defects seem to indicate a functional role in the retina, the specific splicing abnormalities responsible for the disease remain to be discovered.
Alternative Splicing and Aging
Aging is characterized by a general decline of the homeostatic capacity of the organism to reach the normal state, leading to a general decline in the physiological and social functions highly associated with mortality. In this context, aging has been associated with several diseases such as cardiovascular and metabolic disorders, many types of cancer, and neurodegenerative diseases as Alzheimer’s and Parkinson’s diseases, among others [52]. Such age-related decline is associated with tissue damage and several inflammatory processes (inflammaging [53] and immunosenescence [54]); in this context aging seems to be tissue-specific and highly related to genetic expression more than genotype itself. Therefore, AS becomes highly essential since 90% of human protein-coding genes produce multiple transcripts through this process [3], and as previously stated, aberrant splicing events are correlated with age-associated diseases, since most of them lead to aberrant protein formation and, therefore, to misfolded protein accumulation, a process highly related to the molecular pillars of aging [55].
Alternative Splicing, Progeroid, and Age-Related Diseases
Werner syndrome (WS)
It is a well-characterized autosomal recessive progeroid syndrome, in which patients developed normally until they reach puberty and then started to experiment wrong physiological development, the suffering of skin and gonadal atrophy, cataracts, type 2 diabetes, osteoporosis, and loss and graying of hair [56]. Most of WS patients reach 54 years old and die of myocardial infarction; mutations on WRN gene (located at chromosome 8p12 that codified for 34 exons encoding a DNA helicase protein) are responsible for such disease [57]. AS is quite essential in WS since due to exon skipping associated with stop codons, indels, or mutations, most of the pathogenic variants on the WRN gene result in helicase protein truncation; additionally pathogenic variants have also been reported in intronic regions [56].
Hutchinson-Gilford syndrome (HGS)
It is a genetic disease characterized by a dramatic aging phenotype early in childhood [58]. Most of HGS patients die from heart attacks and stroke early in their teens [59]. Interestingly, most cases are characterized by a silent mutation within a single codon of the LMNA gene which enhances the use of an internal 5′ SS in exon 11 and leads to the production of a truncated protein (progerin). Its accumulation could lead to abnormally shaped nuclei, loss of heterochromatin distribution, changes in methylation patterns, and misregulation of nuclear proteins, among other changes [60]. Although it is not clear the relationship between HGS and normal aging, evidence suggests that progerin may be involved in the aging process since the number of (+) progerin protein cells increases with age in samples from healthy individuals [61,62,63].
Bloom syndrome (BS)
It is an autosomal recessive progeroid disease mainly characterized by early growth deficiency, photosensitive skin changes, immune deficiency, insulin resistance, and a substantially increased risk for the development of multiple cancers [64]. Mutations on the BLM gene cause BS; such gene encodes RecQ helicase. Interestingly, such mutations have been found to induce aberrant splicing in which the extra exon 3 s are skipped, and a site mutation on p53 splice acceptor has been found in skin fibroblasts derived from BS patient [65].
Diabetes
It is a chronic disease characterized by hyperglycemia as a result of pancreas not making enough insulin or due to insulin resistance. In this context, insulin sensitivity is associated with insulin receptor isoform, and studies have demonstrated that insulin receptor type B mRNA variant increases in response to bariatric surgery [66] and weight loss for low caloric diet [67] as well as the genetic expression of several splicing factors involved in the insulin receptor [68]. Particularly in the pathogenesis of diabetes type 1, splicing process in T cells and node stromal cells seems to be involved in the modulation of the immunological response against β cells; interestingly , β cells exposed to cytokines activate AS networks that modulate its viability and susceptibility to immune-induced stress [69, 70].
Cancer
It is a broad term to refer to several diseases characterized by abnormal cells that possess a very particular genetic expression related with cell survival, accelerated growth and spreading across the body. Since the incidence of most cancers increases as a person ages, it is considered an age-related disease. Several genome-wide association studies suggest a strong relation between AS and cancer due to the plasticity offered by this process [71]. Splicing aberration related with cancer could be divided in four main categories: those related with alteration on tumor suppressor genes and oncogenes, those related with aberration on spliceosomal components, mutations over splicing factors, and changes in the signaling pathways that regulate splicing process; some of these changes are highly significant to cancer hallmarks [72].
Alzheimer’s disease (AD)
It is characterized by a progressive decline in normal cognitive functions, diminishing the performance of memory, attention, language, and visuospatial skills and in executing tasks [73]. Neuropathology of AD includes the accumulation of β-amyloid deposition and accumulation of Tau-hyperphosphorylated proteins [74]. Mutations occurring in the intronic regions of presenilin 1 and 2 cause missplicing and lead to abnormal expression of β-amyloid [75]. A variant including exon 7 is the dominant splice form of the amyloid precursor protein gene in AD patients and contributes to β-amyloid accumulation; interestingly, RBFox is a trans-acting regulator that leads to the inclusion or exclusion of exon 7 [76]. ApoE4 a major cholesterol transporter in the brain and cholesterol-rich membrane domains increase β-amyloid production affecting β- and γ-secretase complexes; the proteolysis of apoE4 may lead to a loss of function in its ability to remove β-amyloid . Interestingly, E4 isoform is more prone to proteolysis than other APOE [77]. The inclusion of the exon 10 in Tau gene generates an isoform susceptible to microtubule binding involved in the formation of Tau into paired helical filaments [74].
Parkinson’s disease (PD)
It is the second most common neurodegenerative disease, characterized by resting tremor, bradykinesia , stiffness of movement, and postural instability; among its physiopathology is caused by protein aggregation in Lewy bodies and loss of dopamine-containing neurons in the substantia nigra of the midbrain [78]. Six genes, including PARK2, SNCAIP, LRRK2, SNCA, SRRM2, and MAPT, are involved in aberrant AS events in PD patients [79]. For instance, several point mutations in PARK2 splice acceptor or donor sites have been identified in PD patients [80]. On the other side, mutations in LRRK2 are the most common genetic cause of familial late-onset parkinsonism; most of them are on intronic regions highly susceptible to splicing [81]. Interestingly, oxidant generated AS of SNCA plays a central role in dopamine neuron cell death [79]. The MAPT gene which encodes Tau is also susceptible to several mutations present within coding regions and reduces its binding activity and decreases the ability of Tau to promote MT assembly [79].
Conclusions
AS constitutes one of the most important mechanisms for the plasticity of the transcriptome and proteome, since it is not only a spatiotemporal process but it helps to identify critical processes. Growing evidence demonstrates that dysregulation of the AS events is highly implicated in several diseases including those age-related diseases. In this context, it can function to develop biomarkers for such diseases or to develop therapeutic agents. Further research must be performed to improve our understanding of this complicated process.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- AS:
-
Alternative splicing
- BPS:
-
Branch point site
- BS:
-
Bloom syndrome
- CTD:
-
C-terminal domain (CTD)
- DMD:
-
Duchenne muscular dystrophy
- ESE:
-
Exonic splicing enhancer
- ESS:
-
Exonic splicing silencer
- HAT:
-
Histone acetylase
- HDAC:
-
Histone deacetylase
- HGS:
-
Hutchinson-Gilford syndrome
- hnRNP:
-
Heterogeneous nuclear ribonucleoprotein
- ISE:
-
Intronic splicing enhancer
- ISS:
-
Intronic splicing silencer
- NMD:
-
Nonsense-mediated mRNA decay
- PD:
-
Parkinson’s disease
- pre-mRNA:
-
Precursor messenger mRNA
- PTC:
-
Premature termination codons
- RNAPII:
-
RNA polymerase II
- snRNA:
-
Small nuclear RNA
- snRNP:
-
Small nuclear ribonucleoprotein
- SR:
-
Serine-arginine-rich proteins
- SS:
-
Splice sites
- WS:
-
Werner syndrome
References
Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463(7280):457–63.
Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126(1):37–47.
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–6.
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413–5.
Kim H, Klein R, Majewski J, Ott J, Harrington ED, Boue S, et al. Estimating rates of alternative splicing in mammals and invertebrates (multiple letters). Nat Genet. 2004;36(9):915–7.
Kim E, Magen A, Ast G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007;35(1):125–31.
Chen L, Bush SJ, Tovar-Corona JM, Castillo-Morales A, Urrutia AO. Correcting for differential transcript coverage reveals a strong relationship between alternative splicing and organism complexity. Mol Biol Evol. 2014;31(6):1402–13.
Keren H, Lev-Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet. 2010;11(5):345–55.
Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136(4):701–18.
Lee Y, Rio DC. Mechanisms and regulation of alternative Pre-mRNA splicing. Annu Rev Biochem. 2015;84:291–323.
Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15(2):108–21.
Fica SM, Tuttle N, Novak T, Li NS, Lu J, Koodathingal P, et al. RNA catalyses nuclear pre-mRNA splicing. Nature. 2013;503(7475):229–34.
Galej WP, Oubridge C, Newman AJ, Nagai K. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature. 2013;493(7434):638–43.
Naftelberg S, Schor IE, Ast G, Kornblihtt AR. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem. 2015;84(1):165–98.
Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321(5891):956–60.
Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, et al. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell. 2013;50(2):223–35.
Änkö ML. Regulation of gene expression programmes by serine-arginine rich splicing factors. Semin Cell Dev Biol. 2014;32:11–21.
Motta-Mena LB, Heyd F, Lynch KW. Context-dependent regulatory mechanism of the splicing factor hnRNP L. Mol Cell. 2010;37(2):223–34.
Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17(1):19–32.
Hug N, Longman D, Cáceres JF. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2015;44(4):1483–95.
Hillman RT, Green RE, Brenner SE. An unappreciated role for RNA surveillance. Genome Biol. 2004;5(2):R8.
Green RE, Lewis BP, Hillman RT, Blanchette M, Lareau LF, Garnett AT, et al. Widespread predicted nonsense-mediated mRNA decay of alternatively-spliced transcripts of human normal and disease genes. Bioinformatics. 2003;19(Suppl. 1):i118–21.
Pan Q, Saltzman AL, Yoon KK, Misquitta C, Shai O, Maquat LE, et al. Quantitative microarray profiling provides evidence against widespread coupling of alternative splicing with nonsense-mediated mRNA decay to control gene expression. Genes Dev. 2006;20(2):153–8.
Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature. 2007;446(7138):926–9.
Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O’Brien G, et al. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 2007;21(6):708–18.
Saltzman AL, Kim YK, Pan Q, Fagnani MM, Maquat LE, Blencowe BJ. Regulation of multiple core spliceosomal proteins by alternative splicing-coupled nonsense-mediated mRNA decay. Mol Cell Biol. 2008;28(13):4320–30.
Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21(13):1636–52.
De La Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, et al. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell. 2003;12(2):525–32.
Hodges C, Bintu L, Lubkowska L, Kashlev M, Bustamante C. Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science. 2009;325(5940):626–8.
Schwartz S, Meshorer E, Ast G. Chromatin organization marks exon-intron structure. Nat Struct Mol Biol. 2009;16(9):990–5.
de Almeida SF, Carmo-Fonseca M. The CTD role in cotranscriptional RNA processing and surveillance. FEBS Lett. 2008;582(14):1971–6.
McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, et al. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997;385(6614):357–60.
Zhou HL, Luo G, Wise JA, Lou H. Regulation of alternative splicing by local histone modifications: potential roles for RNA-guided mechanisms. Nucleic Acids Res. 2014;42(2):701–13.
Hsin JP, Manley JL. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012;26(19):2119–37.
Luco RF, Misteli T. More than a splicing code: integrating the role of RNA, chromatin and non-coding RNA in alternative splicing regulation. Curr Opin Genet Dev. 2011;21(4):366–72.
Luco R, Pan Q, Tominaga K, Blencowe B, Pereira-Smith O, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327(5968):996–1000.
Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136(4):777–93.
Padgett RA. New connections between splicing and human disease. Trends Genet. 2012;28(4):147–54.
Krawczak M, Thomas NST, Hundrieser B, Mort M, Wittig M, Hampe J, et al. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28(2):150–8.
Lim KH, Ferraris L, Filloux ME, Raphael BJ, Fairbrother WG. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc Natl Acad Sci. 2011;108(27):11093–8.
Stenson PD, Ball EV, Howells K, Phillips AD, Mort M, Cooper DN. The human gene mutation database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum Genomics. 2009;4(2):69–72.
Slaugenhaupt SA, Gusella JF. Familial dysautonomia. Curr Opin Genet Dev. 2002;12(3):307–11.
Sinha R, Kim YJ, Nomakuchi T, Sahashi K, Hua Y, Rigo F, et al. Antisense oligonucleotides correct the familial dysautonomia splicing defect in IKBKAP transgenic mice. Nucleic Acids Res. 2018;46(10):4833–44.
Singh RN, Singh NN. Mechanism of splicing regulation of spinal muscular atrophy genes. Adv Neurobiol. 2018;20:31–61.
Disset A, Bourgeois CF, Benmalek N, Claustres M, Stevenin J, Tuffery-Giraud S. An exon skipping-associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements. Hum Mol Genet. 2006;15(6):999–1013.
Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12(10):715–29.
Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7(9):710–23.
Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VMY. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283(19):13302–9.
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–68.
Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14(4):452–8.
Mordes D, Luo X, Kar A, Kuo D, Xu L, Fushimi K, et al. Pre-mRNA splicing and retinitis pigmentosa. Mol Vis. 2006;12:1259–71.
Laboratories PFG. Epigenetic regulation of ageing. Nat Publ Gr [Internet]. 2015;16(10):593–610. Available from: https://doi.org/10.1038/nrm4048.
Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and Garb-aging. Trends Endocrinol Metab. 2017;28(3):199–212.
Aw D, Silva AB, Palmer DB. Immunosenescence: emerging challenges for an ageing population. Immunology [Internet]. 2007;120(4):435–46. Available from: https://www.ncbi.nlm.nih.gov/pubmed/17313487.
Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, et al. Geroscience: linking aging to chronic disease. Cell [Internet]. 2014;159(4):709–13. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25417146.
Friedrich K, Lee L, Leistritz DF, Nürnberg G, Saha B, Hisama FM, et al. WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum Genet [Internet]. 2010;128(1):103–11. Available from: https://doi.org/10.1007/s00439-010-0832-5.
Rincón A, Mora L, Rojas JA. Case report Werner’s syndrome: understanding the phenotype of premature aging – first case described in Colombia. Case Rep Genet. 2019;2019(8538325):4.
Pollex RL, Hegele RA. Hutchinson–Gilford progeria syndrome. Clin Genet [Internet]. 2004;66(5):375–81. Available from: https://doi.org/10.1111/j.1399-0004.2004.00315.x.
Lopez-Mejia IC, Vautrot V, De Toledo M, Behm-Ansmant I, Bourgeois CF, Navarro CL, et al. A conserved splicing mechanism of the LMNA gene controls premature aging. Hum Mol Genet [Internet]. 2011;20(23):4540–55. Available from: https://doi.org/10.1093/hmg/ddr385.
Rodriguez S, Coppedè F, Sagelius H, Eriksson M. Increased expression of the Hutchinson-Gilford progeria syndrome truncated lamin A transcript during cell aging. Eur J Hum Genet [Internet]. 2009;17(7):928–37. Available from: https://www.ncbi.nlm.nih.gov/pubmed/19172989.
McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, et al. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One [Internet]. 2007;2(12):e1269–e9. Available from: https://www.ncbi.nlm.nih.gov/pubmed/18060063.
Cao K, Capell BC, Erdos MR, Djabali K, Collins FS. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci U S A [Internet]. 2007;104(12):4949–54. Available from: https://www.ncbi.nlm.nih.gov/pubmed/17360355.
Skoczyńska A, Budzisz E, Dana A, Rotsztejn H. New look at the role of progerin in skin aging. Prz Menopauzalny Menopause Rev [Internet]. 2015;14(1):53–8. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26327889.
Cunniff C, Bassetti JA, Ellis NA. Bloom’s syndrome: clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol Syndromol [Internet]. 2017;8(1):4–23. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28232778.
Magnusson KP, Sandström M, Ståhlberg M, Larsson M, Flygare J, Hellgren D, et al. p53 splice acceptor site mutation and increased HsRAD51 protein expression in Bloom’s syndrome GM1492 fibroblasts. Gene [Internet]. 2000;246(1):247–54. Available from: http://www.sciencedirect.com/science/article/pii/S0378111900000688.
Besic V, Shi H, Stubbs RS, Hayes MT. Aberrant liver insulin receptor isoform a expression normalises with remission of type 2 diabetes after gastric bypass surgery. PLoS One [Internet]. 2015;10(3):e0119270. Available from: https://doi.org/10.1371/journal.pone.0119270.
Kaminska D, Hämäläinen M, Cederberg H, Käkelä P, Venesmaa S, Miettinen P, et al. Adipose tissue INSR splicing in humans associates with fasting insulin level and is regulated by weight loss. Diabetologia [Internet]. 2014;57(2):347–51. Available from: https://doi.org/10.1007/s00125-013-3097-4.
Juan-Mateu J, Villate O, Eizirik DL. Mechanisms in endocrinology: alternative splicing: the new frontier in diabetes research. Eur J Endocrinol [Internet]. 2016;174(5):R225–38. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26628584.
Wong C-M, Xu L, MY-C Y. Alternative mRNA splicing in the pathogenesis of obesity. Int J Mol Sci [Internet]. 2018;19(2):632. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29473878.
Dlamini Z, Mokoena F, Hull R. Abnormalities in alternative splicing in diabetes: therapeutic targets. J Mol Endocrinol [Internet]. 2017;59(2):R93–107. Available from: https://jme.bioscientifica.com/view/journals/jme/59/2/JME-17-0049.xml.
David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343–64.
El Marabti E, Younis I. The cancer spliceome: reprograming of alternative splicing in cancer. Front Mol Biosci [Internet]. 2018;5:80. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30246013.
Cummings JL. Alzheimer’s disease. N Engl J Med [Internet]. 2004;351(1):56–67. Available from: https://doi.org/10.1056/NEJMra040223.
Love JE, Hayden EJ, Rohn TT. Alternative splicing in Alzheimer’s disease. J Park Dis Alzheimer’s Dis [Internet]. 2015;2(2):6. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26942228.
Cruts M, Van Broeckhoven C. Presenilin mutations in Alzheimer’s disease. Hum Mutat [Internet]. 1998;11(3):183–90. Available from: https://doi.org/10.1002/(SICI)1098-1004(1998)11:3%3C183::AID-HUMU1%3E3.0.CO.
Alam S, Suzuki H, Tsukahara T. Alternative splicing regulation of APP exon 7 by RBFox proteins. Neurochem Int [Internet]. 2014;78:7–17. Available from: http://www.sciencedirect.com/science/article/pii/S0197018614001843.
Rohn TT. Proteolytic cleavage of apolipoprotein E4 as the keystone for the heightened risk associated with Alzheimer’s disease. Int J Mol Sci [Internet]. 2013;14(7):14908–22. Available from: https://www.ncbi.nlm.nih.gov/pubmed/23867607.
Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science [Internet]. 2003;302(5646):819 LP–822. Available from: http://science.sciencemag.org/content/302/5646/819.abstract.
Fu R-H, Liu S-P, Huang S-J, Chen H-J, Chen P-R, Lin Y-H, et al. Aberrant alternative splicing events in Parkinson’s disease. Cell Transplant [Internet]. 2013;22(4):653–61. Available from: https://doi.org/10.3727/096368912X655154.
Xu L, Lin D, Yin D, Koeffler HP. An emerging role of PARK2 in cancer. J Mol Med [Internet]. 2014;92(1):31–42. Available from: https://doi.org/10.1007/s00109-013-1107-0.
Rideout HJ, Stefanis L. The neurobiology of LRRK2 and its role in the pathogenesis of Parkinson’s disease. Neurochem Res [Internet]. 2014;39(3):576–92. Available from: https://doi.org/10.1007/s11064-013-1073-5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Yustis-Rubio, J.C., Gomez-Verjan, J.C. (2020). Alternative Splicing and Aging. In: Gomez-Verjan, J., Rivero-Segura, N. (eds) Clinical Genetics and Genomics of Aging. Springer, Cham. https://doi.org/10.1007/978-3-030-40955-5_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-40955-5_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-40954-8
Online ISBN: 978-3-030-40955-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)