
Abstract
Atrial fibrillation (AF) is associated with increased cardiovascular morbidity and mortality with projections that it will affect 8–12 million people in the United States by 2050. Obesity has been identified as an important independent risk factor for AF, with weight loss leading to decreased AF burden and improved arrhythmia free survival. The precise mechanisms by which obesity contributes to AF remain poorly understood. However, it has recently been speculated that epicardial adipose tissue (EAT) may be a key mediator between obesity and AF. EAT is a visceral fat depot with anatomic contiguity to the myocardium. Under physiological conditions, EAT plays an important protective role via mechanical, metabolic, and thermogenic functions. However, under pathophysiological conditions, it may contribute to development of AF through various mechanisms including fatty infiltration, fibrosis, inflammation, oxidative stress, atrial remodelling, and genetic factors. EAT has been shown in multiple studies to be a risk factor for development of AF and predictor of recurrence after catheter ablation. The mechanisms directly linking EAT to the pathogenesis of AF also are uncertain. Multiple pharmacologic options have been proposed to target EAT; however, the efficacy of targeted reduction in EAT requires further investigation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara Key Points-
Obesity has been associated with atrial fibrillation, while weight loss decreases atrial fibrillation (AF) burden and recurrence post-ablation.
-
Epicardial adipose tissue is a risk factor for development of AF and predictor of recurrence after catheter ablation.
-
The mechanism by which epicardial adipose tissue contributes to the pathogenesis of atrial fibrillation remains unclear. Various mechanisms have been proposed including fatty infiltration, fibrosis, inflammation, oxidative stress, atrial remodelling, and genetic factors.
-
Peri-atrial epicardial adipose tissue (EAT) may play a more prominent role in the pathogenesis of AF due to its proximity to the atrial myocardium and local paracrine effect.
Introduction
Atrial fibrillation (AF) is linked with increased cardiovascular morbidity and mortality [1, 2]. AF is the most common sustained heart rhythm disorder affecting humans [3]. AF affects 5.2 million Americans as of 2010 with projections that it will affect 12.1 million people in the United States by 2030 [4]. Incremental cost related to AF has been estimated at $6–26 billion per year in the United States alone [5]. This rise in AF prevalence is partially connected to a growing elderly population with AF prevalence doubling with each advancing decade of age >50 years [6, 7]. Besides age, various risk factors predispose to the development of AF, such as coronary artery disease [8, 9], obesity [10, 11], hypertension [6, 12,13,14], diabetes mellitus [15,16,17], heart failure, [6, 18], and sleep apnea [19,20,21].
As these risk factors do not directly affect the atrium, their association with AF is likely mediated by intermediate pathways causing disease in the atrial myocardium. Moreover, as in coronary artery disease , the clinical manifestation of AF is likely preceded by years of subclinical progressive atrial disease (atrial myopathy), which may involve inflammation, oxidative stress, fibrosis, and electrical, structural, and autonomic remodelling [22,23,24,25,26]. Multiple studies show that obesity is associated with the development of AF substrate via various mechanisms [27,28,29,30]. Obesity may contribute to the development of AF indirectly via its effects on comorbidities such as hypertension (activation of renin-angiotensin-aldosterone system) and obstructive sleep apnea (hypoxia and hypercapnia, systemic information). Obesity may also be directly implicated in the development of AF via local pro-inflammatory effects [27,28,29,30].
Obesity is associated with epicardial adipose tissue (EAT) [10, 31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46]. Due to its contiguity to the underlying atrial myocardium, it is possible that epicardial adipose tissue (EAT) is associated with the development of AF. In this chapter, we will explore the nature of the relation between EAT and AF, pathophysiological mechanisms, clinical correlation, and some proposed future therapeutic options targeting EAT.
Obesity and Atrial Fibrillation
The role of adipose tissue in multiple disease states has now been clearly established. Adipose tissue is a heterogeneous tissue that includes mature adipocytes, inflammatory cells of various phenotypic characteristics, fibroblasts, and stem cells as well as vascular cells coming from microvascular structures embedded in the tissue [47]. All cell types contribute to the adipose tissue secretome, which may contain pro-inflammatory and pro-atherogenic adipocytokines (e.g., resistin, leptin, visfatin, and monocyte chemoattractant protein-1) in metabolically unhealthy obesity [48]. The adipocytokines secreted by the adipose tissue can exert paracrine effects on the neighboring vascular wall (i.e., perivascular adipose tissue) [49] or myocardium (i.e., epicardial adipose tissue) [50], as well as endocrine effects (i.e., from “remote” depots like subcutaneous adipose tissue) on the cardiovascular system through the circulation [51].
The most widely used assessment of adiposity is weight/body mass index [52]. The population disease burden due to the obesity epidemic is widely recognized. Obesity rates worldwide have nearly tripled in the last 40 years, with more than a third of the world’s population being obese or overweight [53]. As for the United States, 38% of adults are obese, with a body mass index (BMI) over 30 kg/m2, and an additional 33% are overweight with BMI between 25 and 30 kg/m2 [54]. In recent years, obesity has been shown to be an independent risk factor for development of AF (Table 10.1) [10, 31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46]. Importantly, the Framingham Heart Study revealed a 4% increased risk in incident AF per unit increase in body mass index in both men and women [31]. A meta-analysis of 51 studies involving more than 600,000 individuals evaluated the impact of obesity on AF [36]. It was found that every 5-unit rise in BMI confers an additional 19–29% risk of incident AF, a 10% risk of postoperative AF, and a 13% risk of post-ablation AF [36]. The association of obesity with other coexisting risk factors for AF (such as diabetes and hypertension) raises uncertainty in the independent association between obesity and AF. To account for this, Lee et al. recently conducted a retrospective study with a cohort of nearly 400,000 obese patients free of comorbidities [37]. The association between AF and obesity remained significant with a hazard ratio of 1.3 (95% confidence intervals 1.14–1.48) when compared to healthy nonobese patients [37]. Additionally, one study that followed 3248 patients for 5.1 years found that BMI independently predicted progression of AF from paroxysmal to permanent – hazard ratio of 1.04 (95% CI, 1.03–1.06; P < 0.0001) [38]. BMI was also shown to be an independent predictor of AF relapse post-ablation therapy in a recent multicenter observational study hazard ratio of 1.01 per kg/m2 (95% CI, 1.01–1.02, P = 0.002) [39].
The impact of weight loss on AF is also well established and evident in multiple recent studies [55,56,57,58,59,60]. Abed et al. conducted a randomized, interventional clinical trial in Australia for overweight or obese patients (body mass index >27 kg/m2) with symptomatic AF. Patients were randomized into a structured weight management group (intervention) or general lifestyle advice (control) [55]. The interventional group showed a dramatic reduction in weight, AF symptom burden scores, symptom severity scores, and number of AF episodes when compared to control [55]. The Aggressive Risk Factor Reduction Study for Atrial Fibrillation (ARREST-AF) study [59] included a cohort of patients who underwent a structured risk factor management program following catheter ablation for treatment of AF in one arm, and a control group who did not participate in the risk factor management program in the second arm. Patients in the risk factor management program experienced a 13% decrease in body mass index (versus 1% for the control group) after a median follow-up of 42.8 months with significant decline in blood pressure and LA volume index [59]. Additionally, patients in this program after 2 years of follow-up achieved arrhythmia free survival at final follow-up of 32.9% (P < 0.001) after a single procedure and 87% (P < 0.001) after multiple procedures versus 9.7% (P < 0.001) for a single procedure and 17.8% (P < 0.001) for patients requiring multiple procedures in the control group [59]. The LEGACY study noted that weight loss ≥10% resulted in six times greater probability of AF-free survival (95% confidence interval: 3.4–10.3; P < 0.001) compared to the groups with 3–9% and <3% weight loss [60]. Moreover, the REVERSE-AF study assessed the progression and reversal of AF in the same cohort of the LEGACY study. In patients with ≥10% weight loss, 88% experienced reversal from persistent to paroxysmal or no AF (P < 0.001) at final follow-up after individualized arrhythmia management, a striking difference when compared to 49% and 26% reversal in patients who lost 3–9% and <3% of their body weight, respectively (P < 0.001) [58]. Despite the uncertainty of the exact mechanisms by which weight loss impacts AF, the evidence seems clear and strong. Hence, and for the first time, the 2019 focused update to the AF guidelines [61] introduced a class I recommendation: “For overweight and obese patients with AF, weight loss, combined with risk factor modification, is recommended” based on emerging data [36, 55, 59].
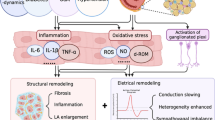
The mechanisms by which obesity impacts AF require investigation. In theory, obesity may directly (in addition to indirect effects through hypertension and diabetes) contribute to AF via systemic effects of visceral adiposity or from direct effects of the cardiac adipose tissue on the adjacent myocardium. The cardiac adipose tissue has been studied as the epicardial adipose tissue (EAT) or pericardial adipose tissue (PAT) and is associated with the development of AF (see Clinical Evidence of Association between EAT and AF) [62,63,64,65,66]. EAT is associated with increased body mass index (BMI) and is a well-documented risk factor for AF initiation and perpetuation [66,67,68]. EAT is a unique visceral adipose depot with anatomic and functional proximity to the myocardium and coronary arteries [69,70,71,72]. Advanced cardiac imaging has paved the way for better characterization of EAT and studying its effect on cardiovascular health [65, 73]. Under physiologic conditions, EAT has cardioprotective metabolic, thermogenic, and mechanical characteristics [70]. The pathophysiological mechanisms that link EAT to AF, however, remain elusive and are not entirely understood. Atrial electrical and structural remodeling, inflammation, oxidative stress, neural mechanisms, and genetic factors may be pathways mediating EAT effects on AF occurrence [22,23,24,25,26]. This will be discussed in the next section.
EAT and Atrial Fibrillation: Anatomy, Physiology, and Pathophysiological Mechanisms
Anatomy
EAT is a peculiar visceral fat depot with anatomical and functional contiguity to the myocardium and coronary arteries [69,70,71,72]. EAT is situated between the myocardium and visceral layer of the pericardium and is present in the atrioventricular and interventricular grooves without a structure or fascia separating it from the myocardium and the epicardial vessels [72, 74,75,76]. This means EAT and the myocardium share common microcirculation, suggesting a close and strong interaction between the two structures [77]. A paracrine process is one possible biological interaction between EAT and its adjacent myocardium. A vasocrine pathway is also likely via the vasa vasorum [72]. In contrast, pericardial adipose tissue is another cardiac fat deposit situated outside the visceral pericardium and on the outer surface of the parietal pericardium [77]. Although PAT and EAT are close in proximity, they are distinct [77]. Unlike EAT, PAT is separated from the myocardium by visceral pericardium [78]. PAT and EAT are also embryologically different [72]. EAT originates from the splanchnopleuric mesoderm, which also gives rise to mesenteric and omental fat [79], while PAT is derived from the primitive thoracic mesenchyme [79]. EAT is perfused by the coronary arteries, while PAT is supplied by a branch of the internal mammary artery, the pericardiophrenic artery [79]. However, it is noteworthy that the effects of PAT rather than EAT on the heart are often reported [63,64,65, 80,81,82,83,84]. This likely relates to the methods of quantitation. A more detailed look at anatomy and terminology is described in Chap. 1.
Moreover, recent findings indicate that regional EAT distribution is important. Left atrial EAT (LAEAT) is significantly increased in patients with AF and may be related to the recurrence of AF after catheter ablation [65, 83, 85,86,87,88,89]. LAEAT displays a unique genetic pattern when compared to peri-coronary and peri-ventricular EAT [90]. LAEAT is enriched in genes involved in oxidative phosphorylation, muscular contraction, and calcium signaling when compared to other EAT areas [90].
Physiology
The functional complexity of human EAT is not yet fully understood. However, under physiological conditions , EAT’s role in the heart is generally distinguished by mechanical, metabolic, thermogenic, and endocrine/paracrine functions [70]. EAT mechanically serves as a cushion protecting the coronary arteries from extreme distortion caused by arterial pulsation and myocardial contraction via its compressibility and elasticity [91]. Metabolically, EAT differs from other visceral fat in its ability to synthesize and break down free fatty acids (FFA) at a higher rate and a lower rate of glucose utilization [75, 92]. EAT contains abundant saturated fatty acids, and this enrichment and increased metabolism of free fatty acids (FFAs) maintain myocardial energy supplies, particularly during periods of high demand [71, 93]. FFA oxidation is responsible for about 50–70% of the energy production of the heart, and it is proposed that EAT functions as a buffer to protect the heart against excessively high levels of FFAs [72].
Moreover, EAT has higher expression of uncoupling protein-1 (UCP1) genes when compared to fat depots in other parts of the body [94]. This may provide EAT the thermogenic ability to produce heat (like brown adipose tissue) to protect the coronary arteries from hypothermia damage similar to what we see in hibernating animals [75]. EAT also acts as a source for paracrine modulators of myocardial inflammation and oxidative stress by producing multiple bioactive cytokines [95]. Under normal physiological conditions, cytokines such as adiponectin and adrenomedullin have antidiabetic, antiatherogenic, antioxidative, and anti-inflammatory roles exerting cardioprotective functions [96,97,98,99]. This is discussed in detail in Chap. 2. However, despite its cardioprotective mechanisms, EAT in the pathological state can be harmful, producing inflammatory factors exacerbating cardiovascular diseases and may participate in the pathogenesis of AF via various mechanisms that will be discussed next.
Pathophysiological Mechanisms
Multiple studies have established an association between EAT and AF (see Clinical Evidence of Association between EAT and AF). However, the exact mechanisms by which EAT contribute to AF development are unresolved and remain an intriguing topic in translational research. Multiple mechanisms have been identified. These mechanisms could include a direct effect of EAT on the atrium (e.g., by the infiltration of adipose tissue into the myocardium leading to altered atrial electrophysiological properties) or indirect mechanisms (e.g., by acting as a source for paracrine modulators of myocardial inflammation and oxidative stress). Altered gene expression may play a role in the contribution of EAT to AF. Alternatively, EAT may influence local atrial and pulmonary vein electrophysiologic properties, such as the refractory period, which interact with a remodeled atrial substrate to sustain AF [100]. These mechanisms are discussed in detail below.
Fatty Infiltration
Given its location and the lack of fascia separating EAT from the underlying myocardium, direct fatty infiltration into the atrium is possible. Multiple previous studies have noted that the abundance of epicardial fat is associated with direct adipocyte infiltration into the underlying atrial myocardium [78, 101]. This may contribute to myocardial functional disorganization and the formation of local arrhythmogenic substrate [102]. Mahajan et al., for example, found an accumulation of epicardial fat with pronounced myocardial infiltration by adipocytes, particularly over the posterior left atrial wall in diet-induced obese sheep [101]. In contrast, non-obese control sheep had less epicardial fat accumulation and minimal adipocyte infiltration into the myocardium. Such direct fatty infiltration can mechanically separate myocytes and could directly result in conduction slowing or could promote side-to-side cell connection loss similar to that seen with microfibrosis [103, 104]. Simultaneous endo-epicardial mapping of atrial electrical activity has shown that breakthrough and micro reentry circuits increase with increasing AF substrate complexity. Losing epicardial layer continuity due to atrial fibrosis appears to be one of the major determinants of the complexity of fibrillatory conduction pathways [105]. In addition to the direct anatomical effect of fatty infiltration, Haemers et al. [106] reported that patients with a history of AF had a higher histological degree of fibrosis of the adipose tissue present in sub-epicardium, with abundance of active inflammatory cells [106]. These observations suggest that this adipose tissue component of the atrial myocardium may not be an innocent bystander.
Inflammation
Inflammation is a potential contributor to the pathogenesis of AF. It may play a role in AF initiation and perpetuation [107]. Notable markers of inflammation, like C-reactive protein (CRP), interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-1b (IL-1b), and tumor necrosis factor-α (TNF-α), have been associated with the incidence, severity, and recurrence of AF [108,109,110,111,112]. These markers are produced by EAT and may have local pro-inflammatory effects on the adjacent atrial myocardium that facilitate arrhythmogenesis. Aviles et al. first demonstrated that elevated CRP predicted increased risk of developing AF in a large population-based prospective cohort including 5806 patients [111]. In another study, it was found that high levels of CRP were a predictor of arrhythmia recurrence after cardioversion [110]. Li et al. demonstrated that serum TNF-α blood levels were higher in patients with AF compared with those in sinus rhythm and in persistent and permanent AF compared with paroxysmal AF [113].
The immunohistochemistry of the left atrial appendage obtained from 16 patients during cardiac surgery showed immunologically active monocytes, and macrophages with abundance of inflammatory proteins like monocyte chemoattractive protein-1, intracellular adhesion molecule-1, and vascular cell adhesion molecule-1 that were more expressed in the endocardium of patients with AF compared to those in sinus rhythm [114]. This supports the hypothesis of a local immunologic inflammatory response in the atrium of patients with AF [114]. Moreover, the neutrophil-to-lymphocyte ratio was also shown to be an independent predictor for non-valvular AF [115]. Interestingly, it was demonstrated that elevated levels of monocyte chemoattractant protein-1 were found to be associated with increased epicardial fat thickness [116]. A significant correlation was found between EAT thickness and neutrophil-to-lymphocyte ratio [117].
In addition, the inflammatory activity of EAT reflected by maximal standardized uptake value (SUV) of 18-fluorodeoxyglucose(FDG)-positron emission tomography (PET) which reflects glucose metabolism was found to be higher in patients with AF compared to controls, and inflammatory activity of EAT adjacent to the LA, atrioventricular groove, and left main artery was greater than in subcutaneous or visceral thoracic tissue [118]. Kusayama et al. [119] reported that inflammation of EAT around the LA, but not subcutaneous adipose tissue is related to the presence of paroxysmal AF.
In addition to pro-inflammatory cytokines, adipose tissue also secretes adiponectin, an adipocytokine with anti-inflammatory effects [120]. Kourliouros et al. conducted a study to investigate the role of adiponectin in the pathogenesis of postoperative AF. Ninety patients undergoing coronary artery bypass graft (CABG) surgery had adiponectin level measured in serum and EAT. Increased levels of adiponectin were associated with sinus rhythm (SR) following surgery, thus reinforcing the inflammatory hypothesis in the pathogenesis of postoperative AF [121]. Moreover, an inverse relationship between adiponectin and LA size, independent of age, sex, insulin resistance, and left ventricular mass, has been reported, suggesting that adiponectin could influence atrial remodeling [122]. Taken together, if inflammatory pathways in peri-atrial adipose tissue are implicated in the pathogenesis of AF, this could be a potential future target for the prevention of AF. For example, intraoperative dexamethasone administration demonstrated significant protection against AF after cardiac surgery in one large clinical study [123]. A recent meta-analysis demonstrated that steroid therapy reduces the incidence of postoperative AF and the length of stay in the intensive care unit or in the hospital [124]. It is possible that the greatest benefit of anti-inflammatory interventions is limited to settings of inflammatory reaction due to tissue damage such as in cardiac surgery and catheter ablation. More studies are needed to elucidate the role of inflammation and the benefit of anti-inflammatory interventions in patients with AF.
Atrial Remodelling
Electrical Remodelling
Left atrial EAT can directly modulate electrical properties and ion currents of LA myocytes. When LA myocytes were incubated with epicardial adipocytes, they had longer action potential durations, a more-positive resting membrane potential, larger late sodium currents, L-type calcium currents, and transient outward potassium currents (Ito), but smaller delayed rectifier potassium and inward rectifier potassium (Kir) currents than control LA myocytes [125]. In the clinical setting, epicardial fat was independently associated with atrial conduction time in a large population study as indicated by P-wave indices [126]. Moreover, left atrial EAT was associated with lower bipolar voltage and electrogram fractionation in 30 patients assessed in sinus rhythm before AF ablation. Multivariate analysis for the potential nonlinear association between EAT and low voltage zones was significant with OR = 1.60 (P < 0.001) [127]. Another small study using a 3D merge process and dominant frequency left atrial map identified EAT locations to correspond to high dominant frequency during AF [128, 129]. These data suggest a possible role of EAT on AF electrophysiological substrate that requires further research.
Structural Remodeling
Another potential mechanism is the role of EAT in atrial structural remodeling . Atrial fibrosis is the hallmark of structural remodeling. Interstitial fibrosis leads to heterogeneity of conduction, which provides a substrate for AF [130]. Atrial fibrosis is the primary pathologic abnormality seen in AF-related structural remodeling, and the degree of fibrosis has clinical implications [116, 131]. EAT may contribute to atrial fibrosis by exerting a paracrine effect on the atrium secreting profibrotic cytokines [132] such as activin A and matrix metalloproteinases (MMP) [72, 78, 121, 133, 134].
Activin A is a member of the TGF-ß superfamily. First recognized as an inducer of follicle-stimulating hormone release, activin A is a multifunctional cytokine expressed in various tissue types. A profibrotic effect of activin A has already been described for liver fibrosis [135,136,137]. In rats, the supplementation of culture media with recombinant human activin A-induced marked rat atrial myocardial fibrosis, while anti-activin A antibody neutralized these profibrotic effects [138]. Both EAT-conditioned medium and activin A induced the expression of TGF-ß1 and -β2 in the atria, which could indirectly mediate the profibrotic effect of activin A [138]. Activin A-induced cardiac effects other than fibrosis have also been described. For instance, this cytokine has anti-hypertrophic and anti-apoptotic properties on the myocardium when it is exposed to ischemia, reperfusion, and pressure overload injuries [139, 140]. Activin A causes a negative inotropic effect on isolated guinea pig cardiac myocytes, suggesting a direct effect of this cytokine on the excitation-contraction coupling process [141].
MMPs are important regulators of extracellular matrix homeostasis, including the various collagen fibers and basement membrane components. During AF, it has been demonstrated that upregulated activity of several MMPs, notably MMP2 and 7, contribute to the accumulation of interstitial fibrosis [142]. Levels of serum MMP2, which has been implicated in atrial remodeling, were higher in patients with greater EAT volume [143]. MMP8, which is abundantly expressed in EAT, is known to be involved in the formation of atherosclerotic plaques [144, 145], whereas little is known of its role in myocardial fibrosis [146]. Venteclef et al. found that the level of both activin A and MMP8 are enhanced in patients with heart failure. Moreover, Greulich et al. [139] report that activin A is more abundantly expressed in the EAT of obese patients with type 2 diabetes.
In a recent study, Wang et al. [147] showed that connective tissue growth factor (cTGF) expression is significantly higher in EAT than in subcutaneous or pericardial adipose tissue from patients with AF and in EAT from patients with sinus rhythm. They concluded that cTGF is associated with atrial fibrosis and can be an essential risk factor for AF [147].
EAT may also affect the atrial cellular components by altering the proliferation of myofibroblasts and the number of dedifferentiated and dystrophic myocytes [148, 149]. In fact, adipose tissue contains abundant stem cells located in the stroma fraction [150, 151] that are capable of differentiating not only into adipocytes but also into cardiomyocytes [152] or myofibroblasts [153]. Therefore, cardiac fatty tissue may constitute a source of precursor cells that can differentiate into myofibroblasts, contributing to atrial structural remodeling.
EAT may contribute to structural remodeling by promoting ventricular diastolic dysfunction. Patients with left ventricular diastolic dysfunction have a significant increase in EAT volume [154]. Suffee et al. [155] demonstrated that adult atrial epicardial progenitor cells can transition to adipocytes and provided evidence that this process is driven by atrial natriuretic peptide (ANP) secreted by the myocardium. EAT accumulation in adult atria is a slow process that could occur in response to chronic alterations of atrial myocardium workload and metabolic conditions. Hence, these data indicate a possible crosstalk between EAT expansion and mechanical function of the atrial myocardium [155].
Oxidative Stress
EAT is known to be a source of reactive oxygen species that could contribute to the pathogenesis of AF [156]. Recently, Antonopoulos et al. [50] demonstrated that products of oxidation , such as 4-hydroxynonenal which is generated in the human cardiac tissue under conditions of increased oxidative stress, can act as signaling molecules and result in adiponectin upregulation in neighboring EAT. Adiponectin has a cardioprotective paracrine effect, and its upregulation is evidence of local defensive response of EAT against oxidative stress. Previous data have shown that the production of reactive oxygen species is higher in human EAT than it is in subcutaneous adipose tissue [156]. Ascorbat, an antioxidant and peroxynitrite decomposition catalyst, inhibited reactive oxygen species in dogs and appeared to attenuate atrial remodeling induced by rapid pacing [157].
Neural Mechanism
The autonomic nervous system plays an important role in the initiation and maintenance of AF. EAT is the anatomic site of the intrinsic cardiac autonomic nervous system, namely, ganglionated plexi (GP) and interconnecting nerves, especially in the posterior wall around pulmonary vein ostia [70]. These ganglia are a critical element responsible for the initiation and maintenance of AF [158, 159]. Sympathovagal imbalance detected by both impaired heart rate variability and heart rate turbulence parameters has been demonstrated to be related to EAT thickness [160]. Activation of the ganglionated plexi (GP) located within the epicardial fat tissue can cause both parasympathetic and sympathetic stimulation, resulting in shortening of action potential duration and increases in calcium transient, respectively, in the atrial myocardium [161]. Recent evidence also suggests that it is the ganglionated plexi activity to the PVs that is important in the pathogenesis of AF [162]. One study demonstrated that stimuli applied to PVs do not induce AF, unless there is simultaneous stimulation of the adjacent GP that does not excite the atrial myocardium [163]. In isolated canine PV preparations, simultaneous administration of acetylcholine plus norepinephrine [164] or local autonomic (both parasympathetic and sympathetic) nerve electrical stimulation [165] has been shown to induce early after depolarizations (EADs) and short surges of triggered firing from the PVs, similar to the pattern recorded from the PVs in patients with paroxysmal AF [164].
Multiple clinical studies have investigated the role that neuromodulation may have in controlling AF, particularly by ablation of ganglionated plexi of the intrinsic cardiac nervous system [166]. It is possible that the encasing epicardial fat influences the ganglionated plexi and thus contributes to arrhythmogenesis. It is interesting to note that botulinum toxin injection into epicardial fat pads during surgery may reduce cardiac autonomic nervous activity and have long-term effects on AF, potentially by suppressing ganglionated plexi [167].
Genetic Factors
Gene expression represents a new perspective in understanding the precise role of EAT in AF pathogenesis. In an experimental study that utilized human and pig atrial samples, both AF and rapid pacing were associated with significant changes in atrial gene expression consistent with the induction of an adipocyte-related expression profile [168]. These genes favor the development of important risk factors for AF like obesity and diabetes mellitus and may facilitate AF substrate formation by increasing atrial ectopic fat and fat infiltration [168].
The peri-atrial EAT transcriptome profile is notable for the expression of genes implicated in oxidative phosphorylation, muscular contraction, and Ca2+ signaling [90]. The SERCA1 gene codes for a Ca2+/ATP-dependent intracellular pump that translocates the cytosolic Ca2+ into the lumen of the sarcoplasmic reticulum and promotes excitation-contraction coupling [169]. Moreover, EAT secretome was described as a possible substrate for postoperative AF [170]. It encompasses various proteins differentially expressed in patients who later develop postoperative AF. Among those, gelsolin reduction, a protein involved in inflammation and ion channel regulation, was associated with postoperative AF [170].
Clinical Evidence of Association Between EAT and Atrial Fibrillation
Several studies have described the association between cardiac adipose tissue and AF (Table 10.2) [64, 65, 80,81,82, 85, 86, 102, 171,172,173,174] and AF recurrence post-catheter ablation (Table 10.3) [65, 83, 85, 87,88,89, 175,176,177,178,179]. One of the largest studies that examined the relationship between pericardial fat and AF is from the Framingham Heart study. In this study involving 3217 participants, the investigators characterized pericardial fat (defined as adipose tissue within the pericardial sac) with computed tomography (CT) and observed that pericardial fat volume predicted AF risk independent of other measures of adiposity OR = 1.28 (95% CI, 1.05–1.6; P = 0.03) for every standard deviation increase in PAT volume [64]. This relation remained significant despite adjustment for common AF risk factors, including age, sex, myocardial infarction, heart failure, BMI, and other regional fat deposits [64]. Importantly, the study did not show similar association of AF with other fat deposits such as intrathoracic or abdominal visceral fat. This suggests that contiguity of adipose tissue rather than generalized increases in fat depots may be essential in the pathogenesis of AF [64].
Another study showed a correlation of pericardial adipose tissue with the severity of AF subtypes. Al Chekakie et al. [80] demonstrated that pericardial fat correlated with paroxysmal (OR = 1.11; 95% CI: 1.01–1.23, P = 0.04) and persistent AF (OR = 1.18; 95% CI: 1.05–1.33, P = 0.004), independent of traditional AF risk factors including age, hypertension, sex, left atrial (LA) enlargement, valvular heart disease, left ventricular ejection fraction, diabetes mellitus, and BMI. This correlation was further confirmed in a recent meta-analysis that reported that EAT volume is associated with increased risk of AF – one standard deviation was associated with 2.2 higher odds of persistent AF compared to paroxysmal AF (OR = 2.19, 95% CI: 1.66–2.88) [66, 68, 180]. Noteworthy, the strength of associations of AF with EAT was greater than for measures of abdominal or overall adiposity [66]. Increased EAT has also been independently associated with AF after coronary artery bypass grafting [82, 181], electrical cardioversion [182], or ablation [65, 83, 85,86,87,88,89, 176, 177, 183]. Moreover, pericardial fat, when measured by MRI in 130 patients, showed a strong dose-response association with LA volume (P < 0.001), AF chronicity (P < 0.05), symptom burden (P < 0.05), and recurrence of AF after ablation (P = 0.035) [65]. However, systemic measures of obesity, including body mass index and body surface area, were not associated with these outcomes. These associations remained valid despite multivariate adjustment and adjustment for body weight.
Recently, Shin et al. also reported that epicardial fat volumes and peri-atrial fat thickness measured by CT scan were significantly associated with the prevalence and persistence of AF [172]. Moreover, a multivariate analysis revealed that only total EAT (P = 0.004) and thickness of intraatrial septum (P = 0.016) were independently associated with left atrial volume in patients with AF even after adjusting for BMI. These studies have shown that EAT volume may be more predictive of the presence and severity of AF than the conventional measures of obesity.
It is important to note that EAT distribution is often asymmetric and may exert different effects according to its location [184, 185]. As mentioned earlier, it was found that peri-atrial epicardial fat has distinct characteristics and genetic profile compared to peri-ventricular or peri-coronary epicardial fat [90]. Hence, due to its proximity and unique genetic profile, it follows that peri-atrial epicardial fat may be more directly related to the pathogenesis of AF. This was observed in many studies that showed left atrial EAT was significantly increased in patients with AF and may be related to the recurrence of AF after catheter ablation [65, 83, 85,86,87,88,89, 171]. Batal et al. [171] examined 169 patients who underwent CT scans for AF or CAD, measuring EAT thickness. The study found posterior LA fat thickness was a statistically significant predictor of AF burden even after adjusting for age, BMI, or LA area OR = 5.30 (95% CI, 1.39–20.24; P = 0.015). On the other hand, what stands true for the left atrial EAT is not necessarily valid for right atrial EAT. Hasebe et al. [186] compared two groups of patients with paroxysmal AF referred for catheter ablation. They reported that EAT volumes around both the RA and LA in the patients whose AF was thought to originate from the RA were significantly smaller than those in the patients whose AF was thought to originate from the LA. Thus, EAT may be more involved in the pathogenesis of AF whose source is the LA. It is important to note that there was no statistical difference in the total volume of EAT surrounding the whole heart between the two groups.
Despite the inability of these studies to demonstrate causation, there is ample evidence of a strong association/correlation between EAT/PAT and AF. This opens the door for questioning about its future clinical implications.
Clinical Implications of EAT in Atrial Fibrillation and Possible Therapeutic Options
EAT as Risk Predictive Biomarker for Atrial Fibrillation
Epicardial adipose tissue is a novel risk prediction marker , and it may have multiple clinical applications in the future. EAT has been linked with various acute coronary syndrome risk prediction scores [187,188,189,190]. These include the GRACE (Global Registry of Acute Coronary Events) score, the Syntax score for the severity of coronary lesions, and the TIMI score for the prediction of adverse coronary events. They all have shown positive correlations with increased epicardial fat volume or thickness [187, 189, 191]. Similarly, an increased quantity of adipose tissue surrounding the heart has been shown to predict major adverse cardiac events, including myocardial infarction and death in subjects with acute coronary syndromes, as well as in those with suspected coronary artery disease [192,193,194]. Like in acute coronary syndromes, multiple studies have established an association between atrial fibrillation and EAT independent of coronary atherosclerosis and classical cardiovascular risk factors [195]. The pericardial adipose tissue was shown to be independently associated with the incidence, severity, recurrence of AF after catheter ablation, and poor ablation outcomes [65, 83].
EAT volume is also associated with stroke and adverse cardiovascular outcomes in AF patients. Akdag et al. [196] calculated the CHA2DS2-VASc score and baseline EAT thickness in 96 consecutive patients with AF and 52 age- and sex-matched controls and found that the group with high CHA2DS2-VASc score had higher EAT thickness compared to the group with low-intermediate CHA2DS2-VASc score; the CHA2DS2-VASc score was positively correlated with EAT. Tsao et al. [197] evaluated whether EAT situated in the vicinity of the LA and the LA appendage (LAA) is correlated with atrial function and the subsequent development of AF-related stroke. They reported that increased accumulation of EAT (OR = 1.12, P < 0.001) around the LA was independently associated with stroke in AF patients [197]. Moreover, peri-atrial EAT was negatively correlated with the mechanical function of the LA (LAEF r = −0.369, P < 0.001). Total EAT was negatively correlated with active EF of the LAA (r = −0.464, P < 0.001). This contractile dysfunction and the circulatory stasis of the LAA may account for the pathophysiological association of EAT- and AF-related stroke [197]. Similarly, Chu et al. [198] demonstrated that increased EAT thickness per 1 mm is associated with adverse cardiovascular events (cardiovascular mortality, hospitalization for heart failure, myocardial infarction, and stroke) OR = 1.224 (95% CI, 1.096–1.368; P < 0.001). Moreover, the addition of EAT thickness to a model containing CHA2DS2-VASc score, LA volume index, and left ventricular systolic and diastolic function significantly improved the predictive value for cardiovascular events [198].
Potential Therapeutic Options Targeting EAT
Weight Loss
Weight loss has been recognized to reduce the pericardial fat burden [199]. Weight loss after bariatric surgery in obese subjects has also been associated with a decrease in EAT [200]. A recent meta-analysis showed a significant reduction in EAT with diet and bariatric surgery, but not with exercise [201]. Furthermore, in severe obesity, successful long-term weight reduction was associated with improved left ventricular diastolic function and exercise capacity, while reduction of epicardial fat thickness predicted improved diastolic function [202]. Sustained weight loss, as described in the ARREST-AF study [59] and LEGACY study [60], showed improved arrhythmia free survival and substantial AF burden reduction. Whether a reduction in EAT mediates this effect, at least in part, remains unknown and requires further research.
Medications
Interestingly, statin therapy has been shown to decrease EAT. In hyperlipidemic post-menopausal women, statin therapy caused EAT regression, and intensive therapy was more effective than moderate-intensity therapy [203]. This, however, does not seem linked to low-density lipoprotein lowering and may be secondary to other actions of statins such as their anti-inflammatory effects [203]. A recent meta-analysis reported that perioperative statin therapy in patients with sinus rhythm undergoing cardiac surgery was associated with reduction in the development of postoperative AF, hospital length of stay, and CRP level [204]. This meta-analysis showed that the beneficial effects on AF and CRP were more marked in patients receiving atorvastatin compared to other statins [204]. Another meta-analysis showed that the use of statins is significantly associated with decreased risk of AF in patients with sinus rhythm, but did not examine changes in EAT [205]. Notably, the greatest benefit for statins was seen for postoperative AF prevention, as well as for secondary prevention of the arrhythmia [205]. Whether this effect is partially mediated by a decrease in EAT remains to be seen.
Antidiabetic medications may play a role in targeting EAT, leading to its reduction. In a study performed in type 2 diabetes patients, insulin replacement therapy resulted in a reduction of epicardial fat thickness [206]. EAT was recently collected from 9 patients during cardiac surgery, and it was found that human EAT expresses the GLP-1 receptor (GLP-1R) [207, 208]. Liraglutide is a glucagon-like peptide-1 (GLP-1) receptor agonist; in an interventional case-controlled study, there was a rapid and substantial reduction in EAT thickness when liraglutide was given in addition to metformin vs metformin as monotherapy [209]. In the liraglutide plus metformin group, EAT thickness decreased by 29% and 36% (P < 0.001) at 3 and 6 months, respectively, whereas there was no EAT reduction in the metformin group [209]. Liraglutide is currently being investigated as a potential pharmaceutical therapeutic option as adjunctive therapy to catheter ablation in the Liraglutide Effect in Atrial Fibrillation (LEAF) clinical trial (NCT03856632, https://clinicaltrials.gov/). It is proposed that liraglutide reduces EAT and may thereby stabilize the atrial substrate.
Conclusion
AF is a complex arrhythmia. The initiation and maintenance of AF are dependent on the presence of both trigger and substrate, including electrical and structural atrial remodelling. Obesity has been identified as an important modifiable risk factor for AF, and recent evidence showed that weight loss has a positive impact on AF. Given the growing prevalence of obesity and the expected rise in AF incidence in the future, it is essential to establish a better understanding of the pathogenesis of AF and find alternative therapy that can address the underlying substrate. EAT is also clearly associated with AF. It is reasonable to speculate that the association between obesity and AF may be mediated, at least in part, by EAT. Under pathological condition, EAT could contribute to the pathogenesis of AF via various mechanisms: fatty infiltration, inflammation, oxidative stress, atrial remodelling (electrical and structural), alterations in gene expression, and neuronal mechanisms. EAT could serve as a new risk predictive biomarker of atrial fibrillation and a potential therapeutic target in the future.
References
Stewart S, Hart CL, Hole DJ, McMurray JJ. A population-based study of the long-term risks associated with atrial fibrillation: 20-year follow-up of the Renfrew/Paisley study. Am J Med. 2002;113:359–64.
Beyerbach DM, Zipes DP. Mortality as an endpoint in atrial fibrillation. Heart Rhythm. 2004;1:8–19.
Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–52.
Colilla S, Crow A, Petkun W, Singer DE, Simon T, Liu X. Estimates of current and future incidence and prevalence of atrial fibrillation in the U.S. adult population. Am J Cardiol. 2013;112:1142–7.
Kim MH, Johnston SS, Chu B-C, Dalal MR, Schulman KL. Estimation of total incremental health care costs in patients with atrial fibrillation in the United States. Circ Cardiovasc Qual. 2011;4:313–20.
Kannel WB, Wolf PA, Benjamin EJ, Levy D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol. 1998;82:2n–9n.
Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution, and gender of patients with atrial fibrillation: analysis and implications. Arch Intern Med. 1995;155:469–73.
Eldar M, Canetti M, Rotstein Z, Boyko V, Gottlieb S, Kaplinsky E, et al. Significance of paroxysmal atrial fibrillation complicating acute myocardial infarction in the thrombolytic era. Circulation. 1998;97:965–70.
Crenshaw BS, Ward SR, Granger CB, Stebbins AL, Topol EJ, Califf RM. Atrial fibrillation in the setting of acute myocardial infarction: the GUSTO-I experience. J Am Coll Cardiol. 1997;30:406–13.
Frost L, Hune LJ, Vestergaard P. Overweight and obesity as risk factors for atrial fibrillation or flutter: the Danish Diet, Cancer, and Health Study. Am J Med. 2005;118:489–95.
Dublin S, French B, Glazer NL, Wiggins KL, Lumley T, Psaty BM, et al. Risk of new-onset atrial fibrillation in relation to body mass index. Arch Intern Med. 2006;166:2322–8.
Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med. 1982;306:1018–22.
Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba Follow-Up Study. Am J Med. 1995;98:476–84.
Conen D, Tedrow UB, Koplan BA, Glynn RJ, Buring JE, Albert CM. Influence of systolic and diastolic blood pressure on the risk of incident atrial fibrillation in women. Circulation. 2009;119:2146.
Dublin S, Glazer NL, Smith NL, Psaty BM, Lumley T, Wiggins KL, et al. Diabetes mellitus, glycemic control, and risk of atrial fibrillation. J Gen Intern Med. 2010;25:853–8.
Benjamin EJ, Levy D, Vaziri SM, D’Agostino RB, Belanger AJ, Wolf PA. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA. 1994;271:840–4.
Watanabe H, Tanabe N, Watanabe T, Darbar D, Roden DM, Sasaki S, et al. Metabolic syndrome and risk of development of atrial fibrillation: the Niigata preventive medicine study. Circulation. 2008;117:1255.
Maisel WH, Stevenson LW. Atrial fibrillation in heart failure: epidemiology, pathophysiology, and rationale for therapy. Am J Cardiol. 2003;91:2–8.
Mehra R, Benjamin EJ, Shahar E, Gottlieb DJ, Nawabit R, Kirchner HL, et al. Association of nocturnal arrhythmias with sleep-disordered breathing: the Sleep Heart Health Study. Am J Respir Crit Care Med. 2006;173:910–6.
Mooe T, Gullsby S, Rabben T, Eriksson P. Sleep-disordered breathing: a novel predictor of atrial fibrillation after coronary artery bypass surgery. Coron Arter Dis. 1996;7:475–8.
Tanigawa T, Yamagishi K, Sakurai S, Muraki I, Noda H, Shimamoto T, et al. Arterial oxygen desaturation during sleep and atrial fibrillation. Heart. 2006;92:1854–5.
Goldberger JJ, Arora R, Green D, Greenland P, Lee DC, Lloyd-Jones DM, et al. Evaluating the atrial myopathy underlying atrial fibrillation: identifying the arrhythmogenic and thrombogenic substrate. Circulation. 2015;132:278–91.
Goudis CA, Vasileiadis IE, Liu T. Epicardial adipose tissue and atrial fibrillation: pathophysiological mechanisms, clinical implications, and potential therapies. Curr Med Res Opin. 2018;34:1933–43.
Wong CX, Ganesan AN, Selvanayagam JB. Epicardial fat and atrial fibrillation: current evidence, potential mechanisms, clinical implications, and future directions. Eur Heart J. 2017;38:1294–302.
Ng Arnold CT, Strudwick M, van der Geest RJ, Ng Austin CC, Gillinder L, Goo Shi Y, et al. Impact of epicardial adipose tissue, left ventricular myocardial fat content, and interstitial fibrosis on myocardial contractile function. Circ Cardiovasc Imaging. 2018;11:e007372.
Mazurek T, Zhang L, Zalewski A, Mannion John D, Diehl James T, Arafat H, et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation. 2003;108:2460–6.
Abed HS, Wittert GA. Obesity and atrial fibrillation. Obes Rev. 2013;14:929–38.
Abed HS, Samuel CS, Lau DH, Kelly DJ, Royce SG, Alasady M, et al. Obesity results in progressive atrial structural and electrical remodeling: implications for atrial fibrillation. Heart Rhythm. 2013;10:90–100.
Baek YS, Yang PS, Kim TH, Uhm JS, Park J, Pak HN, et al. Associations of abdominal obesity and new-onset atrial fibrillation in the general population. J Am Heart Assoc. 2017;6:e004705.
Goudis CA, Korantzopoulos P, Ntalas IV, Kallergis EM, Ketikoglou DG. Obesity and atrial fibrillation: a comprehensive review of the pathophysiological mechanisms and links. J Cardiol. 2015;66:361–9.
Wang TJ, Parise H, Levy D, D’Agostino RB Sr, Wolf PA, Vasan RS, et al. Obesity and the risk of new-onset atrial fibrillation. JAMA. 2004;292:2471–7.
Gami AS, Hodge DO, Herges RM, Olson EJ, Nykodym J, Kara T, et al. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol. 2007;49:565–71.
Tedrow UB, Conen D, Ridker PM, Cook NR, Koplan BA, Manson JE, et al. The long- and short-term impact of elevated body mass index on the risk of new atrial fibrillation the WHS (women’s health study). J Am Coll Cardiol. 2010;55:2319–27.
Knuiman M, Briffa T, Divitini M, Chew D, Eikelboom J, McQuillan B, et al. A cohort study examination of established and emerging risk factors for atrial fibrillation: the Busselton Health Study. Eur J Epidemiol. 2014;29:181–90.
Zacharias A, Schwann TA, Riordan CJ, Durham SJ, Shah AS, Habib RH. Obesity and risk of new-onset atrial fibrillation after cardiac surgery. Circulation. 2005;112:3247–55.
Wong CX, Sullivan T, Sun MT, Mahajan R, Pathak RK, Middeldorp M, et al. Obesity and the risk of incident, post-operative, and post-ablation atrial fibrillation: a meta-analysis of 626,603 individuals in 51 studies. JACC Clin Electrophysiol. 2015;1:139–52.
Lee H, Choi EK, Lee SH, Han KD, Rhee TM, Park CS, et al. Atrial fibrillation risk in metabolically healthy obesity: a nationwide population-based study. Int J Cardiol. 2017;240:221–7.
Tsang TS, Barnes ME, Miyasaka Y, Cha SS, Bailey KR, Verzosa GC, et al. Obesity as a risk factor for the progression of paroxysmal to permanent atrial fibrillation: a longitudinal cohort study of 21 years. Eur Heart J. 2008;29:2227–33.
Providencia R, Adragao P, de Asmundis C, Chun J, Chierchia G, Defaye P, et al. Impact of body mass index on the outcomes of catheter ablation of atrial fibrillation: a European Observational Multicenter Study. J Am Heart Assoc. 2019;8:e012253.
Huxley RR, Lopez FL, Folsom AR, Agarwal SK, Loehr LR, Soliman EZ, et al. Absolute and attributable risks of atrial fibrillation in relation to optimal and borderline risk factors: the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 2011;123:1501–8.
Long MJ, Jiang CQ, Lam TH, Xu L, Zhang WS, Lin JM, et al. Atrial fibrillation and obesity among older Chinese: the Guangzhou Biobank Cohort Study. Int J Cardiol. 2011;148:48–52.
Karasoy D, Bo Jensen T, Hansen ML, Schmiegelow M, Lamberts M, Gislason GH, et al. Obesity is a risk factor for atrial fibrillation among fertile young women: a nationwide cohort study. Europace. 2013;15:781–6.
Berkovitch A, Kivity S, Klempfner R, Segev S, Milwidsky A, Erez A, et al. Body mass index and the risk of new-onset atrial fibrillation in middle-aged adults. Am Heart J. 2016;173:41–8.
Foy AJ, Mandrola J, Liu G, Naccarelli GV. Relation of obesity to new-onset atrial fibrillation and atrial flutter in adults. Am J Cardiol. 2018;121:1072–5.
Lim YM, Yang PS, Jang E, Yu HT, Kim TH, Uhm JS, et al. Body mass index variability and long-term risk of new-onset atrial fibrillation in the general population: a Korean Nationwide Cohort Study. Mayo Clin Proc. 2019;94:225–35.
Sandhu RK, Conen D, Tedrow UB, Fitzgerald KC, Pradhan AD, Ridker PM, et al. Predisposing factors associated with development of persistent compared with paroxysmal atrial fibrillation. J Am Heart Assoc. 2014;3:e000916.
Badimon L, Cubedo J. Adipose tissue depots and inflammation: effects on plasticity and resident mesenchymal stem cell function. Cardiovasc Res. 2017;113:1064–73.
Guzik TJ, Skiba DS, Touyz RM, Harrison DG. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc Res. 2017;113:1009–23.
Margaritis M, Antonopoulos AS, Digby J, Lee R, Reilly S, Coutinho P, et al. Interactions between vascular wall and perivascular adipose tissue reveal novel roles for adiponectin in the regulation of endothelial nitric oxide synthase function in human vessels. Circulation. 2013;127:2209–21.
Antonopoulos AS, Margaritis M, Verheule S, Recalde A, Sanna F, Herdman L, et al. Mutual regulation of epicardial adipose tissue and myocardial redox state by ppar-gamma/adiponectin signalling. Circ Res. 2016;118:842–55.
Antonopoulos AS, Margaritis M, Coutinho P, Digby J, Patel R, Psarros C, et al. Reciprocal effects of systemic inflammation and brain natriuretic peptide on adiponectin biosynthesis in adipose tissue of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 2014;34:2151–9.
Menke A, Muntner P, Wildman RP, Reynolds K, He J. Measures of adiposity and cardiovascular disease risk factors. Obesity. 2007;15:785–95.
NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet. 2016;387:1377–96.
Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of obesity among adults and youth: United States, 2015–2016. CHS Data Brief. 2017;10(288):1–8.
Abed HS, Wittert GA, Leong DP, Shirazi MG, Bahrami B, Middeldorp ME, et al. Effect of weight reduction and cardiometabolic risk factor management on symptom burden and severity in patients with atrial fibrillation: a randomized clinical trial. JAMA. 2013;310:2050–60.
Alonso A, Bahnson JL, Gaussoin SA, Bertoni AG, Johnson KC, Lewis CE, et al. Effect of an intensive lifestyle intervention on atrial fibrillation risk in individuals with type 2 diabetes: the Look AHEAD randomized trial. Am Heart J. 2015;170:770–7.e5.
Jamaly S, Carlsson L, Peltonen M, Jacobson P, Sjostrom L, Karason K. Bariatric surgery and the risk of new-onset atrial fibrillation in Swedish obese subjects. J Am Coll Cardiol. 2016;68:2497–504.
Middeldorp ME, Pathak RK, Meredith M, Mehta AB, Elliott AD, Mahajan R, et al. PREVEntion and regReSsive Effect of weight-loss and risk factor modification on Atrial Fibrillation: the REVERSE-AF study. Europace. 2018;20:1929–35.
Pathak RK, Middeldorp ME, Lau DH, Mehta AB, Mahajan R, Twomey D, et al. Aggressive risk factor reduction study for atrial fibrillation and implications for the outcome of ablation: the ARREST-AF cohort study. J Am Coll Cardiol. 2014;64:2222–31.
Pathak RK, Middeldorp ME, Meredith M, Mehta AB, Mahajan R, Wong CX, et al. Long-term effect of goal-directed weight management in an atrial fibrillation cohort: a long-term follow-up study (LEGACY). J Am Coll Cardiol. 2015;65:2159–69.
January CT, Wann LS, Calkins H, Chen LY, Cigarroa JE, Cleveland JC Jr, et al. 2019 AHA/ACC/HRS focused update of the 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2019;74:104–32.
Hatem SN, Redheuil A, Gandjbakhch E. Cardiac adipose tissue and atrial fibrillation: the perils of adiposity. Cardiovasc Res. 2016;109:502–9.
Al-Rawahi M, Proietti R, Thanassoulis G. Pericardial fat and atrial fibrillation: epidemiology, mechanisms and interventions. Int J Cardiol. 2015;195:98–103.
Thanassoulis G, Massaro JM, O’Donnell CJ, Hoffmann U, Levy D, Ellinor PT, et al. Pericardial fat is associated with prevalent atrial fibrillation: the Framingham Heart Study. Circ Arrhythm Electrophysiol. 2010;3:345–50.
Wong CX, Abed HS, Molaee P, Nelson AJ, Brooks AG, Sharma G, et al. Pericardial fat is associated with atrial fibrillation severity and ablation outcome. J Am Coll Cardiol. 2011;57(17):1745–51.
Wong CX, Sun MT, Odutayo A, Emdin CA, Mahajan R, Lau DH, et al. Associations of epicardial, abdominal, and overall adiposity with atrial fibrillation. Circ Arrhythm Electrophysiol. 2016;9(12):e004378.
Rabkin SW. The relationship between epicardial fat and indices of obesity and the metabolic syndrome: a systematic review and meta-analysis. Metab Syndr Relat Disord. 2014;12:31–42.
Zhu W, Zhang H, Guo L, Hong K. Relationship between epicardial adipose tissue volume and atrial fibrillation: a systematic review and meta-analysis. Herz. 2016;41:421–7.
McAninch EA, Fonseca TL, Poggioli R, Panos AL, Salerno TA, Deng Y, et al. Epicardial adipose tissue has a unique transcriptome modified in severe coronary artery disease. Obesity (Silver Spring). 2015;23:1267–78.
Iacobellis G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat Rev Endocrinol. 2015;11:363–71.
Iacobellis G, Bianco AC. Epicardial adipose tissue: emerging physiological, pathophysiological and clinical features. Trends Endocrinol Metab. 2011;22:450–7.
Iacobellis G, Corradi D, Sharma AM. Epicardial adipose tissue: anatomic, biomolecular and clinical relationships with the heart. Nat Clin Pract Cardiovasc Med. 2005;2:536–43.
Iacobellis G, Willens HJ. Echocardiographic epicardial fat: a review of research and clinical applications. J Am Soc Echocardiogr. 2009;22:1311–9; quiz 417–8.
Bertaso AG, Bertol D, Duncan BB, Foppa M. Epicardial fat: definition, measurements and systematic review of main outcomes. Arq Bras Cardiol. 2013;101:e18–28.
Marchington JM, Mattacks CA, Pond CM. Adipose tissue in the mammalian heart and pericardium: structure, foetal development and biochemical properties. Comp Biochem Physiol B. 1989;94:225–32.
Corradi D, Maestri R, Callegari S, Pastori P, Goldoni M, Luong TV, et al. The ventricular epicardial fat is related to the myocardial mass in normal, ischemic and hypertrophic hearts. Cardiovasc Pathol. 2004;13:313–6.
Iacobellis G. Epicardial and pericardial fat: close, but very different. Obesity (Silver Spring). 2009;17:625; author reply 6–7.
Hatem SN, Sanders P. Epicardial adipose tissue and atrial fibrillation. Cardiovasc Res. 2014;102:205–13.
Sacks HS, Fain JN. Human epicardial adipose tissue: a review. Am Heart J. 2007;153:907–17.
Al Chekakie MO, Welles CC, Metoyer R, Ibrahim A, Shapira AR, Cytron J, et al. Pericardial fat is independently associated with human atrial fibrillation. J Am Coll Cardiol. 2010;56:784–8.
Greif M, von Ziegler F, Wakili R, Tittus J, Becker C, Helbig S, et al. Increased pericardial adipose tissue is correlated with atrial fibrillation and left atrial dilatation. Clin Res Cardiol. 2013;102:555–62.
Drossos G, Koutsogiannidis C-P, Ananiadou O, Kapsas G, Ampatzidou F, Madesis A, et al. Pericardial fat is strongly associated with atrial fibrillation after coronary artery bypass graft surgery. Eur J Cardiothorac Surg. 2014;46:1014–20.
Kim TH, Park J, Park JK, Uhm JS, Joung B, Lee MH, et al. Pericardial fat volume is associated with clinical recurrence after catheter ablation for persistent atrial fibrillation, but not paroxysmal atrial fibrillation: an analysis of over 600-patients. Int J Cardiol. 2014;176:841–6.
Liu J, Taylor HA, Fox CS, Carr JJ, Ding J. Pericardial adipose tissue, atherosclerosis, and cardiovascular disease risk factors: The Jackson Heart Study. Response to Iacobellis and Malavazos. Diabetes Care. 2010;33:e128-e.
Tsao HM, Hu WC, Wu MH, Tai CT, Lin YJ, Chang SL, et al. Quantitative analysis of quantity and distribution of epicardial adipose tissue surrounding the left atrium in patients with atrial fibrillation and effect of recurrence after ablation. Am J Cardiol. 2011;107:1498–503.
Nagashima K, Okumura Y, Watanabe I, Nakai T, Ohkubo K, Kofune T, et al. Association between epicardial adipose tissue volumes on 3-dimensional reconstructed CT images and recurrence of atrial fibrillation after catheter ablation. Circ J. 2011;75:2559–65.
Chao TF, Hung CL, Tsao HM, Lin YJ, Yun CH, Lai YH, et al. Epicardial adipose tissue thickness and ablation outcome of atrial fibrillation. PLoS One. 2013;8:e74926.
Kocyigit D, Gurses KM, Yalcin MU, Turk G, Evranos B, Yorgun H, et al. Periatrial epicardial adipose tissue thickness is an independent predictor of atrial fibrillation recurrence after cryoballoon-based pulmonary vein isolation. J Cardiovasc Comput Tomogr. 2015;9:295–302.
Masuda M, Mizuno H, Enchi Y, Minamiguchi H, Konishi S, Ohtani T, et al. Abundant epicardial adipose tissue surrounding the left atrium predicts early rather than late recurrence of atrial fibrillation after catheter ablation. J Interv Card Electrophysiol. 2015;44:31–7.
Gaborit B, Venteclef N, Ancel P, Pelloux V, Gariboldi V, Leprince P, et al. Human epicardial adipose tissue has a specific transcriptomic signature depending on its anatomical peri-atrial, peri-ventricular, or peri-coronary location. Cardiovasc Res. 2015;108:62–73.
Prati F, Arbustini E, Labellarte A, Sommariva L, Pawlowski T, Manzoli A, et al. Eccentric atherosclerotic plaques with positive remodelling have a pericardial distribution: a permissive role of epicardial fat? A three-dimensional intravascular ultrasound study of left anterior descending artery lesions. Eur Heart J. 2003;24:329–36.
Pezeshkian M, Noori M, Najjarpour-Jabbari H, Abolfathi A, Darabi M, Darabi M, et al. Fatty acid composition of epicardial and subcutaneous human adipose tissue. Metab Syndr Relat Disord. 2009;7:125–31.
Iacobellis G, Malavazos AE, Corsi MM. Epicardial fat: from the biomolecular aspects to the clinical practice. Int J Biochem Cell Biol. 2011;43:1651–4.
Sacks HS, Fain JN, Holman B, Cheema P, Chary A, Parks F, et al. Uncoupling protein-1 and related messenger ribonucleic acids in human epicardial and other adipose tissues: epicardial fat functioning as brown fat. J Clin Endocrinol Metab. 2009;94:3611–5.
Sacks HS, Fain JN. Human epicardial fat: what is new and what is missing? Clin Exp Pharmacol Physiol. 2011;38:879–87.
Turer AT, Scherer PE. Adiponectin: mechanistic insights and clinical implications. Diabetologia. 2012;55:2319–26.
Fang X, Palanivel R, Cresser J, Schram K, Ganguly R, Thong FS, et al. An APPL1-AMPK signaling axis mediates beneficial metabolic effects of adiponectin in the heart. Am J Physiol Endocrinol Metab. 2010;299:E721–9.
Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97.
Wong HK, Cheung TT, Cheung BM. Adrenomedullin and cardiovascular diseases. JRSM Cardiovasc Dis. 2012;1:1–7.
Munger TM, Dong YX, Masaki M, Oh JK, Mankad SV, Borlaug BA, et al. Electrophysiological and hemodynamic characteristics associated with obesity in patients with atrial fibrillation. J Am Coll Cardiol. 2012;60:851–60.
Mahajan R, Lau DH, Brooks AG, Shipp NJ, Manavis J, Wood JP, et al. Electrophysiological, electroanatomical, and structural remodeling of the atria as consequences of sustained obesity. J Am Coll Cardiol. 2015;66:1–11.
Yorgun H, Canpolat U, Aytemir K, Hazirolan T, Sahiner L, Kaya EB, et al. Association of epicardial and peri-atrial adiposity with the presence and severity of non-valvular atrial fibrillation. Int J Cardiovasc Imaging. 2015;31:649–57.
Wong CX, Stiles MK, John B, Brooks AG, Lau DH, Dimitri H, et al. Direction-dependent conduction in lone atrial fibrillation. Heart Rhythm. 2010;7:1192–9.
Wong CX, John B, Brooks AG, Chandy ST, Kuklik P, Lau DH, et al. Direction-dependent conduction abnormalities in the chronically stretched atria. Europace. 2012;14:954–61.
Eckstein J, Zeemering S, Linz D, Maesen B, Verheule S, van Hunnik A, et al. Transmural conduction is the predominant mechanism of breakthrough during atrial fibrillation: evidence from simultaneous endo-epicardial high-density activation mapping. Circ Arrhythm Electrophysiol. 2013;6:334–41.
Haemers P, Hamdi H, Guedj K, Suffee N, Farahmand P, Popovic N, et al. Atrial fibrillation is associated with the fibrotic remodelling of adipose tissue in the subepicardium of human and sheep atria. Eur Heart J. 2017;38:53–61.
Guo Y, Lip GY, Apostolakis S. Inflammation in atrial fibrillation. J Am Coll Cardiol. 2012;60:2263–70.
Wu N, Xu B, Xiang Y, Wu L, Zhang Y, Ma X, et al. Association of inflammatory factors with occurrence and recurrence of atrial fibrillation: a meta-analysis. Int J Cardiol. 2013;169:62–72.
Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, et al. C-reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001;104:2886–91.
Malouf JF, Kanagala R, Al Atawi FO, Rosales AG, Davison DE, Murali NS, et al. High sensitivity C-reactive protein: a novel predictor for recurrence of atrial fibrillation after successful cardioversion. J Am Coll Cardiol. 2005;46:1284–7.
Aviles RJ, Martin DO, Apperson-Hansen C, Houghtaling PL, Rautaharju P, Kronmal RA, et al. Inflammation as a risk factor for atrial fibrillation. Circulation. 2003;108:3006–10.
Rotter M, Jais P, Vergnes MC, Nurden P, Takahashi Y, Sanders P, et al. Decline in C-reactive protein after successful ablation of long-lasting persistent atrial fibrillation. J Am Coll Cardiol. 2006;47:1231–3.
Li J, Solus J, Chen Q, Rho YH, Milne G, Stein CM, et al. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm. 2010;7:438–44.
Yamashita T, Sekiguchi A, Iwasaki YK, Date T, Sagara K, Tanabe H, et al. Recruitment of immune cells across atrial endocardium in human atrial fibrillation. Circ J. 2010;74:262–70.
Acet H, Ertas F, Akil MA, Oylumlu M, Polat N, Yildiz A, et al. New inflammatory predictors for non-valvular atrial fibrillation: echocardiographic epicardial fat thickness and neutrophil to lymphocyte ratio. Int J Cardiovasc Imaging. 2014;30:81–9.
Malavazos AE, Ermetici F, Coman C, Corsi MM, Morricone L, Ambrosi B. Influence of epicardial adipose tissue and adipocytokine levels on cardiac abnormalities in visceral obesity. Int J Cardiol. 2007;121:132–4.
Chang SL, Tuan TC, Tai CT, Lin YJ, Lo LW, Hu YF, et al. Comparison of outcome in catheter ablation of atrial fibrillation in patients with versus without the metabolic syndrome. Am J Cardiol. 2009;103:67–72.
Mazurek T, Kiliszek M, Kobylecka M, Skubisz-Gluchowska J, Kochman J, Filipiak K, et al. Relation of proinflammatory activity of epicardial adipose tissue to the occurrence of atrial fibrillation. Am J Cardiol. 2014;113:1505–8.
Kusayama T, Furusho H, Kashiwagi H, Kato T, Murai H, Usui S, et al. Inflammation of left atrial epicardial adipose tissue is associated with paroxysmal atrial fibrillation. J Cardiol. 2016;68:406–11.
Sun Y, Xun K, Wang C, Zhao H, Bi H, Chen X, et al. Adiponectin, an unlocking adipocytokine. Cardiovasc Ther. 2009;27:59–75.
Kourliouros A, Karastergiou K, Nowell J, Gukop P, Tavakkoli Hosseini M, Valencia O, et al. Protective effect of epicardial adiponectin on atrial fibrillation following cardiac surgery. Eur J Cardiothorac Surg. 2011;39:228–32.
Ybarra J, Resmini E, Planas F, Navarro-López F, Webb S, Pou JM, et al. Relationship between adiponectin and left atrium size in uncomplicated obese patients: adiponectin, a link between fat and heart. Obes Surg. 2009;19:1324–32.
van Osch D, Dieleman JM, van Dijk D, Jacob KA, Kluin J, Doevendans PA, et al. Dexamethasone for the prevention of postoperative atrial fibrillation. Int J Cardiol. 2015;182:431–7.
Liu C, Wang J, Yiu D, Liu K. The efficacy of glucocorticoids for the prevention of atrial fibrillation, or length of intensive care unit or hospital stay after cardiac surgery: a meta-analysis. Cardiovasc Ther. 2014;32:89–96.
Lin YK, Chen YC, Chen JH, Chen SA, Chen YJ. Adipocytes modulate the electrophysiology of atrial myocytes: implications in obesity-induced atrial fibrillation. Basic Res Cardiol. 2012;107:293.
Friedman DJ, Wang N, Meigs JB, Hoffmann U, Massaro JM, Fox CS, et al. Pericardial fat is associated with atrial conduction: the Framingham Heart Study. J Am Heart Assoc. 2014;3:e000477.
Zghaib T, Ipek EG, Zahid S, Balouch MA, Misra S, Ashikaga H, et al. Association of left atrial epicardial adipose tissue with electrogram bipolar voltage and fractionation: electrophysiologic substrates for atrial fibrillation. Heart Rhythm. 2016;13:2333–9.
Takahashi Y, Sanders P, Jais P, Hocini M, Dubois R, Rotter M, et al. Organization of frequency spectra of atrial fibrillation: relevance to radiofrequency catheter ablation. J Cardiovasc Electrophysiol. 2006;17:382–8.
Atienza F, Calvo D, Almendral J, Zlochiver S, Grzeda KR, Martinez-Alzamora N, et al. Mechanisms of fractionated electrograms formation in the posterior left atrium during paroxysmal atrial fibrillation in humans. J Am Coll Cardiol. 2011;57:1081–92.
Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999;100:87–95.
Schnabel RB, Larson MG, Yamamoto JF, Kathiresan S, Rong J, Levy D, et al. Relation of multiple inflammatory biomarkers to incident atrial fibrillation. Am J Cardiol. 2009;104:92–6.
Kremen J, Dolinkova M, Krajickova J, Blaha J, Anderlova K, Lacinova Z, et al. Increased subcutaneous and epicardial adipose tissue production of proinflammatory cytokines in cardiac surgery patients: possible role in postoperative insulin resistance. J Clin Endocrinol Metab. 2006;91:4620–7.
Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802–9.
Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96:1180–4.
Patella S, Phillips DJ, de Kretser DM, Evans LW, Groome NP, Sievert W. Characterization of serum activin-A and follistatin and their relation to virological and histological determinants in chronic viral hepatitis. J Hepatol. 2001;34:576–83.
De Bleser PJ, Niki T, Xu G, Rogiers V, Geerts A. Localization and cellular sources of activins in normal and fibrotic rat liver. Hepatology. 1997;26:905–12.
Werner S, Alzheimer C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev. 2006;17:157–71.
Venteclef N, Guglielmi V, Balse E, Gaborit B, Cotillard A, Atassi F, et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur Heart J. 2015;36:795–805a.
Greulich S, Maxhera B, Vandenplas G, de Wiza DH, Smiris K, Mueller H, et al. Secretory products from epicardial adipose tissue of patients with type 2 diabetes mellitus induce cardiomyocyte dysfunction. Circulation. 2012;126:2324–34.
Oshima Y, Ouchi N, Shimano M, Pimentel David R, Papanicolaou Kyriakos N, Panse Kalyani D, et al. Activin A and follistatin-like 3 determine the susceptibility of heart to ischemic injury. Circulation. 2009;120:1606–15.
Greulich S, de Wiza DH, Preilowski S, Ding Z, Mueller H, Langin D, et al. Secretory products of guinea pig epicardial fat induce insulin resistance and impair primary adult rat cardiomyocyte function. J Cell Mol Med. 2011;15:2399–410.
Boixel C, Fontaine V, Rucker-Martin C, Milliez P, Louedec L, Michel JB, et al. Fibrosis of the left atria during progression of heart failure is associated with increased matrix metalloproteinases in the rat. J Am Coll Cardiol. 2003;42:336–44.
Nagashima K, Okumura Y, Watanabe I, Nakai T, Ohkubo K, Kofune M, et al. Does location of epicardial adipose tissue correspond to endocardial high dominant frequency or complex fractionated atrial electrogram sites during atrial fibrillation? Circ Arrhythm Electrophysiol. 2012;5:676–83.
Laxton RC, Hu Y, Duchene J, Zhang F, Zhang Z, Leung KY, et al. A role of matrix metalloproteinase-8 in atherosclerosis. Circ Res. 2009;105:921–9.
Xiao Q, Zhang F, Lin L, Fang C, Wen G, Tsai TN, et al. Functional role of matrix metalloproteinase-8 in stem/progenitor cell migration and their recruitment into atherosclerotic lesions. Circ Res. 2013;112:35–47.
Felkin LE, Birks EJ, George R, Wong S, Khaghani A, Yacoub MH, et al. A quantitative gene expression profile of matrix metalloproteinases (MMPS) and their inhibitors (TIMPS) in the myocardium of patients with deteriorating heart failure requiring left ventricular assist device support. J Heart Lung Transplant. 2006;25:1413–9.
Wang Q, Xi W, Yin L, Wang J, Shen H, Gao Y, et al. Human epicardial adipose tissue cTGF expression is an independent risk factor for atrial fibrillation and highly associated with atrial fibrosis. Sci Rep. 2018;8:3585.
Aimé-Sempé C, Folliguet T, Rücker-Martin C, Krajewska M, Krajewski S, Heimburger M, et al. Myocardial cell death in fibrillating and dilated human right atria. J Am Coll Cardiol. 1999;34:1577–86.
Ausma J, Wijffels M, Van Eys G, Koide M, Ramaekers F, Allessie M, et al. Dedifferentiation of atrial cardiomyocytes as a result of chronic atrial fibrillation. Am J Pathol. 1997;151:985.
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, et al. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13:4279–95.
Fraser JK, Wulur I, Alfonso Z, Hedrick MH. Fat tissue: an underappreciated source of stem cells for biotechnology. Trends Biotechnol. 2006;24:150–4.
Rangappa S, Fen C, Lee EH, Bongso A, Wei ESK. Transformation of adult mesenchymal stem cells isolated from the fatty tissue into cardiomyocytes. Ann Thorac Surg. 2003;75:775–9.
Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7:211–28.
Vural M, Talu A, Sahin D, Elalmis OU, Durmaz HA, Uyanik S, et al. Evaluation of the relationship between epicardial fat volume and left ventricular diastolic dysfunction. Jpn J Radiol. 2014;32:331–9.
Suffee N, Moore-Morris T, Farahmand P, Rücker-Martin C, Dilanian G, Fradet M, et al. Atrial natriuretic peptide regulates adipose tissue accumulation in adult atria. Proc Natl Acad Sci U S A. 2017;114:E771–E80.
Salgado-Somoza A, Teijeira-Fernandez E, Fernandez AL, Gonzalez-Juanatey JR, Eiras S. Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am J Physiol Heart Circ Physiol. 2010;299:H202–9.
Carnes CA, Chung MK, Nakayama T, Nakayama H, Baliga RS, Piao S, et al. Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ Res. 2001;89:E32–8.
Po SS, Nakagawa H, Jackman WM. Localization of left atrial ganglionated plexi in patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2009;20:1186–9.
Coumel P. Paroxysmal atrial fibrillation: a disorder of autonomic tone? Eur Heart J. 1994;15(Suppl A):9–16.
Balcioglu AS, Cicek D, Akinci S, Eldem HO, Bal UA, Okyay K, et al. Arrhythmogenic evidence for epicardial adipose tissue: heart rate variability and turbulence are influenced by epicardial fat thickness. Pacing Clin Electrophysiol. 2015;38:99–106.
Nakagawa H, Scherlag BJ, Patterson E, Ikeda A, Lockwood D, Jackman WM. Pathophysiologic basis of autonomic ganglionated plexus ablation in patients with atrial fibrillation. Heart Rhythm. 2009;6(12 Suppl):S26–34.
Stavrakis S, Nakagawa H, Po SS, Scherlag BJ, Lazzara R, Jackman WM. The role of the autonomic ganglia in atrial fibrillation. JACC Clin Electrophysiol. 2015;1:1–13.
Scherlag BJ, Yamanashi W, Patel U, Lazzara R, Jackman WM. Autonomically induced conversion of pulmonary vein focal firing into atrial fibrillation. J Am Coll Cardiol. 2005;45:1878–86.
Patterson E, Lazzara R, Szabo B, Liu H, Tang D, Li YH, et al. Sodium-calcium exchange initiated by the Ca2+ transient: an arrhythmia trigger within pulmonary veins. J Am Coll Cardiol. 2006;47:1196–206.
Patterson E, Po SS, Scherlag BJ, Lazzara R. Triggered firing in pulmonary veins initiated by in vitro autonomic nerve stimulation. Heart Rhythm. 2005;2:624–31.
Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res. 2014;114:1500–15.
Pokushalov E, Kozlov B, Romanov A, Strelnikov A, Bayramova S, Sergeevichev D, et al. Long-term suppression of atrial fibrillation by botulinum toxin injection into epicardial fat pads in patients undergoing cardiac surgery: one-year follow-up of a randomized pilot study. Circ Arrhythm Electrophysiol. 2015;8:1334–41.
Chilukoti RK, Giese A, Malenke W, Homuth G, Bukowska A, Goette A, et al. Atrial fibrillation and rapid acute pacing regulate adipocyte/adipositas-related gene expression in the atria. Int J Cardiol. 2015;187:604–13.
Chami M, Gozuacik D, Lagorce D, Brini M, Falson P, Peaucellier G, et al. SERCA1 truncated proteins unable to pump calcium reduce the endoplasmic reticulum calcium concentration and induce apoptosis. J Cell Biol. 2001;153:1301–14.
Viviano A, Yin X, Zampetaki A, Fava M, Gallagher M, Mayr M, et al. Proteomics of the epicardial fat secretome and its role in post-operative atrial fibrillation. Europace. 2018;20:1201–8.
Batal O, Schoenhagen P, Shao M, Ayyad AE, Van Wagoner DR, Halliburton SS, et al. Left atrial epicardial adiposity and atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:230–6.
Shin SY, Yong HS, Lim HE, Na JO, Choi CU, Choi JI, et al. Total and interatrial epicardial adipose tissues are independently associated with left atrial remodeling in patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2011;22:647–55.
Nakanishi K, Fukuda S, Tanaka A, Otsuka K, Sakamoto M, Taguchi H, et al. Peri-atrial epicardial adipose tissue is associated with new-onset nonvalvular atrial fibrillation. Circ J. 2012;76:2748–54.
Kanazawa H, Yamabe H, Enomoto K, Koyama J, Morihisa K, Hoshiyama T, et al. Importance of pericardial fat in the formation of complex fractionated atrial electrogram region in atrial fibrillation. Int J Cardiol. 2014;174:557–64.
Murakami C, Nagai T, Akira F, Kido T, Nishimura K, Inoue K, et al. Total epicardial fat volume is associated with early recurrence of atrial fibrillation after catheter ablation. J Am Coll Cardiol. 2012;59(13 Suppl):E691.
Soucek F, Covassin N, Singh P, Ruzek L, Kara T, Suleiman M, et al. Epicardial adipose tissue volume predicts atrial fibrillation recurrence after pulmonary vein isolation. Eur Heart J. 2014;35:431.
Canpolat U, Aytemir K, Yorgun H, Asil S, Dural M, Özer N. The impact of echocardiographic epicardial fat thickness on outcomes of cryoballoon-based atrial fibrillation ablation. Echocardiography. 2016;33:821–9.
Ciuffo L, Nguyen H, Marques MD, Aronis KN, Sivasambu B, et al. Periatrial fat quality predicts atrial fibrillation ablation outcome. Circ Cardiovasc Imaging. 2019;12:e008764.
Mirolo A, Viart G, Savouré A, Godin B, Raitière O, Eltchaninoff H, et al. Epicardial fat thickness predicts atrial fibrillation recurrence after a first pulmonary vein isolation procedure using a second-generation cryoballoon. Arch Cardiovasc Dis. 2019;112:314–22.
Gaeta M, Bandera F, Tassinari F, Capasso L, Cargnelutti M, Pelissero G, et al. Is epicardial fat depot associated with atrial fibrillation? A systematic review and meta-analysis. Europace. 2017;19:747–52.
Opolski MP, Staruch AD, Kusmierczyk M, Witkowski A, Kwiecinska S, Kosek M, et al. Computed tomography angiography for prediction of atrial fibrillation after coronary artery bypass grafting: proof of concept. J Cardiol. 2015;65:285–92.
Cho KI, Kim BJ, Cha TJ, Heo JH, Kim HS, Lee JW. Impact of duration and dosage of statin treatment and epicardial fat thickness on the recurrence of atrial fibrillation after electrical cardioversion. Heart Vessel. 2015;30:490–7.
Kawakami H, Satomi K, Nakajima I, Miyamoto K, Yamada Y, Okamura H, et al. Total epicardial adipose tissue volume is associated with outcome of pulmonary vein isolation for atrial fibrillation. Europace. 2013;15:ii218.
Gorter PM, van Lindert AS, de Vos AM, Meijs MF, van der Graaf Y, Doevendans PA, et al. Quantification of epicardial and peri-coronary fat using cardiac computed tomography; reproducibility and relation with obesity and metabolic syndrome in patients suspected of coronary artery disease. Atherosclerosis. 2008;197:896–903.
Abbara S, Desai JC, Cury RC, Butler J, Nieman K, Reddy V. Mapping epicardial fat with multi-detector computed tomography to facilitate percutaneous transepicardial arrhythmia ablation. Eur J Radiol. 2006;57:417–22.
Hasebe H, Yoshida K, Iida M, Hatano N, Muramatsu T, Nogami A, et al. Differences in the structural characteristics and distribution of epicardial adipose tissue between left and right atrial fibrillation. Europace. 2017;20:435–42.
Altun B, Colkesen Y, Gazi E, Tasolar H, Temiz A, Simsek HY, et al. Could epicardial adipose tissue thickness by echocardiography be correlated with acute coronary syndrome risk scores. Echocardiography. 2013;30:1130–4.
Gul I, Zungur M, Aykan AC, Gokdeniz T, Kalaycioğlu E, Turan T, et al. The relationship between GRACE score and epicardial fat thickness in non-STEMI patients. Arq Bras Cardiol. 2016;106:194–200.
Özcan F, Turak O, Canpolat U, Kanat S, Kadife İ, Avcı S, et al. Association of epicardial fat thickness with TIMI risk score in NSTEMI/USAP patients. Herz. 2014;39:755–60.
Zencirci E, Zencirci AE, Değirmencioğlu A, Karakuş G, Uğurlucan M, Özden K, et al. The relationship between epicardial adipose tissue and ST-segment resolution in patients with acute ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Heart Vessel. 2015;30:147–53.
Wang T, Liu Q, Liu C, Sun L, Li D, Liu A, et al. Correlation of echocardiographic epicardial fat thickness with severity of coronary artery disease in patients with acute myocardial infarction. Echocardiography. 2014;31:1177–81.
Tanindi A, Erkan AF, Ekici B. Epicardial adipose tissue thickness can be used to predict major adverse cardiac events. Coron Artery Dis. 2015;26:686–91.
Hajsadeghi F, Nabavi V, Bhandari A, Choi A, Vincent H, Flores F, et al. Increased epicardial adipose tissue is associated with coronary artery disease and major adverse cardiovascular events. Atherosclerosis. 2014;237:486–9.
Cheng VY, Dey D, Tamarappoo B, Nakazato R, Gransar H, Miranda-Peats R, et al. Pericardial fat burden on ECG-gated noncontrast CT in asymptomatic patients who subsequently experience adverse cardiovascular events. JACC Cardiovasc Imaging. 2010;3:352–60.
Bos D, Vernooij MW, Shahzad R, Kavousi M, Hofman A, van Walsum T, et al. Epicardial fat volume and the risk of atrial fibrillation in the general population free of cardiovascular disease. JACC Cardiovasc Imaging. 2017;10:1405–7.
Akdag S, Simsek H, Sahin M, Akyol A, Duz R, Babat N. Association of epicardial adipose tissue thickness and inflammation parameters with CHA2DS2-VASc score in patients with nonvalvular atrial fibrillation. Ther Clin Risk Manag. 2015;11:1675.
Tsao HM, Hu WC, Tsai PH, Lee CL, Liu FC, Wang HH, et al. The abundance of epicardial adipose tissue surrounding left atrium is associated with the occurrence of stroke in patients with atrial fibrillation. Medicine (Baltimore). 2016;95:e3260.
Chu CY, Lee WH, Hsu PC, Lee MK, Lee HH, Chiu CA, et al. Association of increased epicardial adipose tissue thickness with adverse cardiovascular outcomes in patients with atrial fibrillation. Medicine (Baltimore). 2016;95:e2874.
Nakazato R, Rajani R, Cheng VY, Shmilovich H, Nakanishi R, Otaki Y, et al. Weight change modulates epicardial fat burden: a 4-year serial study with non-contrast computed tomography. Atherosclerosis. 2012;220:139–44.
Gaborit B, Jacquier A, Kober F, Abdesselam I, Cuisset T, Boullu-Ciocca S, et al. Effects of bariatric surgery on cardiac ectopic fat: lesser decrease in epicardial fat compared to visceral fat loss and no change in myocardial triglyceride content. J Am Coll Cardiol. 2012;60:1381–9.
Rabkin S, Campbell H. Comparison of reducing epicardial fat by exercise, diet or bariatric surgery weight loss strategies: a systematic review and meta-analysis. Obes Rev. 2015;16:406–15.
Fenk S, Fischer M, Strack C, Schmitz G, Loew T, Lahmann C, et al. Successful weight reduction improves left ventricular diastolic function and physical performance in severe obesity. Int Heart J. 2015;56:196–202.
Alexopoulos N, Melek BH, Arepalli CD, Hartlage G-R, Chen Z, Kim S, et al. Effect of intensive versus moderate lipid-lowering therapy on epicardial adipose tissue in hyperlipidemic post-menopausal women: a substudy of the BELLES trial (Beyond Endorsed Lipid Lowering with EBT Scanning). J Am Coll Cardiol. 2013;61:1956–61.
Rezaei Y, Gholami-Fesharaki M, Dehghani MR, Arya A, Haghjoo M, Arjmand N. Statin antiarrhythmic effect on atrial fibrillation in statin-naive patients undergoing cardiac surgery: a meta-analysis of randomized controlled trials. J Cardiovasc Pharmacol Ther. 2016;21:167–76.
Fauchier L, Clementy N, Babuty D. Statin therapy and atrial fibrillation: systematic review and updated meta-analysis of published randomized controlled trials. Curr Opin Cardiol. 2013;28:7–18.
Elisha B, Azar M, Taleb N, Bernard S, Iacobellis G, Rabasa-Lhoret R. Body composition and epicardial fat in type 2 diabetes patients following insulin detemir versus insulin glargine initiation. Horm Metab Res. 2016;48:42–7.
Iacobellis G, Camarena V, Sant DW, Wang G. Human epicardial fat expresses glucagon-like peptide 1 and 2 receptors genes. Horm Metab Res. 2017;49:625–30.
Iacobellis G. Letter to the Editor: “GLP-1 receptor expression within the human heart”. Endocrinology. 2018;159:1964–5.
Iacobellis G, Mohseni M, Bianco SD, Banga PK. Liraglutide causes large and rapid epicardial fat reduction. Obesity. 2017;25:311–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Zaatari, G., Goldberger, J.J. (2020). Atrial Fibrillation and Epicardial Adipose Tissue. In: Iacobellis, G. (eds) Epicardial Adipose Tissue. Contemporary Cardiology. Humana, Cham. https://doi.org/10.1007/978-3-030-40570-0_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-40570-0_10
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-40569-4
Online ISBN: 978-3-030-40570-0
eBook Packages: MedicineMedicine (R0)