
Abstract
The Materials Genome Initiative has brought about a paradigm shift in the design and discovery of novel materials. In a growing number of applications, the materials innovation cycle has been greatly accelerated as a result of insights provided by data-driven materials informatics platforms. High-throughput computational methodologies, data descriptors, and machine learning are playing an increasingly invaluable role in research development portfolios across both academia and industry. Polymers, especially, have long suffered from a lack of data on electronic, mechanical, and dielectric properties across large chemical spaces, causing a stagnation in the set of suitable candidates for various applications. The nascent field of polymer informatics seeks to provide tools and pathways for accelerated polymer property prediction (and materials design) via surrogate machine learning models built on reliable past data. With this goal in mind, we have carefully accumulated a dataset of organic polymers whose properties were obtained either computationally (bandgap, dielectric constant, refractive index, and atomization energy) or experimentally (glass transition temperature, solubility parameter, and density). A fingerprinting scheme that captures atomistic to morphological structural features was developed to numerically represent the polymers. Machine learning models were then trained by mapping the polymer fingerprints (or features) to their respective properties. Once developed, these models can rapidly predict properties of new polymers (within the same chemical class as the parent dataset) and can also provide uncertainties underlying the predictions. Since different properties depend on different length-scale features, the prediction models were built on an optimized set of features for each individual property. Furthermore, these models are incorporated in a user friendly online platform named Polymer Genome (www.polymergenome.org). Systematic and progressive expansion of both chemical and property spaces are planned to extend the applicability of Polymer Genome to a wide range of technological domains.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Introduction: Applications of Machine Learning in Materials Science
The past few years have been witness to a surge in the application of data-driven techniques to a broad spectrum of research and development fields. The discipline of machine learning [1], responsible for bringing such techniques to light, has seen multiple breakthroughs over the past two decades. One of the factors responsible for such rapid advancements in the field is the development of novel algorithms and quantitative approaches capable of learning any arbitrary mapping between a given input and the corresponding output. The increased availability of vast amounts of data and the reduction in the cost of fast computational resources are other reasons that have abetted in the preeminence of the field of machine learning.
The materials science and chemistry communities have greatly benefited from machine learning approaches over the past few years. In these communities, there have been many large-scale efforts to curate accurate and reliable databases of materials properties (both computational [2,3,4] and experimental). Large-scale programs such as the Materials Genome Initiative [5] (in the USA), NOMAD [6] (in Europe), and MARVEL [7] (in Switzerland) have contributed to the development of novel database infrastructures tailored to materials science challenges and have also resulted in high-throughput frameworks capable of leveraging the power of modern high-performance computing facilities [8, 9].
In materials science, the increasing availability of large amounts of data (both computational and experimental) has led to the prominent field of materials informatics [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24]. The overarching goal of the field materials informatics is to accelerate the development of novel materials for specific applications. To this end, the materials science community has used machine learning to accelerate various stages of the materials discovery pipeline. For example, a variety of machine learning force-fields [25,26,27,28] have been developed to provide rapid predictions of energies and forces with quantum-mechanical accuracy. Other approaches involve the utilization of machine learning approaches to bypass the Kohn–Sham equations to directly obtain important electronic properties such as the charge density [29].
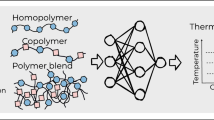
This chapter, however, focuses on the ability of surrogate models to directly predict higher length-scale properties of materials. As shown in Fig. 18.1, the measurement of materials properties has traditionally involved computationally expensive quantum-mechanical simulations or perhaps the utilization of a time-consuming or laborious experimental technique. A novel paradigm has emerged in recent years wherein the properties of materials can be directly and rapidly obtained using predictive frameworks employing machine learning methodologies.

The top two workflows indicate how the physical properties of materials can be obtained using traditional computational or experimental pipelines. Recent efforts, such as the Polymer Genome paradigm, seeks to accelerate the prediction of materials properties using machine learning approaches
A specific example where such a paradigm has been of great utility is the nascent field of polymer informatics. Polymers form an important (and challenging) materials class and they are pervasive with applications ranging from daily products, e.g., plastic packaging and containers, to state-of-the-art technological components, e.g., high-energy density capacitors, electrolytes for Li-ion batteries, polymer light-emitting diodes, and photovoltaic materials. Their chemical and morphological spaces are immensely vast and complex [30], leading to fundamental obstacles in polymer discovery. Some recent successes in rationally designing polymer dielectrics via experiment-computation synergies [10, 11, 19, 23, 31,32,33,34,35,36,37,38] indicate that there may be opportunities for machine learning and informatics approaches in this challenging research and development area.
We have created an informatics platform capable of predicting a variety of important polymer properties on-demand. This platform utilizes surrogate (or machine learning) models, which link key features of polymers to properties, trained on high-throughput DFT calculations and experimental data from literature and existing databases. The main elements of the polymer property prediction pipeline are summarized in the lowermost pipeline of Fig. 18.1.
In the following sections, we explain in detail the various stages of abovementioned pipeline [39], starting from the curation of the dataset all the way up to the machine learning algorithms that we have employed.
2 Dataset
Two strategic tracks were followed for the creation of our dataset (see Fig. 18.2): (1) via high-throughput computation using density functional theory (DFT) as presented earlier [31, 40, 41] and (2) by utilizing experimentally measured properties from literature and data collections [42, 43]. The overall dataset includes 854 polymers made up of a subset of the following species: H, C, N, O, S, F, Cl, Br, and I. Seven different properties were included in the present study. The bandgap, dielectric constant, refractive index, and atomization energy were determined using DFT computations whereas the Tg, solubility parameter, and density were obtained from experimental measurements.

Overview of our polymer dataset used for development of property prediction models [39]. The dataset consists of 854 polymers spanning a chemical space of nine elements and comprises properties obtained using computations as well as experiments
All the computational data were generated through a series of studies related to advanced polymer dielectrics [31, 40, 41]. The computational dataset includes polymers containing the following building blocks, CH2, CO, CS, NH, C6H4, C4H2S, CF2, CHF, and O [19, 22, 40, 41, 44]. Repeat units contained 4–8 building blocks, and 3D structure prediction algorithms were used to determine their structure [31, 40, 41]. The building blocks considered in the dataset are found in common polymeric materials including polyethylene (PE), polyesters, and polyureas, and could theoretically produce an enormous variety of different polymers. The bandgap was computed using the hybrid Heyd–Scuseria–Ernzerhof (HSE06) electronic exchange-correlation functional [45]. Dielectric constant and refractive index (the square root of the electronic part of the dielectric constant) were computed using density functional perturbation theory (DFPT) [46]. The atomization energy was computed for all the polymers following previous work [33,34,35,36, 41, 44, 47,48,49,50,51,52]. The DFT computed properties and associated 3D structures are available from Khazana [53](https://khazana.gatech.edu).
The Tg, solubility parameter, and density data were obtained from the existing databases of experimental measurements [42, 43]. Tg, which is an indication of the transition point between the glassy and supercooled liquid phases in an amorphous polymer, is important in many polymer applications because the structural characteristics (and, consequently, other properties) of the polymer changes dramatically at this point. The solubility parameter of a polymer is typically used to determine a suitable solvent to use during polymer synthesis. In this particular study we consider the Hildebrand solubility parameter.
We have determined the chemical formula and the associated topological structure from the name of polymers listed in the literature. The dataset contains a total of 854 organic polymers composed of 9 frequently found atomic species, i.e., C, H, O, N, S, F, Cl, Br, and I with properties listed in the right side panel of Fig. 18.2. Figure 18.3 shows a summary of the property space for the polymer dataset, including the range of property values, distribution, standard deviation, and the number of polymers associated with each property.

Property space of Polymer Genome dataset [39]. The seven properties considered in this study were the (a) bandgap, (b) dielectric constant, (c) refractive index, (d) atomization energy, (e) Tg, (f) solubility parameter, and (g) density. The histograms represent the distribution of each individual property. The solid line depicts the mean of the distribution whereas the distance between the solid line and dashed line represents the standard deviation. (h) Table detailing the number of data-points, range, and mean of each individual property considered
3 Hierarchical Fingerprinting
Fingerprinting is a crucial step of our data-driven property prediction pipeline. In this step, the geometric and chemical information of the polymers is converted to a numerical representation. This numerical representation, more often than not, is a vector of fixed number of dimensions that can be provided as an input to any given machine learning algorithm. The different dimensions of this vector would represent different characteristics of the polymer repeat unit. Such numerical descriptors of organic molecules have been utilized extensively in the past in the form quantitative structure-property relationship (QSPR) or quantitative structure-activity relationship (QSAR) models. In the current work, we go beyond existing QSPR/QSAR descriptors in order to systematically capture different length-scale features that are specific to polymeric materials built up of very long polymer chains. In essence, given a particular repeat unit, we assume that the polymer chain constructed from that repeat unit is infinitely long and therefore the descriptors that we construct must take into account this “one-dimensional” periodicity.
To comprehensively capture the key features that may control the diversity of properties of interest, we consider three hierarchical levels of descriptors spanning different length scales. At the atomic-scale, the number of times that a fixed set of atomic fragments (or motifs) occur are counted [54]. An example of such a fragment is O1-C3-C4, made up of three contiguous atoms, namely, a one-fold coordinated oxygen, a three-fold coordinated carbon, and a four-fold coordinated carbon, in this order. For a given polymer repeat unit, we count the number of times the O1-C3-C4 fragment occurs and then proceed to normalize this value by the number of atoms in polymer repeat unit (to account for the abovementioned one-dimensional periodicity). Such a series of predefined “triplets” has been shown to be a good fingerprint for a diverse range of organic materials [23, 54]. A vector of such triplets form the fingerprint components at the lowest hierarchy. For the polymer class under study, there are 108 such components.
Next in the hierarchy of fingerprint components are larger length-scale descriptors of the quantitative structure-property relationship (QSPR) type mentioned earlier. A detailed description of such descriptors can be found in the RDKit Python library [55,56,57] that was used for the current work. Examples of such descriptors are van der Waals surface area [58], the topological polar surface area (TPSA) [59, 60], the fraction of atoms that are part of rings (i.e., the number of atoms associated with rings divided by the total number of atoms in the formula unit), and the fraction of rotatable bonds. TPSA is the sum of surfaces of polar atoms in the molecule and we observed this descriptor to be strongly correlated to the solubility. Descriptors such as the fraction of ring atoms and fraction of rotatable bonds strongly influenced properties such as Tg and density. Such descriptors, 99 in total, form the next set of components of our overall fingerprint vector.
The highest length-scale fingerprint components we considered may be classified as “morphological descriptors.” These include features such as the shortest topological distance between rings, fraction of atoms that are part of side chains, and the length of the largest side-chain. Properties such as Tg strongly depend on such features which influence the way the chains are packed in the polymer. For instance, if two rings are very close, the stiffness of the polymer backbone is much higher than if the rings were separated by a larger topological distance. Both the number and the length of the side chains strongly influence the amount of free volume in the polymeric material and therefore directly influence Tg. The larger the free volume, the lower the Tg. We include 22 such morphological descriptors in our overall fingerprint.
Figure 18.4a shows the hierarchy of polymer fingerprints, including atomic level, QSPR and morphological descriptors. The overall fingerprint of a polymer is constructed by concatenating the three classes of fingerprint components. In total, this leads to a fingerprint with 229 components. Since certain descriptors are more relevant for certain properties, in the next section, we outline a methodology to discard irrelevant descriptors for every target property. Moreover, during performance assessment, we use different combinations of the three fingerprint hierarchies. For clarity of the ensuing discussion, we introduce some nomenclature. The atom triples fingerprint, QSPR descriptors, and morphological descriptors are denoted by “A,” “Q,” and “M,” respectively. Therefore, “AQ” implies a combination of just the atom triples and QSPR descriptors.

Hierarchy of descriptors used to fingerprint the polymers, and an example demonstration for the systematic improvement of model performance depending on the type of fingerprint considered. (a) Classification of descriptors according to the physical scale and chemical characteristics are shown with representative examples. Dimension of the fingerprint in each level can be reduced by a recursive feature elimination (RFE) process. In the “+RFE” panel, N, Ω, and E min are total number of features in fingerprint, optimal number of features determined by RFE, and minimum error of prediction model, respectively. Plots at the bottom panel show the performance of machine learning prediction models for glass transition temperature (Tg) with (b) only atomic level descriptors, (c) atomic level and QSPR descriptors, and (d) entire fingerprint components including morphological descriptors. (e) Shows how the optimal subset selected by RFE improves the prediction model for Tg [39]
In order to visualize the chemical diversity of polymers considered here, we have performed principal component analysis (PCA) of the complete fingerprint vector. PCA identifies orthogonal linear combinations of the original fingerprint components that provide the highest variance; the first few principal components account for much of the variability in the data [13]. Figure 18.5 displays the dataset with the horizontal and vertical axes chosen as the first two principal components, PC1 and PC2. Molecular models of some common polymers are shown explicitly, and symbol color, symbol size, and symbol type are used to represent the fraction of sp 3 bonded C atoms, fraction of rings, and TPSA of polymers, respectively. As an example from the figure, PE is composed of only sp 3 bonded C without any rings in the chain, while poly(1,4-phenylene sulfide) contains no sp 3 bonded C atoms, and more than 90% of its atoms are part of rings. As a result, these two polymers are situated far from each other in 2D principal component space.

Graphical summary of chemical space of polymers considered. 854 chemically unique organic polymers generated by structure prediction method (minima-hopping [61]) and experimental sources [42, 43] distributed in 2D principal component space. Two leading components, PC1 and PC2, are produced by principal component analysis, and assigned to axes of the plot. Fraction of sp 3 bonded C atoms, fraction of rings, and normalized TPSA per atoms in a formula unit are used for color code, size, and symbol of each polymer. A few representative structures with various number of aromatic and/or aliphatic rings and their position on the map are shown [39]
4 Surrogate (Machine Learning) Model Development
4.1 Recursive Feature Elimination
As alluded to earlier, our general fingerprint is rather high in dimensionality, and not all of the components may be relevant for describing a particular property. In fact, irrelevant features often lead to a poor prediction capability. On the practical side, large fingerprint dimensionality also implies longer training times. There is thus a need to determine the optimal subset of the complete fingerprint necessary for the prediction of a particular property (i.e., different properties may require different subsets of the fingerprint vector). Rather than manually deciding which fingerprint components to use, one may utilize a wide variety of dimensionality reduction techniques to automatically select a set of features that best represent a particular property. In the current work, we utilize the recursive feature elimination (RFE) algorithm to sequentially eliminate the least important features for a given property [62]. First, linear regression is performed using the complete fingerprint vector via support vector regression. Through this process, each of the features are weighted by certain coefficients and are then ranked based on the square of these coefficients [62]. The feature with the lowest rank is subsequently eliminated and the iteration is repeated to remove the next least-important-feature. As shown in right-most panel of Fig. 18.4, the optimal number of features for a given property can be obtained by plotting the cross-validated root mean square error (RMSE) as a function of the number of descriptors. The final set of features is passed forward to the non-linear machine learning algorithm described next in Sect. 18.4.2. These features can also be used to obtain an intuitive understanding of how certain key fingerprint components influence particular materials properties.
4.2 Gaussian Process Regression
In our past work [12, 19, 31], we have successfully utilized kernel ridge regression (KRR) [63] to learn the non-linear relationship between a polymer’s fingerprint and its properties. However, in this work we utilize Gaussian process regression (GPR) because of two key benefits. Firstly, GPR learns a generative, probabilistic model of the target property and thus provides meaningful uncertainties/confidence intervals for the prediction. Secondly, the optimization of the model hyperparameters is relatively faster in GPR because one may perform gradient-ascent on the marginal likelihood function as opposed to the cross-validated grid-search which is required for KRR. We use a radial basis function (RBF) kernel defined as
where σ, l, and σ n are hyperparameters to be determined during the training process (in the machine learning parlance, these hyperparameters are referred to as signal variance, length-scale parameter, and noise level parameter, respectively). x i and x j are the fingerprint vectors for two polymers i and j. (x i is an m dimensional vector with components \(x^1_i\), \(x^2_i\), \(x^3_i, \ldots , x^m_i\), determined and optimized by the RFE step described above). Performance of the model was evaluated based on the root mean square error (RMSE) and the coefficient of determination (R 2). 80% of the data was used for training and the remaining 20% was set aside as a test set.
5 Model Performance Validation
The final machine learning models for each of the properties under consideration here were constructed using the entire polymer dataset for each property. To avoid overfitting the data, and to ensure that the models are generalizable, we employed five-fold cross-validation, wherein the dataset is divided into 5 different subsets and one subset was used for testing while remaining sets were employed for training. Table 18.1 summarizes the best fingerprint, dimension of fingerprint vector, and performance based on RMSE for the entire dataset. As shown in the table, the best machine learning model for the atomization energy can be constructed using just the atom triples and QSPR descriptors (i.e., “AQ”) whereas most of the other properties necessitate the inclusion of morphological descriptors (i.e., ‘AQM”). In Fig. 18.6, we demonstrate the sensitivity of the bandgap and dielectric constant models to the size of the training set. We see a convergence in the train and test errors as the training set size increases. Therefore, the accuracy of the ML models may be systematically improved as more polymer property values are added to the dataset.

Learning curves constructed from the RMSE of the machine learning models for (a) bandgap and (b) dielectric constant. For each model, data was obtained from 100 independent runs with different selection of train and test set
Parity plots in Fig. 18.7 are shown to compare experimental or DFT computed properties with respect to machine learning predicted values with percentage relative error distribution. Several error metrics, such as RMSE, mean absolute error (MAE), mean absolute relative error (MARE), and 1 − R 2 were considered to evaluate the performance of these models, and shown together in Fig. 18.7h.

The performance of the cross-validated machine learning models developed by GPR with combination of RBF and white noise kernels [39]. Comparison of DFT computed (a) bandgap, (b) dielectric constant, (c) refractive index, (d) atomization energy, experimental (e) Tg, (f) Hildebrand solubility parameter, and (g) density for the predicted values are shown with inset of distribution of % relative error, (y − Y )∕Y × 100 where Y is DFT computed or experimental value, and y is machine learning predicted value. The error bars in the parity plots represent uncertainties (standard deviations) obtained using GPR. Other error metrics including RMSE, mean absolute error (MAE), mean absolute relative error (MARE), and 1 − R 2 are summarized in (h)
As mentioned earlier, the utilization of GPR provides meaningful uncertainties associated with each prediction. Moreover, the noise parameter of the GPR kernel gives insights into the overall errors and uncertainties associated with the prediction of that particular property for a given dataset. These uncertainties could arise as a result of variation in measurement techniques (in the case of Tg, for example) or it may even arise as a result of limitations of our representation technique. For example, we are providing estimates of the bandgap through purely the SMILES string rather than the 3D crystal structure of the polymer. Therefore, the representation technique itself results in partial loss of information and this underlying uncertainty can be estimated statistically using the GPR noise parameter.
6 Polymer Genome Online Platform
For easy access and use of the prediction models developed here, an online platform called Polymer Genome has been created. This platform is available at www.polymergenome.org [64]. The Polymer Genome application was developed using Python and standard web languages such as Hypertext Preprocessor (php) and Hypertext Markup Language (HTML). As user input, the repeat unit of a polymer or its SMILES string may be used (following a prescribed format described in the Appendix). One may also use an integrated drawing tool to sketch the repeat unit of the polymer.
Once the user input is delivered to Polymer Genome by the user, property predictions (with uncertainty) are made, and the results are shown in an organized table. The names of polymers (if there are more than one meeting the search criteria) with SMILES and repeat unit are provided with customizable collection of properties. Upon selection of any polymer from this list, comprehensive information is reported. This one-page report provides the name and class of the polymer, 3D visualization of the structure with atomic coordinates (if such is available), and properties determined using our machine learning models. A typical user output of Polymer Genome is captured in Fig. 18.8.

Overview of Polymer Genome online platform available at www.polymergenome.org. Keyword PE is used as an example user input to show resulting Polymer details page [39]
7 Conclusions and Outlook
The Materials Genome Initiative and similar other initiatives around the world have provided the impetus for data-centric informatics approaches in several subfields of materials research. Such informatics approaches seek to provide tools and pathways for accelerated property prediction (and materials design) via surrogate models built on reliable past data. Here, we have presented a polymer informatics platform capable of predicting a variety of important polymer properties on-demand. This platform utilizes surrogate (or machine learning) models that link key features of polymers (i.e., their “fingerprint”) to properties. The models are trained on high-throughput DFT calculations (of the bandgap, dielectric constant, refractive index, and atomization energy) and experimental data from polymer data handbooks (on the glass transition temperature, solubility parameter, and density). Certain properties, like the atomization energy, depend mainly on the atomic constituents and short-range bonding, whereas other properties, such as the glass transition temperature, are strongly influenced by morphological characteristics like the chain-stiffness and branching. Our polymer fingerprinting scheme is thus necessarily hierarchical and captures features at multiple length scales ranging from atomic connectivity to the size and density of side chains. The property prediction models are incorporated in a user friendly online platform named Polymer Genome (www.polymergenome.org), which utilizes a custom Python-based machine learning and polymer querying framework.
Polymer Genome, including the dataset, fingerprinting scheme, and machine learning models, remains in early stages. Coverage of the polymer chemical space needs to be progressively increased, and further developments on the fingerprinting scheme are necessary to adequately capture conformational (e.g., cis versus trans, tacticity, etc.) and morphological features (e.g., copolymerization, crystallinity, etc.). Systematic pathways to achieve such expansion are presently being examined to extend the applicability of the polymer informatics paradigm to a wide range of technological domains. Moreover, looking to the future, the ability of our informatics platform to automatically suggest polymers that are likely to possess a given set of properties would be of tremendous value within the context of “inverse design” [65]. Approaches involving Bayesian active learning techniques [66] and variational autoencoders [67] will allow the automated search of chemical and morphological space for materials with desired properties at a significantly accelerated pace.
References
M.I. Jordan, T.M. Mitchell, Science 349(6245), 255 (2015)
M. Rupp, A. Tkatchenko, K.R. Müller, O.A. von Lilienfeld, Phys. Rev. Lett. 108, 058301 (2012)
L. Ruddigkeit, R. van Deursen, L.C. Blum, J.L. Reymond, J. Chem. Inf. Model. 52(11), 2864 (2012). https://doi.org/10.1021/ci300415d. PMID: 23088335
R. Ramakrishnan, P.O. Dral, M. Rupp, O.A. Von Lilienfeld, Sci. Data 1, 140022 (2014)
Materials Genome Initiative. https://www.mgi.gov/
The Novel Materials Discovery (nomad) Laboratory. https://nomad-coe.eu/
National Center for Competence in Research - Marvel. nccr-marvel.ch
G. Pizzi, A. Cepellotti, R. Sabatini, N. Marzari, B. Kozinsky, Comput. Mater. Sci. 111, 218 (2016)
K. Mathew, J.H. Montoya, A. Faghaninia, S. Dwarakanath, M. Aykol, H. Tang, I.h. Chu, T. Smidt, B. Bocklund, M. Horton, et al., Comput. Mater. Sci. 139, 140 (2017)
A. Mannodi-Kanakkithodi, A. Chandrasekaran, C. Kim, T.D. Huan, G. Pilania, V. Botu, R. Ramprasad, Mater. Today 21, 785–796 (2017). https://doi.org/10.1016/j.mattod.2017.11.021
R. Ramprasad, R. Batra, G. Pilania, A. Mannodi-Kanakkithodi, C. Kim, npj Comput. Mater. 3, 54 (2017). https://doi.org/10.1038/s41524-017-0056-5
A. Mannodi-Kanakkithodi, G. Pilania, R. Ramprasad, Comput. Mater. Sci. 125, 123 (2016). https://doi.org/10.1016/j.commatsci.2016.08.039
T. Mueller, A.G. Kusne, R. Ramprasad, Machine Learning in Materials Science: Recent Progress and Emerging Applications, vol. 29 (Wiley, Hoboken, 2016), pp. 186–273
G. Hautier, C.C. Fischer, A. Jain, T. Mueller, G. Ceder, Chem. Mater. 22(12), 3762 (2010)
A.O. Oliynyk, E. Antono, T.D. Sparks, L. Ghadbeigi, M.W. Gaultois, B. Meredig, A. Mar, Chem. Mater. 28(20), 7324 (2016)
P. Pankajakshan, S. Sanyal, O.E. de Noord, I. Bhattacharya, A. Bhattacharyya, U. Waghmare, Chem. Mater. 29(10), 4190 (2017). https://doi.org/10.1021/acs.chemmater.6b04229
C. Kim, G. Pilania, R. Ramprasad, Chem. Mater. 28(5), 1304 (2016). https://doi.org/10.1021/acs.chemmater.5b04109
A. Jain, Y. Shin, K.A. Persson, Nat. Rev. Mater. 1, 15004 (2016). https://doi.org/10.1038/natrevmats.2015.4
A. Mannodi-Kanakkithodi, G. Pilania, T.D. Huan, T. Lookman, R. Ramprasad, Sci. Rep. 6, 20952 (2016). https://doi.org/10.1038/srep20952
L. Ghadbeigi, J.K. Harada, B.R. Lettiere, T.D. Sparks, Energy Environ. Sci. 8, 1640 (2015). https://doi.org/10.1039/C5EE00685F
J. Hattrick-Simpers, C. Wen, J. Lauterbach, Catal. Lett. 145(1), 290 (2015). https://doi.org/10.1007/s10562-014-1442-y
J. Hill, A. Mannodi-Kanakkithodi, R. Ramprasad, B. Meredig, Materials Data Infrastructure and Materials Informatics (Springer International Publishing, Cham, 2018), pp. 193–225. https://doi.org/10.1007/978-3-319-68280-8_9
A. Mannodi-Kanakkithodi, T.D. Huan, R. Ramprasad, Chem. Mater. 29(21), 9001 (2017). https://doi.org/10.1021/acs.chemmater.7b02027
C. Kim, T.D. Huan, S. Krishnan, R. Ramprasad, Sci. Data 4, 170057 (2017). https://doi.org/10.1038/sdata.2017.57
J. Behler, M. Parrinello, Phys. Rev. Lett. 98(14), 146401 (2007)
V. Botu, R. Batra, J. Chapman, R. Ramprasad, J. Phys. Chem. C 121(1), 511 (2017). https://doi.org/10.1021/acs.jpcc.6b10908
T.D. Huan, R. Batra, J. Chapman, S. Krishnan, L. Chen, R. Ramprasad, npj Comput. Mater. 3(1), 37 (2017). https://doi.org/10.1038/s41524-017-0042-y
V. Botu, J. Chapman, R. Ramprasad, Comput. Mater. Sci. 129, 332 (2017). https://doi.org/10.1016/j.commatsci.2016.12.007
F. Brockherde, L. Vogt, L. Li, M.E. Tuckerman, K. Burke, K.R. Müller, Nat. Commun. 8(1), 872 (2017)
L. Chen, T.D. Huan, R. Ramprasad, Sci. Rep. 7(1), 6128 (2017)
A. Mannodi-Kanakkithodi, G.M. Treich, T.D. Huan, R. Ma, M. Tefferi, Y. Cao, G.A. Sotzing, R. Ramprasad, Adv. Mater. 28(30), 6277 (2016). https://doi.org/10.1002/adma.201600377
G.M. Treich, M. Tefferi, S. Nasreen, A. Mannodi-Kanakkithodi, Z. Li, R. Ramprasad, G.A. Sotzing, Y. Cao, IEEE Trans. Dielectr. Electr. Insul. 24(2), 732 (2017). https://doi.org/10.1109/TDEI.2017.006329
A.F. Baldwin, T.D. Huan, R. Ma, A. Mannodi-Kanakkithodi, M. Tefferi, N. Katz, Y. Cao, R. Ramprasad, G.A. Sotzing, Macromolecules 48, 2422 (2015)
Q. Zhu, V. Sharma, A.R. Oganov, R. Ramprasad, J. Chem. Phys. 141(15), 154102 (2014). https://doi.org/10.1063/1.4897337
R. Lorenzini, W. Kline, C. Wang, R. Ramprasad, G. Sotzing, Polymer 54(14), 3529 (2013). https://doi.org/10.1016/j.polymer.2013.05.003
A.F. Baldwin, R. Ma, T.D. Huan, Y. Cao, R. Ramprasad, G.A. Sotzing, Macromol. Rapid Commun. 35, 2082 (2014)
A. Mannodi-Kanakkithodi, G. Pilania, R. Ramprasad, T. Lookman, J.E. Gubernatis, Comput. Mater. Sci. 125, 92 (2016). https://doi.org/10.1016/j.commatsci.2016.08.018
T.D. Huan, S. Boggs, G. Teyssedre, C. Laurent, M. Cakmak, S. Kumar, R. Ramprasad, Prog. Mater. Sci. 83, 236 (2016). https://doi.org/10.1016/j.pmatsci.2016.05.001
C. Kim, A. Chandrasekaran, T.D. Huan, D. Das, R. Ramprasad, J. Phys. Chem. C 122(31), 17575 (2018). https://doi.org/10.1021/acs.jpcc.8b02913
T.D. Huan, A. Mannodi-Kanakkithodi, C. Kim, V. Sharma, G. Pilania, R. Ramprasad, Sci. Data 3, 160012 (2016). https://doi.org/10.1038/sdata.2016.12
V. Sharma, C.C. Wang, R.G. Lorenzini, R. Ma, Q. Zhu, D.W. Sinkovits, G. Pilania, A.R. Oganov, S. Kumar, G.A. Sotzing, S.A. Boggs, R. Ramprasad, Nat. Commun. 5, 4845 (2014)
J. Bicerano, Prediction of Polymer Properties (Dekker, New York, 2002)
A.F.M. Barton, Handbook of Solubility Parameters and Other Cohesion Parameters (CRC Press, Florida, 1983)
C.C. Wang, G. Pilania, S.A. Boggs, S. Kumar, C. Breneman, R. Ramprasad, Polymer 55, 979 (2014)
J. Heyd, G.E. Scuseria, M. Ernzerhof, J. Chem. Phys. 118(18), 8207 (2003)
S. Baroni, S. de Gironcoli, A. Dal Corso, P. Giannozzi, Rev. Mod. Phys. 73(2), 515 (2001). https://doi.org/10.1103/RevModPhys.73.515
T.D. Huan, M. Amsler, V.N. Tuoc, A. Willand, S. Goedecker, Phys. Rev. B 86, 224110 (2012)
H. Sharma, V. Sharma, T.D. Huan, Phys. Chem. Chem. Phys. 17, 18146 (2015)
T.D. Huan, V. Sharma, G.A. Rossetti, R. Ramprasad, Phys. Rev. B 90, 064111 (2014)
T.D. Huan, M. Amsler, R. Sabatini, V.N. Tuoc, N.B. Le, L.M. Woods, N. Marzari, S. Goedecker, Phys. Rev. B 88, 024108 (2013)
A.F. Baldwin, R. Ma, A. Mannodi-Kanakkithodi, T.D. Huan, C. Wang, J.E. Marszalek, M. Cakmak, Y. Cao, R. Ramprasad, G.A. Sotzing, Adv. Matter. 27, 346 (2015)
R. Ma, V. Sharma, A.F. Baldwin, M. Tefferi, I. Offenbach, M. Cakmak, R. Weiss, Y. Cao, R. Ramprasad, G.A. Sotzing, J. Mater. Chem. A 3, 14845 (2015). https://doi.org/10.1039/C5TA01252J
Khazana, a Computational Materials Knowledgebase. https://khazana.gatech.edu
T.D. Huan, A. Mannodi-Kanakkithodi, R. Ramprasad, Phys. Rev. B 92(014106), 14106 (2015). https://doi.org/10.1103/PhysRevB.92.014106
C. Nantasenamat, C. Isarankura-Na-Ayudhya, T. Naenna, V. Prachayasittikul, EXCLI J. 8, 74 (2009)
C. Nantasenamat, C. Isarankura-Na-Ayudhya, V. Prachayasittikul, Expert Opin. Drug Discov. 5(7), 633 (2010). https://doi.org/10.1517/17460441.2010.492827. PMID: 22823204
Rdkit, Open Source Toolkit for Cheminformatics. http://www.rdkit.org/
P. Labute, J. Mol. Graph. Model. 18(4), 464 (2000). https://doi.org/10.1016/S1093-3263(00)00068-1
P. Ertl, B. Rohde, P. Selzer, J. Med. Chem. 43(20), 3714 (2000). https://doi.org/10.1021/jm000942e. PMID: 11020286
S. Prasanna, R. Doerksen, Curr. Med. Chem. 16, 21 (2009)
M. Sicher, S. Mohr, S. Goedecker, J. Chem. Phys. 134(4), 044106 (2011)
I. Guyon, J. Weston, S. Barnhill, V. Vapnik, Mach. Learn. 46(1), 389 (2002). https://doi.org/10.1023/A:1012487302797
K. Vu, J.C. Snyder, L. Li, M. Rupp, B.F. Chen, T. Khelif, K.R. Müller, K. Burke, Int. J. Quantum Chem. 115(16), 1115 (2015). https://doi.org/10.1002/qua.24939
Polymer Genome. http://www.polymergenome.org
B. Sanchez-Lengeling, A. Aspuru-Guzik, Science 361(6400), 360 (2018)
D.A. Cohn, Z. Ghahramani, M.I. Jordan, J. Artif. Intell. Res 4, 129 (1996)
H. Dai, Y. Tian, B. Dai, S. Skiena, L. Song (2018, preprint). arXiv:1802.08786
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Chandrasekaran, A., Kim, C., Ramprasad, R. (2020). Polymer Genome: A Polymer Informatics Platform to Accelerate Polymer Discovery. In: Schütt, K., Chmiela, S., von Lilienfeld, O., Tkatchenko, A., Tsuda, K., Müller, KR. (eds) Machine Learning Meets Quantum Physics. Lecture Notes in Physics, vol 968. Springer, Cham. https://doi.org/10.1007/978-3-030-40245-7_18
Download citation
DOI: https://doi.org/10.1007/978-3-030-40245-7_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-40244-0
Online ISBN: 978-3-030-40245-7
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)