
Abstract
An abnormality of cystic fibrosis transmembrane conductance regulator (CFTR) gene is known to be one of the etiologies of male infertility. CFTR gene mutations are associated with cystic fibrosis (CF-severe phenotype) to congenital bilateral absence of the vas deferens (CBAVD-mild phenotype). CF is the most common autosomal recessive disorders in the Caucasians, characterized by chronic lung disease, pancreatic insufficiency, rise in sweat chloride, and obstructive azoospermia. The milder phenotype is classified as congenital absence of the vas deferens (bi- or unilateral) (CBAVD or CUAVD) or ejaculatory duct obstruction (EDO). Some of these CAVD cases are associated with unilateral renal anomalies (URA). The role of CFTR gene in this subtype of CBAVD-URA is still not understood clearly. The utility of advanced assisted reproductive technologies such as intracytoplasmic sperm injection (ICSI) helps CBAVD males to become biological fathers. If female partner is CF carrier, there is a risk of having a child with CF or CF-related disorders. The currently available CFTR mutation panels cover the most common mutations of Caucasians. Recent studies conducted in South Asian population suggested different spectrum of CFTR mutations than Caucasians. There is a need to develop population-specific CFTR gene mutation panels especially for South Asians where CF or CF-related disorders were once considered rare.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
Key Points
-
Male infertility is associated with both cystic fibrosis (CF) and congenital bilateral absence of the vas deferens (CBAVD).
-
CF and CBAVD are two distinct spectrums of CFTR gene abnormalities.
-
CBAVD men carry different CFTR gene mutations than classic CF.
-
Renal anomalies are associated with ~11% men having CBAVD, more common in individuals having congenital unilateral absence of the vas deferens.
-
PESE-ICSI is the widely preferred and accepted treatment for men having CBAVD.
-
If female partner is CF carrier, there is a risk of having a child with CF or CF-related disorders such as CBAVD.
-
CFTR gene testing should be offered to both the partners before planning ICSI.
-
Population-specific mutation panels are required for accurate diagnosis and calculation of genetic risk in CBAVD men.
1 Introduction
Cystic fibrosis (CF) affects multiple organs of the body, and it is associated with mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. CF is considered as the most common autosomal recessive disorder in Caucasians with a frequency of 1/2000 (Nielson et al. 1988). Earlier, it was assumed that CF or CF-related disorders (CFTR-RD) are rare in African and Indian populations. However, recently with the advances in the diagnostic techniques as well as due to the increased awareness, CF and CFTR-RDs are increasingly detected in these populations. However, the incidence is still underestimated in Indian and black South African populations. Moreover, studies have reported that the prevalence of CF varies with geographical location (Casals et al. 1992).
CFTR gene is located on chromosome 7q31.2 and contains 27 exons (~250 kb of DNA). More than 1800 CF-causing CFTR gene mutations have been reported in the CFTR gene mutation database so far. Following are the databases of CFTR gene mutations:
There is limited information on exact pathogenicity of the CFTR gene mutations reported in different populations. Through CFTR 2 project, functional analysis of identified mutations is being investigated. Six functional classes of CF mutations are described (Fig. 9.1):
-
Class I mutations: CFTR production is stopped early and the protein is defective resulting into nonfunctioning CFTR chloride channels. Accounts for ~10% of CFTR gene mutations causing CF worldwide. Mutation leads to premature stop codon, which causes translation of mRNA to stop prematurely.
-
Class II mutations: No proper processing of CFTR and proteins is destroyed within the cell. F508del (absence of phenylalanine at position 508) is the most commonly reported Class II mutation. F508del occurs in around 88.5% of CF patients worldwide as per the CF registry database.
-
Class III mutations: CFTR reaches cell surface but it does not open properly to transport chloride. Only a small percentage CF (2–3%) cases have this mutation.
-
Class IV mutations: Defective conduction of chloride through the channel. These are uncommon mutations and lead to disease ~2% of patients with CF.
-
Class V mutations: The least common mutations. Splicing defects resulting into improper processing of mRNA are the etiology for Class V mutation.
-
Class VI mutations: Although function CFTR protein but unstable at cell surface.

Classes of CFTR gene mutations
The cystic fibrosis transmembrane conductance regulator (CFTR) protein is expressed throughout the epithelial cells in the airways, gastrointestinal tract, and reproductive organs (Quinton 2007). As a result, CF patients manifest symptoms related to multiple organs that include repeated and chronic lung infection, insufficiency of the pancreas, and male infertility. CFTR gene mutations are the main etiological factors due to defective electrolyte and fluid transport (Welsh and Fick 1987; Welsh and Smith 1993; Quinton 2007).
In addition to regulating the chloride ion channel in the epithelial cells, CFTR is involved in the following functions: (a) sodium transport through the sodium ion channel, (b) regulation of the chloride flow outside the cell membrane, (c) regulation of the ATP channels, (d) intracellular vesicle transport, (e) acidification of intracellular organelles, (f) inhibition of endogenous calcium-activated chloride channels, and (g) efficient bicarbonate–chloride exchange.
2 Pathogenesis
The CFTR protein is an epithelial membrane protein, an ATP-binding cassette (ABC)-transporter-class ion channel. It regulates the chloride ions across epithelial cell membranes. The CFTR protein is made of five domains: two membrane-spanning domains (MSDs) that form the channel pores; two nucleotide-binding domains (NBDs), which control channel gating; and one regulatory domain (R domain), which determines the phosphorylation activity.
There are different hypotheses to explain the role of CFTR abnormalities in developing CF or CFTR-RD. Following are the most relevant hypothesis; it may be possible that the combination of these aspects could contribute to the pathogenesis of the CF or CFTR-RD:
-
1.
Low-volume hypothesis: Due to the CFTR dysfunction, there is loss of inhibition of epithelial sodium channels leading to excess sodium and water reabsorption ultimately resulting in dehydration of airway surface materials (Matsui et al. 1998). The low airway surface water volume is not corrected by the epithelium due to the associated loss of chloride. Reduction in periciliary water leads to decrease in the lubricating layer between epithelium and mucus and compresses the cilia by mucus causing inhibition of normal ciliary movement and cough clearance of the mucus. According to this hypothesis, bacteria such as Pseudomonas aeruginosa can grow due to the mucus on the epithelium that leads to plaque formation with hypoxic niches (Boucher 2007).
-
2.
High-salt hypothesis: Absence of functional CFTR protein leads to retention of excess of sodium and chloride in airway surface liquid. The higher levels of chloride in the periciliary layer then disrupt the function of innate antibiotic molecules such as human β-defensin 1 and thereby allow the growth of bacteria that are normally cleared by normal airways to persist in the lungs (Goldman et al. 1997).
-
3.
Dysregulation of the host inflammatory response: Cystic fibrosis cell cultures and uninfected ex vivo tissue samples contain higher concentrations of inflammatory mediators (Freedman et al. 2004). Inflammatory mediators were detected in the lung lavage samples of children as young as 4 weeks of age. The pro-inflammatory molecules (Interleukin 8, Interleukin 6, TNFα, and arachidonic acid metabolites) were detected CF (Freedman et al. 2004). Studies also reported the activation of NFκB pathway, platelet hyperreactivity, and neutrophil apoptosis abnormalities (Carrabino et al. 2006).
-
4.
Primary predisposition to infection: Normally, P. aeruginosa binds to functional CFTR, and rapid and self-limiting innate immune response is initiated. In CF, increase in asialo-GM1 in apical cell membranes allows binding of P aeruginosa and Staphylococcus aureus to the airway epithelium, without CFTR-mediated immune response. The self-limiting response that eliminates P. aeruginosa from the airways is lost in CF and at the same time as there is enhanced attachment of bacteria to the epithelial surface.
3 Epidemiology
Incidence of CF is reported to be 1 in 2000–3000 in Caucasians with a carrier frequency of 1 in 22–28. High prevalence of CF is reported in North America, Europe, and Australia. Recent studies generated evidence of increased number of CF and CF-related disorders in other ethnic populations residing in Africa, South America, Middle East, and Asia (WHO 2004 and Cystic Fibrosis Foundation Patient Registry 2012 Annual Data Report). It has been reported that there is a variation in birth prevalence due to CF worldwide with different ethnic backgrounds. Prevalence of CF was reported as 1 in 3000 in white Americans, 1 in 4000–10,000 in Latin Americans, and 1 in 15,000–20,000 in African Americans (Walters and Mehta al. 2007). Cystic fibrosis was earlier reported to be a rare disorder in Africa and Asia, with a frequency of 1 in 350,000 in Japan (Yamashiro et al. 1997). Frequency F508del mutation was higher in northwest region of Europe than southeast. Similarly, Trp1282X is the most common mutation reported in Israel (O’Sullivan and Freedman 2009).
There could be multiple reasons for lower reporting of CF and CF-related disorders in developing countries in South Asian subcontinent. Majority among them is the lack of awareness about CF and CF-related disorders, limited clinical expertise for diagnosis and management, and limited molecular diagnostic facilities. There has been a good progress in the past few years, and evidence is emerging on CFTR gene mutations in CF and CFTR-RD from the South Asian population. A heterogeneous spectrum of CFTR gene variants was identified in Asians (Fig. 9.2), with a lower frequency of F508del in Asians as compared to Caucasians (Sharma et al. 2009). Evidence is very limited to prove whether low incidence of CF in Asian populations is due to genetic drift or it is due to misdiagnosis. This needs to be thoroughly investigated especially in South Asian countries. Earlier reports indicated the incidence of CF in immigrant Asians residing in Canada as 1/9200, 1/10,000 in the UK, and 1/40,000 in the USA (Powers et al. 1996; Mei-Zahav et al. 2005). Researchers now hypothesize that India may hold the largest CF and CFTR-RDs population in world with up to 1,00,000 undiagnosed CF patients (CFRI News 2013).

Map showing the most common CFTR gene mutations identified across different populations
4 Diagnosis
The preliminary diagnosis of CF is based on elevated sweat chloride level (>60 mmol/L). In a more classical condition, CF is diagnosed if the sweat chloride levels are in the intermediate range (for infants >6 months, 30–59 mmol/L and for old individuals, 40–59 mmol/L) and two severe disease-causing mutations are identified in an individual (Rosenstein et al. 1998; De Boeck et al. 2006; Welsh et al. 2001). Patients with intermediate range (30–60 mmol/L) of sweat chloride levels might have CFTR genotype combining two CF-causing mutations. The American College of Medical Genetics recommended a panel of 23 CF-causing mutations. More than 1800 CFTR gene mutations have been reported and included in the CF registries. The number of novel mutations is also exponentially increasing (Table 9.1). The diagnosis of CF becomes more problematic when sweat chloride levels are intermediate and patient still has symptoms suggestive of CF. A more severe lung disease is observed among the patients with abnormalities in NPD measurement or two CFTR gene mutations (Goubau et al. 2009). However, their disease symptoms are milder as compared to those with a sweat chloride levels above 60 mmol/L. In children with multiple organ involvement with marginal levels of sweat chloride concentration and/or presence of at least one CFTR gene mutation of unknown clinical significance, a terminology of “nonclassical” or “atypical” CF is applicable (Rosenstein et al. 1998). Due to the varied spectrum of clinical phenotypes, now the new terminology of “CFTR-related disorders” (CFTR-RDs) is gaining wider acceptance (Dequeker et al. 2009; Castellani et al. 2008). It is very essential to understand the complete clinical phenotype along with biochemical and molecular tests for reaching out the correct diagnosis of CF or CFTR-RD.
5 Fertility in Men Having CF
Spermatogenesis is a well-orchestrated process by which the totipotent primordial spermatogonia undergo meiosis to produce daughter cells called spermatozoa. In order to form a mature sperm, the spermatozoa undergoes a series of morphological and functional differentiation processes under the influence of hormones including follicle-stimulating hormone (FSH), luteinizing hormone (LH), and testosterone. These processes occur within the seminiferous tubules, which are supported by Sertoli cells that are in close contact with the germ cells. Defects at any stage of spermatogenesis may cause male infertility including azoospermia, oligospermia, and teratospermia. However, the importance of CFTR in spermatogenesis is still controversial, even after its experimental evidence of expression in the testis (Trezíse and Buchwald 1991). Histological studies with testicular tissues of men with CF and CBAVD tried to resolve this controversy but resulted in contradictory findings such as normal spermatogenesis (Tuerlings et al. 1998) to severely decreased spermatogenesis with abnormal sperm and a reduced sperm count (Larriba et al. 1998).
Puberty in men having classic CF and chronic lung disease, malnutrition is usually delayed due to lower levels of follicle-stimulating hormone (FSH) and luteinizing hormones (LH). In spite of the delayed onset of puberty, majority of CF patients
(>90%) achieve normal height. Around 2–5% of CF men are fertile. There is a normal production of immature sperm in testes. Bilateral vas deferens is either atrophied or absent in approximately 95% of CF males. Seminal vesicles are hypoplastic or absent and normal maturation of sperm is impaired. As a result of this, there is reduced seminal volume, no mature sperm, and high acid content, absent or low fructose in semen.
6 CFTR-Related Disorders Associated with Male Infertility
A CFTR-related disorder (CFTR-RD) is a separate clinical condition associated with CFTR gene abnormalities and does not fulfill the diagnostic criteria of CF. Four main clinical entities illustrate these phenotypes:
-
Congenital bilateral absence of the vas deferens (CBAVD) with CFTR dysfunction
-
CBAVD having renal anomalies
-
Congenital unilateral absence of the vas deferens (CUAVD)
-
Ejaculatory duct obstruction (EDO)
6.1 Congenital Bilateral Absence of the Vas Deferens (CBAVD)
CBAVD is a condition in which there is a complete or partial failure of development of vasa deferens before birth. CBAVD in otherwise healthy men also known as isolated CBAVD accounts for ~3% of male infertility. The incidence of CBAVD is ~1:1000 men (Holsclaw et al. 1971; Oates and Amos 1993; Mak and Jarvi 1996). Isolated CBAVD (MIM#277180) is an autosomal recessive genetic disorder known to be associated with CFTR gene abnormalities. Milder phenotype such as CBAVD is due to the CFTR gene variants that retain the CFTR function to its minimum. CBAVD is either due to the one inherited CFTR gene mutation (Dumur et al. 1990; Anguiano et al. 1992; Patrizio et al. 1993) or due to the inheritance of mutations in both the copies of the CFTR gene (70–90% of cases) (Bombieri et al. 2011). CBAVD and CF are now considered as two different spectrums of CFTR due to the distinct genotype and phenotype (Colin et al. 1996).
The diagnosis of CBAVD is based on scrotal examination—bilateral absence of the vas deferens and normal testicular volume (>15 mL) and absence of body and tail of epididymis. Semen analysis is very important in diagnosis as it reveals azoospermia with low seminal volume (<1.0 mL), low pH (average <6.8), and low or absent fructose levels (Casals et al. 1995, Holsclaw et al. 1971). The abnormal CFTR protein could affect the multiple organs including reproductive tract. Transrectal ultrasonography (TRUS) reveals the morphology and size of the seminal vesicles, prostate, and ejaculatory ducts. In CBAVD, body and tail of the epididymis are either atrophic or absent or the epididymis remnants are distended, whereas the head or caput of the epididymis is usually present (McCallum et al. 2000a, b). The sweat chloride levels are usually normal, and testicular biopsy shows normal spermatogenesis in majority of CBAVD cases.
Due to the compound heterozygosity (either one severe and one mild mutation or two mild mutations) in CBAVD, the spectrum of CFTR gene mutations differs from that of the classical CF. In CF patients two severe CFTR gene mutations (88%) or one severe and one mild or variable CFTR mutations (12%) are detected. In CBAVD, one severe and one mild or variable (88%) or two mild CFTR gene mutations (12%) are detected (Bombieri et al. 2011). p.F508del along with IVS8-5T (28%) and p.F508del in trans with p.R117H (6%) are the most common compound heterozygous genotypes found in CBAVD. A significant difference in the frequency is found in the most CF-causing mutations. The frequency of p.F508del is found to be 21–33% in the USA, Canada, and Northern Europe (Oates and Amos 1993; Jarvi et al. 1998; Dork et al. 1997; Claustres et al. 2000; Jarvi et al. 1998) and 12–18% in Southern Europe and India (Kanavakis et al. 1998; Grangeia et al. 2004; Sharma et al. 2009). However, p.F508del is found in lower frequencies in CBAVD men from non-European populations. The IVS8-5T allele is found in similar frequency in Indian (25%) and Japanese (30%) (Sharma et al. 2009; Anzai et al. 2000) or higher frequencies in Egyptians (44%) and Taiwanese (44%) population (Lissens et al. 1995; Wu et al. 2004). IVS8-5T is seen in 5% of general population and is reported in many countries where CF was once considered as a rare disorder. Due to the limited studies in South Asian populations, many of the common CFTR gene mutations are yet to be reported in these populations. IVS8-5T allele is 5–8 times higher in CBAVD men than the general population. Hence, it is the most common “mild” CFTR allele, present in at least 5% of general population worldwide (Bombieri et al. 2011). Studies have found that 34% of CBAVD men from European descent inherit at least one IVS8-5T allele (Casals et al. 1992). However, due to mild pathogenicity, IVS8-5T allele alone or in combination with other CFTR gene mutation cannot result in severe CF phenotype. IVS8-5T causes alternative splicing of exon 9 of the CFTR gene and leads to decreased levels of functional CFTR protein to develop isolated CBAVD phenotype (Casals et al. 1992). It has been reported that Wolffian tissues are the most prone tissues to splicing of exon 9, resulting in reduced full-length CFTR mRNAs as compared to other tissues. IVS8-5T splicing variant also produces low transcript level of full-length CFTR protein which is necessary for normal Wolffian tissues phenotype (Teng et al. 1997). The vas deferens is most sensitive to reduced functional CFTR protein due to the above mentioned mechanisms.
The IVS8-5T allele is known as a genetic modifier of p.R117H mutation when associated in cis position. The IVS8-5T allele is considered a CBAVD mutation with partial or incomplete penetrance. The efficiency of exon 9 splicing is influenced by the (TG)m repeat which lies immediately upstream of the IVS8-Tn tract (Cuppens et al. 1998). Thus, chances of exon 9 skipping is higher in the presence of longer IVS8-TGm and shorter IVS8-Tn repeats leading to misfolded and/or nonfunctional CFTR protein (Cuppens et al. 1998). It has been found that CBAVD men have longer IVS8-TG repeats (12 or 13) as compared to healthy men, who have shorter IVS8-TG repeats (10 or 11) (Cuppens et al. 1998). Longer IVS8-TG repeats (IVS8-TG12 or TG13) in cis with IVS8-5T were found to correlate with CBAVD or CFTR-RD disease status. Therefore, the polymorphic dinucleotide (TG)m repeats could be the reliable predictor for the penetrance of IVS8-5T as a disease-causing allele. So far, the pathogenicity of TG12-5T and TG13-5T is much higher than that of TG11-5T allele. The TGmTn allele represents a model of CBAVD “polyvariant mutant CFTR.”
Point mutations are extensively identified in the CFTR gene of CBAVD men. Often, large rearrangements such as deletions or duplications within the CFTR locus are also identified in 6–10% CBAVD cases, which is lower than the rearrangements found in CF patients (15–25%). Overall, large rearrangements (null mutations, classified as “severe”) represent <1% of CBAVD alleles, a lower proportion than in CF, which reflects the higher contribution of severe alleles to the pathogenesis of CF (Bombieri et al. 2011).
6.2 CBAVD Having Renal Anomalies (CBAVD-URA)
CBAVD is associated with congenital malformations or agenesis of the upper urinary tract in 12–21% of cases. The association of CFTR gene mutations with CBAVD-URA is controversial as majority of cases failed to detect CFTR gene mutation (Anguiano et al. 1992; Augarten et al. 1994; Casals et al. 1995; Mickle et al. 1995; Schlegel et al. 1996; Dörk et al. 1997; de la Taille et al. 1998; Claustres et al. 2000; McCallum et al. 2001). As a result, CBAVD with renal malformation was considered as a distinct clinical phenotype termed “CBAVD-URA” (McCallum et al. 2001). There was no statistically significant difference in physical, laboratory, and radiographic findings of the reproductive derivatives as well as in fertilization and pregnancy rates between CF/CBAVD and CBAVD-URA (Robert et al. 2002). The hypothesis that CBAVD-URA could be a separate clinical disorder is further supported by the marked difference between the renal portions of the mesonephric duct in the two cohorts. The physical separation between the two mesonephric duct derivatives (seminal and renal) occurs by week 7 of gestation (Oates and Amos 1993). During embryonic development, the mesonephric duct gives rise to the vas deferens, seminal vesicle, ejaculatory duct, and distal two-thirds of the epididymis, while the ureteric part induces renal development. The genital ridge extends to form the caput of the epididymis (which is present in men with CBAVD or CF) and the testis. Any abnormalities at the embryonic developmental phase before week 7 could lead to abnormal development of the entire mesonephric duct resulting in CBAVD-URA phenotype (Hall and Oates 1993; McCallum et al. 2001) or CUAVD-URA phenotype (Donohue and Fauver 1989). By contrast, the genetic defect in CBAVD-URA appears to affect the embryo after the division of the mesonephric parts in the seventh week of gestation, so that only the seminal tract will be altered. A few number of patients with CBAVD and URA have now been reported to be heterozygous for a CFTR gene mutations (Mak and Jarvi 1996); the significance of these mutations is undetermined as it could be in conjunction with the IVS8-5T carrier status found in the general population and the lack of investigations in large number of CBAVD-URA patients. Hence, a complete family studies are required in both the CBAVD and CBAVD-URA cohort to determine the genetic causes, the mode of inheritance, and the penetrance of genetic factors in CBAVD and nephrogenesis. More studies are required to prove or disprove the association of CFTR gene with CBAVD and renal anomalies.
6.3 Congenital Unilateral Absence of the Vas Deferens (CUAVD)
Congenital unilateral absence of the vas deferens (CUAVD) occurs in less than 1/1000 men and hence is a rare condition. Mickle et al. (1995) defined CUAVD as the absence of one of the scrotal vasa deferentia and considered as a clinically and genetically distinct phenotype. The frequency of ipsilateral renal agenesis is higher (40–80%) in CUAVD and no CFTR gene mutations were detected (Mickle et al. 1995; Mak and Jarvi 1996; Weiske et al. 2000; McCallum et al. 2001; Kolettis and Sandlow 2002). A large variation is observed in the clinical presentation of CUAVD. Surprisingly, patients could be diagnosed of CUAVD during a clinical evaluation for vasectomy or other urologic conditions. Others may be diagnosed due to infertility and azoospermia because of contralateral testicular or Wolffian duct abnormalities. CUAVD exists as two different forms with and without renal anomalies suggesting different pathophysiological processes.
6.4 Ejaculatory Duct Obstruction
It was suggested that azoospermia not related to vas aplasia may be in some cases associated with CFTR gene mutations, including idiopathic forms of epididymal obstruction (Jarvi et al. 1998; Mak and Jarvi 1996). Bilateral ejaculatory duct obstruction (BEDO) was associated with a higher frequency of CFTR gene mutations (Meschede et al. 1997; Mak and Jarvi 1996). In a study involving 16 men with isolated anomalies of the seminal vesicles (IASV), only one was found to be heterozygous for a missense mutation and one for the 5T allele, with a frequency not different from the general population, so that IASV was not considered a CFTR-related entity (Meschede et al. 1997). The association of chronic bronchopulmonary disease with azoospermia due to a complete bilateral obstruction of the epididymis characterize Young’s syndrome, but, in contrast to CBAVD or CF, there is no anatomical malformation of the seminal ducts.
7 Infertility Management in CBAVD
7.1 Assisted Reproduction
Obstructive azoospermia (OA) is due to the blockage in sperm delivery pathway occurring anywhere in the reproductive tract including the vas deferens, epididymis, and ejaculatory duct. The most common etiology of OA is CBAVD, vasectomy, failed vasoepididymostomy, post-infective epididymitis, and other irreparable obstructions (Chen et al. 1995; Mansour et al. 1997). Intracytoplasmic sperm injection (ICSI) with percutaneous epididymal sperm aspiration (PESA) is the treatment of choice in men with OA due to CBAVD (Celikten et al. 2013).
It is a well-established fact that spermatogenesis is usually normal in majority of CBAVD men (Meng et al. 2001). There are various techniques of sperm retrieval such as microsurgical epididymal sperm aspiration (MESA) and testicular sperm extraction (TESE) allowing biological paternity to CBAVD patients.
There is an increased risk of having a child with CF or CFTR-RD if female partner of CBAVD is CF carrier. The percentage of 5T alleles in intron 8 of CFTR gene was reported as 26.25% in CBAVD, 20% in CUAVD, and 5% in controls in Indian population (Sharma et al. 2009). Thus, a male with CBAVD and F508del/7T alleles, if partnered with a female of normal phenotype possessing the 9T/5T, according to Mendelian expectation, provides a ratio of one in four embryos with F508del/5T genotype, which would result in CF phenotype. The other three predictions would be F508del/9T male having same phenotype as their father, i.e., CBAVD, 7T/9T offspring of normal phenotype, and 7T/5T offspring having normal phenotype (in females) and CBAVD (in males) (Persson et al. 1996). The evidence from follow-up study of children born after ICSI in CBAVD couples suggested 16% increased risk of CF or CBAVD suggesting the mandatory screening for CFTR gene mutations in both the partners prior to ICSI (Bonduelle et al. 1998). The first pregnancy for a couple in which the male partner was having CBAVD was reported in 1987 (Silber et al. 1988). The initial IVF cycles yielded poor oocyte fertilization rates. Since 1993, ICSI is the treatment of choice for CBAVD patients. Although lower fertilization (Patrizio et al. 1993) or lower embryo implantation (Hirsh et al. 1994) rates have been reported in couples with CBAVD, the presence of CFTR mutations in men with CBAVD does not seem to affect sperm function during IVF with micromanipulation (Schlegel et al. 1996; Silber et al. 1995). The success rate of ICSI in CBAVD was reported to be around 31% per cycle and a “take-home baby rate” was 23% (Silber et al. 1990). The meta-analysis of the ICSI outcome suggested that ICSI outcome is independent of whether retrieved spermatozoon is fresh, frozen, epididymal, or testicular. However, it suggested a lower fertilization rate and high miscarriage in CBAVD–CFTR as compared to acquired causes of obstructive azoospermia (Nicopoullos et al. 2004). Liu et al. (1994) reported the first successful PGD for a couple with CBAVD (both partners F508del heterozygous). Three carrier embryos were transferred and a healthy boy was born. The data suggested that the presence of CF- or CBAVD-causing CFTR gene mutations in CBAVD does not compromise significantly in fertilization rates, embryo implantation rates, or the successful delivery of asymptomatic child after PGD (McCallum et al. 2000; Phillipson et al. 2000).
7.2 Genetic Counseling
Genetic counseling prior to ICSI provides an estimated risk of transmitting the CF mutation from each of the parents. The probable CF or CFTR-RD phenotype of the offspring is calculated based upon the female partner’s genotype, the severity of the mutation identified in the male partner, and the presence of intron 8 splice site variant. Even if the female partner is not detected to be a CF carrier by available CF mutation panels, the risk of being a carrier of a missed mutation is 0.1%. The genetic risk for couples having CFTR gene mutations to have a CF child is 1/4000 and 1/2000. The main rationale for CFTR testing in CBAVD, irrespective of the fact that they will be using their sperm for ICSI, is that this information is important from the point of genetic counseling regarding future health impacts of CFTR mutations as well as counseling of the siblings regarding their risk of being CF carriers. Therefore, men with CBAVD should be offered genetic counseling and CFTR testing. The CFTR screening should also be carried out in female partner before undergoing ICSI that utilizes the sperm of CBAVD partner.
7.3 Sperm Collection Techniques
7.3.1 Percutaneous Epididymal Sperm Aspiration (PESA)
PESA is used in obstructive azoospermia due to CBAVD. A small needle is inserted in the scrotum and sperm are collected from the epididymis. Obstructive azoospermia cases can be greatly benefitted from PESA as it is a useful technique to find sperm in the male partner.
This can be done by two methods: (1) testicular sperm extraction (TESE), surgical biopsy of the testis, or (2) testicular sperm aspiration (TESA), sticking a needle in the testis and aspirating fluid and tissue with negative pressure.
7.3.2 Microsurgical Epididymal Sperm Aspiration (MESA)
MESA is a highly advanced sperm retrieval technique. The optimal area of the epididymis is selected using operating microscope. The retrieved sperm are used for intracytoplasmic sperm injection (ICSI). MESA is now considered as a gold standard for sperm retrieval obstructive azoospermia cases. High fertilization and pregnancy rates and low risk of complications are some of the advantages of MESA (Bernie et al. 2013).
8 Our Experience
8.1 CFTR Gene Variants in Isolated CBAVD in Indian Population
Due to the limited information in Indian population, studies were initiated through NIRRH-ICMR, Mumbai. The andrology clinic at NIRRH is providing regular clinical and laboratory services to males with obstructive azoospermia due to vas aplasia. Currently, the clinic has one of the largest cohorts of obstructive azoospermia cases due to congenital absence of the vas deferens in India. Studies in Indian population observed heterogeneous spectrum of CFTR gene mutations suggesting the need to develop population-specific CFTR gene mutation panel. We detected ten novel and nine reported CFTR gene mutations in Indian CBAVD men (Gajbhiye et al., unpublished data). Further studies are ongoing to carry out screening of larger cohorts of CAVD representing different ethnic groups in India. Studies are also being undertaken to functionally characterize the novel CFTR gene mutations reported in Indian CBAVD.
8.2 CBAVD-URA
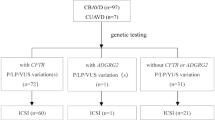
At NIRRH-ICMR, Mumbai, out of 85 CBAVD men, ten patients (11.76%) were found to have unilateral renal anomalies (URA). We detected CFTR gene variants in CBAVD having renal malformations. Congenital bilateral absence of seminal vesicles (CASV) and CBAVD are uncommon anomalies, and such patients usually have normal kidneys. Direct DNA sequencing of the CFTR gene in five CBAVD-URA men detected c.1210-12[5] (IVS8-5T) mutation in four out of five CBAVD males having renal anomalies with an allelic frequency of 40%. Four novel CFTR gene variants (c.2751+85_88delTA, c.2752+106A>T, c.3120+529InsC, c.4375-69C>T); four coding SNPs, V470M, T854T, P1290P, and Q1463Q; and ten previously reported CFTR gene variants were also detected in CBAVD males having renal anomalies (Gajbhiye et al. 2016). Normally, in addition to prostatic secretions, seminal vesicular secretions also contribute to the alkalinity of the ejaculate and make up approximately 90% of fluid in ejaculate. Thus, patients having CASV and CBAVD present with history of infertility, and usually patients with URA remain undiagnosed until there is some pathology in the contralateral kidney.
Two CBAVD-URA patients in our study were found to have longer IVS8-TG repeats (TG12 or TG13) in cis with 5T and M470V polymorphism. Previous studies reported that M470V along with short poly-T (5T) and long TG-repeat tracks (TG12, TG13) may contribute to CBAVD risk. This genotype was not detected in normal male participants suggesting that longer TG-short T repeats in association with M470V and other variants might be responsible for CBAVD-URA phenotype.
9 Future Perspectives
Evidence suggests that CFTR-related male infertility is now well established. The epidemiologic data also suggested variation in CF and CBAVD incidence by ethnic groups indicating that population-specific CFTR gene mutation database and mutation panels should be used for CF or CBAVD men undergoing ICSI. The major challenge is to identify disease-causing CFTR gene mutations in CFTR-related male infertility. This would help us to understand the genotype–phenotype correlation and provide accurate genetic counseling to the CF or CBAVD men undergoing ICSI. Further research should be focused on screening large number of infertile men due to vas aplasia and also to detect the CF carrier frequency in populations where CF or CF-related disorders (CFTR-RDs) were considered to be low. Studies are also required to functionally characterize the novel ethnic-specific mutations. There is a great need to create awareness about the CF and CFTR-RD worldwide. The genetic screening and counseling should be made available through public health care, especially in low- and middle-income countries. The global network of clinicians, scientists, and policy makers shall be established to provide standard care to patients having CF and CFTR-RD. The international experts and NGOs should come forward and empower the health-care providers in developing countries to diagnose and provide treatment to CF and CFTR-RD patients.
References
Anguiano A, Oates RD, Amos JA, Dean M, Gerrard B, Stewart C, Maher TA, White MB, Milunsky A (1992) Congenital bilateral absence of the vas deferens: a primarily genital form of cystic fibrosis. JAMA 267(13):1794–1797
Augarten A, Yahav Y, Laufer J, Szeinberg A, Dor J, Mashiach S, Madgar I, Halle D, Gazit E, Kerem BS (1994) Congenital bilateral absence of vas deferens in the absence of cystic fibrosis. Lancet 344(8935):1473–1474
Anzai C, Morokawa N, Okada H, Kamidono S, Eto Y, Yoshimura K (2003) CFTR gene mutations in Japanese individuals with congenital bilateral absence of the vas deferens. J Cyst Fibros 2:14–18.
Bonduelle M, Aytoz A, Van Assche E, Devroey P, Liebaers I, Van Steirteghem A (1998) Incidence of chromosomal aberrations in children born after assisted reproduction through intracytoplasmic sperm injection. Hum Reprod 13(4):781–782
Bombieri C, Claustres M, De Boeck K, Derichs N, Dodge J, Girodon E, et al. (2011) Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros, 10, pp.86–102.
Boucher RC (2007) Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med 58:157–170
Carrabino S, Carpani D, Livraghi A, Di Cicco M, Costantini D, Copreni E, Colombo C, Conese M (2006) Dysregulated interleukin-8 secretion and NF-κB activity in human cystic fibrosis nasal epithelial cells. J Cyst Fibros 5(2):113–119
Casals T, Vazquez C, Lazaro C, Girbau E, Gimenez FJ, Estivill X (1992) Cystic fibrosis in the Basque country: high frequency of mutation delta F508 in patients of Basque origin. Am J Hum Genet 50(2):404
Casals T, Bassas L, Ruiz-Romero J, Chillon M, Gimenez J, Ramos MD, Tapia G, Narvaez H, Nunes V, Estivill X (1995) Extensive analysis of 40 infertile patients with congenital absence of the vas deferens: in 50% of cases only one CFTR allele could be detected. Hum Genet 95(2):205–211
Castellani C, Cuppens H, Macek M, Cassiman JJ, Kerem E, Durie P, Tullis E, Assael BM, Bombieri C, Brown A, Casals T (2008) Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cyst Fibros 7(3):179–196
Celikten A, Batioglu S, Gungor ANC, Ozdemir E (2013) Intracytoplasmic sperm injection outcomes of obstructive and nonobstructive azoospermic men. Arch Gynecol Obstet 288(3):683–686
CFRI news 2013. http://www.cfri.org/pdf/2013SpringCFRInews.pdf
Chen CS, Chu SH, Soong YK, Lai YM (1995) Andrology: epididymal sperm aspiration with assisted reproductive techniques: difference between congenital and acquired obstructive azoospermia? Hum Reprod 10(5):1104–1108
Chu CS, Trapnell BC, Curristin S, Cutting GR, Crystal RG (1993a) Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat Genet 3(2):151–156
Chu CS, Trapnell BC, Curristin S, Cutting GR, Crystal RG (1993b) Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat Genet 3(2):151–156
Claustres M, Guittard C, Bozon D, Chevalier F, Verlingue C, Ferec C, Girodon E, Cazeneuve C, Bienvenu T, Lalau G, Dumur V (2000) Spectrum of CFTR mutations in cystic fibrosis and in congenital absence of the vas deferens in France. Hum Mutat 16(2):143
Colin AA, Sawyer SM, Mickle JE, Oates RD, Milunsky A, Amos JA (1996) Pulmonary function and clinical observations in men with congenital bilateral absence of the vas deferens. Chest 110:440–445
Cuppens H, Lin W, Jaspers M, et al (1998) Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes. The polymorphic (Tg)m locus explains the partial penetrance of the T5 polymorphism as a disease mutation. J Clin Invest 101:487–496.
Dumur V, Gervais R, Rigot JM, Lafitte JJ, Manouvrier S, Biserte J, Mazeman E, Roussel P (1990) Abnormal distribution of CF delta F508 allele in azoospermic men with congenital aplasia of epididymis and vas deferens. Lancet, pp. 336:512
De Boeck K, Wilschanski M, Castellani C, Taylor C, Cuppens H, Dodge J, Sinaasappel M (2006) Cystic fibrosis: terminology and diagnostic algorithms. Thorax 61(7):627–635
de la Taille A (1998) Correlation between genito-urinary anomalies, semen analysis and CFTR genotype in patients with congenital bilateral absence of the vas deferens. Br J Urol 81(4):614–619
Dequeker E, Stuhrmann M, Morris MA, Casals T, Castellani C, Claustres M, Cuppens H, Des Georges M, Ferec C, Macek M, Pignatti PF (2009) Best practice guidelines for molecular genetic diagnosis of cystic fibrosis and CFTR-related disorders–updated European recommendations. Eur J Hum Genet 17(1):51–65
Donohue RE, Fauver HE (1989) Unilateral absence of the vas deferens: a useful clinical sign. JAMA 261(8):1180–1182
Dörk T, Dworniczak B, Aulehla-Scholz C, Wieczorek D, Böhm I, Mayerova A, Seydewitz HH, Nieschlag E, Meschede D, Horst J, Pander HJ (1997) Distinct spectrum of CFTR gene mutations in congenital absence of vas deferens. Hum Genet 100(3–4):365–377
Estivill, X., Bancells, C. and Ramos, C., 1997. Geographic distribution and regional origin of 272 cystic fibrosis mutations in European populations. Human mutation, 10(2), p. 135.
Freedman SD, Blanco PG, Zaman MM, Shea JC, Ollero M, Hopper IK, Weed DA, Gelrud A, Regan MM, Laposata M, Alvarez JG (2004) Association of cystic fibrosis with abnormalities in fatty acid metabolism. N Engl J Med 350(6):560–569
Gajbhiye R, Kadam K, Khole A, Gaikwad A, Kadam S, Shah R, Kumaraswamy R, Khole V (2016) Cystic fibrosis transmembrane conductance regulator (CFTR) gene abnormalities in Indian males with congenital bilateral absence of vas deferens & renal anomalies. Indian J Med Res 143(5):616
Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM (1997) Human β-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 88(4):553–560
Goubau C, Wilschanski M, Skalická V, Lebecque P, Southern KW, Sermet I, Munck A, Derichs N, Middleton PG, Hjelte L, Padoan R (2009) Phenotypic characterisation of patients with intermediate sweat chloride values: towards validation of the European diagnostic algorithm for cystic fibrosis. Thorax 64(8):683–691
Grangeia A, Niel F, Carvalho F, et al (2004) Characterization of cystic fibrosis conductance transmembrane regulator gene mutations and IVS8 poly(T) variants in Portuguese patients with congenital absence of the vas deferens. Hum Reprod 19:2502–2508
Hall S, Oates RD (1993) Unilateral absence of the scrotal vas deferens associated with contralateral mesonephric duct anomalies resulting in infertility: laboratory, physical and radiographic findings, and therapeutic alternatives. J Urol 150(4):1161–1164
Hirsh AV, Mills C, Bekir J, Dean N, Yovich JL, Tan SL (1994) Factors influencing the outcome of in-vitro fertilization with epididymal spermatozoa in irreversible obstructive azoospermia. Hum Reprod 9(9):1710–1716
Holsclaw DS, Perlmutter AD, Jockin H, Shwachman H (1971) Genital abnormalities in male patients with cystic fibrosis. J Urol 106(4):568
Jarvi K, McCallum S, Zielenski J, Durie P, Tullis E, Wilchanski M, Margolis M, Asch M, Ginzburg B, Martin S, Buckspan MB (1998) Heterogeneity of reproductive tract abnormalities in men with absence of the vas deferens: role of cystic fibrosis transmembrane conductance regulator gene mutations. Fertil Steril 70(4):724–728
Kanavakis E, Tzetis M, Antoniadi T, Pistofidis G, Milligos S, Kat- tamis C (1998) Cystic fibrosis mutation screening in CBAVD patients and men with obstructive azoospermia or severe oligozoospermia. Mol Hum Reprod 4:333–337.
Kolettis PN, Sandlow JI (2002) Clinical and genetic features of patients with congenital unilateral absence of the vas deferens. Urology 60(6):1073–1076
Larriba S, Bassas L, Gimenez J, Ramos MD, Segura A, Nunes V, Estivill X, Casals T (1998) Testicular CFTR splice variants in patients with congenital absence of the vas deferens. Hum Mol Genet 7(11):1739–1744
Lissens W, Mahmoud KZ, El-Gindi E, et al. (1999) Molecular analysis of the cystic fibrosis gene reveals a high frequency of the intron 8 splice variant 5T in Egyptian males with congenital bilateral absence of the vas deferens. Mol Hum Reprod 5: 10–13
Liu J, Lissens W, Silber SJ et al. (1994) Birth after preimplantation diagnosis of the cystic fibrosis AF508 mutation by polymerase chain reaction in human embryos resulting from intracytoplasmic sperm injection with epididymal sperm. J. Am. Med. Assoc 272:1858–1860
Mak V, Jarvi KA (1996) The genetics of male infertility. J Urol 156(4):1245–1257
Mansour RT, Kamal A, Fahmy I, Tawab N, Serour GI, Aboulghar MA (1997) Intracytoplasmic sperm injection in obstructive and non-obstructive azoospermia. Hum Reprod 12(9):1974–1979
Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC (1998) Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95(7):1005–1015
McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD (2000) Fertility in men with cystic fibrosis: an update on current surgical practices and outcomes. Chest 118: 1059–1062
McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD (2000a) Fertility in men with cystic fibrosis: an update on current surgical practices and outcomes. Chest J 118(4):1059–1062
McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD (2000b) Fertility in men with cystic fibrosis: an update on current surgical practices and outcomes. Chest J 118(4):1059–1062
Mei-Zahav M, Durie P, Zielenski J, Solomon M, Tullis E, Tsui LC, Corey M (2005) The prevalence and clinical characteristics of cystic fibrosis in south Asian Canadian immigrants. Arch Dis Child 90(7):675–679
Meng MV, Black LD, Cha I, Ljung BM, Pera RAR, Turek PJ (2001) Impaired spermatogenesis in men with congenital absence of the vas deferens. Hum Reprod 16(3):529–533
Meschede D, Dworniczak B, Behre HM, Kliesch S, Claustres M, Nieschlag E, Horst J (1997) CFTR gene mutations in men with bilateral ejaculatory-duct obstruction and anomalies of the seminal vesicles. Am J Hum Genet 61(5):1200
Mickle J, Milunsky A, Amos JA, Oates RD (1995) Immunology: congenital unilateral absence of the vas deferens: a heterogeneous disorder with two distinct subpopulations based upon aetiology and mutational status of the cystic fibrosis gene. Hum Reprod 10(7):1728–1735
Nicopoullos JDM, Gilling-Smith C and Ramsay JWA. 2004. Does the cause of obstructive azoospermia affect the outcome intracytoplasmic sperm injection: a meta-analysis. BJU Int 93:1282–1286
Nicopoullos J, Gilling-Smith C, Almeida P, Ramsay J (2005) The predictive value of sperm chromatin structure assay. Hum Reprod 20(3):839–839
Nielsen OH, Thomsen BL, Green A, Andersen PK, Hauge M, Schiøtz PO (1988) Cystic fibrosis in Denmark 1945 to 1985. Acta Paediatr 77(6):836–841
Oates RD, Amos JA (1993) Congenital bilateral absence of the vas deferens and cystic fibrosis. World J Urol 11(2):82–88
O'Sullivan BP, Freedman SD (2009) Cystic fibrosis. Lancet 373(9678):1891–1904
Patrizio P, Asch RH, Handelin B, Silber SJ (1993) Aetiology of congenital absence of vas deferens: genetic study of three generations. Hum Reprod 8(2):215–220
Persson J, Peters G, Saunders D (1996) Genetic consequences of ICSI. Hum Reprod 11:921–932
Phillipson GTM, Petrucco OM, Matthews CD (2000) Congenital bilateral absence of the vas deferens, cystic fibrosis mutation analysis and intracytoplasmic sperm injection. Hum Reprod 15(2):431–435
Powers CA, Potter EM, Wessel HU, Lloyd-Still JD (1996) Cystic fibrosis in Asian Indians. Arch Pediatr Adolesc Med 150(5):554–555
Quinton PM (1999) Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev 79(1):S3–S22
Quinton PM (2007) Cystic fibrosis: lessons from the sweat gland. Physiology (Bethesda) 22(3):212–225
Reddy MM, Light MJ, Quinton PM (1999) Activation of the epithelial Na+ channel (ENaC) requires CFTR Cl-channel function. Nature 402(6759):301–304
Robert F, Bey-Omar F, Rollet J, Lapray JF, Morel Y (2002) Relation between the anatomical genital phenotype and cystic fibrosis transmembrane conductance regulator gene mutations in the absence of the vas deferens. Fertil Steril 77:889–896
Rosenstein BJ, Cutting GR (1998) The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 132(4):589–595
Schlegel PN, Shin D, Goldstein M (1996) Urogenital anomalies in men with congenital absence of the vas deferens. J Urol 155(5):1644–1648
Sharma N, Acharya N, Singh SK, Singh M, Sharma U, Prasad R (2009) Heterogenous spectrum of CFTR gene mutations in Indian patients with congenital absence of vas deferens. Hum Reprod 24(5):1229–1236
Silber SJ, Asch R, Balmaceda J et al. (1988) Pregnancy with sperm aspiration from the proximal head of the epididymis: a new treatment for congenital absence of the vas deferens. Fertil Steril 50:525–528
Silber SJ, Ord T, Balmaceda J et al. (1990) Congenital absence of the vas deferens. The fertilizing capacity of human epididymal sperm. N. Engl. J. Meet 323;788–1792
Silber SJ, Nagy Z, Liu J, Tournaye H, Lissens W, Ferec C, Liebaers I, Devroey P, Van Steirteghem AC (1995) Genetics: the use of epididymal and testicular spermatozoa for intracytoplasmic sperm injection: the genetic implications for male infertility. Hum Reprod 10(8):2031–2043
Teng H, Jorissen M, Van Poppel H, Legius E, Cassiman JJ, Cuppens H (1997) Increased proportion of exon 9 alternatively spliced CFTR tran- scripts in vas deferens compared with nasal epithelial cells. Hum Mol Genet 6:85–90
Tournaye H, Liu J, Nagy PZ, Camus M, Goossens A, Silber S, Van Steirteghem AC, Devroey P (1996) Correlation between testicular histology and outcome after intracytoplasmic sperm injection using testicular spermatozoa. Hum Reprod 11(1):127–132
Trezíse, A.E. and Buchwald, M., 1991. In vivo cell-specific expression of the cystic fibrosis transmembrane conductance regulator.
Tuerlings JH, Mol B, Kremer JA, Looman M, Meuleman EJ, te Meerman GJ, Buys CH, Merkus HM, Scheffer H (1998) Mutation frequency of cystic fibrosis transmembrane regulator is not increased in oligozoospermic male candidates for intracytoplasmic sperm injection. Fertil Steril 69(5):899–903
Walters S, Mehta A (2007) Epidemiology of cystic fibrosis. In: Hodson M, Geddes DM, Bush A (eds) Cystic fibrosis. Edward Arnold, London, pp 21–45
Weiske WH, Sälzler N, Schroeder-Printzen I, Weidner W (2000) Clinical findings in congenital absence of the vasa deferentia. Andrologia 32(1):13–18
Welsh MJ, Fick RB (1987) Cystic fibrosis. J Clin Investig 80(6):1523–1526
Welsh MJ, Smith AE (1993) Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73(7):1251–1254
Welsh MJ, Ramsey BW, Accurso F, Cutting JI (2001) Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular basis of inherited diseases, 8th edn. McGraw-Hill, New York, pp 5121–5188
WHO 2004 and Cystic Fibrosis Foundation Patient Registry 2012 Annual Data Report.
Wu CC, Hsieh-Li HM, Lin YM, Chiang HS (2004). Cystic fibrosis trans- membrane conductance regulator gene screening and clinical correla- tion in Taiwanese males with congenital bilateral absence of the vas deferens. Hum Reprod 19:250–253
Yamashiro Y, Shimizu T, Oguchi S, Shioya T, Nagata S, Ohtsuka Y (1997) The estimated incidence of cystic fibrosis in Japan. J Pediatr Gastroenterol Nutr 24(5):544–547
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Gajbhiye, R., Gaikwad, A. (2017). Cystic Fibrosis, CFTR Gene, and Male Infertility. In: SINGH, R., Singh, K. (eds) Male Infertility: Understanding, Causes and Treatment. Springer, Singapore. https://doi.org/10.1007/978-981-10-4017-7_9
Download citation
DOI: https://doi.org/10.1007/978-981-10-4017-7_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4016-0
Online ISBN: 978-981-10-4017-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)