
Abstract
The administration of vasopressors to trauma patients in hemorrhagic shock is challenged primarily on the conceptual basis of “permissive hypotension” or “hypotensive resuscitation.” The phrase “pop the clot” is also frequently referenced to discourage targeting a physiologically appropriate blood pressure to minimize any disruption of hemostasis that has occurred following hemorrhage. It is crucial to recognize that much of the data to support such concepts are the result of heterogeneous animal studies. In addition, limited retrospective data in humans suggest that early vasopressor administration to trauma patients is associated with increased mortality. From a mechanistic perspective, it is theorized that vasopressor administration in bleeding patients will result in increased vasoconstriction and compromise end-organ function. However, several important concepts should be discussed regarding utilization of vasopressors in patients with traumatic hemorrhagic shock. First, there is growing evidence showing that vasopressor use is not associated with increased mortality after severe trauma. Recent randomized controlled trials have even indicated beneficial effects of vasopressin use after hemorrhagic shock. Potential benefits of vasopressors may be restoration of adequate perfusion blood pressure for vital organs and limitation of aggressive volume administration. Second, trauma is a complex and dynamic pathology that combines several causes of hypotension, including anesthesia-related, injury-related (hypovolemia and/or spinal cord injury), and trauma-induced vasoplegia. In conclusion, vasopressors should not be restricted from the trauma bay. Their prescription should be tailored to the clinical context and to the different time points of severe trauma management.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Management of shock after trauma remains a clinical challenge, in particular if associated with active hemorrhage [1]. Prevailing dogma precludes the use of vasopressors in shock after trauma until hemorrhage is excluded or controlled and advocates a hypotensive strategy [2]. The use of vasopressors is considered deleterious and associated with a risk of increased bleeding and organ damage due to excessive vasoconstriction. Despite the controversy and discouraged use in hemorrhagic shock, particularly in trauma centers in the United States and the United Kingdom [3], vasopressors are part of the recommended therapeutic arsenal and routinely used by clinicians in Europe to manage trauma patients in shock [4].
Mounting evidence suggests that the effect of vasopressors in shock and hemorrhage after trauma justifies a more nuanced position. A differentiated approach appears indicated, because not all cardio- and vasoactive agents are the same when it comes to their inotropic and vasoconstrictive capacities. Among all agents, norepinephrine and vasopressin have emerged as the molecules of choice if any cardio/vasoactive effect is to be achieved. In this chapter, we attempt to provide a balanced perspective on the use of norepinephrine and vasopressin in traumatic shock and hemorrhage, based on recent physiological, epidemiological, and clinical data.
2 Pharmacology
2.1 Cardiovascular Effects of Norepinephrine
Norepinephrine is a neurohormone, released from sympathetic, postganglionic nerve fibers. Norepinephrine is a product of the decarboxylation of dopamine, stored in presynaptic granules that release their content into the synaptic space upon depolarization. In the adrenal gland, a methylene group is added, modifying norepinephrine to epinephrine.
After release, norepinephrine acts on postsynaptic alpha- and, to a lesser extent, beta-receptors [5, 6]. The effects on both receptors are dose dependent and with increasing doses the alpha effect dominates. The intracellular signal transmission is G-protein coupled and activates a cAMP-kinase cycle. This results in (1) contraction of smooth muscle fibers in arterial and venous vessels inducing vasoconstriction and (2) myocardial inotropic and chronotropic stimulation [5, 6].
2.2 Cardiovascular Effects of Vasopressin
The physiology of vasopressin is complex and beyond the scope of this review; readers are referred to the excellent work by Holmes et al. [7, 8]. Vasopressin is a neuroendocrine nonapeptide, produced in the neurons of the paraventricular and supraoptic nuclei in the posterior hypothalamus. Vasopressin acts on multiple G-protein-coupled receptors and uses the phosphatidylinositol pathway [7] to increase Ca2+ influx. Vasopressin 1R (V1R) receptors are densely situated on vascular smooth muscles of the systemic, splanchnic, renal, and coronary circulations; their stimulation leads to potent vasoconstriction [7] and concomitant increase in cardiac output and centralization of blood volume [9]. They are also found on cardiac myocytes and in many other organs, such as liver, brain, and renal medulla. In renal efferent arterioles, this vasoconstriction increases glomerular filtration rate (GFR). In the pulmonary vasculature, vasopressin induces less vasoconstriction than norepinephrine. V1R receptor stimulation on platelets facilitates their aggregation. V2R receptors located in the renal collecting system induce antidiuresis by shuttling aquaporin-2-containing vesicles to the cell surface and stimulation of synthesis of aquaporin-2 mRNA. There is also a complex physiologic interaction of vasopressin on oxytocin and purinergic receptors. Purinergic receptors on cardiac endothelium seem to exert positive inotropic stimulation without concomitant positive chronotropy and increase in oxygen demand [10].
2.3 Metabolic and Immunomodulatory Effects of Norepinephrine and Vasopressin
Apart from the hemodynamic manifestations , norepinephrine and vasopressin exert a number of endocrine, metabolic, and immunomodulatory effects. For example, norepinephrine alters the function of most immune cells, reducing the activity of macrophage, T-helper, and natural killer (NK) cells, and up- or downregulates certain cytokines (e.g., interleukin [IL]-6 and -10, tumor necrosis factor [TNF]-α) [11]. A wealth of research indicates that vasopressin appears to have a beneficial effect on the immune system, such as reduction of mRNA of TNF-α, nuclear factor-kappa B (NF-κB), and IL-1β [12]. Norepinephrine increases glycogenolysis and glucose production and modifies lipid metabolism [13]. Via V3R receptors in the pituitary, vasopressin seems to increase adrenocorticotropic secretion and ultimately influences cortisol secretion. The effects of norepinephrine and vasopressin are part of the highly complex immunologic, endocrine, and metabolic response to trauma, shock, and hemorrhage triggering a systemic inflammatory (SIRS) and compensatory anti-inflammatory (CARS) response described as persistent inflammatory and catabolic syndrome (PICS) [14].
3 The Physiologic Response to Traumatic Shock and Hemorrhage
The complex physiology of the response to hemorrhage and shock cannot be reduced to a simple loss of blood volume. An intricate and coordinated series of adaptive mechanisms and interactions has emerged in our understanding of the pathophysiology of hemorrhagic shock [15]. On a macrovascular level, the organism responds to shock and hemorrhage with an intense sympathetic stimulation propelled by the peripheral and central nervous systems [16]. This neurohormonal response induces intense vasoconstriction, an increase in heart rate, respiratory drive and venous return, improved coronary perfusion, and cardiac contractility [16, 17]. Combined, these augment, or at least maintain, stroke volume, cardiac output, arterial pressure, and oxygen delivery. Norepinephrine and vasopressin play crucial roles in this response at the peripheral and central levels.
If the initial source of hemorrhage is not quickly controlled, this phase of compensated hypovolemic shock may evolve to a state of vasodilatory shock . Ultimately all shock forms are considered to decompensate to a form of distributive shock [18, 19]. This vasodilatory phase is caused by numerous mechanisms such as xanthine oxidase [20], prostanoids (PG-1, thromboxane), reactive oxygen species (ROS), hydrogen sulfide, and, probably the most important, nitric oxide (NO) and potassium channels [21]. NO is increased by augmented activation of inducible NO synthase (iNOS) acting via cyclic guanosine monophosphate (cGMP) to reduce intracellular calcium and activate calcium-sensitive (Kca) and ATP-dependent K-channels. The subsequent hyperpolarization and decreased influx of calcium [8, 22] demonstrate that the synergy between K-channels and reduced Ca2+ influx further exacerbates the hyporesponsiveness to catecholamines and results in profound hypotension. Furthermore, adrenoceptors are desensitized and downregulated [23, 24]. The vasopressin response is also subject to desensitization, receptor internalization , and store depletion [7, 19]. In the decompensation phase, even blood transfusion cannot restore normal intravascular pressures [25].
To understand the process at the microvascular level , it is helpful to consider the endothelium as a whole organ system, with a weight of 1 kg and a surface of 5000 m2 [26]; the role of the endothelium in health and disease has probably been underestimated and it is the target of a number of pathophysiologic alterations. At the microvascular level, the onset of hemorrhagic shock reflects a myriad of physical, chemical, cellular, and genomic interactions [15]. Some interactions are triggered by the macrovascular response and many prompt the measurable and clinically apparent macrovascular and hemodynamic response. In addition to the above-described neurohormonal response, vascular injuries expose the endothelial surface and cells to lower oxygen concentration and acidosis. Profound rheological changes accompany a modified vascular reactivity depending on the affected regional perfusion. A variety of cytokines and messengers alter endothelial surface reactivity and permeability, including leukocyte and platelet adhesion and activation. Leukocyte activation triggers synthesis of ROS. Activated protein-C initiates coagulopathy that is induced by cleavage of plasminogen activator inhibitor and increases tissue plasminogen activator. Thrombomodulin is released and platelets become dysfunctional. Numerous cytokines circulate and initiate transcription of adhesion molecules and other pro- and anti-inflammatory mediators, modifying further vascular permeability and vessel reactivity and reducing functional capillary density; the endothelial surface swells, increasing O2 diffusion distance [15].
A body of experimental and clinical evidence points towards the concept of shock-induced endotheliopathy [27] as the central element to the above-described process. The endothelium is covered with a layer of glycosylated proteins (proteoglycans), called the glycocalyx. The interaction between this layer and albumin is now considered as a surrogate for oncotic pressure. In shock, hypoxia, hypotension, acidosis, cytokines , and neurohormones (epinephrine) contribute to the destruction of the glycocalyx, leading to increased vessel permeability and endothelial and end-organ dysfunction. Glycocalyx degradation at the onset of shock/hemorrhage can be quantified by syndecan-1 levels [28, 29]. In trauma patients, syndecan-1 plasma concentration is correlated to the level of injury, mortality, and epinephrine concentration [30].
4 Vasopressors in Shock and Hemorrhage in Trauma: Experimental Evidence
Considering experimental and animal data about the effects of norepinephrine and vasopressin in the physiopathology of hemorrhagic shock it seems important to acknowledge two aspects. First, the mechanisms in septic shock are far better investigated and understood and some observations about norepinephrine and vasopressin have been extrapolated from studies on septic shock to hemorrhagic shock [21]. Second, it is crucial to consider the type of animal model used and which aspect of the shock response is studied [31].
It is usually considered that vasopressors increase afterload and oxygen consumption and decrease organ perfusion [32]. It is noteworthy that some of these experimental studies used vasopressors without concomitant fluid expansion. When applied to achieve a systolic blood pressure of 80–90 mmHg, a critical rise in afterload cannot be documented with norepinephrine administration in patients with vasoplegic shock [5]. In fact, both norepinephrine and vasopressin seem to improve coronary perfusion pressure, venous return, and cardiac output and subsequently organ perfusion in shock [8]. Norepinephrine and vasopressin are part of the physiological response to hemorrhage and the mechanisms responsible for the vasodilatory phase of prolonged shock when physiological compensation is exhausted (see above) may require exogenous vasopressor administration as neurohormonal augmentation therapy [33].
Another important argument against norepinephrine and vasopressin use in hemorrhagic shock is the danger of intensifying bleeding from noncontrolled sources by increasing hydrostatic pressure (“pop the clot”), the rationale behind permissive hypotension. However, recent evidence indicates that even with fluid-only resuscitation, profound and prolonged hypotension is associated with increased end-organ damage. In a model of controlled hemorrhage and shock of 30% blood loss after blast injury, pigs were randomized to two systolic blood pressure levels, 110 mmHg and 80 mmHg, resuscitated with fluids only [34]. Survival was shorter in the 80 mmHg group associated with important metabolic derangement. This finding was confirmed in another pig model of uncontrolled hemorrhagic shock comparing three mean arterial pressure (MAP) levels of 60, 80, and 100 mmHg obtained with fluid resuscitation alone [35]. Both the 60 and 100 mmHg groups demonstrated more organ dysfunction and histopathological damage.
Several studies have explored the effect of norepinephrine or vasopressin use in animal shock models. In a model of hemorrhagic shock in rodents, the use of norepinephrine with fluid expansion was associated with higher survival [36]. In a rat model of shock, Poloujadoff et al. demonstrated that MAP-targeted resuscitation associating norepinephrine and fluids proved beneficial in terms of survival [37]. Liu et al. successfully tested a combination of norepinephrine and vasopressin and fluids in rats to maintain perfusion pressure until definitive hemorrhage control [38]. These studies have in common that the investigating groups used both vasopressor therapy and fluid expansion. Furthermore, administration of norepinephrine or vasopressin does not necessarily induce end-organ damage in shock models; the effects are organ-, dose-, and time-dependent [7, 34, 35]. In fact, Harrois et al. demonstrated protection of intestinal villi in hemorrhage in mice [39]. Dunberry-Poissant et al. revealed no difference comparing various parameters of organ damage after fluid resuscitation versus fluid and norepinephrine in a controlled rat shock model during resuscitation to MAP levels of 55–60 mmHg and after reperfusion [20]. The administration of norepinephrine in this model was associated with a considerably reduced volume of fluid administration.
Of note, different resuscitation strategies (fluid only versus norepinephrine/vasopressin plus fluid) and varying pressure levels at different times affect organs differently [7, 20, 34, 35]. For example, a MAP of 100 mmHg caused more organ damage in the lungs than in the liver and kidney compared to a pressure level of 60 mmHg [7, 20, 34, 35].
The topic becomes more complicated with regard to vasopressor effects on the endothelial surface and glycocalyx. There seems to be an undeniable association between circulating endogenous epinephrine, endothelial damage, and mortality in animals and patients [27]. Sympathectomy appears to protect against this phenomenon in an animal model [40]. In fact, recently, in a controlled hemorrhagic shock model in rats, there was raised capillary permeability in the lung and higher lactate and base excess in the group resuscitated with fluids and vasopressin compared to groups resuscitated with fluids only and those resuscitated with fluids and blood.
In summary, this wealth of experimental knowledge and observations can be interpreted in favor of or against the use of norepinephrine (Table 32.1) and vasopressin (Table 32.2) in shock and hemorrhage after trauma. The complexity of the adaptive physiological and pathophysiologic patterns demonstrates that the potential beneficial or detrimental effects of both agents are not reduced to macro-hemodynamic effects and are part of the physiological response to injury.
5 Clinical Evidence of Permissive Hypotension and Vasopressor Use
One of the hallmark pathophysiologic signs of advanced hemorrhagic shock is hypotension. Hypotension is a time-sensitive and dose-dependent event such that the following questions remain: “How much hypotension is acceptable in a trauma patient?” “And for how long?” Unfortunately, the answers to these questions remain elusive. Despite traumatic mechanisms of injury typically occurring in younger patients with few medical comorbidities, hypotension is significantly associated with increased mortality. In addition, there is a growing population of older patients with comorbidities, such as hypertension and peripheral vascular disease, who are sustaining traumatic injuries. Does all traumatic hemorrhagic shock require the same approach to hemodynamic resuscitation? And do the same hypotensive thresholds apply across all trauma patients? The seemingly obvious answer would be no. However, while few data are available on the duration of hypotension in critically injured trauma patients, there is substantial evidence in the perioperative and critical care literature to support that longer periods, and even single episodes, of hypotension are associated with organ dysfunction and increased mortality [41]. Yet, as previously discussed, there is a pervasive opinion especially among certain trauma providers that hypotension is potentially beneficial in trauma patients and should be acceptable, especially early in the resuscitation phase following hemorrhage.
The topic of hypotensive resuscitation and permissive hypotension in resuscitation from traumatic hemorrhagic shock has been formulated over the last three decades. While early resuscitation strategies emphasized volume replacement with isotonic crystalloid formulations , Bickell et al. [42] demonstrated in a swine model of vascular injury that initial large volumes of crystalloid administration led to decreased animal survival. In a subsequent prospective, randomized trial of immediate versus delayed fluid resuscitation in hypotensive patients who sustained penetrating traumatic injuries, those who received less prehospital crystalloid (mean volume of 92 ml compared to 870 ml) had greater rates of survival [43]. However, the difference in systolic blood pressure upon arrival to the trauma center was clinically negligible (72 mmHg vs. 79 mmHg, respectively), albeit statistically significant. In recent years, attempts to replicate such findings and support tolerating a lower blood pressure during initial trauma resuscitation have failed to demonstrate an improvement in mortality [44]. Unfortunately, the enthusiasm and support for hypotensive resuscitation, and admonishment for the use of vasopressors, are extrapolated primarily from a clinical study in which a blood pressure target was not the primary outcome nor was the systolic blood pressure clinically different among treatment groups. Therefore, it should be clarified that minimizing excessive volumes of crystalloid, or overall excessive volume in general, does not equate to accepting a lower blood pressure in a bleeding patient.
Blood volume replacement remains the initial primary method of resuscitation from hemorrhagic shock. Recent publications in blood component administration and reinvigoration of whole blood therapy have advanced the science of hemorrhagic shock resuscitation towards a cell-based approach that minimizes excess crystalloid administration and emphasizes restoration of the endothelial glycocalyx [27]. As previously discussed, disruption and damage to the endothelium lining the inner vasculature lumen result in release of anticoagulant molecules that contribute to the trauma-induced coagulopathy and exacerbate acute blood loss. In addition, endothelial dysfunction is characterized by a release of iNO with subsequent vascular hyporesponsiveness, vasoplegia, and loss of vascular tone [21]. Patients with severe and profound hemorrhagic shock may continue to demonstrate hypotension despite replacement of adequate blood volume. In a cyclical fashion, this very hypotension and continued hypoperfusion lead to further endothelial dysfunction and further hypotension unresponsive to volume resuscitation. It would therefore seem logical that administration of vasopressor therapy and restoration of vascular tone would serve a valuable function in such patients.
While there is no level 1 clinical evidence to suggest that early administration of vasopressors or vasopressor use in the initial resuscitation of traumatic hemorrhagic shock results in worse outcomes, evidence from some retrospective studies is conflicting. Some studies suggest that vasopressors are associated with increased in-hospital mortality. Sperry et al. reported that early vasopressor use (i.e., phenylephrine, norepinephrine, or vasopressin), within 12 h of injury, was associated with mortality even after adjusting for volume of crystalloid resuscitation [45]. Of note, patients who survived <48 h were excluded, when in fact it is this very population of patients with early mortality as a result of hemorrhage that are of significant interest with regard to vasopressor therapy (i.e., patients who failed to respond to initial resuscitation efforts as a result of profound shock with subsequent vasoplegia may have been salvageable with vasopressor therapy). Dose and duration of vasopressor use were also not reported and the study excluded patients with traumatic brain injury (TBI) or a spinal cord injury, which represents a population that may also be well served by augmented systemic blood pressure and vasopressor therapy. More recent work by Aoki et al. from the Japan Trauma Databank demonstrated that in a propensity-matched cohort, vasopressor use within 24 h of hospital admission was associated with in-hospital mortality [46]. However, there was no difference in emergency department mortality among patients who received early vasopressor treatment. A limitation of this large investigation was that the type of vasopressor, dose, and duration of treatment were unavailable from the trauma databank.
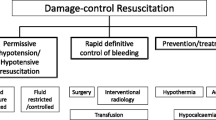
In contrast, there is emerging evidence to suggest that specific vasopressors (i.e., norepinephrine or vasopressin) are not associated with increased mortality in hemorrhagic shock. Gauss et al. performed a retrospective analysis of patients in hemorrhagic shock (defined as patients receiving 4 units of red blood cells [RBCs] within the first 6 h of hospital admission) and after propensity score matching observed that early norepinephrine administration was not associated with in-hospital mortality [47]. A recently published prospective, randomized trial from a single center in the United States has also provided valuable evidence in support of early vasopressor treatment in patients with hemorrhagic shock. The Arginine Vasopressin During the Early Resuscitation of Traumatic Shock (AVERT Shock) [48] trial randomized patients receiving at least 6 units of blood products (packed RBCs, fresh frozen plasma [FFP], and platelets) within 12 h of injury to an intervention that consisted of a bolus of 4 units of vasopressin and then continuous infusion up to 0.04 units/min to achieve a MAP of 65 mmHg versus standard component-based resuscitation . Efforts to achieve hemostatic resuscitation were performed in each study arm and the intervention was continued for 48 h. Patients in the vasopressin group required less total volume of blood products and crystalloid with no overall difference in complication rates, such as acute kidney injury (AKI), acute respiratory distress syndrome (ARDS), prolonged mechanical ventilation, or mortality. These results provide some of the most encouraging clinical evidences that specific vasopressor therapy may potentially be considered as part of the initial resuscitation of trauma patients in hemorrhagic shock (Fig. 32.1).

Suggested practical algorithm for vasopressor use after severe trauma. SAP systolic arterial blood pressure, TBI traumatic brain injury, SCI spinal cord injury
Possible explanations for the utility of vasopressors in hemorrhagic shock resuscitation are the restoration of an adequate perfusing blood pressure in order to maintain vital organ function . Our current understanding of aggressive volume administration, even in the form of blood products, is known to be associated with increased complications such as dilutional coagulopathy, lung injury, and cardiac overload. Therefore, it may be hypothesized that early initiation of vasopressors, in conjunction with appropriate volume administration and correction of coagulation derangements, will reduce overall total resuscitation volumes (both blood products and crystalloid) and minimize post-resuscitation complications. Intrinsic pharmacologic properties of specific vasopressors may target pathophysiologic processes involved in hemorrhagic shock. For example, as previously described, norepinephrine exerts alpha-1 sympathomimetic activity that would be of benefit to a severely injured trauma patient in profound hemorrhagic shock with vasoplegia [37]. Intrinsic beta-1 activity would also augment cardiac contractility and improve oxygen delivery to hypoperfused organs [5, 6]. It is also well described that severely injured and critically ill trauma patients present with endocrine insufficiency, to which vasopressin deficiency may significantly contribute [48]. Administration of exogenous vasopressin may therefore restore components of endothelial integrity, intravascular volume, and platelet function. However, it cannot be overstated that inappropriate use of vasopressors in hemorrhagic shock can result in severe deleterious consequences.
6 A Practical Approach to Vasopressor Use for an Updated Resuscitation Strategy
From the prehospital environment to the intensive care unit (ICU), hemorrhaging trauma patients progress through different phases of shock, defined by the complex physiologic response and the therapeutic strategy. Prohibiting vasopressor use within the first 24 h in patients with shock and hemorrhage may considerably limit the therapeutic arsenal to adapt and shift the response as needed. As exposed in the preceding sections, the available experimental and clinical evidence is insufficient to preclude in principle the use of norepinephrine or vasopressin in the initial 24-h management.
Based on these data, it seems obvious that vasopressor use can only be considered after a fluid challenge, probably between 500 and 1000 ml, fails to achieve hemodynamic stabilization, as recommended. If fluid fails, it is likely that the patient is decompensating into the distributive phase of shock and fluid and blood alone will not be able to prevent further clinical deterioration; vasopressors may be required to prevent prolonged hypotension, associated with an increase in organ damage. Many mature trauma systems manage to increase survival of very critically sick and severely bleeding patients, but even the most efficient systems struggle to obtain hemorrhage control within 1 h [1] and transport times may exceed 60 min. Yet, it seems that the longer the hypotensive phase lasts, the more likely the patient’s risks of organ damage [34]. As much as it is obvious that permissive hypotension should remain a central element of the damage control strategy to limit blood loss, in particular in penetrating trauma, there is an increasingly strong rationale to maintain adequate perfusion pressure even before hemorrhage control is achieved. If the duration of hypotension is too long (>60) or too profound (systolic arterial pressure <60 mmHg), hemodynamic resuscitation may be difficult to control with fluids and blood products only. This constellation is frequently observed in the distributive, decompensated phase of shock and more so in blunt trauma and often associated with a sympatholytic component, such as the required use of induction agents, sedation/anesthesia, brain, or medullary injury. Shock in trauma patients cannot be reduced to bleeding, with bleeding being the main rationale behind the strategy of permissive hypotension. Yet all forms of shock require the restoration of adequate perfusion.
It is true that the definition of adequate perfusion remains a challenge. However, an increase in global trauma mortality has been described for arterial systolic blood pressure lower than 110 mmHg after severe trauma [49]. Recent guidelines from the Trauma Hemostasis and Oxygenation Research Network also highlight this point, raising the systolic arterial blood pressure target from 80–90 to 100 mmHg [49]. These pressure levels cannot be obtained with fluid expansion and blood products only in patients with profound and prolonged traumatic shock. Large volumes of fluid resuscitation are in fact quite harmful. The use of vasopressors becomes mandatory to reach these hemodynamic goals. Higher targets seem crucial, particularly in patients with associate traumatic brain or medullary injury and multisystem trauma [50], to control cerebral and medullary perfusion pressure.
For these reasons, the authors share the assumption that norepinephrine and vasopressin have a place in the therapeutic arsenal to treat trauma patients in shock, including those with active hemorrhage (Fig. 32.1). Both agents should be part of a bridging strategy to maintain tissue perfusion if hypotension is too long or too profound. In no case however should this strategy become a substitute for expedient hemorrhage control. Furthermore, the pressure levels targeted with norepinephrine/vasopressin use require a reasonable trade-off between tissue perfusion and overcorrection , which may increase bleeding by increasing the hydrostatic pressure.
7 Conclusion
Traumatic hemorrhagic shock is a complex disease process that incorporates a dynamic physiologic response to blood loss and tissue injury. While restoration of circulating blood volume is the mainstay of initial treatment of hemorrhagic shock, maintenance of adequate perfusion pressure is essential in order to minimize organ dysfunction. While historically it has been advocated to treat hemorrhagic shock with aggressive blood product transfusion and the use of vasopressors is discouraged, severe injury with profound shock and prolonged hypotension will decompensate to a distributive form of vasoplegia that is unresponsive to further volume administration. Therefore, it appears intuitive that appropriate and targeted use of specific vasopressors has a beneficial contribution to the management of early hemorrhagic shock. We advocate that specific, targeted vasopressor therapy has an integral and necessary role in the early resuscitation of traumatic hemorrhagic shock and that further large-scale scientific and clinical research will more clearly define vasopressor administration in patients with hemorrhagic shock.
References
Alarhayem AQ, Myers JG, Dent D, et al. Time is the enemy: mortality in trauma patients with hemorrhage from torso injury occurs long before the “golden hour”. Am J Surg. 2016;212:1101–5.
Nevin DG, Brohi K. Permissive hypotension for active haemorrhage in trauma. Anaesthesia. 2017;72:1443–8.
Gupta B, Garg N, Ramachandran R. Vasopressors: do they have any role in hemorrhagic shock? J Anaesthesiol Clin Pharmacol. 2017;33:3–8.
Rossaint R, Bouillon B, Cerny V, et al. The European guideline on management of major bleeding and coagulopathy following trauma: fourth edition. Crit Care. 2016;20:100.
Hamzaoui O, Jozwiak M, Geffriaud T, et al. Norepinephrine exerts an inotropic effect during the early phase of human septic shock. Br J Anaesth. 2018;120:517–24.
De Backer D, Pinsky M. Norepinephrine improves cardiac function during septic shock, but why? Br J Anaesth. 2018;120:421–4.
Holmes CL, Landry DW, Granton JT. Science review: vasopressin and the cardiovascular system part 1 – receptor physiology. Crit Care. 2003;7:427–34.
Holmes CL, Landry DW, Granton JT. Science review: vasopressin and the cardiovascular system part 2 - clinical physiology. Crit Care. 2004;8:15–23.
Bown LS, Ricksten SE, Houltz E, et al. Vasopressin-induced changes in splanchnic blood flow and hepatic and portal venous pressures in liver resection. Acta Anaesthesiol Scand. 2016;60:607–15.
Mei Q, Liang BT. P2 purinergic receptor activation enhances cardiac contractility in isolated rat and mouse hearts. Am J Physiol Heart Circ Physiol. 2001;281:H334–41.
Stolk RF, van der Poll T, Angus DC, et al. Potentially inadvertent immunomodulation: norepinephrine use in sepsis. Am J Respir Crit Care Med. 2016;194:550–8.
Russell JA, Walley KR. Vasopressin and its immune effects in septic shock. J Innate Immun. 2010;2:446–60.
Ensinger H, Geisser W, Brinkmann A, et al. Metabolic effects of norepinephrine and dobutamine in healthy volunteers. Shock. 2002;18:495–500.
Lord JM, Midwinter MJ, Chen YF, et al. The systemic immune response to trauma: an overview of pathophysiology and treatment. Lancet. 2014;384:1455–65.
Torres Filho IP, Torres LN, Salgado C, Dubick MA. Novel adjunct drugs reverse endothelial glycocalyx damage after hemorrhagic shock in rats. Shock. 2017;48:583–9.
Schadt JC, Ludbrook J. Hemodynamic and neurohumoral responses to acute hypovolemia in conscious mammals. Am J Phys. 1991;260(2 Pt 2):H305–18.
Hamzaoui O, Georger JF, Monnet X, et al. Early administration of norepinephrine increases cardiac preload and cardiac output in septic patients with life-threatening hypotension. Crit Care. 2010;14:R142.
Barrett LK, Singer M, Clapp LH. Vasopressin: mechanisms of action on the vasculature in health and in septic shock. Crit Care Med. 2007;35:33–40.
Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med. 2001;345:588–95.
Dunberry-Poissant S, Gilbert K, Bouchard C, et al. Fluid sparing and norepinephrine use in a rat model of resuscitated haemorrhagic shock: end-organ impact. Intensive Care Med Exp. 2018;6:47.
Lambden S, Creagh-Brown BC, Hunt J, Summers C, Forni LG. Definitions and pathophysiology of vasoplegic shock. Crit Care. 2018;22:174.
Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–232.
Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–50.
Saito T, Takanashi M, Gallagher E, et al. Corticosteroid effect on early beta-adrenergic down-regulation during circulatory shock: hemodynamic study and beta-adrenergic receptor assay. Intensive Care Med. 1995;21:204–10.
Dalibon N, Schlumberger S, Saada M, Fischler M, Riou B. Haemodynamic assessment of hypovolaemia under general anaesthesia in pigs submitted to graded haemorrhage and retransfusion. Br J Anaesth. 1999;82:97–103.
Aird WC. Endothelium in health and disease. Pharmacol Rep. 2008;60:139–43.
Johansson PI, Stensballe J, Ostrowski SR. Shock induced endotheliopathy (SHINE) in acute critical illness - a unifying pathophysiologic mechanism. Crit Care. 2017;21:25.
Haywood-Watson RJ, Holcomb JB, Gonzalez EA, et al. Modulation of syndecan-1 shedding after hemorrhagic shock and resuscitation. PLoS One. 2011;e23530:6.
Ostrowski SR, Henriksen HH, Stensballe J, et al. Sympathoadrenal activation and endotheliopathy are drivers of hypocoagulability and hyperfibrinolysis in trauma: a prospective observational study of 404 severely injured patients. J Trauma Acute Care Surg. 2017;82:293–301.
Johansson PI, Henriksen HH, Stensballe J, et al. Traumatic endotheliopathy: a prospective observational study of 424 severely injured patients. Ann Surg. 2017;265:597–603.
Tremoleda JL, Watts SA, Reynolds PS, Thiemermann C, Brohi K. Modeling acute traumatic hemorrhagic shock injury: challenges and guidelines for preclinical studies. Shock. 2017;48:610–23.
Beloncle F, Meziani F, Lerolle N, Radermacher P, Asfar P. Does vasopressor therapy have an indication in hemorrhagic shock? Ann Intensive Care. 2013;3:13.
Myburgh J. Norepinephrine: more of a neurohormone than a vasopressor. Crit Care. 2010;14:196.
Garner J, Watts S, Parry C, Bird J, Cooper G, Kirkman E. Prolonged permissive hypotensive resuscitation is associated with poor outcome in primary blast injury with controlled hemorrhage. Ann Surg. 2010;251:1131–9.
Bai X, Yu W, Ji W, Duan K, Tan S, Lin Z, et al. Resuscitation strategies with different arterial pressure targets after surgical management of traumatic shock. Crit Care. 2015;19:170.
Lee JH, Kim K, Jo YH, Kim KS, et al. Early norepinephrine infusion delays cardiac arrest after hemorrhagic shock in rats. J Emerg Med. 2009;37:376–82.
Poloujadoff MP, Borron SW, Amathieu R, et al. Improved survival after resuscitation with norepinephrine in a murine model of uncontrolled hemorrhagic shock. Anesthesiology. 2007;107:591–6.
Liu L, Tian K, Xue M, et al. Small doses of arginine vasopressin in combination with norepinephrine “buy” time for definitive treatment for uncontrolled hemorrhagic shock in rats. Shock. 2013;40:398–406.
Harrois A, Baudry N, Huet O, et al. Norepinephrine decreases fluid requirements and blood loss while preserving intestinal villi microcirculation during fluid resuscitation of uncontrolled hemorrhagic shock in mice. Anesthesiology. 2015;122:1093–102.
Xu L, Yu WK, Lin ZL, et al. Chemical sympathectomy attenuates inflammation, glycocalyx shedding and coagulation disorders in rats with acute traumatic coagulopathy. Blood Coagul Fibrinolysis. 2015;26:152–60.
Wesselink EM, Kappen TH, Torn HM, Slooter AJC, van Klei WA. Intraoperative hypotension and the risk of postoperative adverse outcomes: a systematic review. Br J Anaesth. 2018;121:706–21.
Bickell WH, Bruttig SP, Millnamow GA, O’Benar J, Wade CE. The detrimental effects of intravenous crystalloid after aortotomy in swine. Surgery. 1991;110:529–36.
Bickell WH, Wall MJ Jr, Pepe PE, et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. N Engl J Med. 1994;331:1105–9.
Carrick MM, Morrison CA, Tapia NM, et al. Intraoperative hypotensive resuscitation for patients undergoing laparotomy or thoracotomy for trauma: early termination of a randomized prospective clinical trial. J Trauma Acute Care Surg. 2016;80:886–96.
Sperry JL, Minei JP, Frankel HL, et al. Early use of vasopressors after injury: caution before constriction. J Trauma. 2008;64:9–14.
Aoki M, Abe T, Saitoh D, Hagiwara S, Oshima K. Use of vasopressor increases the risk of mortality in traumatic hemorrhagic shock: a nationwide cohort study in Japan. Crit Care Med. 2018;46:e1145–e51.
Gauss T, Gayat E, Harrois A, et al. Effect of early use of noradrenaline on in-hospital mortality in haemorrhagic shock after major trauma: a propensity-score analysis. Br J Anaesth. 2018;120:1237–44.
Sims CA, Holena D, Kim P, et al. Effect of low-dose supplementation of arginine vasopressin on need for blood product transfusions in patients with trauma and hemorrhagic shock: a randomized clinical trial. JAMA Surg. 2019. Aug 28. https://doi.org/10.1001/jamasurg.2019.2884. [Epub ahead of print].
Woolley T, Thompson P, Kirkman E, et al. Trauma Hemostasis and Oxygenation Research Network position paper on the role of hypotensive resuscitation as part of remote damage control resuscitation. J Trauma Acute Care Surg. 2018;84:S3–S13.
Spaite DW, Hu C, Bobrow BJ, et al. Association of out-of-hospital hypotension depth and duration with traumatic brain injury mortality. Ann Emerg Med. 2017;70:522–30.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Richards, J., Gauss, T., Bouzat, P. (2020). Vasopressors for Post-traumatic Hemorrhagic Shock: Friends or Foe?. In: Vincent, JL. (eds) Annual Update in Intensive Care and Emergency Medicine 2020. Annual Update in Intensive Care and Emergency Medicine. Springer, Cham. https://doi.org/10.1007/978-3-030-37323-8_32
Download citation
DOI: https://doi.org/10.1007/978-3-030-37323-8_32
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-37322-1
Online ISBN: 978-3-030-37323-8
eBook Packages: MedicineMedicine (R0)