
Abstract
The retaining endo-β-D-glucuronidase Heparanase (HPSE) is the primary mammalian enzyme responsible for breakdown of the glycosaminoglycan heparan sulfate (HS). HPSE activity is essential for regulation and turnover of HS in the extracellular matrix, and its activity affects diverse processes such as inflammation, angiogenesis and cell migration. Aberrant heparanase activity is strongly linked to cancer metastasis, due to structural breakdown of extracellular HS networks and concomitant release of sequestered HS-binding growth factors. A full appreciation of HPSE activity in health and disease requires a structural understanding of the enzyme, and how it engages with its HS substrates. This chapter summarizes key findings from the recent crystal structures of human HPSE and its proenzyme. We present details regarding the 3-dimensional protein structure of HPSE and the molecular basis for its interaction with HS substrates of varying sulfation states. We also examine HPSE in a wider context against related β-D-glucuronidases from other species, highlighting the structural features that control exo/endo − glycosidase selectivity in this family of enzymes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Heparanase
- Heparan sulfate
- Glycosaminoglycan
- Extracellular matrix
- Cancer
- Glycoside hydrolase
- Glycosidase
- CAZy
- GH79
1 Introduction
Heparan Sulfate proteoglycans (HSPGs) are ubiquitous components of the extracellular matrix (ECM), where they mediate diverse structural and signaling interactions between cells and proteins of the ECM [1]. HSPGs are comprised of a core transmembrane, membrane-anchored, or extracellular protein attached to one or more chains of the glycosaminoglycan (GAG) polysaccharide heparan sulfate (HS). Interactions between HSPGs and their binding partners primarily occur via the HS chains which decorate the core HSPG protein [2].
The chemical composition of HS is complex and dynamically regulated in response to stimuli via a process of continual turnover [3,4,5]. HS composition has been shown to vary in relation to development [6,7,8], cancer stage [9,10,11], and general age [8, 12]. While biosynthesis of HS is a multistep process involving the concerted action of a host of polymerases, sulfotransferases, and epimerases [13], breakdown of HS in mammals is primarily carried out by a single enzyme – heparanase (HPSE) [14–15].
HPSE is an endo-acting glycoside hydrolase , which cleaves within long HS chains to release product fragments of HS ~5–7 kDa in size [14]. The HS degrading activity of heparanase is essential for ECM remodeling, affecting diverse processes such as inflammation, angiogenesis and cell migration [16,17,18]. HPSE activity can also release growth factors sequestered within networks of HS, which subsequently promote angiogenesis and wound healing [19]. Whilst normal HPSE function is essential for physiological processes which involve ECM remodeling, the HS degrading capability of HPSE can also be co-opted by cancerous cells to promote malignant growth and dissemination. Accordingly, upregulation of heparanase is a hallmark of aggression and metastasis in a wide range of cancers [20,21,22,23,24].
A full summary of the many functions of HPSE in health and disease is beyond the scope of this article, and will be covered elsewhere in this book. Instead, we aim here to provide a structure/function-centric review of HPSE, drawing from insights gained from crystal structures of HPSE and its related proteins. From these, we hope to provide the reader with an appreciation of the structural features that underlie the many biological and biochemical insights obtained from decades of research on HPSE.
2 Heparan Sulfate – The Biochemical Basics
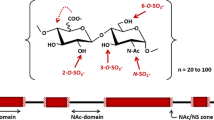
Chemically, HS is a linear glycosaminoglycan polysaccharide comprising of alternating 1,4 linked units of hexuronic acid (HexUA) and glucosamine (GlcN) [25]. HS chains can display high complexity due to the number of permutations possible for the core HexUA and GlcN building blocks. The HexUA of HS can be either β-D-glucuronic acid (GlcUA) or α-L-iduronic acid (IdoUA), and GlcN can be either N-acetyl-α-D-glucosamine (GlcNAc) or N-sulfo-α-D-glucosamine (GlcNS). These core residues are further decorated by varying degrees of O-sulfation (Fig. 5.1a).

(a) Chemical structures of HexUA and GlcNX building blocks of HS, with possible sites of sulfation shown. (b) Representative HSPG illustrating the domain structure of HS chains, and the predominant disaccharide units found within NA and NS domains. Mixed NS/NA domains separating NS and NA domains have not been shown here
Biosynthesis of HS is non-templated, allowing HS composition to vary substantially along a single polysaccharide chain (typical HSPGs contain HS chains between 40–300 sugar units (20–150 nm) in length) [1]. Variations in HS structure occur across broad macromolecular regions, leading to the formation of N-acetyl (NA) domains (characterized by poorly sulfated GlcNAc-GlcUA repeats) and N-sulfo (NS) domains (characterized by highly sulfated GlcNS-IdoUA repeats), separated by mixed NS/NA domains (Fig. 5.1b). This structural heterogeneity is crucial for HS function, enabling a single polysaccharide chain to interact with a host of different binding partners. HS heterogeneity is also of central importance for its breakdown by HPSE. As will be further discussed below (Sect. 5.4.2.), cleavage of HS by HPSE is limited to only certain GlcUA residues within the sugar chain, depending on the local sulfation pattern around the target site.
3 Historical Developments in HPSE Research
3.1 Identification of a Specific Heparan Sulfate Degrading Enzyme
The existence of a specific mammalian HS degrading factor was first demonstrated in 1975 by Ogren and Lindahl [26], and Höök et al [27], who described the isolation of enzyme preparations from mouse mastocytoma and rat liver respectively, which were capable of degrading heparin and HS to low molecular weight fragments (heparin is structurally similar to highly sulfated HS). These studies were closely followed by reports of similar heparan sulfate degrading activities in a number of different cell and tissue types (Vlodavsky et al., Chap. 1 in this volume).
Heparan sulfate degrading activity in platelets was first demonstrated by Wasteson et al., who found that cultured human glial cells exposed to platelet lysates released low molecular weight HS into their culture medium [28]. Similar HS degrading activity was subsequently identified in placental tissue by Klein and von Figura [29]. Nicolson and coworkers demonstrated that B16 mouse melanoma cells utilized a HS degrading enzyme to assist with breakdown of ECM like barriers in vitro [30] , and that the HS degrading capabilities of B16 subpopulations positively correlated with their metastatic potential in vivo [31]. This direct relation between heparanase activity and metastatic potential in cancer cells was further demonstrated by Vlodavsky et al., who showed that the poorly metastatic T-lymphoma cell line Eb and its spontaneous highly metastatic variant ESb differed strongly in their ability to degrade HSPGs [32].
A number of observations from these early studies have since become recognized as hallmarks of HPSE activity. Chemical analysis of enzymatically degraded HS products found that cleavage occurred only at the glucuronic acid of HS, not at the glucosamine, indicating that the responsible enzyme was a glucuronidase [26, 29, 33]. Enzymatic HS cleavage was also found to be limited, leading to the formation of intermediate-sized oligosaccharide products resistant to further degradation, consistent with an endo-glucuronidase that targets specific HS sites [26, 28, 30,31,32]. Although commonalities between these early studies indicated researchers were studying the same enzyme activity, it would take more than a decade for the enzyme responsible to be identified unambiguously.
3.2 Isolation of Heparanase Enzyme and Cloning of the HPSE Gene
The identity of the HS degrading enzyme was controversial for a number of years, with proteins ranging from 8 kDa to 137 kDa mass being reported as possessing HPSE activity [34,35,36]. These discrepancies were resolved in the late 1990s, following several independent reports describing the purification of the same HS degrading protein from various sources. Goshen et al. first reported the purification of a ~50 kDa HS degrading enzyme from human placenta [37], followed by Freeman and Parish, who isolated an enzyme of similar size and biochemical profile from platelets [38]. These reports were closely followed by seminal studies from Toyoshima and Nakajima, Vlodavsky et al., Kussie et al. and Hulett et al., who all carried out peptide sequencing of the isolated HS degrading protein, and used this information to identify and clone the responsible HPSE gene [39,40,41]. These groups all noted the strange observation that whilst the HPSE gene encoded for a ~65 kDa protein; purified HPSE appeared to be ~50 kDa in size, with its N-terminus apparently beginning at Lys158. Furthermore, expression of the full HPSE gene was found to be required for activity, with expression of the sequence corresponding to the ~50 kDa subunit alone failing to endow cells with HS degrading activity [41].
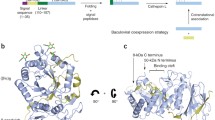
The discrepancy between HPSE gene and protein size was resolved by Fairbanks et al., who demonstrated the existence of a previously undetected 8 kDa subunit in HPSE purified from platelets [42]. This 8 kDa subunit was found to tightly associate with the 50 kDa subunit, only being separable under denaturing conditions, indicating the existence of a non-covalently associated heterodimer. MALDI-TOF analysis identified the 8 kDa subunit of HPSE as Gln36-Glu109, corresponding to an N-terminal fragment encoded by the HPSE gene. Based on these results, Fairbanks et al. proposed the now widely accepted maturation pathway of the HPSE protein. HPSE is initially expressed as a single chain pre-proenzyme (pre-proHPSE), comprising an N-terminal signal peptide (Met1-Ala35), followed by the 8 kDa (Gln36-Glu109) and 50 kDa (Lys158-Ile543) subunit sequences, separated by a 6 kDa linker peptide (Ser110-Gln158). Loss of the signal peptide from pre-proHPSE following signal peptidase cleavage [43] leads to formation of the inactive HPSE proenzyme (proHPSE). Active HPSE is only produced following proteolytic excision of the 6 kDa linker peptide from proHPSE, leading to formation of the mature enzyme, which exists as a non-covalent heterodimer of 50 kDa and 8 kDa subunits (Fig. 5.2).

HPSE biogenesis pathway . Steps pertinent to baculoviral expression of pro- and mature HPSE in insect cells are highlighted in red
3.3 Production of Homogenous Recombinant HPSE
Structural biology studies rely on the availability of large amounts of purified homogenous protein. In this regard, the production of recombinant HPSE presents an unusual challenge, due to the complex process of HPSE maturation. Recombinant expression of HPSE in mammalian cells often leads to a mixture of both 65 kDa proHPSE as well as mature HPSE heterodimer [39, 41], rendering these expression systems unsuitable for structural biology purposes.
Protein production in insect cells using the baculovirus expression vector system (BEVS) [44] has proven an invaluable tool for the study of recombinant HPSE. BEVS is a bipartite gene expression platform utilizing recombinant baculovirus for gene delivery and cultured insect cells for protein production. Because insect cells are eukaryotic animal cells (albeit non-mammalian cells), gene expression using BEVS usually allows for the faithful reproduction of native mammalian protein folds. Serendipitously, insect cells lack the cellular apparatus to carry out proHPSE maturation, thus precluding the production of pro- and mature HPSE mixtures [41]. Although this feature of insect cell protein production provides an obvious route towards proHPSE, the problem of accessing mature HPSE was not addressed until McKenzie et al demonstrated that co-expression of the 50 kDa and 8 kDa subunits under two different promotors led to co-translational association of the two subunits, allowing for direct access to mature HPSE [45] (Fig. 5.2).
A markedly different approach to tackling the HPSE linker problem was reported by Nardella et al [46], who engineered HPSE to replace the linker region with much shorter sequences. Expression of engineered proHPSE in which the linker sequence was replaced by either an artificial (GSGSGS) repeat or the analogous sequence from a Hirudinaria manillensis hyaluronidase (AFKDKTP) gave a single chain variant of HPSE with activity comparable to wild type enzyme. The key role of the 6 kDa linker peptide in controlling (pro)HPSE activity is discussed further below (Sects 5.4.3 and 5.5.1).
4 Heparanase – Insights from Crystal Structures
We reported the crystal structure of mature HPSE in 2015, revealing the overall 3-dimensional protein structure of HPSE, and also (via several ligand complexes), the mode of interaction between HPSE and its substrates [47]. This was followed by the solving of the proHPSE crystal structure in 2017 [48]. In this section of the review, we aim to provide an overview of the main insights into the HPSE structure from these two studies, and how the structural features of HPSE relate to its biochemical and biological properties.
4.1 3-Dimensional Structure of Mature HPSE
Several features were immediately apparent upon initial solving of the HPSE crystal structure (PDB accession code: 5E8M). The HPSE protein comprises two major domains: a predominant (β/α)8 barrel domain, flanked by a smaller β-sandwich domain. The 8 kDa HPSE subunit contributes a single β-sheet towards the β-sandwich domain, as well as the first β-α-β elements of the (β/α)8 domain, with the rest of the protein structure being contributed by the 50 kDa subunit. Such a division of structure between the 8 kDa and 50 kDa subunits of HPSE was postulated by Nardella et al., based upon the predicted secondary structure elements within the HPSE sequence [46]. The (β/α)8 barrel domain is commonly found in glycoside hydrolases, and usually contains the active site of these enzymes [49]. Visual inspection of the (β/α)8 barrel of HPSE revealed a clear cleft in the domain, spanning ~10 Å in diameter, suggesting a binding site for chains of HS. This cleft was lined with a number of basic Arg and Lys residues, which are commonly found in HS interacting protein domains [50,51,52,53,54,55]. Notably, HS binding “domains” (HBDs) I (Lys158-Asp162) and II (Pro271-Met278), previously identified by Levy-Adam et al [56], were found to lie around the HPSE binding cleft, supporting a role for these two domains in facilitating HPSE-HS interactions (Fig. 5.3).

Three-dimensional structure of unliganded human HPSE, showing ‘top’ (left) and ‘side’ (right) views. The 50 kDa subunit is colored in blue, and the 8 kDa subunit is colored in yellow (colors correspond with Fig. 5.2). Two domains can be discerned in the HPSE structure, a (β/α)8 barrel domain containing the HS-binding cleft, and a smaller β-sandwich domain. HBDs I and II identified by Levy-Adam et al [56] are highlighted in pink, other basic residues around the binding cleft are highlighted in cyan. A putative NLS sequence in the β-sandwich domain is highlighted in red. N-glycans are shown in green
N-glycosylation of HPSE is known to be essential for its proper cellular trafficking, and its secretion by cells into the extracellular space [57]. Of the six N-glycosylation sites predicted by analysis of the HPSE sequence, five were visible in the crystal structure of unliganded HPSE, albeit endoglycosidase H digestion carried out prior to protein crystallization meant that most of these were only visible as a single N-linked GlcNAc.
One of the more curious findings of HPSE biology has been the discovery of HPSE in cell nuclei, where it appears to co-localise with highly transcribed euchromatin regions of the genome [58]. Nuclear HPSE can alter the expression of tumor-promoting genes such as matrix metalloproteinase 9 (MMP-9), vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) [58,59,60,61]. Two putative nuclear import signals were noted by Schubert et al. in the HPSE sequence: residues 271–277 (PRRKTAK) and residues 427–430 (KRRK) [62]. While the Pro271-Lys277 sequence forms an alpha helix (and also corresponds to HBD II), Lys427-Lys430 appears in the HPSE crystal structure as a disordered loop near the β-sandwich domain of HPSE. Lack of secondary structure renders this loop-free to interact with the importin machinery involved in nuclear trafficking [63] and is thus consistent with a role for Lys427-Lys430 as a nuclear import signal .
4.2 Structural Insights into HPSE Substrate Interactions
The defining feature of HPSE mediated HS cleavage is the high degree of sequence discrimination displayed by HPSE, rendering only certain sites in a HS chain susceptible to enzymatic attack. This behavior is in marked contrast to the bacterial heparin lyases, which carry out a much more complete breakdown of HS with little regard for the sequence of the sugar chain [64,65,66]. The high specificity of HPSE cleavage was noted as early as the 1970s, with observations that HPSE mediated cleavage of HS produces products intermediate in size between the initial substrate and fully depolymerized HS, and that these oligosaccharide products are resistant to further hydrolysis by HPSE [26, 28, 30,31,32].
Although sulfation of HS substrates had long been suspected to be important for recognition and cleavage by HPSE, early studies on this topic were hampered by a lack of pure enzyme preparations and chemically defined HS substrates. In this light, the pioneering 1999 study by Pikas et al. on HPSE cleavage site specificity stands as an impressive milestone given the state of HPSE research at the time [67]. Subsequent advances in cloning and recombinant expression of purified HPSE, and the advent of chemoenzymatic HS synthesis have contributed to several reassessments on this topic (summarized in Table 5.1) [68,69,70,71]. Whilst studies of HPSE cleavage site specificity agree on a few central points: that HPSE is an endo-β-D-glucuronidase, and that sulfation around the HPSE target site is essential for cleavage, the finer details of substrate recognition, especially regarding the specific sulfation patterns required for cleavage, have been a source of disagreement.
Structural biology can help to address such questions regarding enzyme/substrate specificities, via the direct visualization of enzyme-substrate complexes. Given the heterogeneous nature of HS itself, the development of specific chemoenzymatically driven HS oligosaccharide synthesis by Petersen and Liu was a crucial foundation for our work to characterize well-defined HPSE-HS complexes [70].
We utilized 3 distinct synthetic, commercially-available, HS oligomers with different sulfation states to probe HPSE-HS interactions with both non-sulfated and sulfated substrates. M04 S00a and M04 S02a contain no sulfates and 1 N-sulfate, respectively and are not HPSE substrates. In contrast, M09 S05a contains 4 N-sulfates and 1 O6-sulfate, endowing this oligosaccharide with a consensus HPSE cleavage site (Fig. 5.4a). Soaking crystals of HPSE with these defined HS oligosaccharides enabled the capture of HPSE-HS complexes in crystallo, allowing the molecular basis for interactions between HPSE and its substrates to be mapped (Fig. 5.4b).

(a) HS and heparin oligosaccharides used to obtain ligand complexes with HPSE. Carbohydrate symbol nomenclature as in Fig. 5.1. M09 S05a contains a consensus HPSE cleavage site – highlighted in the red box, with the cleaved bond indicated by the red arrow. pNP - para-nitrophenol. (b) Ribbon and surface figure of an M04 S00a oligosaccharide bound within the active site cleft of HPSE (grey sticks). HBDs and other basic residues around the HPSE binding cleft are highlighted pink and cyan respectively
4.2.1 HPSE Interactions at the −1 Subsite
The −1 subsite of a glycosidase enzyme is the position occupied by the sugar which directly undergoes glycosidic bond cleavage by the enzyme [72]. In all HPSE-HS complexes we obtained (PDB accession codes: 5E97, 5E98, 5E9B; complexes with M04 S00a, M04 S02a and M09 S05a respectively), the −1 subsite was occupied by a GlcUA, making identical interactions to the enzyme active site in all cases, illustrating the invariant nature of GlcUA binding at this position. Glycosidases such as HPSE utilize two key catalytic residues to facilitate substrate hydrolysis, a nucleophile and a general acid/base (detailed reviews of glycosidase mechanisms can be found in Refs. 73,74,75,76,77). GlcUA at the −1 subsite of HPSE positions its anomeric center proximal to the catalytic residues Glu343 (nucleophile) and Glu225 (acid/base), in a position ready to undergo attack by the enzyme.
The HPSE −1 enzyme subsite is also characterized by a dense network of H-bonding interactions, made to the C6 carboxylate of the GlcUA from Gly349, Gly350, and Tyr391. These H-bonding interactions appear to be highly conserved amongst HPSE and its homologs [78–79], and likely function as a specificity filter to recognize and bind GlcUA over superficially similar sugars such as glucose (Fig. 5.5).

HPSE-HS interactions at the −1 enzyme subsite with M04 S00a. For clarity, only the −1 subsite ligand atoms have been shown
4.2.2 HPSE Interactions at the −2 Subsite
Whilst −1 subsite interactions in HPSE were observed to be invariant between M04 S00a, M04 S02a and M09 S05a complexes, differences at the −2 subsite of HPSE could be discerned, highlighting the interactions employed by HPSE to recognize different HS sulfation patterns. M04 S00a, which contains no sulfation and is not cleaved by HPSE, places its −2 GlcNAc N-acetyl moiety near residues Ala388-Tyr391 and Asn64, making direct H-bonds to Tyr391, Asn64, and an ordered water molecule. M04 S02a, which differs from M04 S00a by the presence of an N-sulfate, places its −2 GlcNS in the same orientation as the GlcNAc of M04 S00A. However, the larger N-sulfate of GlcNS can make an additional H-bonding interaction to the backbone amide of Gly389, thus rationalizing the preferred interaction of HPSE with GlcNS at the −2 subsite.
The role of O6 sulfation at the −2 subsite was probed by the M09 S05a complex, which showed that the O6 sulfate of GlcNS(6S) was placed towards the ‘upper’ portion of the −2 subsite, proximal to some of the basic residues lining the substrate binding cleft (Lys158 and Lys159). Although we could not observe ordered interactions between O6 sulfate and these basic residues, non-directional electrostatic interactions likely play a role in stabilizing the −2 subsite complex between HPSE and O6 sulfated HS substrates (Fig. 5.6).

HPSE-HS interactions at the −2 enzyme subsite with M04 S00a, M04 S02a, and M09 S05a. For clarity, only the −2 subsite ligand atoms have been shown
4.2.3 HPSE Interactions at the +1 Subsite
The primary disadvantage of employing HS oligosaccharides to generate HPSE complexes is the propensity of the enzyme to turn over substrates that match the requirements for HS cleavage. Thus whilst M04 S00a and M04 S02a were observed in crystallo to place non-hydrolysed pNP groups at the +1 subsite of HPSE, the presence of a consensus HPSE cleavage site in M09 S05a led to the +1 subsite of the enzyme in this complex to be poorly occupied, due to enzymatic cleavage of the aglycon fragment.
To circumvent this problem, we turned to heparin, a close structural analog of HS, and a known inhibitor of HPSE activity. Soaking HPSE crystals with a heterogeneous heparin dp4 oligosaccharide (obtained through heparin lyase cleavage of polymeric heparin; Fig. 5.4a) yielded a structure with interpretable heparin dp4 electron density within the HPSE active site cleft (PDB accession code: 5E9C). This observed density likely corresponded to a minor component of the heparin dp4 mix, as it was substantially weaker than the electron density observed for the pure HS oligosaccharides. However, this heparin dp4 density spanned the −2, −1 and (crucially) +1 positions of the HPSE active site, thus providing insight into the nature of HPSE +1 subsite interactions.
HPSE +1 subsite interactions with heparin dp4 were broadly similar to those observed at the −2 subsite with M09 S05a, except the helical nature of HS and heparin substrates reversed the roles of N- and O6 sulfates at the +1 subsite. Analogous to the role of −2 subsite N-sulfate, we observed H-bonds from +1 subsite O6 sulfate to the sidechain and backbone amide of Gln270. Electron density for the +1 subsite N-sulfate of dp4 was too poor to be observed directly. However, a + 1 subsite N-sulfate could only plausibly be modeled towards the ‘top’ of the HPSE binding cleft, in position to make electrostatic contacts with the basic residues lining this region (Arg303 and Arg232) (Fig. 5.7).

HPSE-HS interactions at the +1 enzyme subsite with heparin dp4. For clarity, only the +1 subsite ligand atoms have been shown
Taken together, the combined structural data from complexes with M04 S00a, M04 S02a, M09 S05a, and heparin dp4 indicate that HS sulfates in the ‘upper’ portion of the HPSE binding cleft (−2 subsite O6 sulfate, +1 subsite N sulfate) electrostatically interact with the basic residues around the cleft. In contrast, sulfates ‘lower’ in the HS-binding cleft appear to make direct H-bonding interactions with HPSE residues and ordered water molecules. Our structures indicate a ‘dual mode’ of interaction between HPSE and its substrates, with the ‘lower’ H-bonds likely acting as specificity filters for sulfation (due to the directional nature of H-bonding), while the ‘upper’ electrostatic interactions stabilize the binding of HS within the active site cleft (Fig. 5.8).

HPSE-HS interactions across the −2, −1 and + 1 subsites of the HPSE binding cleft, as mapped by complexes with HS oligosaccharides and heparin dp4. The site of enzymatic cleavage is shown (//). H-bonding interactions between HPSE and HS are illustrated in blue. Basic residues in place to make electrostatic interactions with HS substrates are shown in red. Nuc. – catalytic nucleophile (Glu343). (a./b.) – catalytic acid/base (Glu225)
4.2.4 Beyond the +1 Subsite
Although the crystal structures of HS oligosaccharide complexes point to a maximally favored trisaccharide cleavage site, they do not rationalize all findings from biochemical studies of HPSE cleavage specificity. Observations that HS hexasaccharides are preferentially cleaved by HPSE over shorter tetrasaccharides [69], and the ability of HPSE to cleave substrates lacking −2 or + 1 subsite O-sulfation, but containing −3 or + 2 sulfation [71], hint at interactions beyond the core −2, −1 and + 1 subsites that were not captured in our HPSE crystal structure complexes.
The most likely candidates for mediating additional interactions between HPSE and HS are the HBDs postulated by Levy-Adam et al., which may help to bind sulfates outside of the core trisaccharide cleavage site. Modeling studies of HPSE with either the highly sulfated HPSE inhibitor SST0001 [80], or the synthetic HS pentasaccharide fondaparinux [81], suggest that a +2 subsite GlcUA(2S) could interact with basic residues in HPSE HBD2, and thus contribute toward enzyme-substrate binding [82]. It may be the case that HPSE-HS interactions under native contexts are less strictly defined than those captured by static crystal structures, with more distant sulfates potentially being able to compensate for lack of sulfation around the core trisaccharide cleavage site.
4.3 3-Dimensional Structure of proHPSE
Proteolytic excision of the 6 kDa linker peptide of proHPSE is required for its maturation to HPSE, indicating a role for this peptide in inactivating proHPSE towards HS substrates. Based on the positions of the 8 kDa and 50 kDa subunit C- and N-termini (respectively) in mature HPSE, we postulated that the 6 kDa linker peptide of proHPSE would likely lie near the HPSE substrate binding cleft, implying a steric occlusion mechanism for inactivation. This steric mechanism for proHPSE inactivation by its 6 kDa linker peptide was confirmed by the 2017 crystal structure of proHPSE (PDB accession code: 5LA4). Broadly speaking, the proHPSE 6 kDa linker peptide forms a predominantly α-helical domain which sits ‘atop’ the HPSE binding cleft, thereby preventing the HPSE active site from binding HS [48]. Occlusion of HS-binding appears to be the only mechanism whereby the 6 kDa linker peptide inactivates HPSE (Fig. 5.9). Indeed, protein engineering to ‘shrink’ the proHPSE linker peptide produces an enzyme with HS degrading activity similar to wild type HPSE [46].

Three-dimensional structure of human proHPSE, showing ‘top’ (left) and ‘side’ (right) views. The 6 kDa linker peptide of HPSE (green) sterically occludes the HS-binding cleft (compare with Fig. 5.3). HBDs are highlighted in pink. Tyr156 and Gln157, which form part of the CTSL cleavage site involved in HPSE maturation, are highlighted in red. CTSL cleavage occurs between Gln157 and Lys158 (part of HBD I). A ‘binding pocket’ structure can be discerned on the surface of proHPSE, and is shown here in complex with a glucuronidase specific activity-based probe (grey sticks)
ProHPSE readily binds to cell surface HSPGs and undergoes internalization and trafficking to the lysosome, whereupon it undergoes processing to produce mature HPSE [83,84,85]. This process of proHPSE sequestration has been proposed to contribute to aggression and metastasis in cancer cells, by providing a mechanism for these cells to capture extracellular proHPSE and increase their own stores of mature HPSE. Although the substrate binding cleft is occluded in proHPSE, HBD1 and HBD2 remain freely accessible on the surface of proHPSE, and electrostatic interactions between these HBDs and cell surface HSPGs may facilitate proHPSE binding to cell surfaces, with subsequent internalization and processing. Proteolytic processing of proHPSE is mediated by cathepsin L (CTSL), with one key cleavage occurring at Gln157−/−Lys158, directed by CTSL recognition of the nearby Tyr156 residue [86–87]. Tyr156-Lys158 in the proHPSE structure reside within a highly disordered turn towards the end of the 6 kDa linker sequence, where they would be freely accessible for interaction with CTSL (or another protease).
One of the most surprising discoveries upon solving of the proHPSE structure was that the 6 kDa linker peptide only obscures part of the HPSE binding cleft, with a substantial ‘binding pocket’ still remaining on the protein surface. Whilst large HS substrates are occluded from proHPSE, the ‘binding pocket’ of proHPSE renders its active site residues fully accessible to smaller ‘monosaccharide’ like molecules. We confirmed the catalytic competency of proHPSE (at least towards artificial substrates) by labeling the protein with aziridine based activity-based probes, which are highly activated substrate mimics we have previously utilized to study many classes of glycoside hydrolase (PDB accession code: 5LA7; Fig. 5.9) [88,89,90,91]. It remains to be seen whether there are biologically relevant substrates in vivo which are turned over by the proHPSE ‘binding pocket’, or whether this motif is an evolutionary relic from an ancestral enzyme (discussed further in Sect. 5.5.1.).
5 HPSE Within the Broader CAZy Classification
As an enzyme which catalyzes the hydrolytic breakdown of a carbohydrate substrate, HPSE falls within the general enzyme class known as the glycoside hydrolases (or glycosidases). Glycoside hydrolases are a diverse group of enzymes, which facilitate the hydrolytic breakdown of carbohydrate-containing biomolecules (e.g., glycoproteins, polysaccharides, small molecule glycoconjugates) in varied contexts across all domains of life [92].
Reflecting the central importance of carbohydrate-containing molecules in biology, it has been estimated that ~1–3% of the protein-coding genome of a (non-archael) organism corresponds to enzymes involved in carbohydrate processing (both for synthesis and breakdown) [93]. The Carbohydrate Active enZymes (CAZy) classification aims to classify these carbohydrate processing enzymes into sequence-based families [93,94,95,96,97,98,99,100,101,102,103,104]. Given that protein sequence largely dictates structure and function, CAZy families typically contain enzymes with similar structural folds and enzyme mechanisms, although the specific substrates processed by enzymes within a family can vary. Under the CAZy classification, HPSE belongs to the GH79 family, itself further classified into the broader GH-A clan (clans are based on groupings of GH families with similar overall topologies and conservation of active site residues) [100, 105]. The GH79 family primarily consists of retaining β-D-glucuronidases, although the substrate contexts of these glucuronic acid residues are diverse, including HS [47, 79], but also chondroitin sulfate [79], hyaluronic acid [46], β-D-glucuronides linked to plant arabinogalactan proteins [106–107], and small molecule β-D-glucuronide glycoconjugates [108].
5.1 Structural Determinants of exo vs. endo-Glycosidase Activity in the GH79 Family
The GH79 family contains representatives of both endo-acting and exo-acting β-D-glucuronidases, raising the question of how a single ‘scaffold’ can be adapted to process substrates in either an exo- or endo- acting fashion. To date, 3 GH79 enzymes have been structurally characterized: HPSE [47–48], AcGH79 from Acidobacterium capsulatum [78], and BpHep from Burkholderia Pseudomallei [79]. In keeping with the scope of CAZy classification, there is substantial sequence and structural homology between these three enzymes and all three act as β-D-glucuronidases, although the natural substrates of the two bacterial enzymes are not known.
One area of major variability in GH79 family enzymes is a loop which we have termed the ‘exo-pocket’ loop, which connects the 2nd β-sheet of the (β/α)8 barrel domain to the 2nd α-helix. Comparison of the three structurally characterized GH79 enzymes demonstrates that the ‘exo-pocket loop ’ can vary dramatically in size, and appears to act as a key structural determinant of whether an enzyme of the GH79 family behaves as an exo- or an endo- acting glycosidase.
5.1.1 AcGH79
The crystal structure of the exo-acting β-D-glucuronidase AcGH79 was reported by Michikawa et al. in 2012, and was the first enzyme of the GH79 family to be structurally characterized (PDB accession code: 3VNY) [78]. Although the function of AcGH79 in its native biological context is not well understood, the authors determined that this enzyme could not hydrolyze 4-O-methyl GlcUA containing substrates. AcGH79 may be involved in the catabolism of plant arabinogalactan proteins, which typically contain both GlcUA and 4-O-methyl GlcUA substitutions on the main arabinogalactan polymer [106–107].
The ‘exo-pocket’ loop of AcGH79 is 23 residues long, extending from Phe86 to His108 (limits defined on the basis of homology to BpHep and proHPSE; Fig. 5.10). This sequence adopts an extended turn that occludes the ‘rear’ face of the AcGH79 active site, delimiting an exo-acting substrate binding pocket that can only accommodate a single GlcUA residue. Discrimination of 4O-methyl GlcUA vs. GlcUA is facilitated by Glu45, Pro104, and His327, which together form a tight binding pocket around O4 of GlcUA, which does not allow for further methyl substitution .

(a) ‘Exo-pocket’ loop structures for AcGH79 , BpHep and proHPSE (highlighted green) showing their role in delineating an exo-acting binding pocket structure in AcGH79 and proHPSE, or alternatively, an endo-acting binding cleft structure in BpHep. Proteolytic removal of the ‘exo-pocket’ loop of proHPSE (i.e. the 6 kDa linker peptide), reveals the endo-acting binding cleft of mature HPSE. (b) Clustal ω [149] alignments of AcGH79, BpHep, and HPSE showing the variation in ‘exo-pocket’ loop lengths between these three proteins
5.1.2 BpHep
BpHep was the first endo-acting GH79 enzyme to be structurally characterized, and the second structurally characterized GH79 enzyme overall (PDB accession code: 5BWI) [79]. BpHep is an endo-β-D-glucuronidase which can degrade both heparan sulfate and chondroitin sulfate, suggesting it may be a general glycosaminoglycan breakdown enzyme. Saturation-transfer difference NMR binding experiments using defined HS oligomers suggest that BpHep prefers to interact with HS cleavage sites rich in GlcNAc rather than GlcNS, indicating a different HS substrate specificity to that displayed by HPSE.
Compared to AcGH79, the ‘exo-pocket’ loop of BpHep is substantially shorter in length (Gly92 to Asp99; 8 residues long; Fig. 5.10), and is not long enough to occlude any part of the enzyme active site. Instead, this very short ‘exo-pocket’ loop of BpHep results in opening of the enzyme active site, revealing an extended endo-acting binding cleft, well suited for interaction with glycosaminoglycan substrates .
5.1.3 (pro)HPSE
ProHPSE to HPSE maturation provides the most direct example of the role of the ‘exo pocket’ loop in controlling exo−/endo- activity in GH79 family enzymes. The ‘exo-pocket’ loop of (pro)HPSE (110 to 157; 48 residues long) directly corresponds to the 6 kDa linker peptide and is substantially increased in size compared to corresponding ‘exo-pocket’ loop sequences in AcGH79 and BpHep (Fig. 5.10). As mentioned in Sect. 5.4.3., the 6 kDa linker of proHPSE forms an alpha-helical domain that acts as a direct steric block ‘above’ the HPSE binding cleft, preventing interaction of the enzyme with HS substrates. Removal of the proHPSE linker is required for unmasking of the mature HPSE binding cleft, and can be considered analogous to the effect of the minimal ‘exo-pocket’ loop sequences of BpHep [79] and engineered single chain HPSE mutants [46].
Comparison of proHPSE with AcGH79 and BpHep places the exoglycosidase like binding pocket of proHPSE into an understandable evolutionary context (see Sect. 5.4.3.). We hypothesize that expansion of the ‘exo-pocket’ loop sequence from an ancestral exo-acting GH79 enzyme led to formation of the 6 kDa proHPSE linker, without formal loss of the exo-acting binding pocket architecture. The retention of such exo-glycosidase like structural features on proHPSE warrants consideration whether there are genuine substrates that are processed by this protein species in vivo. Supporting this hypothesis, mature HPSE has been demonstrated to possess exo-glycosidase activity against terminal glucuronides within certain HS contexts [109]. There is no structural reason why proHPSE would not also possess such activity, and thus it may play a role in, e.g. trimming terminal glucuronides from certain HS chains .
6 Concluding Remarks and Future Challenges
HPSE has captured the interest of researchers for over four decades, with efforts to understand its function ranging from fundamental biochemical studies on HPSE enzymatic activity to complex biomedical characterizations of its role in cancer and other diseases. Structural studies of HPSE and proHPSE provide a framework on which to place these biochemical and biomedical insights, allowing them to be related to features on the HPSE protein itself.
There are still a number of unresolved challenges in the HPSE field, which will likely be the subject of substantial research efforts in the coming years.
Most pressingly, despite intense interest in HPSE as an anti-cancer target, there are few effective HPSE inhibitors known, and none in use clinically. Various small molecule HPSE inhibitors have been reported, based on benzoxazole, furanylthiazole [110], isoindole [111], benzimidazole [112–113], and other scaffolds. However, none of these small molecule inhibitors appear to have progressed beyond initial enzyme inhibition and invasion/angiogenesis studies. More recently, four HPSE inhibitors have entered clinical trials: PI-88 [114,115,116,117,118,119], SST0001 [82, 120,121,122], M402 [123–124] and PG545 [125,126,127,128,129], although an interim analysis of PI-88 phase III clinical trials showed a failure to meet its primary endpoint (disease-free survival) [130] (Chhabra & Ferro; Noseda et al., Hammond & Dredge, Chaps. 19, 21 and 22 in this volume). All HPSE inhibitors currently under clinical trials are highly sulfated oligosaccharide molecules, and of these only PG545 possesses a well-defined molecular structure. Such oligosaccharide-derived molecules are less likely to possess desirable pharmacokinetic properties, and a renewal of efforts to develop novel small molecule HPSE inhibitors may be timely.
The HPSE field also lacks a reliable, sensitive, and facile method for quantitation of HPSE enzymatic activity [131]. The development of routine activity assays, often relying on artificial chromogenic and fluorogenic substrates, has been essential for enzyme discovery and enzyme characterization efforts in the glycosidase field [132,133,134,135,136]. Robust assays are vital for effective inhibitor development since a potential inhibitor cannot be quantitatively characterized if there is no suitable assay available to inhibit. The lack of ‘gold standard’ assays for HPSE probably reflects the complex nature of its interaction with HS, which may be difficult to recapitulate in artificial substrates.
Finally, the discovery of a close homolog of HPSE, termed HPSE2, which can bind HS but lacks glycosidase activity [137], raises questions regarding the biological functions of HPSE2, and how they might relate to HPSE. HPSE2 expression inversely correlates with the size and grade of tumours [138–139], and it appears to act as an anti-tumorigenic factor [140], possibly through antagonism of HPSE activity [141]. Biallelic mutations in HPSE2 have been linked to the rare genetic condition urofacial syndrome (UFS; also known as Ochoa syndrome) [142,143,144,145], a disease characterized by symptoms of facial grimacing, coupled to loss of adequate urinary voiding [146]. Such symptoms may indicate that HPSE2 plays a role in urinary tract and/or neurological development [147–148]. (Mckenzie; Roberts and Woolf, Chaps. 34 and 35 in this volume).
We anticipate that meeting the above (and other) challenges in the HPSE field will greatly benefit from an improved understanding of HPSE structure/function relationships. Structure-guided development of new methods to assess and modulate HPSE activity will doubtless lead to improvements in our ability to treat HPSE driven cancers and other HPSE related diseases. More fundamentally, improved molecular understanding of HPSE activity will also help us better understand the many varied roles of this enzyme in the regulation of HSPGs and the ECM.
References
Sarrazin, S., Lamanna, W. C., & Esko, J. D. (2011). Heparan sulfate proteoglycans. Cold Spring Harbor Perspectives in Biology, 3(7).
Xu, D., & Esko, J. D. (2014). Demystifying heparan sulfate-protein interactions. Annual Review of Biochemistry, 83, 129–157.
Turnbull, J., Powell, A., & Guimond, S. (2001). Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends in Cell Biology, 11(2), 75–82.
Tang, J. P., Julian, J., Glasser, S. R., & Carson, D. D. (1987). Heparan sulfate proteoglycan synthesis and metabolism by mouse uterine epithelial cells cultured in vitro. The Journal of Biological Chemistry, 262(26), 12832–12842.
Schmidt, A., Skaletz-Rorowski, A., & Buddecke, E. (1995). Basic fibroblast growth factor controls the expression and molecular structure of heparan sulfate in corneal endothelial cells. European Journal of Biochemistry, 234(2), 479–484.
David, G., Bai, X. M., Van der Schueren, B., Cassiman, J. J., & Van den Berghe, H. (1992). Developmental changes in heparan sulfate expression: In situ detection with mAbs. The Journal of Cell Biology, 119(4), 961–975.
Brickman, Y. G., Ford, M. D., Gallagher, J. T., Nurcombe, V., Bartlett, P. F., & Turnbull, J. E. (1998). Structural modification of fibroblast growth factor-binding heparan sulfate at a determinative stage of neural development. Journal of Biological Chemistry, 273(8), 4350–4359.
Ghadiali, R. S., Guimond, S. E., Turnbull, J. E., & Pisconti, A. (2017). Dynamic changes in heparan sulfate during muscle differentiation and ageing regulate myoblast cell fate and FGF2 signalling. Matrix Biology, 59, 54–68.
Jayson, G. C., Lyon, M., Paraskeva, C., Turnbull, J. E., Deakin, J. A., & Gallagher, J. T. (1998). Heparan sulfate undergoes specific structural changes during the progression from human colon adenoma to carcinoma in vitro. The Journal of Biological Chemistry, 273(1), 51–57.
Knelson, E. H., Nee, J. C., & Blobe, G. C. (2014). Heparan sulfate signaling in cancer. Trends in Biochemical Sciences, 39(6), 277–288.
Kleeff, J., Ishiwata, T., Kumbasar, A., Friess, H., Buchler, M. W., Lander, A. D., & Korc, M. (1998). The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. The Journal of Clinical Investigation, 102(9), 1662–1673.
Feyzi, E., Saldeen, T., Larsson, E., Lindahl, U., & Salmivirta, M. (1998). Age-dependent modulation of heparan sulfate structure and function. The Journal of Biological Chemistry, 273(22), 13395–13398.
Kreuger, J., & Kjellen, L. (2012). Heparan sulfate biosynthesis: Regulation and variability. The Journal of Histochemistry and Cytochemistry, 60(12), 898–907.
Fux, L., Ilan, N., Sanderson, R. D., & Vlodavsky, I. (2009). Heparanase: Busy at the cell surface. Trends in Biochemical Sciences, 34(10), 511–519.
Griffin, L. S., & Gloster, T. M. (2017). The enzymatic degradation of Heparan Sulfate. Protein Peptide Letters, 24(8), 710–722.
Goldberg, R., Meirovitz, A., Hirshoren, N., Bulvik, R., Binder, A., Rubinstein, A. M., & Elkin, M. (2013). Versatile role of heparanase in inflammation. Matrix Biology, 32(5), 234–240.
Elkin, M., Ilan, N., Ishai-Michaeli, R., Friedmann, Y., Papo, O., Pecker, I., & Vlodavsky, I. (2001). Heparanase as mediator of angiogenesis: mode of action. The FASEB Journal, 15(7), –1661.
Cheng, C. C., Lee, Y. H., Lin, S. P., HuangFu, W. C., & Liu, I. H. (2014). Cell-autonomous heparanase modulates self-renewal and migration in bone marrow-derived mesenchymal stem cells. Journal of Biomedical Science, 21, 21.
Whitelock, J. M., Murdoch, A. D., Iozzo, R. V., & Underwood, P. A. (1996). The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. Journal of Biological Chemistry, 271(17), 10079–10086.
Arvatz, G., Weissmann, M., Ilan, N., & Vlodavsky, I. (2016). Heparanase and cancer progression: New directions, new promises. Human Vaccines & Immunotherapeutics, 12(9), 2253–2256.
Maxhimer, J. B., Quiros, R. M., Stewart, R., Dowlatshahi, K., Gattuso, P., Fan, M., Prinz, R. A., & Xu, X. L. (2002). Heparanase-1 expression is associated with the metastatic potential of breast cancer. Surgery, 132(2), 326–333.
Rohloff, J., Zinke, J., Schoppmeyer, K., Tannapfel, A., Witzigmann, H., Mossner, J., Wittekind, C., & Caca, K. (2002). Heparanase expression is a prognostic indicator for postoperative survival in pancreatic adenocarcinoma. British Journal of Cancer, 86(8), 1270–1275.
Nobuhisa, T., Naomoto, Y., Ohkawa, T., Takaoka, M., Ono, R., Murata, T., Gunduz, M., Shirakawa, Y., Yamatsuji, T., Haisa, M., Matsuoka, J., Tsujigiwa, H., Nagatsuka, H., Nakajima, M., & Tanaka, N. (2005). Heparanase expression correlates with malignant potential in human colon cancer. Journal of Cancer Research and Clinical Oncology, 131(4), 229–237.
Gohji, K., Okamoto, M., Kitazawa, S., Toyoshima, M., Dong, J., Katsuoka, Y., & Nakajima, M. (2001). Heparanase protein and gene expression in bladder cancer. The Journal of Urology, 166(4), 1286–1290.
Rabenstein, D. L. (2002). Heparin and heparan sulfate: Structure and function. Natural Product Reports, 19(3), 312–331.
Ogren, S., & Lindahl, U. (1975). Cleavage of macromolecular heparin by an enzyme from mouse mastocytoma. The Journal of Biological Chemistry, 250(7), 2690–2697.
Hook, M., Wasteson, A., & Oldberg, A. (1975). A heparan sulfate-degrading endoglycosidase from rat liver tissue. Biochemical and Biophysical Research Communications, 67(4), 1422–1428.
Wasteson, A., Hook, M., & Westermark, B. (1976). Demonstration of a platelet enzyme, degrading heparan sulphate. FEBS Letters, 64(1), 218–221.
Klein, U., & Von Figura, K. (1976). Partial purification and characterization of heparan sulfate specific endoglucuronidase. Biochemical and Biophysical Research Communications, 73(3), 569–576.
Kramer, R. H., Vogel, K. G., & Nicolson, G. L. (1982). Solubilization and degradation of subendothelial matrix glycoproteins and proteoglycans by metastatic tumor cells. The Journal of Biological Chemistry, 257(5), 2678–2686.
Nakajima, M., Irimura, T., Di Ferrante, D., Di Ferrante, N., & Nicolson, G. L. (1983). Heparan sulfate degradation: Relation to tumor invasive and metastatic properties of mouse B16 melanoma sublines. Science, 220(4597), 611–613.
Vlodavsky, I., Fuks, Z., Bar-Ner, M., Ariav, Y., & Schirrmacher, V. (1983). Lymphoma cell-mediated degradation of sulfated proteoglycans in the subendothelial extracellular matrix: Relationship to tumor cell metastasis. Cancer Research, 43(6), 2704–2711.
Oldberg, A., Heldin, C. H., Wasteson, A., Busch, C., & Hook, M. (1980). Characterization of a platelet endoglycosidase degrading heparin-like polysaccharides. Biochemistry, 19(25), 5755–5762.
Hoogewerf, A. J., Leone, J. W., Reardon, I. M., Howe, W. J., Asa, D., Heinrikson, R. L., & Ledbetter, S. R. (1995). CXC chemokines connective tissue activating peptide-III and neutrophil activating peptide-2 are heparin/heparan sulfate-degrading enzymes. The Journal of Biological Chemistry, 270(7), 3268–3277.
Oosta, G. M., Favreau, L. V., Beeler, D. L., & Rosenberg, R. D. (1982). Purification and properties of human platelet heparitinase. The Journal of Biological Chemistry, 257(19), 11249–11255.
Graham, L. D., & Underwood, P. A. (1996). Comparison of the heparanase enzymes from mouse melanoma cells, mouse macrophages, and human platelets. Biochemistry and Molecular Biology International, 39(3), 563–571.
Goshen, R., Hochberg, A. A., Korner, G., Levy, E., Ishai-Michaeli, R., Elkin, M., de Groot, N., & Vlodavsky, I. (1996). Purification and characterization of placental heparanase and its expression by cultured cytotrophoblasts. Molecular Human Reproduction, 2(9), 679–684.
Freeman, C., & Parish, C. R. (1998). Human platelet heparanase: Purification, characterization and catalytic activity. The Biochemical Journal, 330(Pt 3), 1341–1350.
Kussie, P. H., Hulmes, J. D., Ludwig, D. L., Patel, S., Navarro, E. C., Seddon, A. P., Giorgio, N. A., & Bohlen, P. (1999). Cloning and functional expression of a human heparanase gene. Biochemical and Biophysical Research Communications, 261(1), 183–187.
Toyoshima, M., & Nakajima, M. (1999). Human heparanase. Purification, characterization, cloning, and expression. The Journal of Biological Chemistry, 274(34), 24153–24160.
Vlodavsky, I., Friedmann, Y., Elkin, M., Aingorn, H., Atzmon, R., Ishai-Michaeli, R., Bitan, M., Pappo, O., Peretz, T., Michal, I., Spector, L., & Pecker, I. (1999). Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nature Medicine, 5(7), 793–802.
Fairbanks, M. B., Mildner, A. M., Leone, J. W., Cavey, G. S., Mathews, W. R., Drong, R. F., Slightom, J. L., Bienkowski, M. J., Smith, C. W., Bannow, C. A., & Heinrikson, R. L. (1999). Processing of the human heparanase precursor and evidence that the active enzyme is a heterodimer. The Journal of Biological Chemistry, 274(42), 29587–29590.
Paetzel, M., Karla, A., Strynadka, N. C., & Dalbey, R. E. (2002). Signal peptidases. Chemical Reviews, 102(12), 4549–4580.
van Oers, M. M., Pijlman, G. P., & Vlak, J. M. (2015). Thirty years of baculovirus-insect cell protein expression: From dark horse to mainstream technology. The Journal of General Virology, 96, 6–23.
McKenzie, E., Young, K., Hircock, M., Bennett, J., Bhaman, M., Felix, R., Turner, P., Stamps, A., McMillan, D., Saville, G., Ng, S., Mason, S., Snell, D., Schofield, D., Gong, H. P., Townsend, R., Gallagher, J., Page, M., Parekh, R., & Stubberfield, C. (2003). Biochemical characterization of the active heterodimer form of human heparanase (Hpa1) protein expressed in insect cells. Biochemical Journal, 373, 423–435.
Nardella, C., Lahm, A., Pallaoro, M., Brunetti, M., Vannini, A., & Steinkuhler, C. (2004). Mechanism of activation of human heparanase investigated by protein engineering. Biochemistry, 43(7), 1862–1873.
Wu, L., Viola, C. M., Brzozowski, A. M., & Davies, G. J. (2015). Structural characterization of human heparanase reveals insights into substrate recognition. Nature Structural & Molecular Biology, 22(12), 1016–1022.
Wu, L., Jiang, J., Jin, Y., Kallemeijn, W. W., Kuo, C. L., Artola, M., Dai, W., van Elk, C., van Eijk, M., van der Marel, G. A., Codee, J. D. C., Florea, B. I., Aerts, J., Overkleeft, H. S., & Davies, G. J. (2017). Activity-based probes for functional interrogation of retaining beta-glucuronidases. Nature Chemical Biology, 13(8), 867–873.
Rigden, D. J., Jedrzejas, M. J., & de Mello, L. V. (2003). Identification and analysis of catalytic TIM barrel domains in seven further glycoside hydrolase families. FEBS Letters, 544(1–3), 103–111.
Belov, A. A., & Mohammadi, M. (2013). Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harbor Perspectives in Biology, 5(6).
Dementiev, A., Petitou, M., Herbert, J. M., & Gettins, P. G. W. (2004). The ternary complex of antithrombin-anhydrothrombin heparin reveals the basis of inhibitor specificity. Nature Structural & Molecular Biology, 11(9), 863–867.
Li, W., Johnson, D. J. D., Esmon, C. T., & Huntington, J. A. (2004). Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nature Structural & Molecular Biology, 11(9), 857–862.
Xue, Y., Lee, S., & Ha, Y. (2011). Crystal structure of amyloid precursor-like protein 1 and heparin complex suggests a dual role of heparin in E2 dimerization. Proceedings of the National Academy of Sciences of the United States of America, 108(39), 16229–16234.
DiGabriele, A. D., Lax, I., Chen, D. I., Svahn, C. M., Jaye, M., Schlessinger, J., & Hendrickson, W. A. (1998). Structure of a heparin-linked biologically active dimer of fibroblast growth factor. Nature, 393(6687), 812–817.
Faham, S., Hileman, R. E., Fromm, J. R., Linhardt, R. J., & Rees, D. C. (1996). Heparin structure and interactions with basic fibroblast growth factor. Science, 271(5252), 1116–1120.
Levy-Adam, F., Abboud-Jarrous, G., Guerrini, M., Beccati, D., Vlodavsky, I., & Ilan, N. (2005). Identification and characterization of heparin/heparan sulfate binding domains of the endoglycosidase heparanase. The Journal of Biological Chemistry, 280(21), 20457–20466.
Simizu, S., Ishida, K., Wierzba, M. K., & Osada, H. (2004). Secretion of heparanase protein is regulated by glycosylation in human tumor cell lines. Journal of Biological Chemistry, 279(4), 2697–2703.
He, Y. Q., Sutcliffe, E. L., Bunting, K. L., Li, J., Goodall, K. J., Poon, I. K. A., Hulett, M. D., Freeman, C., Zafar, A., McInnes, R. L., Taya, T., Parish, C. R., & Rao, S. (2012). The endoglycosidase heparanase enters the nucleus of T lymphocytes and modulates H3 methylation at actively transcribed genes via the interplay with key chromatin modifying enzymes. Transcription, 3(3), 130–145.
Ramani, V. C., Yang, Y., Ren, Y., Nan, L., & Sanderson, R. D. (2011). Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. The Journal of Biological Chemistry, 286(8), 6490–6499.
Purushothaman, A., Hurst, D. R., Pisano, C., Mizumoto, S., Sugahara, K., & Sanderson, R. D. (2011). Heparanase-mediated loss of nuclear Syndecan-1 enhances histone Acetyltransferase (HAT) activity to promote expression of genes that drive an aggressive tumor phenotype. Journal of Biological Chemistry, 286(35), 30377–30383.
Yang, Y., Gorzelanny, C., Bauer, A. T., Halter, N., Komljenovic, D., Bauerle, T., Borsig, L., Roblek, M., & Schneider, S. W. (2015). Nuclear heparanase-1 activity suppresses melanoma progression via its DNA-binding affinity. Oncogene, 34(47), 5832–5842.
Schubert, S. Y., Ilan, N., Shushy, M., Ben-Izhak, O., Vlodavsky, I., & Goldshmidt, O. (2004). Human heparanase nuclear localization and enzymatic activity. Laboratory Investigation, 84(5), 535–544.
Freitas, N., & Cunha, C. (2009). Mechanisms and signals for the nuclear import of proteins. Current Genomics, 10(8), 550–557.
Tripathi, C. K., Banga, J., & Mishra, V. (2012). Microbial heparin/heparan sulphate lyases: Potential and applications. Applied Microbiology and Biotechnology, 94(2), 307–321.
Dong, W., Lu, W., McKeehan, W. L., Luo, Y., & Ye, S. (2012). Structural basis of heparan sulfate-specific degradation by heparinase III. Protein & Cell, 3(12), 950–961.
Han, Y. H., Garron, M. L., Kim, H. Y., Kim, W. S., Zhang, Z., Ryu, K. S., Shaya, D., Xiao, Z., Cheong, C., Kim, Y. S., Linhardt, R. J., Jeon, Y. H., & Cygler, M. (2009). Structural snapshots of heparin depolymerization by heparin lyase I. The Journal of Biological Chemistry, 284(49), 34019–34027.
Pikas, D. S., Li, J. P., Vlodavsky, I., & Lindahl, U. (1998). Substrate specificity of heparanases from human hepatoma and platelets. Journal of Biological Chemistry, 273(30), 18770–18777.
Mao, Y., Huang, Y., Buczek-Thomas, J. A., Ethen, C. M., Nugent, M. A., Wu, Z. L. L., & Zaia, J. (2014). A liquid chromatography-mass spectrometry-based approach to characterize the substrate specificity of mammalian Heparanase. Journal of Biological Chemistry, 289(49), 34141–34151.
Okada, Y., Yamada, S., Toyoshima, M., Dong, J., Nakajima, M., & Sugahara, K. (2002). Structural recognition by recombinant human heparanase that plays critical roles in tumor metastasis - hierarchical sulfate groups with differential effects and the essential target disulfated trisaccharide sequence. Journal of Biological Chemistry, 277(45), 42488–42495.
Peterson, S., & Liu, J. (2012). Deciphering mode of action of Heparanase using structurally defined oligosaccharides. Journal of Biological Chemistry, 287(41), 34836–34843.
Peterson, S. B., & Liu, J. (2010). Unraveling the specificity of Heparanase utilizing synthetic substrates. Journal of Biological Chemistry, 285(19), 14504–14513.
Davies, G. J., Wilson, K. S., & Henrissat, B. (1997). Nomenclature for sugar-binding subsites in glycosyl hydrolases. The Biochemical Journal, 321(Pt 2), 557–559.
Vasella, A., Davies, G. J., & Bohm, M. (2002). Glycosidase mechanisms. Current Opinion in Chemical Biology, 6(5), 619–629.
Davies, G. J., Planas, A., & Rovira, C. (2012). Conformational analyses of the reaction coordinate of glycosidases. Accounts of Chemical Research, 45(2), 308–316.
Speciale, G., Thompson, A. J., Davies, G. J., & Williams, S. J. (2014). Dissecting conformational contributions to glycosidase catalysis and inhibition. Current Opinion in Structural Biology, 28, 1–13.
Zechel, D. L., & Withers, S. G. (2000). Glycosidase mechanisms: Anatomy of a finely tuned catalyst. Accounts of Chemical Research, 33(1), 11–18.
Rye, C. S., & Withers, S. G. (2000). Glycosidase mechanisms. Current Opinion in Chemical Biology, 4(5), 573–580.
Michikawa, M., Ichinose, H., Momma, M., Biely, P., Jongkees, S., Yoshida, M., Kotake, T., Tsumuraya, Y., Withers, S. G., Fujimoto, Z., & Kaneko, S. (2012). Structural and biochemical characterization of glycoside hydrolase family 79 beta-glucuronidase from Acidobacterium capsulatum. The Journal of Biological Chemistry, 287(17), 14069–14077.
Bohlmann, L., Tredwell, G. D., Yu, X., Chang, C. W., Haselhorst, T., Winger, M., Dyason, J. C., Thomson, R. J., Tiralongo, J., Beacham, I. R., Blanchard, H., & von Itzstein, M. (2015). Functional and structural characterization of a heparanase. Nature Chemical Biology, 11(12), 955–957.
Naggi, A., Casu, B., Perez, M., Torri, G., Cassinelli, G., Penco, S., Pisano, C., Giannini, G., Ishai-Michaeli, R., & Vlodavsky, I. (2005). Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. Journal of Biological Chemistry, 280(13), 12103–12113.
Choay, J., Petitou, M., Lormeau, J. C., Sinay, P., Casu, B., & Gatti, G. (1983). Structure-activity relationship in heparin: A synthetic pentasaccharide with high affinity for antithrombin III and eliciting high anti-factor Xa activity. Biochemical and Biophysical Research Communications, 116(2), 492–499.
Pala, D., Rivara, S., Mor, M., Milazzo, F. M., Roscilli, G., Pavoni, E., & Giannini, G. (2016). Kinetic analysis and molecular modeling of the inhibition mechanism of roneparstat (SST0001) on human heparanase. Glycobiology, 26(6), 640–654.
Gingis-Velitski, S., Zetser, A., Kaplan, V., Ben-Zaken, O., Cohen, E., Levy-Adam, F., Bashenko, Y., Flugelman, M. Y., Vlodavsky, I., & Ilan, N. (2004). Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. Journal of Biological Chemistry, 279(42), 44084–44092.
Zetser, A., Levy-Adam, F., Kaplan, V., Gingis-Velitski, S., Bashenko, Y., Schubert, S., Flugelman, M. Y., Vlodavsky, I., & Ilan, N. (2004). Processing and activation of latent heparanase occurs in lysosomes. Journal of Cell Science, 117(Pt 11), 2249–2258.
Nadav, L., Eldor, A., Yacoby-Zeevi, O., Zamir, E., Pecker, I., Ilan, N., Geiger, B., Vlodavsky, I., & Katz, B. Z. (2002). Activation, processing and trafficking of extracellular heparanase by primary human fibroblasts. Journal of Cell Science, 115(10), 2179–2187.
Abboud-Jarrous, G., Atzmon, R., Peretz, T., Palermo, C., Gadea, B. B., Joyce, J. A., & Vlodavsky, I. (2008). Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. Journal of Biological Chemistry, 283(26), 18167–18176.
Abboud-Jarrous, G., Rangini-Guetta, Z., Aingorn, H., Atzmon, R., Elgavish, S., Peretz, T., & Vlodavsky, I. (2005). Site-directed mutagenesis, proteolytic cleavage, and activation of human proheparanase. The Journal of Biological Chemistry, 280(14), 13568–13575.
Jiang, J., Kallemeijn, W. W., Wright, D. W., van den Nieuwendijk, A., Rohde, V. C., Folch, E. C., van den Elst, H., Florea, B. I., Scheij, S., Donker-Koopman, W. E., Verhoek, M., Li, N., Schurmann, M., Mink, D., Boot, R. G., Codee, J. D. C., van der Marel, G. A., Davies, G. J., Aerts, J., & Overkleeft, H. S. (2015). In vitro and in vivo comparative and competitive activity-based protein profiling of GH29 alpha-l-fucosidases. Chemical Science, 6(5), 2782.
Jiang, J., Kuo, C. L., Wu, L., Franke, C., Kallemeijn, W. W., Florea, B. I., van Meel, E., van der Marel, G. A., Codee, J. D., Boot, R. G., Davies, G. J., Overkleeft, H. S., & Aerts, J. M. (2016). Detection of active mammalian GH31 alpha-Glucosidases in health and disease using in-class, broad-spectrum activity-based probes. ACS Central Science, 2(5), 351–358.
Kwan, D. H., Jin, Y., Jiang, J., Chen, H. M., Kotzler, M. P., Overkleeft, H. S., Davies, G. J., & Withers, S. G. (2016). Chemoenzymatic synthesis of 6-phospho-cyclophellitol as a novel probe of 6-phospho-beta-glucosidases. FEBS Letters, 590(4), 461–468.
Witte, M. D., Kallemeijn, W. W., Aten, J., Li, K. Y., Strijland, A., Donker-Koopman, W. E., van den Nieuwendijk, A. M., Bleijlevens, B., Kramer, G., Florea, B. I., Hooibrink, B., Hollak, C. E., Ottenhoff, R., Boot, R. G., van der Marel, G. A., Overkleeft, H. S., & Aerts, J. M. (2010). Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nature Chemical Biology, 6(12), 907–913.
Kötzler, M.P., Hancock, S.M. and Withers, S.G. (2014). Glycosidases: Functions, Families and Folds. In eLS, John Wiley & Sons, Ltd (Ed.). https://doi.org/10.1002/9780470015902.a0020548.pub2
Davies, G. J., Gloster, T. M., & Henrissat, B. (2005). Recent structural insights into the expanding world of carbohydrate-active enzymes. Current Opinion in Structural Biology, 15(6), 637–645.
Levasseur, A., Drula, E., Lombard, V., Coutinho, P. M., & Henrissat, B. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnology for Biofuels, 6, 41.
Biely, P. (2012). Microbial carbohydrate esterases deacetylating plant polysaccharides. Biotechnology Advances, 30(6), 1575–1588.
Garron, M. L., & Cygler, M. (2010). Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology, 20(12), 1547–1573.
Lombard, V., Bernard, T., Rancurel, C., Brumer, H., Coutinho, P. M., & Henrissat, B. (2010). A hierarchical classification of polysaccharide lyases for glycogenomics. The Biochemical Journal, 432(3), 437–444.
Coutinho, P. M., Deleury, E., Davies, G. J., & Henrissat, B. (2003). An evolving hierarchical family classification for glycosyltransferases. Journal of Molecular Biology, 328(2), 307–317.
Campbell, J. A., Davies, G. J., Bulone, V., & Henrissat, B. (1997). A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. The Biochemical Journal, 326(Pt 3), 929–939.
Henrissat, B., & Bairoch, A. (1996). Updating the sequence-based classification of glycosyl hydrolases. The Biochemical Journal, 316(Pt 2), 695–696.
Henrissat, B., & Bairoch, A. (1993). New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. The Biochemical Journal, 293(Pt 3), 781–788.
Henrissat, B. (1991). A classification of glycosyl hydrolases based on amino acid sequence similarities. The Biochemical Journal, 280(Pt 2), 309–316.
Henrissat, B., Claeyssens, M., Tomme, P., Lemesle, L., & Mornon, J. P. (1989). Cellulase families revealed by hydrophobic cluster analysis. Gene, 81(1), 83–95.
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., & Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Research, 42(Database issue), D490–D495.
Durand, P., Lehn, P., Callebaut, I., Fabrega, S., Henrissat, B., & Mornon, J. P. (1997). Active-site motifs of lysosomal acid hydrolases: Invariant features of clan GH-A glycosyl hydrolases deduced from hydrophobic cluster analysis. Glycobiology, 7(2), 277–284.
Kuroyama, H., Tsutsui, N., Hashimoto, Y., & Tsumuraya, Y. (2001). Purification and characterization of a beta-glucuronidase from Aspergillus Niger. Carbohydrate Research, 333(1), 27–39.
Eudes, A., Mouille, G., Thevenin, J., Goyallon, A., Minic, Z., & Jouanin, L. (2008). Purification, cloning and functional characterization of an endogenous beta-glucuronidase in Arabidopsis thaliana. Plant & Cell Physiology, 49(9), 1331–1341.
Sasaki, K., Taura, F., Shoyama, Y., & Morimoto, S. (2000). Molecular characterization of a novel beta-glucuronidase from Scutellaria baicalensis georgi. Journal of Biological Chemistry, 275(35), 27466–27472.
Gong, F., Jemth, P., Escobar Galvis, M. L., Vlodavsky, I., Horner, A., Lindahl, U., & Li, J. P. (2003). Processing of macromolecular heparin by heparanase. The Journal of Biological Chemistry, 278(37), 35152–35158.
Courtney, S. M., Hay, P. A., Buck, R. T., Colville, C. S., Phillips, D. J., Scopes, D. I. C., Pollard, F. C., Page, M. J., Bennett, J. M., Hircock, M. L., McKenzie, E. A., Bhaman, M., Felix, R., Stubberfield, C. R., & Turner, P. R. (2005). Furanyl-1,3-thiazol-2-yl and benzoxazol-5-yl acetic acid derivatives: Novel classes of heparanase inhibitor. Bioorganic & Medicinal Chemistry Letters, 15(9), 2295–2299.
Courtney, S. M., Hay, P. A., Buck, R. T., Colville, C. S., Porter, D. W., Scopes, D. I., Pollard, F. C., Page, M. J., Bennett, J. M., Hircock, M. L., McKenzie, E. A., Stubberfield, C. R., & Turner, P. R. (2004). 2,3-Dihydro-1,3-dioxo-1H-isoindole-5-carboxylic acid derivatives: A novel class of small molecule heparanase inhibitors. Bioorganic & Medicinal Chemistry Letters, 14(12), 3269–3273.
Pan, W. T., Miao, H. Q., Xu, Y. J., Navarro, E. C., Tonra, J. R., Corcoran, E., Lahiji, A., Kussie, P., Kiselyov, A. S., Wong, W. C., & Liu, H. (2006). 1-[4-(1H-benzoimidazol-2-yl)-phenyl]-3-[4-(1H-benzoimidazol-2-yl)-phenyl]-urea derivatives as small molecule heparanase inhibitors. Bioorganic & Medicinal Chemistry Letters, 16(2), 409–412.
Xu, Y. J., Miao, H. Q., Pan, W. T., Navarro, E. C., Tonra, J. R., Mitelman, S., Camara, M. M., Deevi, D. S., Kiselyov, A. S., Kussie, P., Wong, W. C., & Liu, H. (2006). N-(4-{4-(1H-benzoimidazol-2-yl)-arylaminol-methyl}-phenyl)-benzamide derivatives as small molecule heparanase inhibitors. Bioorganic & Medicinal Chemistry Letters, 16(2), 404–408.
Liu, C. J., Chang, J. L. N., Lee, P. H., Lin, D. Y., Wu, C. C., Jeng, L. B., Lin, Y. J., Mok, K. T., Lee, W. C., Yeh, H. Z., Ho, M. C., Yang, S. S., Yang, M. D., Yu, M. C., Hu, R. H., Peng, C. Y., Lai, K. L., Chang, S. S. C., & Chen, P. J. (2014). Adjuvant heparanase inhibitor PI-88 therapy for hepatocellular carcinoma recurrence. World Journal of Gastroenterology, 20(32), 11384–11393.
Liao, B. Y., Wang, Z., Hu, J., Liu, W. F., Shen, Z. Z., Zhang, X., Yu, L., Fan, J., & Zhou, J. (2016). PI-88 inhibits postoperative recurrence of hepatocellular carcinoma via disrupting the surge of heparanase after liver resection. Tumor Biology, 37(3), 2987–2998.
Liang, X. J., Yuan, L., Hu, J., Yu, H. H., Li, T., Lin, S. F., & Tang, S. B. (2012). Phosphomannopentaose sulfate (PI-88) suppresses angiogenesis by downregulating heparanase and vascular endothelial growth factor in an oxygen-induced retinal neovascularization animal model. Molecular Vision, 18(169–73), 1649–1657.
Khasraw, M., Pavlakis, N., McCowatt, S., Underhill, C., Begbie, S., de Souza, P., Boyce, A., Parnis, F., Lim, V., Harvie, R., & Marx, G. (2010). Multicentre phase I/II study of PI-88, a heparanase inhibitor in combination with docetaxel in patients with metastatic castrate-resistant prostate cancer. Annals of Oncology, 21(6), 1302–1307.
Elli, S., Stancanelli, E., Handley, P. N., Carroll, A., Urso, E., Guerrini, M., & Ferro, V. (2018). Structural and conformational studies of the heparan sulfate mimetic PI-88. Glycobiology, 28(10), 731–740.
Parish, C. R., Freeman, C., Brown, K. J., Francis, D. J., & Cowden, W. B. (1999). Identification of sulfated oligosaccharide-based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Research, 59(14), 3433–3441.
Ritchie, J. P., Ramani, V. C., Ren, Y. S., Naggi, A., Torri, G., Casu, B., Penco, S., Pisano, C., Carminati, P., Tortoreto, M., Zunino, F., Vlodavsky, I., Sanderson, R. D., & Yang, Y. (2011). SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the Heparanase/Syndecan-1 Axis. Clinical Cancer Research, 17(6), 1382–1393.
Cassinelli, G., Lanzi, C., Tortoreto, M., Cominetti, D., Petrangolini, G., Favini, E., Zaffaroni, N., Pisano, C., Penco, S., Vlodavsky, I., & Zunino, F. (2013). Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochemical Pharmacology, 85(10), 1424–1432.
Galli, M., Chatterjee, M., Grasso, M., Specchia, G., Magen, H., Einsele, H., Celeghini, I., Barbieri, P., Paoletti, D., Pace, S., Sanderson, R. D., Rambaldi, A., & Nagler, A. (2018). Phase I study of the heparanase inhibitor roneparstat: An innovative approach for multiple myeloma therapy. Haematologica, 103(10), E469–E472.
Schultes, B. C., Lolkema, M. P. J. K., Chu, C. L., Zhou, H., Long, A., Lockley, M., Avery, W., Kurtagic, E., Galcheva-Gargova, Z., Miller, P., Duffner, J., Maschek, B. J., Jarlenski, D., Tuveson, D. A., Roach, J., Venkataraman, G., Kishimoto, T. K., & Flaherty, K. T. (2012). M402, a heparan sulfate mimetic and novel candidate for the treatment of pancreatic cancer. Journal of Clinical Oncology, 30(15).
Zhou, H., Roy, S., Cochran, E., Zouaoui, R., Chu, C. L., Duffner, J., Zhao, G. L., Smith, S., Galcheva-Gargova, Z., Karlgren, J., Dussault, N., Kwan, R. Y. Q., Moy, E., Barnes, M., Long, A., Honan, C., Qi, Y. W., Shriver, Z., Ganguly, T., Schultes, B., Venkataraman, G., & Kishimoto, T. K. (2011). M402, a Novel Heparan Sulfate Mimetic, Targets Multiple Pathways Implicated in Tumor Progression and Metastasis. Plos One, 6(6).
Dredge, K., Brennan, T. V., Hammond, E., Lickliter, J. D., Lin, L. W., Bampton, D., Handley, P., Lankesheer, F., Morrish, G., Yang, Y. P., Brown, M. P., & Millward, M. (2018). A phase I study of the novel immunomodulatory agent PG545 (pixatimod) in subjects with advanced solid tumours. British Journal of Cancer, 118(8), 1035–1041.
Ostapoff, K. T., Awasthi, N., Cenik, B. K., Hinz, S., Dredge, K., Schwarz, R. E., & Brekken, R. A. (2013). PG545, an angiogenesis and Heparanase inhibitor, reduces primary tumor growth and metastasis in experimental pancreatic cancer. Molecular Cancer Therapeutics, 12(7), 1190–1201.
Ferro, V., Liu, L. G., Johnstone, K. D., Wimmer, N., Karoli, T., Handley, P., Rowley, J., Dredge, K., Li, C. P., Hammond, E., Davis, K., Sarimaa, L., Harenberg, J., & Bytheway, I. (2012). Discovery of PG545: A highly potent and simultaneous inhibitor of angiogenesis, tumor growth, and metastasis. Journal of Medicinal Chemistry, 55(8), 3804–3813.
Dredge, K., Hammond, E., Handley, P., Gonda, T. J., Smith, M. T., Vincent, C., Brandt, R., Ferro, V., & Bytheway, I. (2011). PG545, a dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models. British Journal of Cancer, 104(4), 635–642.
Dredge, K., Hammond, E., Davis, K., Li, C. P., Liu, L., Johnstone, K., Handley, P., Wimmer, N., Gonda, T. J., Gautam, A., Ferro, V., & Bytheway, I. (2010). The PG500 series: Novel heparan sulfate mimetics as potent angiogenesis and heparanase inhibitors for cancer therapy. Investigational New Drugs, 28(3), 276–283.
Chen, P. J., Lee, P. H., Han, K. H., Fan, J., Cheung, T. T., Hu, R. H., Paik, S. W., Lee, W. C., Chau, G. Y., Jeng, L. B., Wang, H. J., Choi, J. Y., Chen, C. L., Cho, M., Ho, M. C., Wu, C. C., Lee, K. S., Mao, Y., Hu, F. C., & Lai, K. L. (2017). A phase III trial of muparfostat (PI-88) as adjuvant therapy in patients with hepatitis virus related hepatocellular carcinoma (HV-HCC) after resection. Annals of Oncology, 28.
Chhabra, M., & Ferro, V. (2018). The Development of Assays for Heparanase Enzymatic Activity: Towards a Gold Standard. Molecules, 23(11).
Chen, H. M., Armstrong, Z., Hallam, S. J., & Withers, S. G. (2016). Synthesis and evaluation of a series of 6-chloro-4-methylumbelliferyl glycosides as fluorogenic reagents for screening metagenomic libraries for glycosidase activity. Carbohydrate Research, 421, 33–39.
Goldbarg, J. A., Tsou, K. C., Rutenburg, S. H., Rutenburg, A. M., & Seligman, A. M. (1958). A method for the colorimetric determination of alpha-D-Glucosidase with a chromogenic substrate. Archives of Biochemistry and Biophysics, 75(2), 435–442.
Yu-Bin, C., Glew, R. H., Diven, W. F., & Lee, R. E. (1980). Comparison of various β-glucosidase assays used to diagnose gaucher’s disease. Clinica Chimica Acta, 105(1), 41–50.
Salafsky, I. S., & Nadler, H. L. (1973). A fluorometric assay of alpha-glucosidase and its application in the study of Pompe’s disease. The Journal of Laboratory and Clinical Medicine, 81(3), 450–454.
Ball, A. L., Chambers, K. A., Hewinson, M., Navaratnarajah, S., Samrin, L., Thomas, N., Tyler, A. E., Wall, A. J., & Lloyd, M. D. (2008). A microtitre plate assay for measuring glycosidase activity. Journal of Enzyme Inhibition and Medicinal Chemistry, 23(1), 131–135.
McKenzie, E., Tyson, K., Stamps, A., Smith, P., Turner, P., Barry, R., Hircock, M., Patel, S., Barry, E., Stubberfield, C., Terrett, J., & Page, M. (2000). Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochemical and Biophysical Research Communications, 276(3), 1170–1177.
Gross-Cohen, M., Feld, S., Doweck, I., Neufeld, G., Hasson, P., Arvatz, G., Barash, U., Naroditsky, I., Ilan, N., & Vlodavsky, I. (2016). Heparanase 2 attenuates head and neck tumor vascularity and growth. Cancer Research, 76(9), 2791–2801.
Gross-Cohen, M., Feld, S., Naroditsky, I., Nativ, O., Ilan, N., & Vlodavsky, I. (2016). Heparanase 2 expression inversely correlates with bladder carcinoma grade and stage. Oncotarget, 7(16), 22556–22565.
Vlodavsky, I., Gross-Cohen, M., Weissmann, M., Ilan, N., & Sanderson, R. D. (2018). Opposing functions of Heparanase-1 and Heparanase-2 in Cancer progression. Trends in Biochemical Sciences, 43(1), 18–31.
Levy-Adam, F., Feld, S., Cohen-Kaplan, V., Shteingauz, A., Gross, M., Arvatz, G., Naroditsky, I., Ilan, N., Doweck, I., & Vlodavsky, I. (2010). Heparanase 2 interacts with heparan sulfate with high affinity and inhibits heparanase activity. The Journal of Biological Chemistry, 285(36), 28010–28019.
Mahmood, S., Beetz, C., Tahir, M. M., Imran, M., Mumtaz, R., Bassmann, I., Jahic, A., Malik, M., Nurnberg, G., Hassan, S. A., Rana, S., Nurnberg, P., & Hubner, C. A. (2012). First HPSE2 missense mutation in urofacial syndrome. Clinical Genetics, 81(1), 88–92.
Al Badr, W., Al Bader, S., Otto, E., Hildebrandt, F., Ackley, T., Peng, W., Xu, J., Li, J., Owens, K. M., Bloom, D., & Innis, J. W. (2011). Exome capture and massively parallel sequencing identifies a novel HPSE2 mutation in a Saudi Arabian child with Ochoa (urofacial) syndrome. Journal of Pediatric Urology, 7(5), 569–573.
Pang, J., Zhang, S., Yang, P., Hawkins-Lee, B., Zhong, J., Zhang, Y., Ochoa, B., Agundez, J. A., Voelckel, M. A., Fisher, R. B., Gu, W., Xiong, W. C., Mei, L., She, J. X., & Wang, C. Y. (2010). Loss-of-function mutations in HPSE2 cause the autosomal recessive urofacial syndrome. American Journal of Human Genetics, 86(6), 957–962.
Daly, S. B., Urquhart, J. E., Hilton, E., McKenzie, E. A., Kammerer, R. A., Lewis, M., Kerr, B., Stuart, H., Donnai, D., Long, D. A., Burgu, B., Aydogdu, O., Derbent, M., Garcia-Minaur, S., Reardon, W., Gener, B., Shalev, S., Smith, R., Woolf, A. S., Black, G. C., & Newman, W. G. (2010). Mutations in HPSE2 cause urofacial syndrome. American Journal of Human Genetics, 86(6), 963–969.
Ochoa, B. (2004). Can a congenital dysfunctional bladder be diagnosed from a smile? The Ochoa syndrome updated. Pediatric Nephrology, 19(1), 6–12.
Roberts, N. A., Woolf, A. S., Stuart, H. M., Thuret, R., McKenzie, E. A., Newman, W. G., & Hilton, E. N. (2014). Heparanase 2, mutated in urofacial syndrome, mediates peripheral neural development in Xenopus. Human Molecular Genetics, 23(16), 4302–4314.
Guo, C. M., Kaneko, S., Sun, Y., Huang, Y. C., Vlodavsky, I., Li, X. K., Li, Z. R., & Li, X. (2015). A mouse model of urofacial syndrome with dysfunctional urination. Human Molecular Genetics, 24(7), 1991–1999.
Sievers, F., & Higgins, D. G. (2018). Clustal omega for making accurate alignments of many protein sequences. Protein Science, 27(1), 135–145.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Wu, L., Davies, G.J. (2020). An Overview of the Structure, Mechanism and Specificity of Human Heparanase. In: Vlodavsky, I., Sanderson, R., Ilan, N. (eds) Heparanase. Advances in Experimental Medicine and Biology, vol 1221. Springer, Cham. https://doi.org/10.1007/978-3-030-34521-1_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-34521-1_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-34520-4
Online ISBN: 978-3-030-34521-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)