
Abstract
Myogenesis is the formation of skeletal muscle. Pioneering findings addressing the control of myogenesis are paradigmatic for numerous molecular control processes in developmental and cell biology, including stem cell biology and tissue regeneration. Skeletal muscle consists of terminally differentiated cells that have fused to form multinucleated myofibres ensheathed by several hierarchies of connective tissue housing blood vessels and nerves, as well as stem cells. In vertebrates, myogenesis occurs during the embryonic and fetal period, as well as during adulthood upon training or injury, as well as during normal replacement of aged myonuclei. Adult myogenesis relies mainly on satellite cells, however, other cells have also been shown to participate in regeneration, at least under experimental conditions. Specific Muscle Regulatory Factors (MRFs) and surface markers are well characterized in all steps of myogenesis including muscle stem cells. We introduce the mechanisms of stem cell activation and the cross-talk between cells contributing to the stem cell niche. Finally, the basic underlying causes of the most frequent myopathies are introduced and stem cell-based therapy approaches for myopathies and muscle aging as well as the advantages and problems of these approaches are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

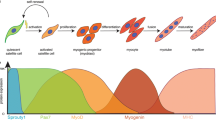

To understand the biology of skeletal muscle stem cells involved in muscle regeneration, you have to understand myogenesis in the embryo. In this chapter, the steps and regulators of myogenesis are introduced. You will learn about the sources of muscle progenitors in the mesoderm and their distribution in the embryo. During this process, muscle stem cells are set aside which attach to the muscle fibres as undifferentiated quiescent satellite cells representing the main source for muscle growth and regeneration. In addition to satellite cells, pericytes, endothelial and interstitial cells, mesoangioblasts and side-population cells possess myogenic potential. You will learn about skeletal muscle specific transcription factors (MRFs) and their functions. Finally, you will learn about muscle wasting, which occurs during aging and muscle dystrophies. In this context, novel stem cell-based approaches involving reprogramming will be explained.
5.1 Myogenesis in the Embryo
Skeletal muscles, the most abundant tissue in the human body forms about 40% of the total body mass. It is involved in the control of movement, posture, breathing as well as control of whole-body metabolism (Frontera and Ochala 2015). The skeletal muscle tissue is made up of long terminally differentiated multinucleated cells (myofibres) that are ensheathed in several hierarchies of connective tissues containing blood vessels, nerves and stem cells. These myofibres contain specialized proteins; actin and myosin that enable the muscle to perform its contractile function to bring about various movements in the body as well as maintenance of posture of the body (Sambasivan and Tajbakhsh 2015). These muscle fibres are formed throughout the body and during the entire life of vertebrates. The progenitor cells of these elongated multinucleated cells in the vertebrate embryo originate from distinct mesoderm populations. The muscles of the trunk and its appendages are derived from the somites, bilateral paired blocks of paraxial mesoderm that form along both sides of the notochord and the neural tube (Yusuf and Brand-Saberi 2012).
The somites are initially epithelial spheres filled by losely-packed mesenchymal cells and are developed in a cranio-caudal sequence to form sequential portions of paraxial mesoderm (Christ and Ordahl 1995). With progress of the developmental processes, the somites are transformed into more complex structures: The dorsally located dermomyotome which yields epaxial and hypaxial skeletal muscle (Christ et al. 2000) together with other derivatives such as angioblasts, dermis and smooth muscle (Kalcheim et al. 1999; Ben-Yair and Kalcheim 2008), and the ventral sclerotome, which differentiates into axial cartilages of the vertebral column and ribs (Christ and Ordahl 1995).
The head and neck muscles are heterogeneous in origin. The head muscles such as the extraocular muscles, muscles of mastication and muscles of facial expression are derived from cells of the pre-otic paraxial head mesoderm and the prechordal mesoderm. The pre-otic paraxial mesoderm cells migrate into the first and second pharyngeal arches, respectively. The other muscles of the head such as the tongue muscles, hypobranchial muscles and the posterior pharyngeal muscles arise from the occipital somites that migrate into the third pharyngeal arch. The pharyngeal mesoderm forms the inner core of the pharyngeal arches and is made up of cells from the paraxial mesoderm and the splanchnic mesoderm which are almost not separable. The regulatory networks governing the development of the craniofacial muscles and the trunk muscles are distinct, both at signaling level and the level of the transcription factors (Tzahor 2015).
5.2 Molecular Regulation of Embryonic Myogenesis
Skeletal muscle development in the embryo is controlled both by intrinsic and extrinsic regulatory pathways. For example, Myf5; MyoD double knockout embryos in which MRF4 expression is not compromised fail to develop limb and craniofacial muscles whereas some trunk muscles are developed (Kassar-Duchossoy et al. 2004). On the other hand, mice lacking both Pax3/Myf5 (and MRF4) are unable to develop trunk muscles but are able to develop normal head muscles (Tajbakhsh et al. 1997), an indication that different molecular pathways are involved in the development of the craniofacial, trunk and limb muscles. Thus, Pax3 is required for the expression of MyoD in the trunk and not the head, which is consistent with the absence of expression of Pax3 in the muscle progenitors of the head (Hacker and Guthrie 1998; Harel et al. 2009).
Specification of skeletal myoblasts develops in the somites in response to signaling molecules from the neighbouring tissues such as the neural tube, notochord and dorsal ectoderm (Fan and Tessier-Lavigne 1994). These signaling molecules include the Wnt family, sonic hedgehog (SHH) and noggin as activators, and Bone morphogenic protein 4 (BMP4) as inhibitor (Hirsinger et al. 1997). In the trunk, expression of SHH and noggin by the notochord and the floor of the neural tube cause the ventral part of the somite to form the Pax1 and Pax9-positive sclerotome for vertebral column formation (Huang and Christ 2000). The ectoderm overlying the somite and the dorsal aspect of the neural tube express Wnts which in conjunction with the low levels of SHH causes the dorsal portion of the somite to form the dermomyotome; a sheet-like pseudostratified epithelium with ventrally curved lips. The cells of the dermomyotome express the paired- and homeodomain-containing transcription factors Pax3 (the first skeletal muscle-relevant myogenesis regulator) and Pax7 (Bober et al. 1994). Also, an interaction between the activating Wnts and inhibitory BMPs directs the dorsomedial portion of the dermomyotome to form the Myf5-positive muscle precursor cells of the primary myotome. The latter consists of elongated unit-length muscle pioneer cells spreading from the medial to the lateral extent throughout the myotome (Kahane et al. 2007). According to studies of mutant mice, the induction of Myf5 in the epaxial myotome relies on SHH (Borycki et al. 1999; ◘ Fig. 5.1).

Signals and genes controlling somite compartment formation. WNTs from the dorsal neural tube and ectoderm activate Pax3, which is initially expressed throughout the epithelial somite. It is subsequently maintained only in the dermomyotome, which contains muscle stem cells/progenitors. The myotome is formed from the dermomyotome in two waves: First, Myf5-positive pioneer cells arise from the dorsomedial lip of the dermomyotome. In a second wave, myotome cells are recruited from all four edges of the dermomyotome. The combined influence of activating WNT proteins and inhibitory BMP4 protein controls MyoD expression in the ventrolateral region to create the hypaxial muscle cell precursors. Noggin (BMP inhibitor), secreted by the notochord counteracts the BMPs from the lateral plate. SHH is produced by the notochord and also by the floor plate of the neural tube. It is essential for the expression of Myf5 in the DML (low levels), and also causes the ventral part of the somite to form the sclerotome (high levels). Pax1 and Pax9 are induced in the sclerotome, which control chondrogenesis and vertebra formation. (Adapted from Yusuf and Brand-Saberi (2012))
The Wnt/β-catenin signaling pathway also regulates multiple steps of myogenesis by regulating step-specific targets (Suzuki et al. 2015). During the organization of the mesodermal epithelia to form somites, Wnt6 signaling from the overlying ectoderm maintains the epithelial structure of the dermomyotome of the somite. Transduction of Wnt6 signaling by its receptor molecule frizzled7 (Linker et al. 2005) is mediated by paraxis (bHLH transcription factor; Burgess et al. 1996) and leads to activation of β- catenin required for the maintenance of the epithelial structure of the somite (Linker et al. 2005). Indeed, mouse embryos deficient in Wnt/β-catenin signaling are embryonic lethal by E8.5 with increased cell death (Haegel et al. 1995; Girardi and Le Grand 2018), while those with conditional depletion of β-catenin in the muscle precursor Pax7+ cell lineage show reduced muscle mass and slow myofibres (Hutcheson et al. 2009). Moreover, upregulation of Dkk1/4, a Wnt/β-catenin antagonist (Zorn 2001; Hirata et al. 2011) inhibits muscle development. Hence Wnt/β-catenin signaling plays a crucial role in skeletal muscle development and homeostasis (Suzuki et al. 2015), because the proliferation of adult skeletal muscle stem/precursor cells is also regulated by Wnt/β-catenin signaling.
The myogenic regulatory factors (MRFs) were the first tissue-specific regulators of differentiation (Weintraub et al. 1991). They comprise four distinct muscle-specific transcription factors (MyoD, myogenin, Myf5 and MRF4) that are involved in the regulation of myogenesis in the embryo and in vitro. They belong to the basic-Helix-Loop-Helix superfamily that is involved in establishing as well as maintaining the myogenic lineage (Naidu et al. 1995). Traditionally, the MRFs were classified in those responsible for myogenic specification („early group“: Myf5 and MyoD), myogenin and MRF4 were considered as control factors of muscle differentiation (“late group”). MRFs share many common features and it was later found that they exert overlapping functional activities, e.g. in the absence of Myf5, MRF4 carries out myogenic determination activity, although it was initially described to be involved in myotube differentiation (Summerbell et al. 2002; Kassar-Duchossoy 2004; Moncaut et al. 2013). Despite the overlapping functional activity of this gene family, the temporal and spatial expression patterns of individual members suggest that during normal myogenesis, each plays a unique role in controlling aspects of skeletal muscle myogenesis (Naidu et al. 1995). Indeed, gene ablation studies of the MRFs of this gene family showed their involvement in different aspects of myogenesis (Hasty et al. 1993; Naidu et al. 1995). For instance, Myf-5 and MyoD act upstream of myogenin to specify myoblasts for terminal differentiation while myogenin and MRF4 are directly involved in the differentiation process and trigger the expression of myotube-specific genes (Bentzinger et al. 2012; Ganassi et al. 2018).
5.3 Skeletal Muscle Regeneration
Skeletal muscles have extensive metabolic and functional plasticity as well as a robust regenerative capacity (Tajbakhsh 2009), which enables them to generate new myofibres when they are damaged by injuries or diseases (Carlson 1973). This striking regenerative capacity of skeletal muscle makes it a good tool (Fry et al. 2015) for the study and application of regenerative medicine (Church et al. 1966; Zouraq et al. 2013). The satellite cells between the sarcolemma and the basal lamina of the skeletal muscle syncytium are the main players in the regeneration of skeletal muscles (Tedesco et al. 2010). The satellite cells also contribute to the postnatal growth of the myofibre, which is evident by the higher number (approximately 6–8 times) of nuclei in the adult myofibre as compared to that of the neonate (Mauro 1961). In addition to the satellite cells, other progenitor cells including pericytes, endothelial and interstitial cells located outside the basal lamina have shown some myogenic potential in vitro and after transplantation (Cossu and Biressi 2005). For some time now, there has been much interest in understanding the cellular and molecular mechanisms underlying the regeneration of skeletal muscles in different contexts as such knowledge might contribute to further development of therapies for diseases such as muscular dystrophy; this will be described in more detail in subsequent paragraphs.
Skeletal muscle regeneration employs essential aspects of embryonic myogenesis, and it is a very important homeostatic process in the adult muscle, which allows for repair of damaged muscle fibres. When a muscle fibre is damaged, the satellite cells respond to the injury by activation and re-entry into the cell cycle. The vast majority of the satellite cell- derived progenitors exits the cell cycle after one or more rounds of proliferation and enters a terminal (G0) phase that leads to differentiation, followed by either fusion to one another to generate new muscle fibres or to repair existing muscle fibres (Olguin and Olwin 2004; Olguín and Pisconti 2012; ◘ Fig. 5.2).

Skeletal muscle regeneration. During skeletal muscle injury, the satellite cell expressing Pax7 becomes activated (expressing Pax7, MyoD and Myf5) to enter into the cell cycle. It then divides asymmetrically giving rise to two daughter cells, one of which strongly expresses Pax7 and hence re-enters the quiescent state to replenish the satellite cell stock, while the other one expressing MyoD and Myf5 becomes a myoblast. The myoblast undergoes several cell divisions before cells fuse with each other and differentiate to form myotubes expressing myogenin. The myotube undergoes remodeling and maturation to either fuse with existing myofibres to repair the damaged fibre or form an entirely new myofibre to replace a completetly damaged fibre
There are two concepts to understand the replenishment of the satellite cells in the regenerating myofibres; first, the activated satellite cells have been shown to divide asymmetrically giving rise to a daughter cell that has self-renewal capabilities and another daughter cell that becomes a myoblast (Kuang et al. 2007; Troy et al. 2012; Dumont et al. 2015). Simultaneously, the proliferating myoblasts are induced to upregulate Pax7, which inhibits myogenin expression and promotes the entry of the cell into a mitotically quiescent state (Olguin and Olwin 2004; Wen et al. 2012).
5.4 Stages of Skeletal Muscle Regeneration
Skeletal muscle regeneration proceeds through three sequential but overlapping stages: inflammatory reaction, satellite cell activation and formation of myofibres, and remodeling of the newly formed myofibres (Charge and Rudnicki 2004; Ciciliot and Schiaffino 2010).
During the inflammatory stage, there is an influx of calcium that leads to activation of calcium-dependent proteases such as calpains that disintegrate the myofibril and other cell constituents as a result of the damaged sarcolemma. This together with the entry of plasma proteins and activation of complement cascades induce chemotactic recruitment of neutrophils and macrophages (Tidball 2008). The early macrophages (called inflammatory macrophages) secrete pro-inflammatory cytokines such as tumor necrosis factor α (TNFα) and interleukin 1β (IL-1β) and are responsible for the removal of necrotic tissues from the damaged muscle. At about 24 hours after the onset of injury, these early invading macrophages expressing CD68 (a marker for late endosomes and lysosomes) reach their highest numbers and begin to be replaced by a second type of macrophages expressing CD163 (involved in the removal of proinflammatory ligands), possibly due to a phenotypic switch from CD68+/CD163- to CD68-/CD163+. These late macrophages, also called anti-inflammatory macrophages, secrete anti-inflammatory cytokines such as interleukin 10 (IL-10) that contribute to the termination of the inflammation and release factors that promote myogenic precursor proliferation, growth and differentiation (Cantini et al. 2002; Sonnet et al. 2006; Arnold et al. 2007). Thus, macrophages play a central role in skeletal muscle response to injury by removing necrotic tissues and promoting muscle regeneration (Ciciliot and Schiaffino 2010).
Towards the end of the inflammatory stage, the satellite cells undergo a finely orchestrated cellular and molecular response to regenerate well functional muscle fibres in two ways: By producing myocytes that either fuse with the existing functional fibres to repair them or fuse with each other to form new myofibres to replace the damaged ones (Charge and Rudnicki 2004). Nagata et al. (2006) indicated that multiple signals appear to trigger satellite cell activation, which include sphingosine-1-phosphate synthesized by the plasma membrane, that stimulate the entry of the satellite cells into the cell cycle. Abrogation of the synthesis of sphingosine-1-phosphate renders skeletal muscle regeneration defective. Nitric oxide (NO) has also been found to be necessary for the activation of satellite cells possibly through the activation of matrix metalloproteinases, which induce the production of hepatocyte growth factor (HGF) from the satellite cells. HGF contributes to satellite cell activation (Tatsumi et al. 1998, 2006) and at the same time inhibits satellite cell differentiation (Miller et al. 2000).
The switch of the myoblasts from the proliferation state to the differentiation state, just as in embryonic development, appears to be controlled by the Notch-Wnt signaling pathway with Notch signaling prevalent during the proliferation phase, while Wnt signaling is dominant during the differentiation phase (Conboy and Rando 2002; Brack et al. 2008). After injury, there is sustained Notch signaling, which ensures proper expansion of the satellite cell progeny, while the Wnt signaling drives the differentiation process (Brack et al. 2008).
The regenerated myofibres undergo a variety of remodeling and maturation processes usually based on conditions of the injury such as the type of muscle injury, involvement of blood vessels and re-establishment of neuromuscular and myotendinous connections (Ciciliot and Schiaffino 2010). One of the major factors of muscle regeneration is the successful establishment and maintenance of the basal lamina of the fibres within which satellite cells and myotubes can proliferate and fuse to form normal muscle fibres. In rodents and humans, freshly regenerated muscle fibres are characterized by the presence of their centrally located nuclei (Ciciliot and Schiaffino 2010; Fry et al. 2015). Regenerating muscle fibres may remodel to form different patterns, which include clusters of smaller muscle fibres as a result of non-fusion of myotubes within the same basal lamina or formation of fork fibres as a result of fusion at only one extremity (Schmalbruch 1976). After segmental necrosis, regenerative processes are concentrated at the level of the damaged stump and if the reconstitution of myofibre integrity is prevented by scar tissue that separates the two stumps, then a new myotendinous junctions will be formed (Järvinen et al. 2008) to repair the muscle tissue.
5.5 Types of Muscle Stem/Progenitor Cells
More than 50 years ago, the best described stem cells of skeletal muscle were discovered by Alexander Mauro in transmission electron micrographs: Satellite cells (Mauro 1961). Like most other skeletal muscle progenitors, they arise from the dermomyotomes of the somites within the population of Pax3 expressing cells that somewhat later also express Pax7 (Armand et al. 1983; Gros et al. 2005). In contrast to the postmitotic myonuclei, the satellite cells on muscle fibres are mitotically quiescent and can be activated due to injury or training-stimulated muscle growth. Satellite cells are specified and characterized by the expression of the paired box transcription factor Pax7 that protects them from apoptosis and is essential for the production of fetal myogenic progenitors and myofibres (Seale et al. 2000; Olguin and Olwin 2004; Kassar-Duchossoy et al. 2005; Relaix et al. 2005, 2006; Hutcheson et al. 2009).
Some of the satellite cells also express Myf5 and are thus committed to muscle formation. While the latter are considered as muscle progenitor cells, Pax7+ Myf5- cells are regarded as multipotent stem cells (Asakura et al. 2001). The expression of Myf5 depends on epigenetic changes of the Pax7 as well as the Myf5 locus. First of all, Pax7 has to be methylated at several arginine residues by an arginine methyltransferase called CARM1 (Kawabe et al. 2012). Subsequently, a histone methyltransferase complex is recruited to the Myf5 locus to allow for Myf5 transcription. However, Myf5 is not translated into protein immediately, but sequestered in mRNP granules (Crist et al. 2012). In this way, satellite cells are maintained in the quiescent state, but at the same time are poised for rapid entry into myogenic differentiation upon activation.
The aforementioned chromatin remodelling processes go along with the occurrence of asymmetrical divisions during which two kinds of daughter cells are being generated: the Myf5+ daughter cells poised for muscle differentiation and their Myf5- sisters that will remain quiescent stem cells. These asymmetrical divisions are also controlled by miRNA-489, which maintains stemness and quiescence in one of the daughter cells by inhibiting the translation of the oncogene DEK that leads to the proliferation of committed progenitor cells upon activation (Cheung et al. 2012). Furthermore, miRNA-31 is a component of the mRNP granules in poised daughter cells. It targets Myf5, thus preventing its translation (Crist et al. 2012).
In contrast to the situation in the trunk and limbs, satellite cells in the head and neck region are independent of the Pax3 pathway. Here, satellite cells are derived from the non-somitic cranial paraxial mesoderm and express Mesp1 and Isl1 (Harel et al. 2009). However, Pax7 is activated also in head muscle progenitors during the fetal period and retained in adult satellite cells (Sambasivan et al. 2009; Gnocchi et al. 2009). In addition to the transcription factor Pax7, surface markers have been established in satellite cells, among them c-met, M-cadherin, syndecan3 and 4, Vcam-1, NCAM-1, and CD34, E-cadherin, Vcam1, Icam1, Cldn5 (claudin 5), Esam (endothelial cell-specific adhesion molecule), and Pcdhb9 (Cornelison and Wold 1997; Irintchev et al. 1994; Beauchamp et al. 2000; Fukada et al. 2007). Interestingly, some of these cell surface molecules are shared with hematopoietic stem cells or with endothelial cells pointing to a common derivation within the mesoderm (Kardon et al. 2002), an issue to be kept in mind when reading the following paragraph.
The satellite cell niche is complex and comprises the extracellular matrix and neighbouring cells, which includes the cell contacts and secretome of the latter. The cellular neighbourhood of satellite cells comprises the myofibre, fibroblasts (interstitial cells) and endothelial cells of the capillaries (Christov et al. 2007). During inflammation, it also contains inflammatory cells. The endothelial cells can fuel the proliferation of satellite cells by IGF1, HGF, FGF2, VEGF and PDGFBB (Christov et al. 2007). Interestingly, myoblasts derived from satellite cells and cultivated in dispersed culture employ mechanisms of differentiation that differ from the ones in the presence of their niche, i.e. if they are cultivated together with their myofibre. A key regulator in support of myoblast quiescence in satellite cell–derived myoblast maintained in their niche is the oncogene p53, whereas the majority of dispersed myoblasts are undergoing differentiation upon downregulation of the ERK1 pathway after 3 days in culture (Flamini et al. 2018).
5.5.1 Interstitial Cells: Derived Muscle Stem Cells
Although satellite cells are the main source of muscle stem cells (reviewed by Relaix and Zammit (2012)), several other cells with myogenic potential have been described in the past. However, their topography is less well defined, because with one exception (for blood-derived myogenic stem cells, see below) all are residing outside the basement membrane of the myofibre within the surrounding connective tissue (“interstitium”) in a stricter or broader sense. Some of them cling tightly to vessels. All of these myogenic progenitor cells differ in their potential to contribute to skeletal muscle regeneration by engrafting into preexisting myofibres to a certain extent, which may also partially depend on the assays used by the experimentors. The borders between the subpopulations are floating and the terminology in the literature partially depends on authors and their particular experimental approaches.
Based on their transplantation efficiency, the most important ones next to satellite cells are pericytes (Dellavalle et al. 2007). Fully mature pericytes are highly branched cells accompanying the microvessels in all body tissues. They are regarded as tissue-specific adult stem cells (Dellavalle et al. 2007, 2011) The ancestry of pericytes is diverse and consequently, several subtypes of pericytes can be distinguished on the basis of their markers and differentiation behaviour (Birbrair et al. 2013). Although their developmental origin remains elusive, muscle-derived pericytes share with brain-derived pericytes NG2 proteoglycan and with many others the more ubiquitous PDGFRs (Balabanov et al. 1996) and transitorily the intermediate filament Nestin, the decline of which can be used to distinguish pericytes with a myogenic potential from those with a neurogenic potential (Birbrair et al. 2013). The marker that distinguishes muscle pericytes from other pericytes as well as from satellite cells is Alkaline Phosphatase (AP). AP is neither found on satellite cells nor on myofibres (Dellavalle et al. 2007, 2011). Using AP, it was shown by genetic reporter expression (inducible Alkaline Phosphatase CreERT2) that AP-positive pericytes contributed to developing myofibres as well as to the satellite cell pool. Pericytes have been demonstrated to participate in muscle growth also in vivo (Dellavalle et al. 2011).
For our understanding of muscle dystrophies, it is important to note that pericytes also contribute to the production of adipose tissue and connective tissue within aging or diseased skeletal muscles (fibro-adipogenic progenitors, FAPs). In this context interestingly, two subtypes of muscle pericytes can be distinguished on the basis of PDGFRα: Type-1 muscle pericytes positive for both factors participate in adipogenesis and collagen production, whereas PDGFRα negative type-2 muscle pericytes are involved in myogenesis and angiogenesis (Birbrair et al. 2014; Lemos et al. 2015). Although pericytes have been shown to enter the satellite compartment, it has also been suggested that they exert additional interactive functions that have a positive impact on myogenesis. In this way the reinnervation of skeletal muscle via the recruitment of Schwann cells from pericytes is affected (Birbrair et al. 2013). Secondly, the angiogenic contribution to regenerating muscle is affected (Birbrair et al. 2014) and a third effect has been described via paracrine interactions from pericytes to satellite cells as well as endothelial cells which have been shown to be close neighbours (Christov et al. 2007).
Apart from pericytes, interstitial cells expressing PW1 (PW1+ interstitial cells; PICs) have been described to contribute to myonuclei in regenerating muscle (Relaix et al. 1996; Mitchell et al. 2010; Besson et al. 2011; Pannérec et al. 2013). PW1 is a zinc-finger transcription factor that interacts with the TNF receptor-2 and is also involved in the p53 axis, whereby it regulates the stress response, also in myoblasts (Schwarzkopf et al. 2006). PW1 is encoded by the paternally expressed gene 3 (PEG-3) and has been found to be a reliable marker for adult stem cells that can significantly contribute to the regeneration of tissues of ectodermal, mesodermal and endodermal origin (Besson et al. 2011). In invertebrates (planarians), pluripotent adult stem cells (neoblasts) capable of regenerating the whole body express Piwi, an orthologue of PW1. In vertebrates, PICs are multipotent, but have a strong preference to differentiate towards the mesodermal lineage, especially skeletal and smooth muscle tissue (Mitchell et al. 2010). Those involved in skeletal myogenesis start to express Pax7 in vivo only in response to local stimuli that recruit them to sites of muscle injury and the satellite cell compartment.
Mesenchymal-like cells closely abutting the endothelium of larger vessels have been detected and characterized by FLK1 expression (VEGFR2, KDR, CD309) that can participate in myogenesis under experimental conditions as well (Minasi et al. 2002; Sampaolesi et al. 2003; reviewed by Cossu and Bianco (2003)). These cells are called mesoangioblasts, because of their potential to adopt the angiogenic or the myogenic fate. When they were isolated from healthy dogs and grafted to the dystrophic Golden Retriever, muscle regeneration was significantly improved (Sampaolesi et al. 2006).
Such cell populations in the interstitium resemble mesenchymal (stromal) stem cells (MSCs) in morphology and topography resulting in ongoing discussions about whether or not mesenchymal stem cells can give rise to muscle cell types. First of all, care should be taken to distinguish between skeletal and smooth muscle cells, secondly MSC markers do not overlap with myogenic progenitor markers.
5.5.2 Side-Population
The so-called side-population (SP) represents another group of myogenic progenitors (Gussoni et al. 1999). The term “side population” which is not restricted to muscle stem/progenitor cells refers to the fact that this cell pool is not detected by the usual set of markers applied by flow cytometry for a particular cell population of interest, such as muscle stem/progenitor cells (Golebiewska et al. 2011).
SP cells express the membrane protein ABCG2 (ATP-binding cassette transporters) that is also found in hematopoietic stem cells and has been implied in therapy (multidrug) resistence in cancers. In the case of stem cells bearing this transporter, it is responsible for the exclusion of the dye Hoechst 33342. Thus, the myogenic SP cells are Hoechst-negative (Gussoni et al. 1999; Asakura et al. 2002).
The fact that some of the side population cells also express syndecan 3 and syndecan 4 (regarded as satellite cell markers) along with ABCG2 contributes to the difficulty or even impossibility to characterize muscle progenitor cell subpopulations without any overlap (Tanaka et al. 2009).
5.5.3 Blood-Derived Muscle Stem Cells
In contrast to the aforementioned groups of muscle stem/progenitor cells that reside within complex tissues (muscle resident), a fraction from the peripheral blood consisting of monocytes has been described to have myogenic potencies: The CD133-positive cells (Torrente et al. 2004; Péault et al. 2007; Negroni et al. 2009). CD133 is another name derived from the hematopoietic nomenclature for the multipass transmembrane protein Prominin-1, at first described in murine epithelial tissue (Weigmann et al. 1997) and also a well-known marker of hematopoietic stem and its AC133 epitope is also found on VEGFR2 of endothelial progenitor cells (Shmelkov et al. 2005).
From the clinical angle, it has enormous technical and patient-friendly advantages to obtain such stem cells from the patients’ blood. Thus, the CD133+ cell has received much attention and has successfully been used in gene therapy (Torrente et al. 2007). The coexpression of CD34 allows predictions regarding the proliferative and differentiation behaviour of the CD133+ cells where CD133+/CD34+ cells had a higher myoblast/myotube fusion index after intramuscular injection and even yielded a better outcome in comparison to satellite cells due to a higher migration activity within the host muscle (Negroni et al. 2009).
5.6 Muscular Dystrophies
Muscular dystrophies are inherited diseases usually regarded as a distinct heterogeneous group of diseases that belong to the larger group of myopathies. They are characterized by progressive muscle wasting in an early life phase caused by defects or absence of structural proteins in the muscle tissue that can lead to severe mobility impairment and even a restricted lifespan. They have in common the histological aspect that is mainly characterized by variations in muscle fiber diameter due to different stages of regeneration attempts, and the invasion of macrophages and inflammatory cells. As the disease advances, the skeletal muscles accumulate fibrous and adipose tissue in replacement of the functional muscle tissue (Emery 1993).
We will restrict ourselves here to the brief introduction of three common congenital muscular dystrophies: Duchenne’s muscular dystrophy (DMD), and its milder variant Becker’s muscular dystrophy (BMD), and Limb girdle muscular dystrophy (LGMD) in which the underlying mutations have been identified. DMD is an X-linked disease affecting male newborns with an incidence of about 1 in 3600 per year (Greenberg et al. 1988). Like BMD, it is caused by different kinds of mutations in the largest human gene, encoding the sarcolemma protein dystrophin. Dystrophin is a major component of the dystrophin-dystroglycan complex that combines the cytoskeleton of the myofibre to the extracellular matrix of the endomysium. The gene spans more than two megabases and contains 79 exons and the described mutations comprise frameshifts, deletions and nonsense point mutations resulting in the absence of a functional dystrophin protein. As a consequence, the myofibres are destabilized during contraction and their myonuclei become apoptotic (Meryon 1851, 1852; Duchenne 1868).
Affected boys show weakness in the muscles of their shoulder and pelvic girdles and proximal leg muscles during the first 5 years of life. Patients suffer progressive muscle wasting, which results in the loss of ambulation at 12 or 13 years of age (reviewed by Emery (1993)). The life expectancy has recently slightly increased as a result of improvements in the medical care to about 30 years, however, secondary complications such as infections of the respiratory system and coagulative disorders in the context of surgical interventions may still cause a lethal outcome before the patients reach their twenties. Dystrophin is also an essential component in cardiac muscles; thus, dilatative cardiomyopathy is observed in patients after 10 years and heart failure is one of the inevitable causes of death in DMD patients (Emery 1993).
BMD is less abundant and dystrophin is usually not absent, but present at compromised quantities and qualities, such as shortened dystrophin variants. Thus, it shows typically a later onset at the age of about 12 years and is much less debilitating than DMB.
In contrast to DMD and BMD, Limb Girdle Muscular Dystrophy (LGMD) is an autosomal inherited disease and comprises a somewhat heterogeneous group of muscular dystrophies with 31 different underlying causes (reviewed by Nigro and Savarese (2014)). It can thus be divided into different types depending on the underlying genetic disorders. Thereof, eight mutations are autosomal dominant and 23 are autosomal recessive. The dominant forms (LGMD1) comprise for example myotilin (LGMD1A), lamin A/C (LGMD1B), caveolin 3 (LGMD1C), desmin (LGMD1E); the recessive ones comprise for example calpain (LGMD2A), dysferlin (LGMD2B), γ sarcoglycan (LGMD2C), α sarcoglycan (LGMD2D), β sarcoglycan (LGMD2E), δ sarco- glycan (LGMD2F), telethonin (LGMD2G), titin (LGMD2J), dystroglycan (LGMD2P), and again desmin (LG- MD2R), to name just a few.
In LGMD 1b, the LaminA/C (LMNA) gene is mutated, a feature shared with the Emery-Dreyfuss muscular dystrophy (Morris 2001; Bonne et al. 2000). The lamins are major constituents of the nuclear membrane and loss of function results in compromised cell function and survival. Thus, disruption of cell membrane components is not the only underlying disorder, which causes muscular dystrophies.
After reading the preceeding paragraphs of this chapter, you may wonder why satellite cells are unable to compensate for the loss of myofibres in muscular dystrophies, in particular in the case of defective dystrophin, which was not considered to be expressed in satellite cells (Miranda et al. 1988; Huard et al. 1991). In general, the failure of stem cell therapies was considered to be a result of stem cell exhaustion, similar to the situation in aging muscle (Sousa-Victor and Munoz-Canoves 2016).
However, it has been shown only recently that dystrophin is indeed present also prior to muscle differentiation and that it is critically involved in the asymmetrical divisions occuring during the activation of satellite cells in mdx-mice, a well established murine model for studying Duchenne muscular dystrophy (Dumont et al. 2015; Chang et al. 2016). Since asymmetrical divisions between Myf5+ and Myf5- satellite cells form the basis of generating daughter cells that are competent to enter the differentiation pathway (Myf5+) and to participate in the regeneration of myotubes after satellite cell activation (Kuang et al. 2007), loss of polarity results in a loss of asymmetrical divisions and a failure to produce myogenic satellite cells. Dystrophin associates with the asymmetry regulating proteins Mark2 (Par1b), a Ser/Thr kinase which enables the polarized distribution of Pard3 resulting in the asymmetric activation of p38/α/b and myogenic commitment of daughter cells. Loss of function in the Par complex results in abrogation of asymmetrical division and ensuing absence of myogenic differentiation (Troy et al. 2012). During this process, the dystrophin+ cell is maintained as satellite stem cell, whereas the other daughter cell is the satellite progenitor cell that enters into the myogenic program (◘ Fig. 5.3).

Cell polarity defect in dystrophin deficient satellite cells. a Normal satellite stem cells undergo asymmetric division upon dystrophin-dependent polarization of MARK2 and PARD3 to opposite sides along the apicobasal axis of the dividing cell. This results in asymmetrical distribution of cell fate determinants such as mediators of Notch signaling during mitosis to enforce different cell fates (stem cell self-renewal and myogenic commitment). b Dystrophin-deficiency leads to downregulation of MARK2 in the satellite cells resulting in equal distribution of PARD3 within the dividing cell. The absence of these polarity cues and the abnormal mitotic progression cause the satellite stem cells to undergo cell cycle arrest and they may enter senescence. (Graph redrawn and modified on the basis of Fig. 4 in Chang et al. (2016))
5.7 Stem Cell-Based Therapies
Until now, there has been no cure for the muscular dystrophies. Thus, therapy revolves around surgical interference for contractures, attention to respiratory care and cardiovascular complications. In the past decades, muscular dystrophies have been treated mainly to slow down the process of muscle loss, primarily by interfering with the secondary events such as immune response by administration of glucocorticoids to the patients.
Causative gene-therapies aiming at the restoration of a functional dystrophin by transgenic approaches have faced a number of problems. First of all, dystrophin is the largest gene in the human genome. High-capacity adenoviral vectors can accommodate and transfer full lengh dystrophin (Clemens et al. 1996), however the expression just persists transiently episomal and holds risks for human patients due to likelihood of an immune response or other complications resulting from the viral vectors. To overcome this difficulty, scientists have designed shortened versions of dystrophin comprising only the most essential exons, which could be incorporated into integrating systems like retro/lentiviral vectors or adeno-associated vector (AAV). Alternatively, another related smaller protein linking the cytoskeleton with the extracellular matrix in developing muscle, called utrophin (“dystrophin-related protein”) has been explored to fulfill substitution of dystrophin function (Helliwell et al. 1992; Miura and Jasmin 2006). Adeno-associated vectors that can efficiently transduce satellite cells still have to be explored in more detail, because the commonly used AAV6 and AAV8 serotypes that transduce skeletal muscle successfully fail to transduce satellite cells or at very low efficiencies, rendering the delivery of functional dystrophin genes into satellite cells a challenge (Arnett et al. 2014; Chang et al. 2016).
To date, one of the most promising approaches is exon-skipping. The exon-skipping approach is offered by a few companies uses antisense-oligonucleotides to hide or mask particular defective exons in the gene sequence of the dystrophin gene, in order to avoid the truncation of the dystrophin protein during translation. Instead, a shorter but partially functional gene product will ensue. This will result in a milder manifestation of the disease, comparable to that of Becker’s muscular dystrophy. Depending on the mutation, not all patients can benefit from the exon-skipping method (Fletcher et al. 2010; Aartsma-Rus et al. 2009). Especially frame-shift mutations constituting more than 80% of Duchenne patients are amenable to this approach (Pichavant et al. 2011; Koo and Wood 2013). So far, only a small number of products have been approved by the FDA and the treatment involves regular (weekly) infusions of the patients with the oligonucleotides.
In mdx mice, the CRISPR/Cas approach in vivo combining systemic and local administration of Cas9 and guide RNA, respectively, resulted in a partial rescue of the mdx phenotype (Long et al. 2016; Nelson et al. 2016; Tabebordbar et al. 2016). The immense potential of the CRISPR/Cas approach for genome editing of dystrophin is being exploited and refined successfully (Amoasii et al. 2017).
In order to achieve more lasting effects in human patients, genome editing in skeletal muscle stem cells or induced pluripotent stem cells (iPSC)-derived muscle cells appears to be an approach that should be developed and pursued in future efforts. The difficulty of ex vivo expansion and transducing satellite cells still needs to be solved. On the other hand, the generation of functional satellite-like cells from human iPSCs has turned out challenging. Two-dimensional culture systems with growth factors administrations over several weeks show success in provision of Pax7+ satellite cells from human iPSCs (Chal et al. 2015, 2016). The current challenge is to take the complexity of skeletal muscle development in the near-natural tissue context into account and develop three-dimensional ‘organoid’ differentiation systems (Brand-Saberi and Zaehres 2016).
Recent advances on tissue culture conditions reaching beyond media and soluble factors, have further revealed that a relatively elastic environment enhances the myogenic properties of stem cells (Gilbert et al. 2010; Hosseini et al. 2012). In particular, organoids aiming at disease modeling are rapidly developing also in combination with bioprinting approaches (Brand-Saberi and Zaehres 2016; Kim et al. 2018). In summary, interdisciplinary approaches combining 3D organoid cultures, bioprinting and genome editing appear particularly exciting and promising steps in the process towards understanding and treating skeletal muscle diseases.
Take Home Message
-
1.
Skeletal muscle progenitors of the trunk and limbs arise from the dermomyotomes of the somites. Head and neck muscles are derived from the unsegmented preotic, the prechordal, the paraxial and splanchnic mesoderm.
-
2.
The key regulatory factors of skeletal muscle specification and differentiation belong to the MyoD family of bHLH transcription factors: MyoD, Myf5, Mrf4 and Myogenin, summarized as the muscle regulatory factors (MRFs).
-
3.
The best characterized and most abundant muscle stem cells are the satellite cells, but several other cell types in the interstitium and the blood have been shown to have myogenic competence; they are characterized by a plethora of different marker combinations.
-
4.
Satellite cells depend on the paired box transcription factor Pax7 for their maintenance. They remain in a quiescent state and can undergo asymmetrical divisions upon activation.
-
5.
Muscular dystrophies are inherited muscle wasting diseases still lacking cure; in some of them, cardiac muscle is also affected, for example Duchenne’s muscular dystrophy, an X-linked lethal disease.
-
6.
Stem cell-based approaches are being developed for disease modeling in vitro as well as for patient treatment combining organoid cultures, bioprinting and genome editing in the future.
References (and Further Reading)
Aartsma-Rus, A., Fokkema, I., Verschuuren, J., Ginjaar, I., Van Deutekom, J., van Ommen, G. J., & Den Dunnen, J. T. (2009). Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Human Mutation, 30(3), 293–299.
Amoasii, L., Long, C., Li, H., Mireault, A. A., Shelton, J. M., Sanchez-Ortiz, E., & Hauschka, S. D. (2017). Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Science Translational Medicine, 9(418), eaan8081.
Armand, O., Boutineau, A. M., Mauger, A., Pautou, M. P., & Kieny, M. (1983). Origin of satellite cells in avian skeletal muscles. Archives d’Anatomie Microscopique et de Morphologie Expérimentale, 72(2), 163–181.
Arnett, A. L., Konieczny, P., Ramos, J. N., Hall, J., Odom, G., Yablonka-Reuveni, Z., Chamberlain, J. R., & Chamberlain, J. S. (2014). Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Molecular Therapy: Methods & Clinical Development, 1, 14038.
Arnold, L., Henry, A., Poron, F., Baba-Amer, Y., Van Rooijen, N., Plonquet, A., Gherardi, R. K., & Chazaud, B. (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. The Journal of Experimental Medicine, 204(5), 1057–1069.
Asakura, A., Rudnicki, M. A., & Komaki, M. (2001). Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation, 68(4–5), 245–253.
Asakura, A., Seale, P., Girgis-Gabardo, A., & Rudnicki, M. A. (2002). Myogenic specification of side population cells in skeletal muscle. The Journal of Cell Biology, 159(1), 123–134.
Balabanov, R., Washington, R., Wagnerova, J., & Dore-Duffy, P. (1996). CNS microvascular pericytes express macrophage-like function, cell surface integrin αM, and macrophage marker ED-2. Microvascular Research, 52(2), 127–142.
Beauchamp, J. R., Heslop, L., David, S. W., Tajbakhsh, S., Kelly, R. G., Wernig, A., & Zammit, P. S. (2000). Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. The Journal of Cell Biology, 151(6), 1221–1234.
Bentzinger, C. F., Wang, Y. X., & Rudnicki, M. A. (2012). Building muscle: Molecular regulation of myogenesis. Cold Spring Harbor Perspectives in Biology, 4(2), a008342.
Ben-Yair, R., & Kalcheim, C. (2008). Notch and bone morphogenetic protein differentially act on dermomyotome cells to generate endothelium, smooth, and striated muscle. The Journal of Cell Biology, 180(3), 607–618.
Besson, V., Smeriglio, P., Wegener, A., Relaix, F., Oumesmar, B. N., Sassoon, D. A., & Marazzi, G. (2011). PW1 gene/paternally expressed gene 3 (PW1/Peg3) identifies multiple adult stem and progenitor cell populations. Proceedings of the National Academy of Sciences, 108(28), 11470–11475.
Birbrair, A., Zhang, T., Wang, Z. M., Messi, M. L., Enikolopov, G. N., Mintz, A., & Delbono, O. (2013). Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Research, 10(1), 67–84.
Birbrair, A., Zhang, T., Wang, Z. M., Messi, M. L., Mintz, A., Olson, J. D., & Delbono, O. (2014). Type-2 pericytes participate in normal and tumoral angiogenesis. American Journal of Physiology. Cell Physiology, 307, C25–C38, 2014.
Bober, E., Franz, T., Arnold, H. H., Gruss, P., & Tremblay, P. (1994). Pax-3 is required for the development of limb muscles: A possible role for the migration of dermomyotomal muscle progenitor cells. Development, 120(3), 603–612.
Bonne, G., Mercuri, E., Muchir, A., Urtizberea, A., Becane, H. M., Recan, D., Merlini, L., Wehnert, M., Boor, R., Reuner, U., & Vorgerd, M. (2000). Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Annals of Neurology, 48(2), 170–180.
Borycki, A. G., Brunk, B., Tajbakhsh, S., Buckingham, M., Chiang, C., & Emerson, C. P. (1999). Sonic hedgehog controls epaxial muscle determination through Myf5 activation. Development, 126, 4053–4063.
Boutet, S. C., Cheung, T. H., Quach, N. L., Liu, L., Prescott, S. L., Edalati, A., & Rando, T. A. (2012). Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell, 10(3), 327–336.
Brack, A. S., Conboy, I. M., Conboy, M. J., Shen, J., & Rando, T. A. (2008). A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell, 2(1), 50–59.
Brand-Saberi, B., & Zaehres, H. (2016). The development of anatomy: From macroscopic body dissections to stem cell–derived organoids. Histochemistry and Cell Biology, 146(6), 647–650.
Burgess, R., Rawls, A., Brown, D., Bradley, A., & Olson, E. N. (1996). Requirement of the paraxis gene for somite formation and musculoskeletal patterning. Nature, 384(6609), 570.
Cantini, M., Giurisato, E., Radu, C., Tiozzo, S., Pampinella, F., Senigaglia, D., Zaniolo, G., Mazzoleni, F., & Vitiello, L. (2002). Macrophage-secreted myogenic factors: A promising tool for greatly enhancing the proliferative capacity of myoblasts in vitro and in vivo. Neurological Sciences, 23(4), 189–194.
Carlson, B. M. (1973). The regeneration of skeletal muscle—A review. The American Journal of Anatomy, 137(2), 119–149.
Chal, J., Oginuma, M., Al Tanoury, Z., Gobert, B., Sumara, O., Hick, A., & Tassy, O. (2015). Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nature Biotechnology, 33(9), 962.
Chal, J., Al Tanoury, Z., Hestin, M., Gobert, B., Aivio, S., Hick, A., & Pourquié, O. (2016). Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nature Protocols, 11(10), 1833.
Chang, N. C., Chevalier, F. P., & Rudnicki, M. A. (2016). Satellite cells in muscular dystrophy–lost in polarity. Trends in Molecular Medicine, 22(6), 479–496.
Charge, S. B., & Rudnicki, M. A. (2004). Cellular and molecular regulation of muscle regeneration. Physiological Reviews, 84(1), 209–238.
Cheung, T. H., Quach, N. L., Charville, G. W., Liu, L., Park, L., Edalati, A., Yoo, B., Hoang, P., & Rando, T. A. (2012). Maintenance of muscle stem-cell quiescence by microRNA-489. Nature, 482(7386), 524.
Christ, B., & Ordahl, C. P. (1995). Early stages of chick somite development. Anatomy and Embryology, 191(5), 381–396.
Christ, B., Huang, R., & Wilting, J. (2000). The development of the avian vertebral column. Anatomy and Embryology, 202(3), 179–194.
Christov, C., Chrétien, F., Abou-Khalil, R., Bassez, G., Vallet, G., Authier, F. J., & Gherardi, R. K. (2007). Muscle satellite cells and endothelial cells: Close neighbors and privileged partners. Molecular Biology of the Cell, 18(4), 1397–1409.
Church, J. C., Noronha, R. F., & Allbrook, D. B. (1966). Satellite cells and skeletal muscle regeneration. The British Journal of Surgery, 53(7), 638–642.
Ciciliot, S., & Schiaffino, S. (2010). Regeneration of mammalian skeletal muscle: Basic mechanisms and clinical implications. Current Pharmaceutical Design, 16(8), 906–914.
Clemens, P. R., Kochanek, S., Sunada, Y., Chan, S., Chen, H. H., Campbell, K. P., & Caskey, C. T. (1996 Nov). In vivo muscle gene transfer of full-length dystrophin with an adenoviral vector that lacks all viral genes. Gene Therapy, 3(11), 965–972.
Conboy, I. M., & Rando, T. A. (2002). The regulation of notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Developmental Cell, 3(3), 397–340.
Cornelison, D. D., & Wold, B. J. (1997). Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Developmental Biology, 191(2), 270–283.
Cossu, G., & Bianco, P. (2003). Mesoangioblasts - vascular progenitors for extravascular mesodermal tissues. Current Opinion in Genetics & Development, 13(5), 537–542.
Cossu, G., & Biressi, S. (2005). Satellite cells, myoblasts and other occasional myogenic progenitors: Possible origin, phenotypic features and role in muscle regeneration. Seminars in Cell & Developmental Biology, 16(4–5), 623–631.
Crist, C. G., Montarras, D., & Buckingham, M. (2012). Muscle satellite cells are primed for myogenesis but maintain quiescence with sequestration of Myf5 mRNA targeted by microRNA-31 in mRNP granules. Cell Stem Cell, 11(1), 118–126.
Dellavalle, A., Sampaolesi, M., Tonlorenzi, R., Tagliafico, E., Sacchetti, B., Perani, L., Innocenzi, I., Galvez, B. G., Messina, G., Morosetti, R., Belicchi, M., Peretti, G., Chamberlain SJ Wright, E. W., Torrente, Y., & Li, S. (2007). Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nature Cell Biology, 9(3), 255.
Dellavalle, A., Maroli, G., Covarello, D., Azzoni, E., Innocenzi, A., Perani, L., & Cossu, G. (2011). Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nature Communications, 2, 499.
Duchenne, G. B. (1868). Recherches sur Ie paralysie musculaire pseudohypertrophique ou paralysie myosclerosique. I. Symptomatologie, marche, duree, terminaison. Archives of General Internal Medicine, 11, 179.
Dumont, N. A., Wang, Y. X., Von Maltzahn, J., Pasut, A., Bentzinger, C. F., Brun, C. E., & Rudnicki, M. A. (2015). Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nature Medicine, 21(12), 1455.
Emery, A. E. (1993). Duchenne muscular dystrophy—Meryon’s disease. Neuromuscular Disorders, 3(4), 263–266.
Fan, C. M., & Tessier-Lavigne, M. (1994). Patterning of mammalian somites by surface ectoderm and notochord: Evidence for sclerotome induction by a hedgehog homolog. Cell, 79, 1175–1186.
Flamini, V., Ghadiali, R. S., Antczak, P., Rothwell, A., Turnbull, J. E., & Pisconti, A. (2018). The satellite cell niche regulates the balance between myoblast differentiation and self-renewal via p53. Stem Cell Reports, 10(3), 970–983.
Fletcher, S., Adams, A. M., Johnsen, R. D., Greer, K., Moulton, H. M., & Wilton, S. D. (2010). Dystrophin isoform induction in vivo by antisense-mediated alternative splicing. Molecular Therapy, 18(6), 1218–1223.
Frontera, W. R., & Ochala, J. (2015). Skeletal muscle: A brief review of structure and function. Calcified Tissue International, 96(3), 183–195.
Fry, C. S., Lee, J. D., Mula, J., Kirby, T. J., Jackson, J. R., Liu, F., Yang, L., Mendias, C. L., Dupont-Versteegden, E. E., McCarthy, J. J., & Peterson, C. A. (2015). Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nature Medicine, 21(1), 76.
Fukada, S. I., Uezumi, A., Ikemoto, M., Masuda, S., Segawa, M., Tanimura, N., Yamamoto, H., Miyagoe-Suzuki, Y., & Takeda, S. I. (2007). Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells, 25(10), 2448–2459.
Ganassi, M., Badodi, S., Quiroga, H. P., Zammit, P. S., Hinits, Y., & Hughes, S. M. (2018). Myogenin promotes myocyte fusion to balance fibre number and size. Nature Communications, 9(1), 4232.
Gilbert, P. M., Havenstrite, K. L., Magnusson, K. E., Sacco, A., Leonardi, N. A., Kraft, P., & Blau, H. M. (2010). Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science, 329(5995), 1078–1081.
Girardi, F., & Le Grand, F. (2018). Wnt signaling in skeletal muscle development and regeneration. In J. Larraín & G. Olivares (Eds.), Progress in molecular biology and translational science, Amsterdam: Elsevier Academic Press, 153, 157–179.
Gnocchi, V. F., White, R. B., Ono, Y., Ellis, J. A., & Zammit, P. S. (2009). Further characterisation of the molecular signature of quiescent and activated mouse muscle satellite cells. PLoS One, 4(4), e5205.
Golebiewska, A., Brons, N. H., Bjerkvig, R., & Niclou, S. P. (2011). Critical appraisal of the side population assay in stem cell and cancer stem cell research. Cell Stem Cell, 8(2), 136–147.
Greenberg, C. R., Jacobs, H. K., Nylen, E., Rohringer, M., Averill, N., Van Ommen, G. J. B., & Wrogemann, K. (1988). Gene studies in newborn males with Duchenne muscular dystrophy detected by neonatal screening. The Lancet, 332(8608), 425–427.
Gros, J., Manceau, M., Thomé, V., & Marcelle, C. (2005). A common somitic origin for embryonic muscle progenitors and satellite cells. Nature, 435(7044), 954.
Gussoni, E., Soneoka, Y., Strickland, C. D., Buzney, E. A., Khan, M. K., Flint, A. F., & Mulligan, R. C. (1999). Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature, 401(6751), 390.
Hacker, A., & Guthrie, S. (1998). A distinct developmental programme for the cranial paraxial mesoderm in the chick embryo. Development, 125(17), 3461–3472.
Haegel, H., Larue, L., Ohsugi, M., Fedorov, L., Herrenknecht, K., & Kemler, R. (1995). Lack of beta-catenin affects mouse development at gastrulation. Development, 121(11), 3529–3537.
Harel, I., Nathan, E., Tirosh-Finkel, L., Zigdon, H., Guimarães-Camboa, N., Evans, S. M., & Tzahor, E. (2009). Distinct origins and genetic programs of head muscle satellite cells. Developmental Cell, 16(6), 822–832.
Hasty, P., Bradley, A., Morris, J. H., Edmondson, D. G., Venuti, J. M., Olson, E. N., & Klein, W. H. (1993). Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature, 364(6437), 501.
Helliwell, T. R., Man, N. T., Morris, G. E., & Davies, K. E. (1992). The dystrophin-related protein, utrophin, is expressed on the sarcolemma of regenerating human skeletal muscle fibres in dystrophies and inflammatory myopathies. Neuromuscular Disorders, 2, 177–184.
Hirata, H., Hinoda, Y., Nakajima, K., Kawamoto, K., Kikuno, N., Ueno, K., Yamamura, S., Zaman, M. S., Khatri, G., Chen, Y., & Saini, S. (2011). Wnt antagonist DKK1 acts as a tumor suppressor gene that induces apoptosis and inhibits proliferation in human renal cell carcinoma. International Journal of Cancer, 128(8), 1793–1803.
Hirsinger, E., Duprez, D., Jouve, C., Malapert, P., Cooke, J., & Pourquié, O. (1997). Noggin acts downstream of Wnt and Sonic Hedgehog to antagonize BMP4 in avian somite patterning. Development, 124(22), 4605–4614.
Hosseini, V., Ahadian, S., Ostrovidov, S., Camci-Unal, G., Chen, S., Kaji, H., & Khademhosseini, A. (2012). Engineered contractile skeletal muscle tissue on a microgrooved methacrylated gelatin substrate. Tissue Engineering Part A, 18(23–24), 2453–2465.
Huang, R., & Christ, B. (2000). Origin of the epaxial and hypaxial myotome in avian embryos. Anatomy and Embryology, 202(5), 369–374.
Huard, J., Labrecque, C., Dansereau, G., Robitaille, L., & Tremblay, J. P. (1991). Dystrophin expression in myotubes formed by the fusion of normal and dystrophic myoblasts. Muscle & Nerve: Official Journal of the American Association of Electrodiagnostic Medicine, 14(2), 178–182.
Hutcheson, D. A., Zhao, J., Merrell, A., Haldar, M., & Kardon, G. (2009). Embryonic and fetal limb myogenic cells are derived from developmentally distinct progenitors and have different requirements for β-catenin. Genes & Development, 23(8), 997–1013.
Irintchev, A., Zeschnigk, M., Starzinski-Powitz, A., & Wernig, A. (1994). Expression pattern of M-cadherin in normal, denervated, and regenerating mouse muscles. Developmental Dynamics, 199(4), 326–337.
Järvinen, T. A., Kääriäinen, M., Äärimaa, V., Järvinen, M., & Kalimo, H. (2008). Skeletal muscle repair after exercise-induced injury. In S. Schiaffino & T. Partridge (Eds.), Skeletal muscle repair and regeneration (Vol. 3, pp. 217–242). Dordrecht: Springer.
Kahane, N., Ben-Yair, R., & Kalcheim, C. (2007). Medial pioneer fibers pattern the morphogenesis of early myoblasts derived from the lateral somite. Developmental Biology, 305(2), 439–450.
Kalcheim, C., Cinnamon, Y., & Kahane, N. (1999). Myotome formation: A multistage process. Cell and Tissue Research, 296(1), 161–173.
Kardon, G., Campbell, J. K., & Tabin, C. J. (2002). Local extrinsic signals determine muscle and endothelial cell fate and patterning in the vertebrate limb. Developmental Cell, 3(4), 533–545.
Kassar-Duchossoy, L., Gayraud-Morel, B., Gomès, D., Rocancourt, D., Buckingham, M., Shinin, V., & Tajbakhsh, S. (2004). Mrf4 determines skeletal muscle identity in Myf5: Myod double-mutant mice. Nature, 431(7007), 466.
Kassar-Duchossoy, L., Giacone, E., Gayraud-Morel, B., Jory, A., Gomès, D., & Tajbakhsh, S. (2005). Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes & Development, 19(12), 1426–1431.
Kawabe, Y. I., Wang, Y. X., McKinnell, I. W., Bedford, M. T., & Rudnicki, M. A. (2012). Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell, 11(3), 333–345.
Kim, J. H., Seol, Y. J., Ko, I. K., Kang, H. W., Lee, Y. K., Yoo, J. J., & Lee, S. J. (2018). 3D bioprinted human skeletal muscle constructs for muscle function restoration. Scientific Reports, 8, 12307.
Koo, T., & Wood, M. J. (2013). Clinical trials using antisense oligonucleotides in Duchenne muscular dystrophy. Human Gene Therapy, 24(5), 479–488.
Kuang, S., Kuroda, K., Le Grand, F., & Rudnicki, M. A. (2007). Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell, 129(5), 999–1010.
Lemos, D. R., Babaeijandaghi, F., Low, M., Chang, C. K., Lee, S. T., Fiore, D., & Rossi, F. M. (2015). Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nature Medicine, 21(7), 786.
Linker, C., Lesbros, C., Gros, J., Burrus, L. W., Rawls, A., & Marcelle, C. (2005). β-Catenin-dependent Wnt signalling controls the epithelial organisation of somites through the activation of paraxis. Development, 132(17), 3895–3905.
Long, C., Amoasii, L., Mireault, A. A., McAnally, J. R., Li, H., Sanchez-Ortiz, E., & Olson, E. N. (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science, 351(6271), 400–403.
Mauro, A. (1961). Satellite cell of skeletal muscle fibers. The Journal of Biophysical and Biochemical Cytology, 9(2), 493.
Meryon, E. (1851). On fatty degeneration of the voluntary muscles. Lancet, 2, 588–589.
Meryon, E. (1852). On granular and fatty degeneration of the voluntary muscles. Medico-Chirurgical Transactions, 35, 73.
Miller, K. J., Thaloor, D., Matteson, S., & Pavlath, G. K. (2000). Hepatocyte growth factor affects satellite cell activation and differentiation in regenerating skeletal muscle. American Journal of Physiology. Cell Physiology, 278(1), C174–C181.
Minasi, M. G., Riminucci, M., De Angelis, L., Borello, U., Berarducci, B., Innocenzi, A., & Boratto, R. (2002). The meso-angioblast: A multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development, 129(11), 2773–2783.
Miranda, A. F., Bonilla, E., Martucci, G., Moraes, C. T., Hays, A. P., & Dimauro, S. (1988). Immunocytochemical study of dystrophin in muscle cultures from patients with Duchenne muscular dystrophy and unaffected control patients. The American Journal of Pathology, 132(3), 410.
Mitchell, K. J., Pannérec, A., Cadot, B., Parlakian, A., Besson, V., Gomes, E. R., Marazzi, G., & Sassoon, D. A. (2010). Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nature Cell Biology, 12(3), 257.
Miura, P., & Jasmin, B. J. (2006). Utrophin upregulation for treating Duchenne or Becker muscular dystrophy: How close are we? Trends in Molecular Medicine, 12, 122–129.
Moncaut, N., Rigby, P. W., & Carvajal, J. J. (2013). Dial M (RF) for myogenesis. The FEBS Journal, 280(17), 3980–3990.
Morris, G. E. (2001). The role of the nuclear envelope in Emery–Dreifuss muscular dystrophy. Trends in Molecular Medicine, 7(12), 572–577.
Nagata, Y., Partridge, T. A., Matsuda, R., & Zammit, P. S. (2006). Entry of muscle satellite cells into the cell cycle requires sphingolipid signaling. The Journal of Cell Biology, 174(2), 245–253.
Naidu, P. S., Ludolph, D. C., To RQ, Hinterberger, T. J., & Konieczny, S. F. (1995). Myogenin and MEF2 function synergistically to activate the MRF4 promoter during myogenesis. Molecular and Cellular Biology, 15(5), 2707–2718.
Negroni, E., Riederer, I., Chaouch, S., Belicchi, M., Razini, P., Di Santo, J., Torrente, Y., Butler-Brown, S. G., & Mouly, V. (2009). In vivo myogenic potential of human CD133+ muscle-derived stem cells: A quantitative study. Molecular Therapy, 17(10), 1771–1778.
Nelson, C. E., Hakim, C. H., Ousterout, D. G., Thakore, P. I., Moreb, E. A., Rivera, R. M. C., & Asokan, A. (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science, 351(6271), 403–407.
Nigro, V., & Savarese, M. (2014). Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Acta Myologica, 33(1), 1.
Olguin, H. C., & Olwin, B. B. (2004). Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: A potential mechanism for self-renewal. Developmental Biology, 275(2), 375–388.
Olguín, H. C., & Pisconti, A. (2012). Marking the tempo for myogenesis: Pax7 and the regulation of muscle stem cell fate decisions. Journal of Cellular and Molecular Medicine, 16(5), 1013–1025.
Pannérec, A., Formicola, L., Besson, V., Marazzi, G., & Sassoon, D. A. (2013). Defining skeletal muscle resident progenitors and their cell fate potentials. Development, 140(14), 2879–2891.
Péault, B., Rudnicki, M., Torrente, Y., Cossu, G., Tremblay, J. P., Partridge, T., & Huard, J. (2007). Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Molecular Therapy, 15(5), 867–877.
Pichavant, C., Aartsma-Rus, A., Clemens, P. R., Davies, K. E., Dickson, G., Takeda, S. I., & Tremblay, J. P. (2011). Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Molecular Therapy, 19(5), 830–840.
Relaix, F., & Zammit, P. S. (2012). Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns Centre stage. Development, 139(16), 2845–2856.
Relaix, F., Weng, X., Marazzi, G., Yang, E., Copeland, N., Jenkins, N., & Sassoon, D. (1996). Pw1, a novel zinc finger gene implicated in the myogenic and neuronal lineages. Developmental Biology, 177(2), 383–396.
Relaix, F., Rocancourt, D., Mansouri, A., & Buckingham, M. (2005). A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature, 435(7044), 948.
Relaix, F., Montarras, D., Zaffran, S., Gayraud-Morel, B., Rocancourt, D., Tajbakhsh, S., Mansouri, A., Cumano, A. and Buckingham, M. (2006). Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. Journal of Cell Biology 172, 91–102.
Sambasivan, R., & Tajbakhsh, S. (2015). Adult skeletal muscle stem cells. In B. Brand-Saberi (Ed.), Vertebrate myogenesis. Results and problems in cell differentiation 56 (pp. 191–213). Berlin, Heidelberg: Springer.
Sambasivan, R., Gayraud-Morel, B., Dumas, G., Cimper, C., Paisant, S., Kelly, R. G., & Tajbakhsh, S. (2009). Distinct regulatory cascades govern extraocular and pharyngeal arch muscle progenitor cell fates. Developmental Cell, 16(6), 810–821.
Sampaolesi, M., Torrente, Y., Innocenzi, A., Tonlorenzi, R., D’Antona, G., Pellegrino, M. A., Barresi, R., Bresolin, N., De Angelis, M. G., Campbell, K. P., Bottinelli, R., & Cossu, G. (2003). Cell therapy of α-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science, 301(5632), 487–492.
Sampaolesi, M., Blot, S., D’Antona, G., Granger, N., Tonlorenzi, R., Innocenzi, A., Mognol, P., Thibaud, J. L., Galvez, B. G., Barthelemy, I., Perani, L., Mantero, S., Guttinger, M., Pansarasa, O., Rinaldi, C., Cusella De Angelis, M. G., Torrente, Y., Bordignon, C., Bottinelli, R., & Cossu, G. (2006). Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature, 444(7119), 574.
Schmalbruch, H. (1976). The morphology of regeneration of skeletal muscles in the rat. Tissue & Cell, 8(4), 673–692.
Schwarzkopf, M., Coletti, D., Sassoon, D., & Marazzi, G. (2006). Muscle cachexia is regulated by a p53–PW1/Peg3-dependent pathway. Genes & Development, 20(24), 3440–3452.
Seale, P., Sabourin, L. A., Girgis-Gabardo, A., Mansouri, A., Gruss, P., & Rudnicki, M. A. (2000). Pax7 is required for the specification of myogenic satellite cells. Cell, 102(6), 777–786.
Shmelkov, S. V., Clair, R. S., Lyden, D., & Rafii, S. (2005). AC133/CD133/Prominin-1. The International Journal of Biochemistry & Cell Biology, 37(4), 715–719.
Sonnet, C., Lafuste, P., Arnold, L., Brigitte, M., Poron, F., Authier, F., Chrétien, F., Gherardi, R. K., & Chazaud, B. (2006). Human macrophages rescue myoblasts and myotubes from apoptosis through a set of adhesion molecular systems. Journal of Cell Science, 119(12), 2497–2507.
Sousa-Victor, P., & Munoz-Canoves, P. (2016). Regenerative decline of stem cells in sarcopenia. Molecular Aspects of Medicine, 50, 109–117.
Summerbell, D., Halai, C., & Rigby, P. W. (2002). Expression of the myogenic regulatory factor Mrf4 precedes or is contemporaneous with that of Myf5 in the somitic bud. Mechanisms of Development, 117(1–2), 331–335.
Suzuki, A., Pelikan, R. C., & Iwata, J. (2015). WNT/β-catenin signaling regulates multiple steps of myogenesis by regulating step-specific targets. Molecular and Cellular Biology, 35(10), 1763–1776. https://doi.org/10.1128/MCB.01180-14.
Tabebordbar, M., Zhu, K., Cheng, J. K., Chew, W. L., Widrick, J. J., Yan, W. X., & Cong, L. (2016). In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science, 351(6271), 407–411.
Tajbakhsh, S. (2009). Skeletal muscle stem cells in developmental versus regenerative myogenesis. Journal of Internal Medicine, 266(4), 372–389.
Tajbakhsh, S., Rocancourt, D., Cossu, G., & Buckingham, M. (1997). Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell, 89(1), 127–138.
Tanaka, K. K., Hall, J. K., Troy, A. A., Cornelison, D. D. W., Majka, S. M., & Olwin, B. B. (2009). Syndecan-4-expressing muscle progenitor cells in the SP engraft as satellite cells during muscle regeneration. Cell Stem Cell, 4(3), 217–225.
Tatsumi, R., Anderson, J. E., Nevoret, C. J., Halevy, O., & Allen, R. E. (1998). HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Developmental Biology, 194(1), 114–128.
Tatsumi, R., Liu, X., Pulido, A., Morales, M., Sakata, T., Dial, S., Hattori, A., Ikeuchi, Y., & Allen, R. E. (2006). Satellite cell activation in stretched skeletal muscle and the role of nitric oxide and hepatocyte growth factor. American Journal of Physiology-Cell Physiology, 290(6), C1487–C1494.
Tedesco, F. S., Dellavalle, A., Diaz-Manera, J., Messina, G., & Cossu, G. (2010). Repairing skeletal muscle: Regenerative potential of skeletal muscle stem cells. The Journal of Clinical Investigation, 120(1), 11–19.
Tidball, J. G. (2008). Inflammation in skeletal muscle regeneration. In S. Schiaffino & T. Partridge (Eds.), Skeletal muscle repair and regeneration Vol. 3 (pp. 243–268). Dordrecht: Springer.
Torrente, Y., Belicchi, M., Sampaolesi, M., Pisati, F., Meregalli, M., D’Antona, G., & Pellegrino, M. A. (2004). Human circulating AC133+ stem cells restore dystrophin expression and ameliorate function in dystrophic skeletal muscle. The Journal of Clinical Investigation, 114(2), 182–195.
Torrente, Y., Belicchi, M., Marchesi, C., D’antona, G., Cogiamanian, F., Pisati, F., & Lamperti, C. (2007). Autologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patients. Cell Transplantation, 16(6), 563–577.
Troy, A., Cadwallader, A. B., Fedorov, Y., Tyner, K., Tanaka, K. K., & Olwin, B. B. (2012). Coordination of satellite cell activation and self-renewal by par-complex-dependent asymmetric activation of p38α/β MAPK. Cell Stem Cell, 11(4), 541–553.
Tzahor, E. (2015). Head muscle development. In B. Brand-Saberi (Ed.), Vertebrate Myogenesis. Results and problems in cell differentiation 56 (pp. 123–142). Berlin, Heidelberg: Springer.
Weigmann, A., Corbeil, D., Hellwig, A., & Huttner, W. B. (1997). Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proceedings of the National Academy of Sciences of the United States of America, 94(23), 12425–12430.
Weintraub, H., Davis, R., Tapscott, S., Thayer, M., Krause, M., Benezra, R., Blackwell, T. K., Turner, D., Rupp, R., Hollenberg, S., Zhuang, Y., & Lassar, A. (1991). The myoD gene family: Nodal point during specification of the muscle cell lineage. Science, 251, 761–766.
Wen, Y., Bi, P., Liu, W., Asakura, A., Keller, C., & Kuang, S. (2012). Constitutive notch activation upregulates Pax7 and promotes the self-renewal of skeletal muscle satellite cells. Molecular and Cellular Biology, 32(12), 2300–2311. https://doi.org/10.1128/MCB.06753-11.
Yusuf, F., & Brand-Saberi, B. (2012). Myogenesis and muscle regeneration. Histochemistry and Cell Biology, 138(2), 187–199.
Zhou, B. P., Deng, J., Xia, W., Xu, J., Li, Y. M., Gunduz, M., & Hung, M. C. (2004). Dual regulation of snail by GSK-3β-mediated phosphorylation in control of epithelial–mesenchymal transition. Nature Cell Biology, 6(10), 931.
Zorn, A. M. (2001). Wnt signalling: Antagonistic Dickkopfs. Current Biology, 11(15), R592–R595.
Zouraq, F. A., Stölting, M., & Eberli, D. (2013). Skeletal muscle regeneration for clinical application. In Regenerative medicine and tissue engineering, London: InTech Open, 680–712. https://doi.org/10.5772/55739.
Acknowledgments
At the beginning of 2019, more than 25.000 entries in Pubmed referred to muscle stem cells. The authors apologize to those authors who made significant contributions to this field, but were not mentioned in our textbook chapter due to space restrictions and the prominent teaching purpose of this book, which forced us to make a rigorous selection. The authors wish to thank PD Holm Zaehres, PhD, for discussions and Maximilian Mucke, M.Sc., for technical help. The financial support of the EU’s sixth framework program MYORES (511978), DFG, FoRUM (F647-09, F732N-2011, F873-16), MERCUR (Pr-2012-0058), Deutsche Gesellschaft für Muskelkranke e.V. (DGM Foundation), Freiburg, and Deutsche Duchenne-Stiftung, action benni & co e. V., Bochum, is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Brand-Saberi, B., Offei, E.B. (2020). Skeletal Muscle Stem Cells. In: Brand-Saberi, B. (eds) Essential Current Concepts in Stem Cell Biology. Learning Materials in Biosciences. Springer, Cham. https://doi.org/10.1007/978-3-030-33923-4_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-33923-4_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-33922-7
Online ISBN: 978-3-030-33923-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)