
Abstract
Plant biomass is largely composed of an intricate assembly of polymeric carbohydrates and lignin. Efficient degradation by fungi and other microorganisms requires a suite of extracellular enzymes capable of degrading the polymeric cell wall fractions into monomers, which can then be assimilated by microbes. This chapter describes several spectrophotometric assays to quantify activities of extracellular hydrolytic enzymes required to degrade cellulose and selected hemicellulases. Pure fungal cultures are grown on leaf disks in mineral solution and the culture supernatant is then used as a source of enzymes to determine endohydrolase activities. To this end, supernatants are added to dissolved carbohydrates (e.g., carboxymethylcellulose, mannan, xylan), dinitrosalicylic acid (DNS) is added and the reaction is allowed to proceed for 5 min. Subsequently, enzymatic activity is assessed spectrophotometrically by quantifying the released monomers (glucose, mannose, xylose) at 540 nm. β-Glucosidase activity is measured by using p-nitrophenyl-b-glucoside as substrate, and total cellulase activity is quantified as the release of glucose from filter paper. Knowledge on the amount and catalytic efficiency of extracellular hydrolases degrading polymeric carbohydrates are important in understanding the pattern and extent of microbial litter decomposition.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Carbohydrates
- Cellulases
- Cellulose
- Enzymatic potential
- Hemicellulases
- Hydrolysis
- Litter degradation
- Plant cell walls
- β-Glucosidase
1 Introduction
Plant polysaccharides are the most abundant organic polymers in the biosphere. Microorganisms produce a battery of extracellular hydrolytic and oxidative enzymes to depolymerize them into smaller, more readily useable compounds. These breakdown products are then assimilated and serve as energy sources or precursors in cell biosynthesis.
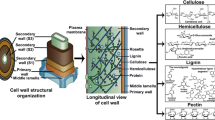
The most abundant biopolymer on earth is cellulose, which is found primarily as a structural component in the cell wall of plants and marine algae. Cellulose consists of long, unbranched homopolymers of D-glucose units linked by β-1,4-glycosidic bonds to form a linear chain of over 10,000 glucose residues (Joshi and Mansfield 2007; Hon and Shiraishi 1991). Individual glucan chains adhere to each other by hydrogen bonding and van der Waals forces, and form insoluble networks. The secondary and tertiary structures of native cellulose are complex and may vary significantly depending on the source and biosynthetic machinery that produces the polymer (e.g. vascular plant or marine algae). Furthermore, the cellulose polymers of higher plants are intricately associated with lignin and hemicellulose moieties, resulting in even more complex morphologies.
Primary cellulose degradation results from either chemical or enzymatic hydrolysis of the polymer into oligomeric and monomeric soluble sugars. Due to the inherent insolubility and physical complexity of polymeric cellulose, several different enzymes are needed for complete solubilization (Mansfield et al. 1999). The current understanding is that enzyme-mediated hydrolysis of native cellulose results primarily from the interaction of extracellular β-1,4-endoglucanases and β-1,4-exoglucanases (cellobiohydrolases) to yield cello-oligosaccharides such as cellobiose, which are subsequently cleaved to glucose by β-glucosidase. The activities of endo- and exoglucanases are synergistic (Mansfield et al. 1999). The general mechanism suggests that the endoglucanases produce free chain ends on the cellulose surface for the cellobiohydrolases to act upon. However, synergy has also been observed between different types of cellobiohydrolases (Nidetzky et al. 1993), as well as between two endoglucanases (Gübitz et al. 1998; Mansfield et al. 1998). Although all cellulolytic enzymes have similar bond specificities (β-1,4), important functional differences are found in their mode of action. More recently, it has been shown that the polysaccharide monooxygenase enzyme AA9 (formerly known as GH61) interacts synergistically with cellulases to enhance the enzymatic hydrolysis of a range of “commercially relevant” pretreated and “model” cellulosic substrates (Hu et al. 2014; Hu et al. 2016).
Generally, the activity of endoglucanases is assayed with a water-soluble substrate, such as carboxymethylcellulose (CMC) or phosphoric acid-swollen cellulose. The assay quantifies the amount of reducing sugars released from the substrate by the interaction with the enzyme (Ghose 1987). In contrast, exoglucanases differ substantially in their substrate specificity and are capable of solubilizing crystalline cellulose substrates, such as Avicel, filter paper, or cotton. Their activity is also usually measured by the amount of reducing ends that are generated (Ghose 1987). An alternative method uses a chromophoric disaccharide derivative and a homologous series of 4-methylumbelliferyl glycosides of cello-oligosaccharides (van Tilbeurgh et al. 1982, Chapter 44).
β-Glucosidases catalyze the hydrolysis of terminal, nonreducing β-D-glucose residues from β-D-glucosides, including cellobiose and cello-oligosaccharides. In some cases, mixed oligosaccharides consisting of mannose and glucose serve as substrates. In the enzymatic conversion of cellulose, it is important that the level of β-glucosidase is in excess, as cellobiose has an inhibitory effect on the cellobiohydrolases (Mansfield et al. 1999).
In addition to cellulose, various hemicelluloses are important polysaccharides in nature. These are low-molecular-weight heteropolymeric polysaccharides composed of a number of different residues, the most common of which are D-xylose, D-mannose, D-galactose, D-glucose, L-arabinose, D-rhamnose, D-galacturonic acid, D-glucuronic acid, and 4-O-methyl-D-glucuonic acid (Fengel and Wegener 1983; Sjöström 1993). The complexity and chemical nature of the hemicelluloses vary both between cell types and species.
The main xylan-derived hemicelluloses are polysaccharides with a backbone of 1,4-linked β-D-xylopyranosyl units, substituted at the carbon 2 and 3 positions. The extent of substitution is dependent on origin (Sjöström 1993): xylans of deciduous trees and conifers carry 4-O-methylglucuronic acid and L-arabinofuranosyl side groups, respectively, while xylans from annual plants may contain only the latter or both side groups. Furthermore, xylans derived from deciduous trees are acetylated, whereas xylans of conifers are not (Sjöström 1993). Xylans from annual plants may, in addition to acetyl groups, carry esterified phenolic hydroxycinnamic acids such as feruloyl and p-coumaroyl moieties (Grabber et al. 2000). The crucial enzyme for xylan depolymerization is endo-β-1,4-xylanase, which preferentially attacks the main xylan chain, generating non-substituted and branched or esterified oligosaccharides. The branching substituents are liberated by corresponding glycosidases or esterases (debranching or accessory enzymes): α-L-arabinofuranosidases and α-glucuronidase. Finally, acetic acid, ferulic acid, and p-coumaric acid residues can be liberated from the xylan by corresponding xylan esterases. β-Xylosidase liberates D-xylose from the nonreducing end of xylo-oligosaccharides.
Mannan-based hemicelluloses are substituted heteropolysaccharides that are widespread in both deciduous and coniferous trees. Their designation is largely dependent on the constituent monomers comprising the backbone and the side chains and can be divided into (1) pure mannans, (2) glucomannans, (3) galactomannans, and (4) galactoglucomannans. The biodegradation of β-mannans occurs by the synergistic action of endo-1,4-β-mannanases, β-D-mannosidases, β-D-glucosidases, α-D-galactosidases, and acetyl mannan esterases (Tenkanen et al. 1993). Endo-β-mannanases cleave polymeric mannans as well as mannooligosaccharides, usually with a degree of polymerization greater than three. Some endomannanases also cleave β-1,4 linkages between mannose and glucose in glucomannans. The degree of substitution and the distribution of the side groups significantly influence the overall capacity for endomannanase to catalyze the degradation of β-1,4 linkages. Thus, the combined actions of endomannanases and accessory enzymes such as α-galactosidase and acetyl esterase are required for total degradation of galactoglucomannan (Tenkanen et al. 1993). β-Mannosidase catalyzes the hydrolysis of terminal, nonreducing β-D-mannose residues in mannans, heteromannans, and mannooligosaccharides. Some β-mannosidases also cleave the 1,4-β-mannose-glucose linkages in glucomannans. β-Mannosidases occur in a wide range of plant and animal tissues and in many microorganisms (Gübitz et al. 1996).
The use of purified enzymes is essential to determine substrate specificities of individual enzymes and to elucidate molecular mechanisms of catalysis. However, simplified assays exist to determine each of the general classes of extracellular cellulolytic and hemicellulolytic enzymes secreted by microorganisms. This chapter presents three types of methods to quantify the major hydrolytic enzymes in fungal cultures, but does not include procedures to determine the debranching enzymes required for total cell wall carbohydrate degradation: (1) The determination of β-1,4-endoglucanases, β-1,4-endoxylanase, and β-1,4-endomannanase (on any mannan-based substrate) follows a variation of Bailey et al. (1992), with appropriate substitution for substrates and corresponding standards. For example, β-1,4-endoglucanases activity is analyzed on carboxymethylcellulose using glucose as a standard. (2) Filter paper activity is a good measure of total cellulase activity. Since exoglucanases are required for the solubilization of crystalline cellulose, this method is also a relatively good indicator of the presence of cellobiohydrolase; however, it does not specifically quantify exoglucanases (Ghose 1987). (3) Finally, β-glucosidase, β-xylosidase, and β-mannosidase are quantified based on an assay by Ghose (1987), with appropriate substitution for substrates and corresponding standards. For example, β-glucosidase activity is determined by using p-nitrophenyl-β-glucoside and glucose.
2 Equipment, Chemicals, and Solutions
2.1 Equipment and Material
-
Analytical balance
-
Cooled centrifuge (4 °C, 20,000 g)
-
Shaking incubator (20 °C)
-
Boiling water bath
-
Water bath (50 °C)
-
Water bath (20 °C)
-
pH meter
-
Magnetic stirrer
-
Spectrophotometer
-
Vortex
-
Laboratory timer or stopwatch
-
Adjustable micropipettors (0.2–1.8 ml)
-
Petri plates
-
Erlenmeyer flasks
-
Centrifuge tubes
-
Cuvettes (disposables are suitable)
-
Test tubes (15 ml)
-
Test tube rack
-
Filter paper (Whatman No. 1)
-
Fungal isolates maintained on 1% malt agar plates at 15–20 °C (to isolate aquatic hyphomycetes, see Chap. 23)
-
Sterilized leaf discs
2.2 Chemicals
-
Agar
-
Malt extract
-
Yeast extract
-
KH2PO4
-
MgSO4∙7H2O
-
NaCl
-
K2HPO4
-
KNO3
-
KCl
-
(NH4)2SO4
-
NaOH
-
3,5-Dinitrosalicylic acid (DNS)
-
KNaC4H4O6·4H2O (Na K tartrate or Rochelle salt)
-
Na2S2O5 (Na metabisulfite)
-
Phenol (melt at 50 °C)
-
Deionized water
-
Glycine
-
Glucose
-
Xylose
-
Mannose
-
Sodium citrate buffer (1 M, pH 4.5)
-
Carboxymethylcellulose (2% w:v in 50 mM sodium citrate buffer)
-
p-Nitrophenyl-β-glucoside
-
p-Nitrophenyl-β-xyloside
-
Birchwood xylan (1% w:v in 50-mM sodium citrate buffer)
-
Mannan (1% w:v in 50-mM sodium citrate buffer)
-
Ivory nut mannan (pure mannan)
-
Konjac mannan (glucomannan)
-
Softwood galactoglucomannan
-
Locust bean mannan or guar gum (galactomannan)
-
p-Nitrophenyl-β-mannoside
2.3 Solutions
-
Mineral solution for fungal growth: 10-mM KNO3, 2.5-mM KH2PO4, 2.5-mM K2HPO4, 3-mN NaCl, 1-mM MgSO4; adjust to pH 7 before autoclaving
-
Solution 1: Dinitrosalicylic acid (DNS) reagent – measure 801.0 ml of deionized water; dissolve 11.2-g NaOH and 6.0-g DNS in about 400 ml of the water in a 1000-ml container; use a powder funnel to add 173.2-g NaK tartrate and 4.7-g Na metabisulfite; use the remaining water to wash all reagents into the 1000-ml container; add 4.3-g phenol melted at 50 °C and stir to dissolve
-
Solution 2: 0.4-M glycine buffer – dissolve 60 g of glycine in 1500 ml of deionized water, add 50% (v:v) NaOH solution until the pH is 10.8, and dilute to exactly 2000 ml
3 Experimental Procedures
3.1 Fungal Growth in Liquid Culture
-
1.
Dispense 150 ml of mineral solution to 500-ml Erlenmeyer flask.
-
2.
Add 1–2 g sterile leaf discs to flask.
-
3.
Inoculate aseptically with a 5-mm plug from a 7–14-day-old culture.
-
4.
Grow isolate as shake flask culture (ca. 140 rpm) at 15–20 °C for 7–21 days.
-
5.
Transfer content of flask to centrifuge tube and recover culture supernatant by centrifugation (20,000 g) for 10 min at 4 °C.
3.2 Endohydrolase Activity
-
1.
Select appropriate standards and substrate.
-
2.
Make up stock solution of standards (i.e., 10 mM).
-
3.
Dilute standard stock to give dilution standards (2, 4, 6, 8, and 10 μM).
-
4.
Dispense 1.5 ml of each standard and unknown substrate in separate 15-ml test tubes.
-
5.
Condition substrate in water bath at 50 °C for at least 5 min.
-
6.
At 1-min intervals, add 0.5 ml of culture filtrate, standards, or buffer (blank) to each test tube containing substrate.
-
7.
Vortex tube and return to water bath.
-
8.
Incubate for exactly 5 min.
-
9.
Stop reaction by adding 3.0-ml DNS reagent.
-
10.
Vortex tube and place directly in boiling water bath for exactly 5 min.
-
11.
Prepare enzyme blank by adding 3.0 ml of DNS to a tube containing substrate, and then add 0.5 ml of culture filtrate and place in the boiling water bath for exactly 5 min.
-
12.
Cool tube down in water bath (20 °C).
-
13.
Zero spectrophotometer with reaction blank at 540 nm.
-
14.
Read absorbance of sample at room temperature at 540 nm.
-
15.
Generate linear standard curve by plotting sugar concentration (μmol ml−1) versus absorbance at 540 nm (correct for dilution of standard concentrations) by forcing line through zero. Obtain slope, intercept, and r2 value, which should be >0.98.
-
16.
Determine net absorbance by subtracting appropriate culture filtrate blanks from hydrolysis samples (averaged value).
-
17.
Determine sugar concentration (μmol ml−1) liberated by hydrolysis from calibration curve.
-
18.
Calculate enzyme activity, determined as nkat ml−1 culture filtrate, by the following equation:
3.3 Filter Paper Activity (Total Cellulase Activity)
-
1.
Prepare glucose standards (6.7, 5.0, 3.3, and 2.0 mg ml−1).
-
2.
Place 50 mg of Whatman No. 1 (1 cm × 6 cm) filter paper into 25-ml test tube.
-
3.
Add 1 ml of 50 mM sodium citrate buffer (pH 4.5).
-
4.
Condition substrate in water bath at 50 °C for at least 5 min.
-
5.
Add 0.5 ml of culture filtrate, sugar stock solutions, and blank (buffer) to individual test tubes and mix.
-
6.
Incubate at 50 °C for exactly 60 min.
-
7.
Terminate reaction by adding 3.0 ml of DNS reagent.
-
8.
Vortex tube and place in boiling water bath for exactly 5 min.
-
9.
Cool tube down in water bath (20 °C).
-
10.
Add 10 ml of deionized water.
-
11.
Zero spectrophotometer with buffer blank at 540 nm.
-
12.
Determine absorbance of sample at 540 nm.
-
13.
Generate linear standard curve by plotting absolute amount of sugar (mg 0.5 ml−1) versus absorbance at 540 nm by forcing line through zero. Obtain slope, intercept, and r2 value, which should be >0.98.
-
14.
Determine net absorbance by subtracting culture filtrate blanks from absorbance of hydrolysis samples.
-
15.
Determine concentration of glucose liberated during the reaction from calibration curve.
-
16.
Calculate total cellulase activity, determined as units ml−1 of culture filtrate, by the following equation:
3.4 β-Glucosidase Activity
-
1.
Add 1 ml of 5 mM p-nitrophenyl -β-glucoside in 50-mM sodium acetate buffer (pH 4.8) to a 10-ml test tube.
-
2.
Add 1.8 ml of 100-mM acetate buffer.
-
3.
Condition substrate in water bath at 50 °C for at least 5 min.
-
4.
To the substrate add 200-μl culture filtrate, and then vortex vigorously.
-
5.
Place in water bath at 50 °C for exactly 30 min.
-
6.
Terminate reaction by adding 4 ml 0.4-M glycine buffer.
-
7.
Cool tube down in water bath (20 °C).
-
8.
Use blank of 200-μl fresh culture media to zero spectrophotometer at 430 nm.
-
9.
Determine absorbance of sample at 430 nm.
-
10.
Generate linear standard curve by plotting sugar concentration (μmol ml−1) versus absorbance at 430 nm (correct for dilution of standard concentrations) by forcing line through zero. Obtain slope, intercept, and r2 value, which should be >0.99.
-
11.
Average duplicate measurements.
-
12.
Determine net absorbance by subtracting appropriate culture filtrate blanks from hydrolysis samples (averaged value).
-
13.
Determine sugar concentration (μmol ml−1) liberated by hydrolysis from calibration curve.
-
14.
Use the following equation to calculate enzyme activity as nkat ml−1 of culture filtrate, where 1 nkat is the activity that releases 1 nmol of p-nitrophenol equivalent per second during the assay:
4 Final Remarks
Instead of culture filtrates, extracts from stream-exposed leaves can be used. However, this generally requires tests to ensure that there is measurable activity.
Activities of hydrolytic enzymes, which are thermostable, have traditionally been measured at 50 °C, even though this is generally far higher than the temperature experienced by microorganisms in the field. These assays thus measure enzymatic potentials. If actual release of sugars under natural conditions is of interest, incubation at ambient stream temperatures is required. This may involve much longer incubation periods, and precautions will have to be taken to prevent bacterial contamination.
In the endocellulase assay, each set of assays has a reagent blank and a set of standards where buffer and standards are added to the reaction instead of culture filtrate. Additionally, each assay has an enzyme blank where DNS is added to the substrate before the enzyme so that enzyme activity is prevented and the reducing sugars in the culture filtrate can be determined.
Enzyme activity is generally expressed in nkat ml−1 of culture filtrate, where 1 nkat is the activity that releases 1 nmol of product (i.e., reducing sugar or p-nitrophenol equivalent) per second during the assay. The international unit (IU) is also often used. It represents the release of 1 μmol of product per minute.
-
1 katal = 1 mol s−1
-
1 nkat = 1 nmol s−1
-
1 IU = 1 μmol min-1 = 16.7 nkat
References
Bailey, M. J., Biely, P., & Poutanen, K. (1992). Interlaboratory testing of methods for assay of xylanase activity. Journal of Biotechnology, 23, 257–270.
Fengel, D., & Wegener, G. (1983). Wood: Chemistry, ultrastructure, reactions. New York: Walter de Gruyter.
Ghose, T. K. (1987). Measurement of cellulase activities. Pure and Applied Chemistry, 59, 257–268.
Grabber, J. H., Ralph, J., & Hatfield, R. D. (2000). Cross-linking of maize walls by ferulate dimerization and incorporation into lignin. Journal of Agriculture and Food Chemistry, 48, 6106–6113.
Gübitz, G. M., Hayn, M., Sommerauer, M., & Steiner, W. (1996). Mannan-degrading enzymes from Sclerotium rolfsii: Characterisation and synergism of two endo β-mannanases and a β-mannosidase. Bioresource Technology, 58, 127–135.
Gübitz, G. M., Mansfield, S. D., Böhm, D., & Saddler, J. N. (1998). Effect of endoglucanases and hemicellulases in magnetic and flotation deinking of xerographic and laser-printed papers. Journal of Biotechnology, 65, 209–215.
Hon, D. N.-S., & Shiraishi, N. (1991). Wood and cellulosic chemistry. New York: Marcel Dekker.
Hu, J., Arantes, V., Pribowo, A., Gourlay, K., & Saddler, J. N. (2014). Substrate factors that influence the synergistic interaction of AA9 and cellulases during the enzymatic hydrolysis of biomass. Energy & Environmental Science, 7, 2308–2315.
Hu, J., Pribowo, A., & Saddler, J. N. (2016). Oxidative cleavage of some cellulosic substrates by auxiliary activity (AA) family 9 enzymes influences the adsorption/desorption of hydrolytic cellulase enzymes. Green Chemistry, 18, 6329–6336.
Joshi, C. P., & Mansfield, S. D. (2007). The cellulose paradox – simple molecule, complex biosynthesis. Current Opinion in Plant Biology, 10, 220–226.
Mansfield, S. D., Saddler, J. N., & Gübitz, G. M. (1998). Characterization of endoglucanases from the brown rot fungi Gloeophyllum sepiarium and Gloeophyllum trabeum. Enzyme and Microbial Technology, 23, 133–140.
Mansfield, S. D., Mooney, C., & Saddler, J. N. (1999). Substrate and enzyme characteristics that limit cellulose hydrolysis. Biotechnology Progress, 15, 804–816.
Nidetzky, B., Hayn, M., Macarron, R., & Steiner, W. (1993). Synergism of Trichoderma reesei cellulases while degrading different celluloses. Biotechnology Letters, 15, 71–76.
Sjöström, E. (1993). Wood chemistry: Fundamentals and applications (2nd ed.). London: Academic.
Tenkanen, M., Puls, J., Rättö, M., & Viikari, L. (1993). Enzymatic deacetylation of galactoglucmannans. Applied Microbiology and Biotechnology, 39, 159–165.
van Tilbeurgh, H., Claeyssens, M., & de Bruyne, C. K. (1982). The use of 4-methylumbelliferyl and other chromophoric glycosides in the study of cellulolytic enzymes. FEMS Letters, 149, 152–156.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mansfield, S.D. (2020). Extracellular Fungal Hydrolytic Enzyme Activity. In: Bärlocher, F., Gessner, M., Graça, M. (eds) Methods to Study Litter Decomposition. Springer, Cham. https://doi.org/10.1007/978-3-030-30515-4_41
Download citation
DOI: https://doi.org/10.1007/978-3-030-30515-4_41
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-30514-7
Online ISBN: 978-3-030-30515-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)