
Abstract
Mammalian frataxin is a small mitochondrial protein involved in iron sulfur cluster assembly. Frataxin deficiency causes the neurodegenerative disease Friedreich’s Ataxia. Valuable knowledge has been gained on the structural dynamics of frataxin, metal-ion-protein interactions, as well as on the effect of mutations on protein conformation, stability and internal motions. Additionally, laborious studies concerning the enzymatic reactions involved have allowed for understanding the capability of frataxin to modulate Fe–S cluster assembly function. Remarkably, frataxin biological function depends on its interaction with some proteins to form a supercomplex, among them NFS1 desulfurase and ISCU, the scaffolding protein. By combining multiple experimental tools including high resolution techniques like NMR and X-ray, but also SAXS, crosslinking and mass-spectrometry, it was possible to build a reliable model of the structure of the desulfurase supercomplex NFS1/ACP-ISD11/ISCU/frataxin. In this chapter, we explore these issues showing how the scientific view concerning frataxin structure-function relationships has evolved over the last years.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Frataxin
- Structural dynamics
- Structure-Function relationships
- Iron binding
- Iron–Sulfur cluster assembly
- Conformational stability
Friedreich’s Ataxia and the History of Frataxin: Why This Chapter is Focused on Frataxin
Friedreich’s Ataxia is a rare autosomal recessive genetic disease (ORPHA95), which is characterized by progressive degeneration and damage of the central and peripheral nervous systems. It has a prevalence of approximately 1:50,000, representing half of all the genetic ataxias and three-quarters of the inherited ataxias in individuals younger than 25 years of age (Pandolfo 2009). The first symptoms occur typically in 8–15-year-olds and some of the most common are ataxia of gait and limbs, absence of tendon reflexes, loss of position sense and dysarthria (alteration in the articulation of words). Remarkably, two-thirds of patients have hypertrophic cardiomyopathy at the time of diagnosis (Durr et al. 1996). The name of this ataxia comes from Dr. Nikolaus Friedreich (1825–1882) who described the disease in five articles published during the late 19th century (Koeppen 2013), presenting the clinical findings of patients with “a severe hereditary disorder of the nervous system”. In these works, Dr. Friedreich identified the main features of the disease and highlighted that it is hereditary and that its onset usually occurs before puberty. It is worthy of mention that Dr. Friedreich described the existence of hypertrophic cardiomyopathy, which is now recognized as the main cause of death in Friedreich’s ataxia.
A hundred years later, in 1988, the genetic alteration responsible for this disease was localized in the long arm of chromosome 9 by Chamberlain and coworkers (Chamberlain et al. 1988, 1989); whereas in 1996, a gene termed X25, was identified in the critical region for the FRDA locus by Campuzano and coworkers (Campuzano et al. 1996). The X25 gene codes for a small protein of 210 amino acid residues that was named frataxin (FXN).
In this seminal work, Campuzano and collaborators described the structure of the FXN gene and found that most patients with Friedreich’s Ataxia (98%) were homozygous for a GAA trinucleotide expansion of 700–800 repeats in most of the cases, localized in the first intron of the gene. Nowadays we also know that the expansion ranges from 70 to more than 1000 GAA triplets in patients, while the first intron of the FXN gene in normal chromosomes contains up to 35 to 40 GAA triplets (Pandolfo 2006; Patel and Isaya 2001). Additionally, close to 2–5% of patients exhibit a compound genotype having an expansion in one allele and a point mutation in the other. The triplet expansion alters FXN gene transcription, decreasing its protein expression, which is essential for viability in mammals. Moreover, FXN has homologs in very distant organisms, like bacteria and yeast. Even if it was soon discovered that FXN is involved in assembly of iron-sulfur clusters, the biological function of FXN have remained elusive for long time. Structural biology and biochemistry concerning this protein have advanced notably in several directions during the last two decades. In particular, during the last 10 years, there have been extremely interesting findings that have changed the paradigm regarding the function of FXN, placing FXN in a new macromolecular context.
The Long Path from Structure to Function: FXN Is Involved in Iron–Sulfur Cluster Assembly
After unambiguously linking the FXN gene to Friedreich’s Ataxia, Campuzano and collaborators were able to predict the secondary structure for the X25-encoded protein: an alpha helical structure for the regions between residues 90–110 and 185–195, with a possible beta sheet region around residues 125–145 and 175–180 (Campuzano et al. 1996). Furthermore, they suggested that the first 22 amino acids might constitute a cleavable N-terminal signal peptide and, furthermore, no transmembrane domain was identified, indicating that FXN is a soluble protein. During the same year, it was shown that FXN has mitochondrial localization (Koutnikova et al. 1997; Priller et al. 1997).
The first experimental structure for FXN was obtained four years later by Dhe-Paganon and coworkers (PDBID: 1EKG). The crystallographic structure (with a 1.8 Å resolution) provided new insights (Dhe-Paganon et al. 2000). First, it revealed a novel fold and confirmed Campuzanos’ secondary structure predictions: two parallel alpha-helical elements supported by a platform, which is provided by a five-stranded, antiparallel beta sheet (Fig. 13.1). By examining the crystallographic structure (PDB ID: 1EKG), one may find that the correct boundaries for the helical stretches are Glu92 to Ala114 for the first and Leu182 to Leu194 for the second alpha helix. Additionally, a C-terminal segment of 15 residues, with non-periodic structure lays over the alpha helices. Throughout this chapter, this segment will be called the C-terminal region (CTR). Second, the authors identified two different protein surfaces, both very conserved through evolution. One of them, the region known as the “acidic ridge”, is formed by several acidic residues of the alpha helix 1, loop-1 and the beta strand 1 (Fig. 13.2) and it comprises ~915 Å2, accounting for ~20% of the total accessible surface area of the protein. The other conserved surface forms an extended patch of ~1000 Å2 involving apolar and positively charged residues from the beta-sheet. Interestingly, considering the amino acid conservation profile, the researchers predicted that this surface must be involved in protein-protein interaction processes. Given that some mutations that result in FRDA are in the anionic (D122Y) or in the apolar/positive (G130 V, W155R, and R165C) surfaces, the authors suggested that these surfaces may be critical for FXN function (Fig. 13.3). Moreover, structure-function-phenotype relationships were suggested because some of these mutations result in specific FRDA phenotypes. Without any doubt, the interpretation of these findings was confirmed some years later when FXN was put in context and its essential interaction with other proteins, by means of these surfaces, was established. We will analyze this issue in detail later in this chapter.

The human FXN globular domain. The ribbon model of PDB ID: 1EKG is shown. Loop-1 (Cyan), as well as α-helices and β-strands are indicated. Additionally, CTR is depicted in red. N and C indicate the N- and the C-terminal residues, respectively

The negative charged surface of the acidic ridge of FXN. a and b are two different views of the representation of the FXN electrostatic surface. c and d are in the same orientation as panels (a) and (b), respectively, but show the topology of the protein. In (c), Glu and Asp residues present in the acidic ridge (alpha helix 1, loop-1 and beta strand 1, in red ribbon) are shown. Electrostatics was calculated using APBS and the color scale ranges from +5 (blue) to −5 kT/e (red)

Crucial Residues for FXN Stability and Function. Panels a, b, c and d show some of the residues involved in FRDA. Mutations that result in the FRDA phenotype affect stability and/or protein function. They are the following: in the region of loop-1 and β-turn β2-β3: G137V, D122Y, Y122D; residues located in the core region of the protein: L106S, I154F, L156P, W173G, L182F, H183R, L182H, L186R, L190P, L198R; residues located on the accessible face of the β-sheet and β-turn β1-β2: G130V, N146K, Q148R, W155R, R165C, R165D, R165N, R165P
Some controversies have emerged regarding the processing of FXN inside the mitochondria and as to which is the functional species. Nevertheless, it was found that FXN has a disordered N-terminal region (Popovic et al. 2015; Prischi et al. 2009; Castro et al. 2018), with a mitochondrial translocation signal that spans the first 41 residues. FXN is processed at least in two steps, the first producing an intermediary form (FXN 42-210) and the second producing the mature (FXN 81-210) and full-functional form (Koutnikova et al. 1998; Schmucker et al. 2008; Cavadini et al. 2000a) (Fig. 13.4). Additionally, the first ~ten residues of the mature form (residues 81 to 90) are disordered, though they may be functionally relevant (Gentry et al. 2013).

The Sequence of the Precursor Form of Human FXN (residues 1–210). MISP is the mitochondrial import signal peptide. In addition, boxes corresponding to α-helices, loops and β-strands are represented accordingly to PDB ID: 1EKG in magenta, cyan and blue, respectively. The C-terminal region (CTR) of FXN is highlighted in red. The amino acid sequence 42–56 is highlighted in gray. Points of processing during the import to the mitochondria: the first cut between residues 41 and 42 yielding the intermediate form and the second cut between residue 80 and 81 yielding the mature form of FXN
It is worthy of mention that at least three different isoforms of FXN (type I, II, III) result from alternative splicing (Xia et al. 2012). Type I is the canonical isoform that is observed in mitochondria and presents 130-aminoacid residues with a molecular weight of 14.2 kDa. Type II is 135-residue long, with an expected molecular mass of 14.9 kDa, meanwhile FXN III has 164 residues with a molecular mass of 18.2 kDa. FXN II and III are located mostly in the cytosol and nucleus, respectively. The expression of these isoforms is tissue-specific. Whereas FXN type II is abundant in the nervous system, type III is abundant in the heart (Xia et al. 2012). Type III showed great iron binding capacity (reducing its toxicity) and it promoted Fe-S production and the increase of aconitase activity (Xia et al. 2012).
Recent reports have described that erythrocytes have a FXN isoform called isoform E (Guo et al. 2018). This protein is 135-amino acids long (similar to type II isoform), lacks the mitochondrial targeting sequence and is N-terminally acetylated, in the first methionine residue. This isoform is three times more abundant in erythrocytes than the mature mitochondrial FXN present in other blood cells (20.9 ± 6.4 ng/mL vs. 7.1 ± 1.0 ng/mL, respectively) (Guo et al. 2018).
It was initially proposed that FXN was involved in iron homeostasis because iron concentrations increase in the mitochondria of FRDA cells. In this context, the presence of the extended acidic ridge suggested that, as for other proteins like Ferritins, iron binding would be mediated by clusters of acidic side-chains of glutamic and aspartic residues. In addition, the presence of acidic residues suggested that FXN might play a role in regulating mitochondrial iron transport. Interestingly, knocking-out the FXN homolog gene in E. coli did not affect cellular iron content and sensitivity to oxidants (Li et al. 1999). This fact indicates that at least bacterial FXN may have a different function than iron binding and/or protection against the oxidative stress. Because of the deficiency in the activity of the aconitase enzyme and the respiratory complexes I, II and III (all of them containing Fe–S clusters), iron–sulfur (Fe–S) proteins were regarded as the targeted proteins in FRDA (Rotig et al. 1997). However, as the Fe–S cluster is highly sensitive to free radicals, FRDA was linked to iron accumulation.
Fe–S clusters are inorganic cofactors of proteins with unusual electronic configurations, critical for electron transfer reactions involving single electrons (Fig. 13.5). Rhombic clusters ([2Fe–2S]) are found in proteins with reductase activity such as ferredoxins and glutaredoxins. The coalescence of two clusters of [2Fe–2S] type yields a cubane-type cluster [4Fe–4S]. This type of cluster is present, for example, in mitochondrial aconitase. The two iron atoms in the upper part of the cubane-type Fe S cluster share a hybrid orbital in which a single electron is delocalized. In the lower plane a similar delocalization occurs yielding reactivity (Rouault 2015).

The atomic structure of clusters. Rhombic iron–sulfur clusters ([2Fe–2S]) from a PDB ID: 2Z7E, mounted on three Cys and one His residue (crystal structure of Aquifex aeolicus ISCU) or mounted on Cys residues (b) from PDB ID: 3P1M (crystal structure of human FDX1). In c the structure of a cubane-type Fe–S cluster from PDB ID: 1C97 is shown (the crystal structure of mutant S642A of mitochondrial aconitase from Bos taurus in complex with isocitrate and oxygen). Iron atoms are represented in green
Wong and collaborators showed that the absence of FXN confers cellular sensitivity to oxidative stress (Wong et al. 1999). Even though iron chelators rescued FRDA fibroblasts from oxidant-induced death (iron is a key reagent in the Fenton reactions), they found only a slight increase of 40% in the mean mitochondrial iron content of FRDA cells.
Remarkably, using a yeast FXN deletant strain as a model and iron chelator in the culture medium, the mitochondrial iron concentration was found to be similar to the one observed for the wild-type yeast, whereas the activity of the respiratory complexes was restored. However, the activity of the mitochondrial aconitase remained low, suggesting that incorporation of the iron–sulfur cluster into some enzymes might be impaired in a way that is not linked to iron toxicity (Foury 1999). Also, in the absence of yeast FXN, the activity of succinate dehydrogenase (another Fe–S-containing enzyme) proved to be diminished, whereas the activity of a non-Fe–S-containing mitochondrial enzymes, the malate dehydrogenase, was unaffected, suggesting a direct role of FXN in the formation of Fe-S clusters (Chen et al. 2002). The direct link between the lack of FXN function and Fe–S cluster assembly deficiency was proved shortly after that observation.
The Gibson’s group studied the phylogenetic distribution of FXN and the co-occurrence of other genes in several genomes and found that FXN has the same distribution as the molecular chaperones hscA and hscB/JAC1, specialized forms of Hsp70 and their J protein co-chaperone, respectively, thus suggesting a direct role in the iron–sulfur cluster protein assembly rather than in iron homeostasis (Huynen et al. 2001). From an experimental viewpoint, but in the same vein, Duby and coworkers showed that even though FXN was not essential for Fe–S protein assembly in yeast, its presence improved the efficiency of the process (Duby et al. 2002). Remarkably, Foury’s group demonstrated, by means of an in vivo screening, that yeast FXN and the ISU1 (which is the mitochondrial scaffold protein for the Fe-S cluster assembly), have strongly linked functions, acting in concert to promote Fe-S cluster assembly (Ramazzotti et al. 2004). Cross-linking experiments in isolated mitochondria provided the evidence for a direct interaction. Meanwhile, by using a mitochondrial glutathione-S transferase fusion protein of yeast FXN, Gerber and collaborators showed that FXN formed a complex with ISU1 and the cysteine desulfurase NFS1, which is the key enzyme that provides the persulfur group, necessary for cluster assembly (Muhlenhoff et al. 2003; Gerber et al. 2003). Moreover, interaction between FXN and these proteins was strikingly increased by iron. This dependence on iron suggested a role for FXN in iron loading of the ISU1.
In fact, it was shown that human FXN is able to bind Fe2+ and Fe3+ metal ions with moderate affinity (10 and 50 µM, respectively) (Yoon and Cowan 2003) and provision of desulfurase as a sulfur donor and holo FXN was sufficient for cluster assembly, obtaining holo ISCU with good yields (Nuth et al. 2002).
More importantly, Foury’s group showed that alteration of the electrostatic properties of the acidic ridge of FXN (substituting two or four acidic residues by Lys or Ala) compromised Fe-S cluster assembly, weakening interaction between FXN and ISU1, suggesting that the acidic ridge is essential for FXN function and is likely to be involved in protein-protein interactions (Foury et al. 2007). In addition, Wang and Craig found that yeast FXN and ISU1 interact through the beta-sheet platform. Interestingly, they reported mutations on residues Asn122, Lys123, Gln124 (Asn151, Lys152 and Gln153 in the human variant) that have detrimental effects on Fe-S cluster biogenesis and on the interaction with ISU1, without altering iron binding (Wang and Craig 2008). In agreement with the involvement of the beta-sheet in the ISU1 binding process, Foury’s group found a significant impact of residues from beta-strands 3 and 4, whereas the aromatic side-chain of Trp131 (Trp155 in the human variant) is one of the major contributors to binding energy (Leidgens et al. 2010). It is worthy of note that mutation of Trp155 to arginine (W155R) results in FRDA.
One important aspect was whether interaction of FXN, ISU1 and NFS1 was cooperative and if mutation of the acidic ridge region may alter interaction with NFS1, thus altering interaction with ISU1. In this regard, Pastore’s group, by means of a combination of biophysical methods, such as ITC, SAXS and NMR, complemented with mutagenesis analysis (Prischi et al. 2010), defined the structural bases of the interaction of the bacterial orthologue of FXN with the IscS/IscU complex (Fig. 13.6, IscS/IscU complex is named NFS1/ISU1 in yeast and NFS1/ISCU in humans). They showed that monomeric FXN binds desulfurase in a region between its active site and the dimer interface. Mutations at the complex interface altered binding and the rates of enzymatic cluster formation. FXN binding increased the affinity of the other members of the protein complex. Furthermore, the triple mutation R220E/R223E/R225E designed to invert the charge of a positively charged patch formed by residues Arg55, Arg67, Arg219, Arg220, Arg223, Arg225 and Arg237 (close to the dimer interface) was enough to abolish interaction with the complementary charged surface of FXN.

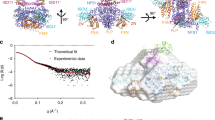
Structural bases of the interaction of FXN from the bacterial orthologue with desulfurase IscS and IscU. a Small-angle X-ray scattering (SAXS) shapes corresponding to the different protein complexes: IscS alone, IscS/IscU, FXN/IscS and FXN/IscS/IscU (bacterial FXN is known as CyaY). In all cases the structure of IscS desulfurase (PDB ID: 1P3W is superimposed. These experiments were carried out by Pastore’s group. Regions with additional densities, which are not explained by IscS alone, in the binary and ternary complexes, are highlighted in yellow or red ovals. The envelopes of the complexes are markedly different from those of the isolated desulfurase, allowing the authors to identify the excess volumes in which monomeric IscU, FXN or both fitted. Molecular modelling based on SAXS (ab initio bead models) of IscU/IscS (b), CyaY/IscS (c) and CyaY/IscS/IscU (d) complexes superimposed with rigid body models. Each model is left-handed rotated around the vertical y axis. e Surface representation of the ternary complex obtained by combining data from SAXS and NMR experiments. IscS subunits are shown in blue and cyan; in red and orange the IscU; subunits and in gold and yellow FXN (CyaY). Reproduced from Figs. 3, 4 and 6 in reference Prischi et al. (2010)
On the other hand, Puccio’s group studied interaction of mammalian FXN with the Fe-S assembly protein complex by means of immunoprecipitation experiments. They found that the main endogenous interactors of a recombinant mature human FXN are ISCU, NFS1 and also the small protein ISD11, the components of the core Fe-S assembly complex. Furthermore, using a heterologous expression system (E. coli) and a series of mutants, they demonstrated that mammalian FXN interacts through the acidic ridge and the beta sheet with the preformed core complex, rather than with the individual components. Mutations W155R and the N146K (two pathogenic mutations) strongly affected interaction with ISCU and NFS1. Moreover, some mutations directed against the residues from the acidic ridge (E108K, E111K and D124K) drastically disrupted interaction of FXN with the complex (Schmucker et al. 2011). By contrast, mutation of residues E96, D104, D115 and D122, as well as G130V (a pathogenic mutation associated with a milder phenotype), had no significant impact on the interaction (Schmucker et al. 2011).
Conformational Stability, Internal Motions and Folding Dynamics
Understanding the Effect of Mutations on Structural Dynamics
Even though in the enormous majority of cases patients with FRDA are homozygotes for the expansion, about 2–8% of them exhibit a point mutation in the FXN coding sequence that alters one or more of FXN’s features needed for its biological function.
Some mutations yield a decrease in the conformational stability of FXN: in equilibrium conditions, the difference in free energy between the folded and unfolded state is lower for these mutants compared to that measured for the wild-type variant. Usually, this information is provided by the analysis of in vitro experiments in which the stability of the isolated FXN is perturbed by denaturants like urea or guanidinium hydrochloride. The unfolded state is stabilized at high denaturant concentrations (Fig. 13.7). Alternatively, the perturbing agent may be by temperature (and much more rarely, pressure or even stretching). The variation in the folded fraction of the protein (and the equilibrium constant KNU) is very commonly monitored by changes in spectroscopic signals like tryptophan fluorescence (330–350 nm) that reports alterations in the environment of the aromatic residues. In particular, tryptophan residues are monitored because they are specifically excited at 295 nm and emit in the range of 310–450 nm and their maximal wavelength and quantum yield are sensitive to the chemical environment. Emission from an apolar environment occurs in the range of 320–340 nm, whereas from a polar and solvated environment it occurs at 350 nm. Additionally, circular dichroism (CD) in the far-UV region (190–240 nm), which reports changes in the secondary structure content (Fig. 13.8), is an important complement.

Characterization of the conformational stability of human FXN. The stability of the wild-type (black), the L198R pathogenic mutant (green) and the 90–195 truncated variant (red) were perturbed by the addition of a chaotropic agent (urea). Two different probes were used to characterize folding under equilibrium, a the circular dichroism (CD) signal at 220 nm that monitors the secondary structure of the protein, and specifically the alpha helical content, and b tryptophan fluorescence intensity that monitors the tertiary structure of the protein, and specifically the environment of tryptophan residues of FXN. For this protein, the fluorescence intensity of the native state is higher than that of the unfolded state, whereas the CD spectrum of native state is characterized by a negative band at 220 nm (it is worthy of note that the sign of the CD signal at 220 nm was changed). c A two-state model was fitted to the data obtaining the difference in free energy of unfolding in the absence of denaturant (ΔG°NU H20, or, for simplicity, ΔG°NU throughout this chapter) and the mNU value, which is the dependence of ΔG°NU on the denaturant concentration (∂ΔG°NU/∂[urea]). Buffer was 20 mM Tris-HCl, 100 mM, 1 mM EDTA, 7.0. d The superimposition of CD and fluorescence data. An adequate superimposition between conformational probes indicates that secondary and tertiary structures are cooperatively stabilized suggesting a two-state mechanism. Experimental details may be found in Faraj et al. (2014)

Characterization of FXN conformation by using spectroscopic probes. A series of FXN mutants (FXN 90-210) were prepared and studied. a Far-UV CD, b Near-UV CD, c Tryptophan fluorescence (7.0 M urea unfolded state in black dashed line), d 1H–15N HSQC NMR and e the chemical shift perturbation per residue respect to the wild-type protein (extracted from the analysis of panel d). Even though the truncated variant and the pathogenic L198R mutant are highly unstable, they exhibit some average conformational features (CD and Trp fluorescence spectra) similar to the wild-type FXN. On the other hand, NMR spectra showed significant differences in chemical shifts for these variants. Panel a–c and d, e are reproduced from Figs. 2 and 3 in reference Faraj et al. (2014)
The biophysical characterization of the protein variants include the change in the hydrodynamic radius, hydrodynamic behavior or compactness, which can be monitored by light scattering (DLS), SEC-FPLC, or NMR (DOSY), and the study of the changes in the tertiary structure that may be followed by monitoring CD in the near-UV region and NMR spectroscopy (e.g., 1H-15N-HSQC, which, by means of the chemical shifts, monitors the environment of backbone amide groups). Figure 13.8 shows some results for wild-type FXN.
In particular, residues Leu106, Tyr123, Ile154, Leu156, Trp173, Leu182, His183, Leu190 and Leu198 conform the hydrophobic core and they are tightly packed, suggesting that their mutations cause complementary destabilization of the fold. In Table 13.1, a list of mutants is presented together with data regarding experimental and predicted conformational stabilities.
This explains the effect of FRDA mutations p.L106S, p.Y123D, p.I154F, p.L156P, p.W173G p.L182F, p.L182H, p.L182F, p.H183R, p.L186R, p.L190P and p.L198R, which alter FXN packing by means of steric clashes and steric strain, packing defects like internal cavities, or result in the location of a charged residue in an unfavorable environment.
Additionally, the processing of some pathological variants G130V and I154F has shown to be affected (Clark et al. 2017). In both cases, it was detected that the levels of mature FXN were reduced in cells due to an accumulation of the intermediate and an enhancing of MPP binding to these protein variants with a less efficient processing (Clark et al. 2017).
Some pathogenic FRDA mutations have drastic consequences on the internal motions of FXN. These variants may exhibit altered conformational ensembles at physiological conditions of pH and ionic strength. Usually relaxation NMR strategies, which cover the time scale from picoseconds to milliseconds, are applied to investigate the molecular dynamics. Typically, R1, R2 and hetero nuclear NOE (hnNOE) experiments are carried out (Fig. 13.9). Relaxation is sensitive to fast-internal motions (picosecond to nanosecond timescale) but are also sensitive to slow motions (microsecond to millisecond timescale) produced by conformational exchanges, protein aggregation and molecular tumbling (this is the case of R2, transverse relaxation). For some pathogenic mutants of FXN, relaxation dispersion (Rex, CPMG) experiments, which focus on the contribution of the conformational exchange to transverse relaxation, were also done, adding the timescale of microseconds to milliseconds (Fig. 13.10). Motions in this range are usually associated with conformational exchanges and function and, therefore, these experiments are very revealing. Wild-type FXN exhibits a pattern of Rex characterized by higher Rex values for residues 94, 96, 100, 103, 130, 131, 145, 146, 147, 151,152, 154, 156 and 166 (Faraj et al. 2014; Noguera et al. 2017). These amino acids are clustered in the three-dimensional structure and most of them establish a network of contacts linking numerous secondary structure elements. As this region of the protein is involved in interactions with ISCU, the scaffold protein in the biosynthesis of iron–sulfur clusters, mutations that alter this network of motions might alter FXN biological function. After the analysis of relaxation dispersion profiles in two different magnetic fields, the populations of each native substate exhibiting conformational exchange may be obtained. In the case of wild-type FXN, a two-state model was fitted to the data, the most abundant native substrate (A) and the excited state (B) have populations of approximately 0.98 and 0.02, respectively. Additionally, the rate coefficients for the exchange processes may be obtained: for wild-type FXN kAB and kBA they are 3.5 and 213 s−1, respectively (Noguera et al. 2017). Moreover, hydrogen-deuterium experiments allowed access to milliseconds or even more (seconds to minutes and hours). Therefore, by complementing these experiments, one may gain access to a broad range of timescales and different processes.

Internal motions studied by NMR relaxation. Internal motions of L198R and the truncated variant were mapped on the PDB structure, a and b, respectively. Fast motions (ps-ns) hnNOE values <0.7 (blue); slow motions (μs-ms): residues showing T1/T2 higher than the mean plus 1SD (red); residues for which we do not have dynamic information are shown in gray. The L198R mutant (c and d) and the 90-195 truncated variant (E and F) show local and long-range effects of mutations on the internal motions, respectively. Loop-1 Mutants: Two variants in which the loop-1 of the KPYTFED sequence was mutated to the GGGYTFGGG sequence (g, h and i, red, variant 1L2) or to the GGGGGGG sequence (j, k and l, green, 1L3 variant). Panels g and j show the chemical shift perturbations (for each variant, each cross-peak was compared to that of the wild-type FXN). Panels H and K correspond to the heteronuclear NOE, whereas panels i and l show T1/T2 ratios. The wild-type is relatively rigid with unvarying hnNOE and relaxation values (black bars). As expected for the 1L2 and 1L3 mutations, which replace a stretch by a poly-Gly, analysis of hnNOE showed that fast motions were locally increased in the mutants’ loop-1. The observed effects of loop-1 mutations on T1 and T2 relaxation rates are subtle and very localized. T1/T2 profiles for the 1L2 and 1L3 mutants are mainly similar to that of the wild-type protein, as expected for a highly conserved tertiary structure. Additionally, the T1/T2 values are sensitive to the rotational diffusion anisotropy of FXN. This is due to the deviation of the globularity of the protein: helical elements (and their amide groups) are oriented to the longer axis whereas the beta strands (and their amide groups) are oriented to the short axis. This Figure is a partial reproduction of Figs. 5 and 7, and supplementary Figure S5 is from reference Noguera et al. (2017) and Fig. 8 from reference Faraj et al. (2014)

Wild-type human FXN conformational exchange studied by NMR relaxation dispersion (CPMG). a The contribution of conformational exchange to transverse relaxation (ΔR2 obs) was estimated from the difference in R2 obs at the lowest and the highest CPMG frequencies and plotted along the FXN sequence. b CPMG relaxation dispersion profiles for the backbone 15N amide of residues E96, E100, L103, G130, V131, K147, K152, L156 and D167, collected at two different magnetic field strengths, 14.1 (■) and 16.5 T (○) (600 and 700 MHz 1H Larmor frequencies, respectively) and 22 ℃. Lines indicate best-fit curves obtained with global values of the exchange rate, kex and state populations pA and pB, according to Eqs. (8–12) in reference Noguera et al. (2017). c Residues mentioned above mapped on the FXN structure. This Figure is a partial reproduction of Figs. 9 and S7 from reference Noguera et al. (2017)
Resistance-to-proteolysis assays are also used to test the flexibility of the protein chain. In this case, the analysis of the peptides and the resulting fragments can be carried out by reverse face HPLC, mass spectrometry, SDS-PAGE, among others. Although wild-type FXN is very resistant to proteolysis, some variants are particularly sensitive to protease treatment. Trypsin that cuts the protein backbone specifically at positively charged residues (Lys or Arg residues) and chymotrypsin, which cuts at aromatic residues (Trp, Phe or Tyr), are commonly used to test flexibility. In fact, when the reaction is carried out using chymotrypsin, only Tyr205, located in the C-terminal region (CTR), is susceptible to proteolysis and this occurs only after long incubation times. However, for some mutants, Tyr205 was a suitable probe for CTR flexibility enhancements (Faraj et al. 2014). Figure 13.11 shows the results of proteolysis experiments carried out by Correia and coworkers for a set of pathogenic variants: D122Y, G130V, I154F and W155R (Correia et al. 2008). The variants exhibit proteolysis profiles reflecting local instabilities (Fig. 13.11a) and, accordingly, the variants show altered proteolysis kinetics (Fig. 13.11b).

Proteolysis experiments. a Peptide maps resulting from controlled proteolysis (90 min at 37 ℃) of FXN at pH8.5 using trypsin for wild-type, D122Y, G130V, I154F and W155R variants. Symbols indicate: D91-R97 (*), L136-K147 (◆), Q153-K164 (■), Y166-K171 (✦), N172-K192 (●) and L198-K208 (+) peptides. b Time-course of controlled proteolysis monitored by the quantification of N172-K192 peptide for wildtype (filled squares) and D122Y (unfilled diamonds), G130 V (unfilled squares), I154F (unfilled circles) and W155R (unfilled triangles) variants. This Figure is a reproduction of Figs. 5 and 6 from reference Correia et al. (2008)
Even though FXN is completely soluble with a very low propensity to forming aggregates, some mutations increase the tendency of FXN to aggregate. Aggregation may be monitored by turbidimetry, SDS-PAGE, ultracentrifugation and light scattering (DLS), among other techniques. The effect of some mutations was studied in detail.
Here we have selected some mutations, which exhibit a broad range of alterations. It is worthy of mention that it is not an exhaustive description of all mutants found and studied so far. A very complete review regarding a complete list of compound heterozygous FXN mutations was recently published (Galea et al. 2016).
Two interesting mutants studied in detail are p.G130V and p.G137V. In both cases, the side chain of residue Gly, a hydrogen, is replaced by the isopropyl group of Val, a non-polar branched-side-chain amino acid.
Mutation G130V
Mutation p.G130V results in a large shift in Tm value (ΔTm = 23 ℃) and in a significant difference of free energy compared with the wild-type variant (ΔΔG°NU ~ 2.9 kcal/mol). Additionally, as proven by proteolysis, the flexibility of this variant is enhanced. G130V is highly flexible in the loop between strands β3 and β4, as shown by the appearance of a peak matching the Q153-K164 fragment (Correia et al. 2008). Conformation of the turn formed by residues Gly128, Ser129 and Gly130 between strands β1 and β2 is disrupted by mutation G130V and, consequently, an increment in the local strain should take place (Correia et al. 2008). As the backbone carbonyl oxygen atom of Gly130 forms a hydrogen bond with the backbone amide hydrogen of residue Lys147, which is involved in binding to ISCU, mutation of Gly130 is predicted to alter FXN function (Galea et al. 2016). Moreover, given that Lys147 is the in vivo ubiquitination site (Rufini et al. 2011) for the protein degradation pathway via proteasome, mutation of Gly by Val might result in higher degradation inside the cell. Residue Gly130 is involved in slow conformational exchanges as determined by relaxation dispersion (CPMG) experiments (Noguera et al. 2017), and motions are typically related to protein function. Altogether, these data make clear that a number of different aspects should be considered in order to understand in detail the effect of a point mutation.
G137V Mutation
In the case of p.G137V, its structure and activity are preserved; however, the variant exhibits significantly reduced conformational stability (Faggianelli et al. 2015). Probably, this mutation causes misfolding with the consequent reduction of protein concentration in the patient. Indicative of the lower folding efficiency compared with the wild-type is the fact that when expressed in E. coli only a low amount of the G137V variant is obtained in the soluble fraction, whereas 100% of the wild-type FXN is obtained in the soluble fraction. Even though all the spectroscopic features are preserved (1H-15N HSQC-NMR chemical shifts, CD, etc.), the melting temperature of the mutant drastically decreases compared to that of the wild-type (ΔTm ~ 20 ℃). On the other hand, this variant is active as an allosteric activator for in vitro cluster biosynthesis, suggesting that this mutation has no direct effect on protein function. Consequently, lower concentrations of FXN found in this case may be the result of low stability of p.G137V and the cause of a particular FRDA form with an onset at the boundary between a classical and a late onset phenotype.
L198R Mutation
Our group studied the L198R mutation in detail because we were interested in understanding the role of CTR (residues 196–210) in FXN’s folding and dynamics. In this case, we observed that p.L198R destabilized the native state in a significant fashion (ΔΔG°NU ~ 4.2 kcal/mol). Other mutations at this position, which only truncate the side chain, were also destabilizing (L198A and L198C), pointing to the relevance of CTR and, in particular, pointing to Leu198 as a key side chain for FXN stability; the stabilities of FXN variants are L198R < L198A < L198C < wild-type. This result is compatible with the fact that the Cys residue can establish interactions via its thiol group, whereas the L198A variant carries a shorter side chain, deleting a number of native contacts. Additionally, kinetic studies of folding dynamics showed that L198A, like other mutations located in this stretch, only alters the stability of the native state but not the stability of the transition, intermediate or unfolded states (Fig. 13.12) (Faraj et al. 2016). As the contribution of the interactions mediated by CTR to the energetics of the transition states and intermediate ensembles are negligible, one can infer that CTR is unstructured in these ensembles but structured in the native state.

Human FXN folding dynamics. The stability of the native state and the CTR. a Chevron plots showing that differences in kinetics among the mutants of CTR of FXN 90-210 come from the unfolding branch (right); on the other hand, the refolding branch (left) among the variants superimposes very well. This effect can be interpreted as a difference in the stability of the native state. b Reaction coordinate for folding of human FXN (90-210): the difference in free energy between Nwild-type and Nmut CTR without the effect of mutation on the transition state ensembles (TSE1 and TSE2) or in the intermediate state (I). In panel (c) a model of FXN structure is shown in which the CTR is depicted in yellow and key residues L198, L200 and L203 that stabilized the native state are modeled using 50% of the van der Waals volumes. For the rest of the protein, the molecular surface is modeled. d Structure-based molecular dynamic simulations using a funneled energy landscape of protein folding (native–centric GO model) allowed the study of the folding state of each one of the secondary structure elements along the reaction coordinate. The fraction of the global number of contacts is plotted. This Figure is a partial reproduction of Figs. 1 and 8 and S1 from reference Faraj et al. (2016)
It is worthy of note that in p.L198R a charged residue is located in an apolar environment and this feature promotes alterations in the native ensemble, with particular altered motions near the mutation site. Although wild-type FXN shows a high resistance to proteolysis, and after 4 h of incubation a single cutting site at the Y205 position is detected, the L198R mutation enhances sensitivity to the chymotrypsin. Even at a very short incubation time (30 s), 50% of the protein is cut at the Y205 position. In agreement with this result, NMR relaxation dispersion (CPMG) experiments showed that L198R exhibits more dynamic behavior in the microsecond/millisecond timescale in localized regions near the mutation (Faraj et al. 2014). On the other hand, the absence of CTR results in a very destabilizing variant with a global effect on the dynamics and a 10% of protein populating the unfolded ensemble at physiological conditions of ionic strength, pH and temperature (Faraj et al. 2014). Remarkably, the finding that a frame shift that produces the truncation of FXN at residue 193 determines FRDA with a rapid disease progression indicates that CTR is crucial for the biological function of the protein (Sacca et al. 2013).
W155R Mutation
As we mentioned above, some mutations linked to FRDA occur in the beta-sheet, the region involved in protein-protein interactions. This is the case of W155R. Tryptophan 155 is an exposed residue that, most likely, participates in the interaction with ISCU. This variant is slightly destabilizing, showing a decrease of only ~5 ℃ in Tm (Correia et al. 2006). Destabilization is expected from the obliteration of a π-cation interaction between a tryptophan residue and the contiguous Arg165 residue, and the electrostatic repulsion that results from the insertion of an additional positively charged residue close to the Arg165. Whereas the 1H–15N HSQC NMR spectrum of W155R is very similar to that of the wild-type FXN, relaxation NMR experiments indicated that this mutant has a significantly larger correlation time (τc) compared to that measured for the wild-type variant (7.9 and 9.2 ns, respectively). This may be indicative of the mutant’s high tendency towards aggregation. Interestingly, the W155R mutant is proteolyzed at a fast rate at the protein C-terminus and at the loop between strands β2 and β3, but also the first α-helix shows a proteolytic site (Correia et al. 2008), which is indicative of a decreased rigidity and some disorder or local conformational exchange. On the contrary, proteolysis within a rigid canonic secondary structure element would be very energetically unfavorable and unlikely, as in the case of the wild-type variant.
N146K Mutation
By contrast with other mutations mentioned above, p.N146K mutation resulted in a significant stabilization of the native conformation, as suggested by the Tm values (64.9 ± 0.2 and 69.4 ± 0.1 ℃ for the wild-type and the mutant variants, respectively, non-published results from our group) and computational predictions (ΔΔG°NU are -0.7 and −1.3 kcal/mol, using FOLDX and Dynamut, respectively, and ΔΔG°NU is defined as ΔG°NU, wild-type − ΔG°NU, mutant). On the other hand, this mutation alters interaction with ISCU and, therefore, NFS1 supercomplex activity. Residue N146 is at van der Waals distance (~5 Å) of Trp155 and a charged residue located at this position may imbalance the electrostatics of the FXN surface. In this regard, using a series of mutants, Barondeau’s group carried out a key work on structure-function relationships revealing determinants of FXN-mediated activation of the Fe–S assembly complex (Bridwell-Rabb et al. 2011). Interestingly, whereas the wild-type FXN exhibits a KD = 0.22 ± 0.05 μM for the complex NFS1/ISD11-ACP/ISCU, the KD for the N146K mutant is significantly higher (6.25 ± 1.40 μM). Moreover, NFS1 activity, cluster formation activity and catalytic efficiency (kcat/KM) are approximately 8, 4 and 3 times higher for wild-type than for this mutant, respectively. Besides, weaker binding to the complex NFS1/ISD11-ACP/ISCU observed for the N146K variant compared to the one observed for N146A (a variant designed in the laboratory to dissect the effect of the charge) is consistent with results from pull-down experiments performed by Puccio’s group (Schmucker et al. 2011). Additionally, Puccio’s laboratory generated murine fibroblast cell lines stably expressing full-length N146K and N146A (among other variants) and observed that N146A clones did not present any gross phenotype or sensitivity to oxidative stress. By contrast, the N146K clone showed strong growth defects and displayed the classical features of FRDA (degenerating mitochondria, electron dense deposits, deficit in Fe–S enzyme activities).
D122Y Mutation
D122Y mutation is located in the acidic ridge, in the loop 1 region (residues Asp115 to Tyr123). Although this mutation is predicted to have little effect on the stability of the native conformation of the protein (Galea et al. 2016), the experimental Tm value for this variant decreased in a significant fashion compared to that of the wild-type (ΔTm ~ 15° C monitored by circular dichroism (Correia et al. 2008), or ΔTm ~ 8 ℃, unfolding monitored by Sypro-orange probe, unpublished results from our group), in agreement with a significant decrease in conformational stability (ΔΔG°NU = 2.1 kcal mol−1, urea-induced unfolding (Correia et al. 2008)). The side-chain of Asp122 establishes two interactions with neighbor residues: one of them with the amine group of the side chain of Lys135 from β2 and the second with the amide group of backbone Gly138, from the β2-β3 turn. These interactions are removed in p.D122Y. In the same way, the pKa for Asp122 is predicted by PROPKA (Olsson et al. 2011) as more acidic than expected for a free aspartic acid side-chain (pKa D122 and pKa model are 2.90 and 3.80, respectively), suggesting its involvement in intramolecular interactions. Variant D122Y exhibited a lower iron binding stoichiometry (Correia et al. 2008).
FXN Degradation Inside the Cell
Studies on FXN degradation inside the cell have shown that precursor concentration may be regulated by the proteasome system. In this context, the transfection of cell lines with the FXN precursor showed that the precursor is ubiquitinated. When HEK-293 cells were treated with proteasome inhibitors such as MG132, the accumulation of the precursor was detected and, more importantly, an increment of 2.5 times the mature form was observed (Rufini et al. 2011; Benini et al. 2017).
It was observed that Lys147 of the FXN precursor is the key residue that is modified by ubiquitination. The K147R mutation showed an increment of the lifetime with higher levels of the precursor and the mature form. Interestingly, Lys147 is located in a cleft where some target molecules might bind blocking degradation (Rufini et al. 2011). It has been detected that this post-translational modification is performed by the RING E3 ligase RNF 126, which specifically recognize and modifies FXN. Its inhibition may be a key point for incrementing FXN levels in Friedreich ataxia patients. Ubiquitin-competing molecules inhibited FXN degradation, promoting its accumulation and rescuing aconitase activity in model cell lines (Benini et al. 2017; Rufini et al. 2015).
Other reports have shown that modulation of FXN degradation could be triggered by phosphorylation (Cherubini et al. 2015). It has been shown that when the FXN precursor is co-expressed with the Src kinase domains, the precursor is modified in a tyrosine residue. To get information about which is the target residue, the eight Tyr residues of FXN were mutated (Tyr74, Tyr95, Tyr118, Tyr123, Tyr143, Tyr166, Tyr175 and Tyr205) to Phe and each variant was transfected into the HEK-293 line in the presence of an active Src-Y527F kinase. As a control, the same was done with an inactive Src-Y527F kinase. A total abrogation of protein modification was detected in the case of the Y118F FXN mutant, suggesting that this residue is the one that is mostly modified (Cherubini et al. 2015). When the ubiquitination of the Y118F variant was evaluated, a decrease in the modification and degradation was observed. To confirm the hypothesis that phosphorylation of the Tyr118 residue triggers FXN degradation, the HEK-293 line expressing the wild-type protein was treated with different Src inhibitors. These drugs promoted FXN accumulation in a dose-dependent form. Interestingly, when these inhibitors were used in a FXN deficient B cell line derived from patients of Friedreich’s ataxia, an accumulation of FXN and a recovery of aconitase activity were detected, suggesting that FXN phosphorylation is a key pathway for regulating the levels of this protein in the cell (Cherubini et al. 2015).
In summary, FXN presents at least three isoforms, being type I the most important in the mitochondrial and it is a process in the 81–210 mature form. Additionally, FXN can suffer several post-translational modifications that modulate its concentration and function inside the cell.
Iron Binding and Function
FXN may have a role in iron sensing and the control of cluster biosynthesis. It is well known that the presence of iron accelerates Cys desulfurase reaction catalyzed by the NFS1 supercomplex. Whether FXN is the protein that donates iron atoms to the cluster assembly is under debate. As we mentioned before, it was determined that FXN binds Fe2+ and Fe3+ with moderated affinity; however, some controversy exists regarding the latter (Cai et al. 2018b; Faraj et al. 2014; Yoon and Cowan 2003), most likely due to the extremely low solubility of the metal ion in a buffer with physiological conditions. Nevertheless, experiments using minimalist peptide models showed that motifs present in the FXN surface are able to bind both (Vazquez et al. 2015). Thus, experiments concerning Fe3+ can be performed under non-physiological conditions like pH 4–5 or under non-equilibrium conditions. The insolubility of Fe3+ was used to separate bound from non-bound metal ions after incubation with the protein, and iron concentration can be accurately measured by the phenanthroline method.
In this context, it is worthy of mention that some pathogenic variants fail to bind these metal ions. Variant D122Y as well as G130V and L198R exhibited a lower iron binding capability (Correia et al. 2008; Faraj et al. 2014).
Chemical shift perturbation and paramagnetic properties of iron allowed monitoring of iron binding to FXN. In particular, 1H-15N cross peaks corresponding to the amide groups of residues that directly interact with metal ions in 1H–15N-HSQC NMR experiments undergo a significant intensity decrease, making the cross peaks undetectable. Therefore, binding sites can be inferred at the residue level (Fig. 13.13). This procedure is performed under anaerobic conditions in order to maintain Fe2+concentrations in solution. In the case of human FXN, after the addition of two equivalents of Fe2+, significant changes in chemical shifts were observed for residues of the acidic ridge: Asp104, Ser105, Asp112, Leu113, Ala114, Asp115, Thr119, Asp122, Asp124 and Val125. After the addition of three equivalents of Fe2+, Asp91, Asp104, and Asp122, the three underwent further perturbations of their chemical shifts (Gentry et al. 2013). Signals corresponding to Asp112, Leu113 and Asp 115 broadened, making them undetectable at three molar equivalents of iron (Gentry et al. 2013). Remarkably, His86 located in the N-terminal disorder stretch of the mature form of human FXN was essential for high affinity iron coordination (Gentry et al. 2013).

Direct interaction between Fe2+ and human FXN monitored by 1H-15N-HSQC NMR experiments. a The change in the normalized amide chemical shift (δNH) and b the change in intensity of 1H–15N-FXN resonances upon addition of 1, 2 and 3 molar equivalents of Fe2+ (gray, cyan, and dark blue, respectively). Asterisks indicate the residues whose resonances broadened beyond detection during the titrations. Protein concentration was 560 μM and samples were prepared anaerobically. This Figure partially reproduces Fig. 5 from reference Gentry et al. (2013)
Whether FXN undergoes conformational changes or changes in the internal motions upon iron binding is a key issue because iron binding may exert an effect on function via the modulation of the interaction between FXN and NFS1 and/or ISCU proteins. Hydrogen-deuterium exchange experiments coupled to mass spectrometry (HDX-MS experiments) were performed by Gentry and coworkers (Gentry et al. 2013) for FXN in the presence of different ratios of protein/metal ion for Co2+, Fe2+ or Fe3+. By means of HDX-MS, they monitored the rate of exchange between H and D as the incorporation of deuterium in the backbone of FXN (Fig. 13.14). The differences in mass between deuterated and non-deuterated proteolytical peptides can be determined by mass spectrometry. The exchange during the first 15 s reports accessibility of the H-N groups to D2O and the amide groups compromised in hydrogen bonding, in secondary structure elements, are protected from the exchange. On the other hand, slower exchanges can be the result of local unfolding events or conformational exchanges, even involving the global unfolded state.

Hydrogen-deuterium exchange of FXN backbone modulated by metal interaction. a Protection determined by the percentage of amide groups exchanging for apo-FXN was subtracted from those values obtained with 2 or 3 equivalents of metal: 2 equivalents of Co2+ (pink); 3 equivalents of Co2+ (red); 2 equivalents of Fe2+ (light purple); 3 equivalents of Fe2+ (purple); 2 equivalents Fe3+ (cyan); 3 equivalents of Fe3+ (blue). Panels b–g show those FXN peptides that exhibited HDX changes greater than 10% at three equivalents of metal. These peptides are mapped to the structure of FXN 90–210 (PDB:1EKG) using the colors as used in panel A. It is worthy of note that this HDX data included peptide 81–91, which is not observed in the structure. This Figure is a reproduction of Fig. 6 from reference Gentry et al. (2013)
Whereas the addition of one equivalent of metal ion to the FXN only produced a slight decrease in deuterium incorporation, the addition of two and three equivalents of metal showed a lower incorporation. On the other hand, no further differences were observed for higher metal-to-protein ratios, suggesting the distribution among three binding sites. Peptides from the N-terminal region (residues 81–91), in strand β1 (residues 122–127) and in the β4–β5 strands (residues 156–172) showed reduced deuterium incorporation for all metal ions in comparison to that observed for the apo form of FXN. Incubation with the third equivalent of metals promotes additional protection of the H-N- amide groups in α1 and β1. These results indicated that interaction with the metal may produce some changes in FXN local stabilities, in some cases in regions near the metal binding sites; however, other amide groups far from these sites are also protected, in particular, amide groups located in strands β4–β5. The authors suggested that this fact may be relevant from a functional viewpoint (Gentry et al. 2013). This region can interact with the ISCU protein (see below).
Taking into account FXN iron capability, and given that yeast cells lacking FXN gene showed very low cytochrome content and cells were defective in iron use by ferrochelatase, a mitochondrial membrane-associated protein that catalyzes the terminal step of heme synthesis, consisting in the insertion of Fe2+ into protoporphyrin IX, it was proposed that FXN may be involved in heme biosynthesis (Lesuisse et al. 2003). In the same fashion, combining mouse and human microarray data for FXN-deficient cells and tissues, Schoenfeld and coworkers detected a significant decrease in coproporphyrinogen oxidase (the enzyme that converts coproporphyrinogen III to protoporphyrinogen IX) and down regulation of ferrochelatase, a result consistent with an observed increase in cellular protoporphyrin IX levels and the reduction of mitochondrial heme (A and C) levels, and indicative that FXN deficiency causes alterations in the heme pathway (Schoenfeld et al. 2005). Additional results coming from a quantitative proteomic analysis of Friedreich’s ataxia B-lymphocytes showed a 2.5 decrease in the expression of the protoporphyrinogen oxidase, the penultimate enzyme in the heme biosynthetic pathway (Telot et al. 2018).
On the other hand, interesting data were obtained in recent work, employing whole cell systems. Steinkellner and coworkers found no significant differences in heme synthesis levels in normal and FRDA-patient progenitor-erythroid cells (Steinkellner et al. 2017). This result is in line with the fact that most FRDA patients do not suffer anemia.
Direct interaction between human FXN and ferrochelatase was studied by Yoon and coworkers by means of isothermal titration calorimetry (KD ~ 17 nM at 25 ℃ and pH 8, (Yoon and Cowan 2004)). Interaction between both proteins only occurred in the presence of iron, suggesting that iron may mediate interaction. Heme production activity (monitored by a change in absorbance at 506 nm) increased with FXN concentration being optimal at a ratio of one FXN molecule per ferrochelatase dimer.
More recently, Söderberg and coworkers studied the interaction between yeast FXN and ferrochelatase by means of cross-linking in combination with mass spectroscopic analysis and single-particle reconstruction from electron microscopic images; these data were used together with computational docking to model the structure of the complex (Soderberg et al. 2016). The analysis of the model suggested that both proteins interact creating a path for iron transfer from the FXN metal binding site to the ferrochelatase reaction center. Additionally, kinetics experiments showed that the rate of heme synthesis was slightly accelerated when iron-loaded yeast FXN was included in the assay (Soderberg et al. 2016).
Remarkably, FXN from plants exhibits ferrochelatase activity. Armas et al. studied possible functions of FXN from Arabidopsis thaliana and found a potential role as an iron donor for heme biosynthesis, suggesting that plant FXN is able to directly catalyze the incorporation of iron into protoporphyrin IX in vitro (Armas et al. 2019). The efficiency of this process significantly increases in the presence of plant desulfurase NFS1/ISD11. Remarkably, the same group found that Arabidopsis thaliana lines deficient in FXN showed a substantial reduction in heme content in flowers and leaves and that it is correlated with a decreased content of several transcripts related to the heme pathway (Maliandi et al. 2011).
It is worthy of mention that at least for FXN from Vibrio cholerae it was shown that the protein exhibits the capability of heme binding with an apparent KD value of 21 ± 6 nM (Uchida et al. 2017). Given that heme binding resulted in a decrease in the affinity of the protein for iron, it may be considered a point of regulation/control of the iron transport dependent on the heme group (Uchida et al. 2017), Possibly, an evidence of a cross-talk between pathways.
Significant evidence is still required to elucidate the details and physiological relevance of the interactions between FXN and ferrochelatase in mammals. In this respect, further kinetic and metabolic analysis regarding FXN, the heme biosynthetic pathway and iron sulfur assembly would shed light over conjectures attempting to eventually find the relationships that might link them.
A Macromolecular Context for FXN in Iron–Sulfur Cluster Biosynthesis
As mentioned before, FXN’s activity depends on the interaction with at least two proteins: ISCU and NFS1, of the supercomplex NFS1/ACP-ISD11/FXN/ISCU. How the supercomplex is stabilized by the subunits and how FXN modulates the kinetics of reactions for persulfur synthesis and cluster assembly are key questions. From a functional point of view, it is clear now that FXN controls the reaction by accelerating the rate-limiting sulfur transfer step in the biosynthesis of [2Fe–2S] clusters, on the mammalian iron–sulfur assembly supercomplex (Fox et al. 2015). From an experimental viewpoint, cluster assembly is typically detected, under anaerobic conditions, by an increase of absorbance at 456 nm and the increase of circular dichroism signals at 330 and 430 nm, which are characteristic of the [2Fe–2S]-ISCU complex formation (Fox et al. 2015).
Currently, we know that the supercomplex is formed by two subunits of the NFS1 desulfurase, which are critically stabilized by two subunits of the ISD11 protein (Fig. 13.15). From an evolutionary perspective, ISD11 is only found in eukaryotic organisms. Richards and coworkers concluded that the origin of this protein occurred during or just after the single endosymbiotic event, giving rise to the mitochondria, and ISD11 evolved as an exclusively eukaryotic addition to the pathway of derived iron–sulfur cluster biogenesis of α-proteobacterium (Richards and van der Giezen 2006).

The X-ray structure of supercomplex NFS1/ACP-ISD11/ISCU (PDB ID: 5WLW, (Boniecki et al. 2017)). A dimer of desulfurase NFS1 (cyan and green) is stabilized by the interaction of two subunits of the SD11 (orange and brown), which is a small tutor protein that, in fact, stabilizes the dimer and active conformation of the desulfurase. Interestingly, each one of the ISD11 subunits is solubilized and stabilized by the mitochondrial acyl carrier protein (ACP, magenta and yellow). Additionally, two ISCU subunits (red and blue), the proteins that will function as the scaffold to assemble the clusters [Fe–S], interact with NFS1 subunits. This complex also interacts with FDX and FXN proteins
Friemel and coworkers studied some ISD11 mutations; among them, a mutation at position Y26A affects the interaction with the NFS1 protein yielding a twofold increase in KM and a threefold decrease in kcat (Friemel et al. 2017). They also showed that NFS1 dimer formation is very sensitive to the Y26A mutation, resulting in an impaired complex formation.
Moreover, we also know that ISD11 folding likely depends on the interaction with the holo-mitochondrial acyl-carrier protein (holo-ACP) (Herrera et al. 2018), the phosphopantetheine (4’PPT) moiety and the fatty acid chain that the latter carries. In fact, the fatty acid attached to the 4’PPT of ACP penetrates the core of ISD11 and extensively interacts with the protein (Fig. 13.16).

The structure of the complex ACP and ISD11. Two different views of E. coli ACP and human ISD11extracted from the supercomplex PDB ID: 5WGB (ribbon representation, ACP in magenta and ISD11 in orange) showing the phosphoSer36 4’PPT and the acyl chain in van der Waals volumes (C, O, N, S and P elements in cyan, red, blue, yellow, and orange respectively)
ISCU may populate two interconverting conformational states: one structured and the other disordered (Markley et al. 2013). Although there are some controversies concerning which is the conformation that forms the supercomplex (Cai et al. 2013), by applying NMR strategies to study large complexes Dr. Annalisa Pastore’s group showed that ISCU binds in its structured form to the desulfurase homolog from E. coli (Yan et al. 2014). Crystallographic data confirmed this result for mammalian ISCU and NFS1 (Fig. 13.15) (Boniecki et al. 2017). These studies carried out by Boniecki and coworkers provided a much more complete picture concerning the structure of the supercomplex and the details at the catalytic center defined by the active-site Cys 381 of NFS1 and conserved Cys, Asp, and His residues of ISCU.
It is worthy of mention that in the supercomplex, electrostatic contribution to protein-protein interactions seems to be critical and coevolving. On the one hand, an extensive positive surface of ISD11 interacts with the large negatively charged surface of ACP. On the other hand, the NFS1 positive surface may interact with the broad negative acidic ridge of FXN.
The acidic ridge of FXN interacts not only with NFS1 but also with iron. The fact that FXN binds iron is very well known; however, much less known is what exactly the role of this interaction is.
As we mentioned above, NMR studies showed, with an amino acid level of resolution, which residues of the acid ridge form the iron-binding site in the FXN surface. At least two different possibilities for the iron binding function have been suggested: (i) that FXN may be the first iron donor for cluster assembly; or, (ii) rather than being the iron donor, that FXN may control the entry of iron to ISCU.
Remarkably, Markley’s group (Cai et al. 2018b) carried out a series of NMR experiments that allowed the study of FXN in the context of the supercomplex, which exhibits a very high correlation time. FXN is labeled with 15N (whereas the rest of the proteins of the supercomplex are not) and the 2D 1H,15N TROSY-HSQC spectra of 15N-labeled FXN are acquired in the presence or in the absence of the supercomplex. It was observed that the complex formed between FXN and Fe2+ binds to the supercomplex without iron release (Cai et al. 2018b). Given the paramagnetic properties that this metal ion displays, iron interaction yields the disappearance of specific cross peaks corresponding to the protein-iron interaction sites, and especially, the absence of these cross peaks persists in the context of the supercomplex. Conversely, the initiation of cluster assembly by addition of L-cysteine and reductant (DTT or Ferredoxin protein) leads to the dissociation of Fe2+ from FXN, the absent peaks reappear, and other chemical shifts perturbations that result from iron binding return to their positions in the spectrum of iron-free FXN (Cai et al. 2018b).
Significant efforts were made to establish, with high resolution, how mammalian FXN interacts in the supercomplex with the rest of the proteins. How FXN is docked is important to understand how the activity of the supercomplex is allosterically controlled by FXN.
Very recently, Markley’s group, by means of chemical crosslinking experiments combined with mass spectrometry, small-angle X-ray scattering and distance constraints from nuclear magnetic resonance, concluded that the supercomplex involves closed NFS1 dimer conformations for both the NFS1/ACP-ISD11/ISCU and the NFS1/ACP-ISD11/ISCU/FXN complexes.
The resulting model showed interactions between the highly conserved Arg-rich loop of NFS1 (the stretch of RRRPRVR sequence) and the conserved acidic ridge of FXN. Additionally, ISCU and FXN interact by means of the β3–β5 strands of the latter with the conserved CLPPVKLH sequence of ISCU, located between helix 4 and helix 5, the C-terminal helix. In this model, FXN fits into a cleft between NFS1 desulfurase and the scaffolding protein ISCU (Fig. 13.17), and Trp155 of FXN is at van der Waals distance (3.6 Å) of His112 of ISCU, two key residues for supercomplex activity. The catalytic center is in fact defined by the Cys381 of NFS1 and the conserved Cys44, Cys70, Cys113, Asp46, and His112 residues of ISCU. Interestingly, this experimental model of the supercomplex suggests that FXN’s Asn146 and Gln148 may modulate the activity of the Fe-S cluster assembly. This model elucidates why W155R, N146K or Q148R (Gomes and Santos 2013; Bridwell-Rabb et al. 2011) have a negative effect on function by placing positive charges at these sites, resulting in pathogenic mutations.

An experimental model of the supercomplex including human FXN. A structural model constructed by Dr. Markley’s group (Cai et al. 2018a). Chemical crosslinking experiments were combined with mass spectrometry, small-angle X-ray scattering and distance constraints from nuclear magnetic resonance of the supercomplex. For clarity, only both NFS1 subunits (green and cyan), one ISCU subunit (blue) and one FXN subunit (red) in ribbon are shown. The interaction between the NFS1 stretch of sequence RRRPRVR (gray) and the acidic ridge of FXN (magenta) is shown in van der Waals. FXN fits into a cleft between desulfurase NFS1 and ISCU. Q148 and Trp155 and (orange) of FXN are very close to the active site of ISCU (Cys44, Cys70, Cys113, Asp46, and His112, brown). Cys 381 (yellow) from Cys-loop in the active site of NFS1 is at van der Waals distance of the active center of ISCU
There are still other proteins that interact with the supercomplex. Among them, ferredoxins (FDX) play a key role as electron donors for cluster biosynthesis. Mitochondrial FDX1 and FDX2 interact with the supercomplex as judged by the analysis of signal broadening and chemical shift perturbations (experiments 1H-15N TROSY-HSQC) for residues Phe11, Asn13, Arg14, Leu57-Ile63, Ile71-Leu90, Leu96, Thr97 and Asn102 and residues Leu38-Ile40, Leu50, Tyr64, Glu81-Leu97 for FDX1 and FDX2, respectively (Cai et al. 2017); most of the mentioned residues are close to the FDX cluster [2Fe–2S]. However, apo-FDXs (FDXs without cluster) do not interact with the supercomplex (Cai et al. 2017).
Additionally, the release of the cluster [2Fe–2S] from ISCU is facilitated by a specific chaperone system formed by a dedicated HSP70 chaperon (mtHSP70) and a J-type co-chaperone (HscB/Jac1) (Wachnowsky et al. 2018; Maio and Rouault 2015). When the chaperon-co-chaperon complex binds to the holo-ISCU, the latter may change its conformation, releasing the cluster to specific [2Fe–2S] cluster carrier proteins (Wachnowsky et al. 2018).
Activator in Eukaryotes, Inhibitor in Prokaryotes: Differences in the Role of FXN in the Fe-S Clusters Biosynthesis
It has been demonstrated that in eukaryotes the fundamental role of FXN is in iron homeostasis through the pathway of biosynthesis of the Fe–S clusters. In contrast, the deletion of the FXN gene in Bacteria essentially has no consequences, even when FXN is a highly conserved protein among these organisms. Additionally, a number of experiments have shown that, while in eukaryotes FXN increases the synthesis speed of Fe–S clusters by NFS1 desulfurase, the FXN homologue in bacteria called CyaY has the opposite effect, that is, in eukaryotes FXN is an activator and in Bacteria a desulfurase inhibitor. In E. coli and S. enterica it was found that CyaY deletion does not lead to a marked phenotype, although this may happen under certain growth conditions or in conjunction with mutations of other genes associated with Fe–S cluster metabolism (Yoon et al. 2012; Lesuisse et al. 2003; Duby et al. 2002; Vivas et al. 2006). It is important to consider that in most bacteria there is an alternative pathway for Fe–S cluster synthesis, known as SUF (sulfur assimilation), which is activated under conditions of oxidative stress or lack of iron.
Thus, several questions arise: Is the role of FXN conserved among these organisms? Why is FXN vital in eukaryotes and unessential in prokaryotes? Why is this protein an activator of the synthesis of Fe-S clusters in eukaryotes and an inhibitor in prokaryotes?
As we will review in this section, the differences in the activity of FXN on the synthesis of Fe-S clusters between eukaryotes and prokaryotes does not seem to reside in FXN itself, but mainly in the desulfurase and in the complexes that the latter forms with other key proteins of the biosynthetic pathway.
Although the three-dimensional structures of the eukaryotic and prokaryotic desulfurase enzymes are very similar (RMSD = 1.9 Å between the human variant and E. coli), as well as their sequences (~60% identity), there are very important differences from the functional viewpoint (Table 13.2): E. coli desulfurase is very active itself (kcat = 7.5–8.5 min−1 (Urbina et al. 2001; Bridwell-Rabb et al. 2012), while eukaryotic desulfurase requires accessory proteins ACP-ISD11 to be stable and exhibits significant lower enzymatic activity (kcat = 0.9 min−1 for the human complex NFS1/ISD11-ACP, (Bridwell-Rabb et al. 2014)), requiring FXN and ISCU to have a comparable enzymatic activity (kcat = 6.7 min−1 for the NFS1/ACP-ISD11/ISCU/FXN complex, (Bridwell-Rabb et al. 2014)) to the prokaryotic variant.
On the other hand, some aspects of the interaction mode between FXNs and desulfurase from eukaryotic or prokaryotic organisms might be similar because both CyaY and human FXN complement yeast strains, in which the FXN gene was deleted (Δyfh1), in terms of the restitution of enzyme activities for Fe-S cluster biosynthesis and iron homeostasis (Bedekovics et al. 2007; Cavadini et al. 2000b). To investigate this fact in detail, in vitro experiments were carried out to measure desulfurase activity and assembly of the Fe-S clusters, where it was found that the effects of these proteins on desulfurase are interchangeable (Bridwell-Rabb et al. 2012). When ISCU (either human or from E. coli) was added to the purified complex ACP-ISD11-NFS1, and also human FXN or CyaY from E. coli was included, the result in all cases was an increase in desulfurase activity, which is revealed by an increase in kcat. On the other hand, when the desulfurase was from E. coli, the activity markedly decreases after adding bacterial ISCU and had a small additional decrease when human FXN or CyaY were added. Regarding the cluster assembly activity, in the absence of FXN the activity of the human NFS1/ACP-ISD11/ISCU complex is essentially null. On the other hand, after the addition of FXN, the activity strongly increases, while the increase is moderate when adding CyaY. The equivalent experiment with bacterial desulfurase and bacterial ISCU showed total inhibition with CyaY and partial inhibition with human FXN.
The three-dimensional structure of the dimeric IscS desulfurase from E. coli was determined by crystallography (PDB ID: 1P3W, (Cupp-Vickery et al. 2003) and 3LVM, (Shi et al. 2010)) and the monomers are arranged so that the active site lies between the two subunits of IscS (closed conformation). On the other hand, a very different dimeric arrangement was found for the human NFS1/ACP-ISD11, where the active sites are exposed (open conformation, PDB ID: 5USR, (Cory et al. 2017)). As we mentioned above in the previous section, ACP was from E. coli.
Almost simultaneously, the structure of the same complex of human NFS1 (without or with ISCU) was independently determined by crystallography (PDB ID: 5WGB, 5WLW and 5WKP), finding a closed conformation, similar to that observed for IscS. Subsequently, the structure of the complex was studied in solution by SAXS and crosslinking coupled to MS for the ACP-ISD11-NFS1-ISCU complex alone and with FXN (Cai et al. 2018a). From XL-MS the authors obtained crosslinks compatible with the closed conformation but incompatible with the open form; also, the profiles and envelopes obtained by SAXS were better adjusted by the structure of NFS/ACP-ISD11/ISCU in the closed conformation than by a model derived from the open conformation docked with ISCU.
Even so, complex 2[NFS1/ACP-ISD11] may exist as two alternative conformations that might interconvert; indeed, this balance might be relevant to explain the functional difference between NFS1 and IscS.
Finally, the differences observed for FXN activity in the eukaryotic and prokaryotic systems may be originated from the transient complexes that the desulfurase enzymes form with other proteins. In E. coli, IscS desulfurase interacts with a number of proteins, including those involved in the Fe-S cluster synthesis pathway: IscU, CyaY, FDX and IscX; and thiolation and the thiamin biosynthesis or molybdenum cofactor, such as TusA (PDB ID: 3LVJ) and ThiI (Hidese et al. 2011; Leimkuhler et al. 2017). Several of the binding sites have been mapped in the structure of IscS, some of these sites are independent and others are overlapping (Kim et al. 2013; Yan et al. 2013). On the other hand, in humans, NFS1 interacts with different proteins in the mitochondrial matrix and in the cytosol. In the mitochondria, its role is the synthesis of the Fe-S clusters, for which it requires interaction with the ISCU, FXN, FDX, and ACP-ISD11 proteins, although it also transfers the persulfur group to TUM1 for tRNA modification (Noma et al. 2009).
In particular, interaction with ferredoxin is different between eukaryotes and prokaryotes. In E. coli, CyaY and ferredoxin (FDX) compete for the same site (Yan et al. 2013; Kim et al. 2013). By contrast, in the human biosynthetic complex, it was found that kinetic experiments carried at different concentrations of the components are better explained when considering that both ferredoxin and FXN are simultaneously bound in a complex with NFS1/ACP-ISD11/ISCU (Webert et al. 2014).
On the other hand, IscX, a small protein of 66 amino acid residues which is absent in almost all eukaryotes (Pastore et al. 2006), is able to bind iron by means of a negatively charged surface and, also, it binds to IscS. Notably, IscX, CyaY and FDX compete for the same site in IscS, as was suggested by pull-down experiments using mutants affecting the interaction surfaces (Shi et al. 2010); although recent studies showed that IscX binds to two different sites in IscS (Adinolfi et al. 2017), binding of IscX to one of them can displace CyaY from the complex.
It has been observed that cluster synthesis inhibition exerted by CyaY increases with iron concentration (Adinolfi et al. 2009). The addition of IscX attenuates this effect at low iron concentrations; however, at higher iron concentrations, IscX has no effect on the inhibition of CyaY. While the interaction of CyaY with IscS is dependent on iron concentration, the interaction of desulfurase with IscX is not. Based on these results, it was suggested that IscX may modulate CyaY such that at low concentrations of iron, the IscX junction displaces CyaY competing for the same site where IscX has higher affinity, but at a higher iron concentration, the affinity between CyaY and IscS increases, contributing to the modulation of cluster biosynthesis.
We conclude that as more insight into the complexes formed by desulfurase is gained, our understanding about the differential effect of FXN between eukaryotes and prokaryotes will be enhanced.
Conflicting results have been obtained about the effect of FXN in the NFS1 persulfide formation step of the cluster assembly. Studying the yeast cluster biosynthetic system using 35S labelling of L-cysteine, it was found that the persulfide is not formed in NFS1 in the absence of ACP-ISD11(Pandey et al. 2013). This fact was used to develop an assay to separately monitor persulfide formation in NFS1 from its transfer to ISCU. They found that FXN is not necessary for persulfide formation in NFS1, but it is strongly enhanced by FXN binding, probably as a result of a conformational change induced by FXN binding, which exposes additional binding sites for cysteine. However, this latter result was challenged by another study with the human proteins, where no effect was observed for FXN in persulfide formation in NFS1 (Parent et al. 2015). These experiments were based in a novel assay of maleimide labeling of both persulfides and thiols. They noted that FXN enhances persulfur transfer from NFS1 to ISCU. These emerging mechanistic insights may probably reveal further differences in FXN regulation within eukaryotes.
FXN as a Scaffold to Form Oligomers and Nanoparticles
It was found that some FXN variants are able to form high molecular weight arrangements. In particular, this behavior was first observed for yeast FXN. In this case, the multimeric form is stabilized in the presence of iron. Even though yeast FXN is a soluble monomeric protein of ~14 kDa, the addition of Fe2+ results in the assembly of a spherical and homogeneous multimer with a molecular mass of ~1.1 × 106 Da and a diameter of 13 ± 2 nm, as judged by Atomic Force Microscopy (AFM) and Electron microscopy (EM) (Adamec et al. 2000). Each multimer is formed by several subunits of FXN and it may bind a large quantity of iron (Gakh et al. 2002). Additionally, size exclusion chromatography results suggest that there is a starting point in which the monomer is assembled first into a trimer and afterward two trimers may form a hexamer to subsequently form a dodecamer and, finally, dodecamers may form the multimer. In addition, the mature form of yeast Y73A FXN mutant assembles into trimers and 24-subunit oligomers, in an iron-independent fashion (Fig. 13.18). Remarkably, the multimer may acquire and store iron. Schagerlöf and coworkers by means of Cryo-EM and single-particle reconstruction techniques obtained important information regarding multimer structure (Schagerlof et al. 2008), showing that packing within the particles is stabilized by weak interactions between trimers. This model added to the information provided by the X-ray structure of the yeast FXN trimer variant (Y73A) (Karlberg et al. 2006) supports a key role for the N-terminal region in the oligomerization process.

FXN as a scaffold for generating small oligomers and nanoparticles. a and b Yeast FXN can form large oligomers (24 subunits) in the presence of iron, cryoEM structures, and single-particle reconstruction. c On the other hand, mutant Y73A forms a trimer, crystallographic structure PDB ID: 2FQL. In (d), the crystallographic structure of the trimeric form is docked into the iron-loaded FXN reconstructions. e E. coli FXN may be stabilized as a dimer (rigid-body modeling for iron-induced CyaY dimer, SAXS results). This Figure is a partial reproduction of Fig. 5 from reference Schagerlof et al. (2008), Fig. 1 from reference Karlberg et al. (2006) and Fig. 4 from reference Fekry et al. (2017)
Oligomerization was also demonstrated for E. coli FXN. In the presence of iron, the protein mostly forms a dimer; nevertheless, it was shown that trimers, tetramers, pentamers and possibly hexamers may exist in solution (Fekry et al. 2017). Modeling of the dimer using data provided by small-angle X-ray scattering (SAXS) experiments confirms a head-to-tail packing arrangement of the monomers (Fekry et al. 2017), as previously assessed by cross-linking, mass spectrometry and docking studies (Ahlgren et al. 2017) (Fig. 13.18d).
It is worthy of mention that some human FXN also forms oligomers (Faraj et al. 2013). This is the case of the variant that includes an N-terminal stretch from residue 56. The overexpression of this form in E. coli yields both the oligomer and the monomeric forms. After size exclusion chromatography, most of the protein was found in the high molecular weight fractions. The N-terminal moiety is an essential element for the assembly of this high molecular weight multimer. Whereas biophysical characterization by fluorescence and circular dichroism showed that subunits are well folded, sharing similar features to the mature form of FXN, controlled proteolysis of the oligomer indicated that the N-terminal segment is labile in the context of the oligomer and the yielded FXN 81-210 that persisted is strongly resistant (Faraj et al. 2013).
In vitro studies have shown that low concentration of guanidinium hydrochloride disrupts intermolecular interactions, shifting the ensemble toward the monomeric form and showing that the protein-protein interactions that stabilize the multimer are easily weakened.
Whereas functionality of the oligomers inside the cell and inside the mitochondria, in the case of eukaryotic FXN variants, remains under debate, all these results suggest FXN is a malleable scaffold and building block that may be used in nanoparticle design to obtain different species from monomer, dimers, trimers to oligomers.
Conclusions
To understand what the relationships between protein structure and function of mammalian FXN are, and with the aim of finding a cure for Friedreich’s Ataxia, it was essential to gain knowledge concerning the tridimensional structure of FXN, its internal motions, interaction with iron, and the effect of mutations of its conformation and stability.
For understanding FXN function, however, the study of its interaction with the mitochondrial enzyme NFS1 and its allosteric modulation, interaction with the scaffolding protein ISCU and involvement of FXN in mitochondrial iron–sulfur cluster assembly were all of great importance.
The combination of multiple experimental tools including high resolution techniques like NMR and X-ray, but also SAXS, crosslinking and mass-spectrometry was necessary to build reliable models of the structure of the desulfurase complex NFS1/ACP-ISD11/ISCU/FXN. Finally, the study of the enzymatic reactions involved will help to understand the capability of FXN to modulate function. Furthermore, knowledge regarding the specific control inside the cell, of conformational stability, protein degradation and protein complex assembly/disassembly dynamics in both normal and pathological contexts will provide new strategies to act on cell function.
Abbreviations
- 4’PPT:
-
4’-phosphopantetheine
- ACP:
-
Acyl carrier protein
- AFM:
-
Atomic Force Microscopy
- CD:
-
Circular dichroism
- CPMG:
-
The Carr-Purcell-Meiboom-Gill pulse sequence
- CTR:
-
C-terminal region
- CyaY:
-
Frataxin from E. coli
- DLS:
-
Dynamic light scattering
- DOSY:
-
Diffusion order spectroscopy
- EM:
-
Electron microscopy
- FDX:
-
Ferredoxin
- Fe–S:
-
Iron–sulfur
- FRDA:
-
Friedreich’s Ataxia
- FXN:
-
Frataxin
- HPLC:
-
High-performance liquid chromatography
- HSQC:
-
Heteronuclear single quantum coherence spectroscopy
- ISCU:
-
Iron–sulfur cluster assembly enzyme
- ISD11:
-
NFS1 interacting protein
- ITC:
-
Isothermal titration calorimetry
- NFS1:
-
Mitochondrial desulfurase enzyme
- NMR:
-
Nuclear magnetic resonance
- NOE:
-
Nuclear Overhauser effect
- PAGE:
-
Polyacrylamide gel electrophoresis
- PDB:
-
Protein Data Bank
- RMSD:
-
Root-mean-square deviation
- SAXS:
-
Small-angle X-ray scattering
- SDS:
-
Sodium dodecyl sulfate
- SEC:
-
Size exclusion chromatography
- SUF:
-
Sulfur assimilation
References
Adamec J, Rusnak F, Owen WG, Naylor S, Benson LM, Gacy AM, Isaya G (2000) Iron-dependent self-assembly of recombinant yeast frataxin: implications for Friedreich ataxia. Am J Hum Genet 67(3):549–562
Adinolfi S, Iannuzzi C, Prischi F, Pastore C, Iametti S, Martin SR, Bonomi F, Pastore A (2009) Bacterial frataxin CyaY is the gatekeeper of iron–sulfur cluster formation catalyzed by IscS. Nat Struct Mol Biol 16(4):390–396
Adinolfi S, Puglisi R, Crack JC, Iannuzzi C, Dal Piaz F, Konarev PV, Svergun DI, Martin S, Le Brun NE, Pastore A (2017) The MOLECULAR bases of the dual regulation of bacterial iron sulfur cluster biogenesis by CyaY and IscX. Front Mol Biosci 4:97
Ahlgren EC, Fekry M, Wiemann M, Soderberg CA, Bernfur K, Gakh O, Rasmussen M, Hojrup P, Emanuelsson C, Isaya G, Al-Karadaghi S (2017) Iron-induced oligomerization of human FXN81-210 and bacterial CyaY frataxin and the effect of iron chelators. PLoS ONE 12(12):e0188937
Armas AM, Balparda M, Terenzi A, Busi MV, Pagani MA, Gomez-Casati DF (2019) Ferrochelatase activity of plant frataxin. Biochimie 156:118–122
Bedekovics T, Gajdos GB, Kispal G, Isaya G (2007) Partial conservation of functions between eukaryotic frataxin and the Escherichia coli frataxin homolog CyaY. FEMS Yeast Res 7(8):1276–1284
Benini M, Fortuni S, Condo I, Alfedi G, Malisan F, Toschi N, Serio D, Massaro DS, Arcuri G, Testi R, Rufini A (2017) E3 Ligase RNF126 Directly ubiquitinates frataxin, promoting its degradation: identification of a potential therapeutic target for Friedreich ataxia. Cell Rep 18(8):2007–2017
Boniecki MT, Freibert SA, Muhlenhoff U, Lill R, Cygler M (2017) Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat Commun 8(1):1287
Bridwell-Rabb J, Fox NG, Tsai CL, Winn AM, Barondeau DP (2014) Human frataxin activates Fe–S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 53(30):4904–4913
Bridwell-Rabb J, Iannuzzi C, Pastore A, Barondeau DP (2012) Effector role reversal during evolution: the case of frataxin in Fe–S cluster biosynthesis. Biochemistry 51(12):2506–2514
Bridwell-Rabb J, Winn AM, Barondeau DP (2011) Structure-function analysis of Friedreich’s ataxia mutants reveals determinants of frataxin binding and activation of the Fe–S assembly complex. Biochemistry 50(33):7265–7274
Cai K, Frederick RO, Dashti H, Markley JL (2018a) Architectural features of human mitochondrial cysteine desulfurase complexes from crosslinking mass spectrometry and small-angle X-ray scattering. Structure 26(8):1127–1136 e 1124
Cai K, Frederick RO, Kim JH, Reinen NM, Tonelli M, Markley JL (2013) Human mitochondrial chaperone (mtHSP70) and cysteine desulfurase (NFS1) bind preferentially to the disordered conformation, whereas co-chaperone (HSC20) binds to the structured conformation of the iron–sulfur cluster scaffold protein (ISCU). J Biol Chem 288(40):28755–28770
Cai K, Frederick RO, Tonelli M, Markley JL (2018b) Interactions of iron-bound frataxin with ISCU and ferredoxin on the cysteine desulfurase complex leading to Fe–S cluster assembly. J Inorg Biochem 183:107–116
Cai K, Tonelli M, Frederick RO, Markley JL (2017) Human mitochondrial ferredoxin 1 (FDX1) and ferredoxin 2 (FDX2) both bind cysteine desulfurase and donate electrons for iron–sulfur cluster biosynthesis. Biochemistry 56(3):487–499
Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271(5254):1423–1427
Castro IH, Ferrari A, Herrera MG, Noguera ME, Maso L, Benini M, Rufini A, Testi R, Costantini P, Santos J (2018) Biophysical characterisation of the recombinant human frataxin precursor. FEBS Open Bio 8(3):390–405
Cavadini P, Adamec J, Taroni F, Gakh O, Isaya G (2000a) Two-step processing of human frataxin by mitochondrial processing peptidase. Precursor and intermediate forms are cleaved at different rates. J Biol Chem 275(52):41469–41475
Cavadini P, Gellera C, Patel PI, Isaya G (2000b) Human frataxin maintains mitochondrial iron homeostasis in Saccharomyces cerevisiae. Hum Mol Genet 9(17):2523–2530
Chamberlain S, Shaw J, Rowland A, Wallis J, South S, Nakamura Y, von Gabain A, Farrall M, Williamson R (1988) Mapping of mutation causing Friedreich’s ataxia to human chromosome 9. Nature 334(6179):248–250
Chamberlain S, Shaw J, Wallis J, Rowland A, Chow L, Farrall M, Keats B, Richter A, Roy M, Melancon S et al (1989) Genetic homogeneity at the Friedreich ataxia locus on chromosome 9. Am J Hum Genet 44(4):518–521
Chen OS, Hemenway S, Kaplan J (2002) Inhibition of Fe–S cluster biosynthesis decreases mitochondrial iron export: evidence that Yfh1p affects Fe–S cluster synthesis. Proc Natl Acad Sci U S A 99(19):12321–12326
Cherubini F, Serio D, Guccini I, Fortuni S, Arcuri G, Condo I, Rufini A, Moiz S, Camerini S, Crescenzi M, Testi R, Malisan F (2015) Src inhibitors modulate frataxin protein levels. Hum Mol Genet 24(15):4296–4305
Clark E, Butler JS, Isaacs CJ, Napierala M, Lynch DR (2017) Selected missense mutations impair frataxin processing in Friedreich ataxia. Ann Clin Transl Neurol 4(8):575–584
Correia AR, Adinolfi S, Pastore A, Gomes CM (2006) Conformational stability of human frataxin and effect of Friedreich’s ataxia-related mutations on protein folding. Biochem J 398(3):605–611
Correia AR, Pastore C, Adinolfi S, Pastore A, Gomes CM (2008) Dynamics, stability and iron-binding activity of frataxin clinical mutants. FEBS J 275(14):3680–3690
Cory SA, Van Vranken JG, Brignole EJ, Patra S, Winge DR, Drennan CL, Rutter J, Barondeau DP (2017) Structure of human Fe–S assembly subcomplex reveals unexpected cysteine desulfurase architecture and acyl-ACP-ISD11 interactions. Proc Natl Acad Sci U S A 114(27):E5325–E5334
Cupp-Vickery JR, Urbina H, Vickery LE (2003) Crystal structure of IscS, a cysteine desulfurase from Escherichia coli. J Mol Biol 330(5):1049–1059
Dhe-Paganon S, Shigeta R, Chi YI, Ristow M, Shoelson SE (2000) Crystal structure of human frataxin. J Biol Chem 275(40):30753–30756
Duby G, Foury F, Ramazzotti A, Herrmann J, Lutz T (2002) A non-essential function for yeast frataxin in iron–sulfur cluster assembly. Hum Mol Genet 11(21):2635–2643
Durr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M (1996) Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med 335(16):1169–1175
Faggianelli N, Puglisi R, Veneziano L, Romano S, Frontali M, Vannocci T, Fortuni S, Testi R, Pastore A (2015) Analyzing the Effects of a G137V Mutation in the FXN Gene. Front Mol Neurosci 8:66
Faraj SE, Gonzalez-Lebrero RM, Roman EA, Santos J (2016) Human frataxin folds via an intermediate state. Role of the C-terminal region. Sci Rep 6:20782
Faraj SE, Roman EA, Aran M, Gallo M, Santos J (2014) The alteration of the C-terminal region of human frataxin distorts its structural dynamics and function. FEBS J 281(15):3397–3419
Faraj SE, Venturutti L, Roman EA, Marino-Buslje CB, Mignone A, Tosatto SC, Delfino JM, Santos J (2013) The role of the N-terminal tail for the oligomerization, folding and stability of human frataxin. FEBS Open Bio 3:310–320
Fekry M, Alshokry W, Grela P, Tchorzewski M, Ahlgren EC, Soderberg CA, Gakh O, Isaya G, Al-Karadaghi S (2017) SAXS and stability studies of iron-induced oligomers of bacterial frataxin CyaY. PLoS ONE 12(9):e0184961
Foury F (1999) Low iron concentration and aconitase deficiency in a yeast frataxin homologue deficient strain. FEBS Lett 456(2):281–284
Foury F, Pastore A, Trincal M (2007) Acidic residues of yeast frataxin have an essential role in Fe–S cluster assembly. EMBO Rep 8(2):194–199
Fox NG, Das D, Chakrabarti M, Lindahl PA, Barondeau DP (2015) Frataxin accelerates [2Fe–2S] cluster formation on the human Fe–S assembly complex. Biochemistry 54(25):3880–3889
Fox NG, Yu X, Feng X, Bailey HJ, Martelli A, Nabhan JF, Strain-Damerell C, Bulawa C, Yue WW, Han S (2019) Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat Commun 10(1)
Friemel M, Marelja Z, Li K, Leimkuhler S (2017) The N-terminus of iron–sulfur cluster assembly factor ISD11 Is crucial for subcellular targeting and interaction with l-cysteine desulfurase NFS1. Biochemistry 56(12):1797–1808
Gakh O, Adamec J, Gacy AM, Twesten RD, Owen WG, Isaya G (2002) Physical evidence that yeast frataxin is an iron storage protein. Biochemistry 41(21):6798–6804
Galea CA, Huq A, Lockhart PJ, Tai G, Corben LA, Yiu EM, Gurrin LC, Lynch DR, Gelbard S, Durr A, Pousset F, Parkinson M, Labrum R, Giunti P, Perlman SL, Delatycki MB, Evans-Galea MV (2016) Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann Neurol 79(3):485–495
Gentry LE, Thacker MA, Doughty R, Timkovich R, Busenlehner LS (2013) His86 from the N-terminus of frataxin coordinates iron and is required for Fe–S cluster synthesis. Biochemistry 52(35):6085–6096
Gerber J, Muhlenhoff U, Lill R (2003) An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep 4(9):906–911
Gomes CM, Santos R (2013) Neurodegeneration in Friedreich’s ataxia: from defective frataxin to oxidative stress. Oxid Med Cell Longev 2013:487534
Guo L, Wang Q, Weng L, Hauser LA, Strawser CJ, Mesaros C, Lynch DR, Blair IA (2018) Characterization of a new N-terminally acetylated extra-mitochondrial isoform of frataxin in human erythrocytes. Sci Rep 8(1):17043
Herrera MG, Pignataro MF, Noguera ME, Cruz KM, Santos J (2018) Rescuing the rescuer: on the protein complex between the human mitochondrial acyl carrier protein and ISD11. ACS Chem Biol 13(6):1455–1462
Hidese R, Mihara H, Esaki N (2011) Bacterial cysteine desulfurases: versatile key players in biosynthetic pathways of sulfur-containing biofactors. Appl Microbiol Biotechnol 91(1):47–61
Huynen MA, Snel B, Bork P, Gibson TJ (2001) The phylogenetic distribution of frataxin indicates a role in iron–sulfur cluster protein assembly. Hum Mol Genet 10(21):2463–2468
Iannuzzi C, Adinolfi S, Howes BD, Garcia-Serres R, Clemancey M, Latour JM, Smulevich G, Pastore A (2011) The role of CyaY in iron sulfur cluster assembly on the E. coli IscU scaffold protein. PLoS One 6 (7):e21992
Karlberg T, Schagerlof U, Gakh O, Park S, Ryde U, Lindahl M, Leath K, Garman E, Isaya G, Al-Karadaghi S (2006) The structures of frataxin oligomers reveal the mechanism for the delivery and detoxification of iron. Structure 14(10):1535–1546
Kim JH, Frederick RO, Reinen NM, Troupis AT, Markley JL (2013) [2Fe–2S]-ferredoxin binds directly to cysteine desulfurase and supplies an electron for iron–sulfur cluster assembly but is displaced by the scaffold protein or bacterial frataxin. J Am Chem Soc 135(22):8117–8120
Koeppen AH (2013) Nikolaus Friedreich and degenerative atrophy of the dorsal columns of the spinal cord. J Neurochem 126(Suppl 1):4–10
Koutnikova H, Campuzano V, Foury F, Dolle P, Cazzalini O, Koenig M (1997) Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat Genet 16(4):345–351
Koutnikova H, Campuzano V, Koenig M (1998) Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase. Hum Mol Genet 7(9):1485–1489
Leidgens S, De Smet S, Foury F (2010) Frataxin interacts with Isu1 through a conserved tryptophan in its beta-sheet. Hum Mol Genet 19(2):276–286
Leimkuhler S, Buhning M, Beilschmidt L (2017) Shared sulfur mobilization routes for tRNA Thiolation and molybdenum cofactor biosynthesis in prokaryotes and eukaryotes. Biomolecules 7(1)
Lesuisse E, Santos R, Matzanke BF, Knight SA, Camadro JM, Dancis A (2003) Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1). Hum Mol Genet 12(8):879–889
Li DS, Ohshima K, Jiralerspong S, Bojanowski MW, Pandolfo M (1999) Knock-out of the cyaY gene in Escherichia coli does not affect cellular iron content and sensitivity to oxidants. FEBS Lett 456(1):13–16
Maio N, Rouault TA (1853) Iron–sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim Biophys Acta 1853(6):1493–1512
Maliandi MV, Busi MV, Turowski VR, Leaden L, Araya A, Gomez-Casati DF (2011) The mitochondrial protein frataxin is essential for heme biosynthesis in plants. FEBS J 278(3):470–481
Markley JL, Kim JH, Dai Z, Bothe JR, Cai K, Frederick RO, Tonelli M (2013) Metamorphic protein IscU alternates conformations in the course of its role as the scaffold protein for iron–sulfur cluster biosynthesis and delivery. FEBS Lett 587(8):1172–1179
Muhlenhoff U, Gerber J, Richhardt N, Lill R (2003) Components involved in assembly and dislocation of iron–sulfur clusters on the scaffold protein Isu1p. EMBO J 22(18):4815–4825
Noguera ME, Aran M, Smal C, Vazquez DS, Herrera MG, Roman EA, Alaimo N, Gallo M, Santos J (2017) Insights on the conformational dynamics of human frataxin through modifications of loop-1. Arch Biochem Biophys 636:123–137
Noma A, Sakaguchi Y, Suzuki T (2009) Mechanistic characterization of the sulfur-relay system for eukaryotic 2-thiouridine biogenesis at tRNA wobble positions. Nucleic Acids Res 37(4):1335–1352
Nuth M, Yoon T, Cowan JA (2002) Iron–sulfur cluster biosynthesis: characterization of iron nucleation sites for assembly of the [2Fe–2S]2 + cluster core in IscU proteins. J Am Chem Soc 124(30):8774–8775
Olsson MH, Sondergaard CR, Rostkowski M, Jensen JH (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput 7(2):525–537
Pandey A, Golla R, Yoon H, Dancis A, Pain D (2012) Persulfide formation on mitochondrial cysteine desulfurase: enzyme activation by a eukaryote-specific interacting protein and Fe–S cluster synthesis. Biochem J 448(2):171–187
Pandey A, Gordon DM, Pain J, Stemmler TL, Dancis A, Pain D (2013) Frataxin directly stimulates mitochondrial cysteine desulfurase by exposing substrate-binding sites, and a mutant Fe–S cluster scaffold protein with frataxin-bypassing ability acts similarly. J Biol Chem 288(52):36773–36786
Pandolfo M (2006) Friedreich ataxia: detection of GAA repeat expansions and frataxin point mutations. Methods Mol Med 126:197–216
Pandolfo M (2009) Friedreich ataxia: the clinical picture. J Neurol 256(Suppl 1):3–8
Parent A, Elduque X, Cornu D, Belot L, Le Caer JP, Grandas A, Toledano MB, D’Autreaux B (2015) Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat Commun 6:5686
Pastore C, Adinolfi S, Huynen MA, Rybin V, Martin S, Mayer M, Bukau B, Pastore A (2006) YfhJ, a molecular adaptor in iron–sulfur cluster formation or a frataxin-like protein? Structure 14(5):857–867
Patel PI, Isaya G (2001) Friedreich ataxia: from GAA triplet-repeat expansion to frataxin deficiency. Am J Hum Genet 69(1):15–24
Popovic M, Sanfelice D, Pastore C, Prischi F, Temussi PA, Pastore A (2015) Selective observation of the disordered import signal of a globular protein by in-cell NMR: the example of frataxins. Protein Sci 24(6):996–1003
Priller J, Scherzer CR, Faber PW, MacDonald ME, Young AB (1997) Frataxin gene of Friedreich’s ataxia is targeted to mitochondria. Ann Neurol 42(2):265–269
Prischi F, Giannini C, Adinolfi S, Pastore A (2009) The N-terminus of mature human frataxin is intrinsically unfolded. FEBS J 276(22):6669–6676
Prischi F, Konarev PV, Iannuzzi C, Pastore C, Adinolfi S, Martin SR, Svergun DI, Pastore A (2010) Structural bases for the interaction of frataxin with the central components of iron–sulphur cluster assembly. Nat Commun 1:95
Ramazzotti A, Vanmansart V, Foury F (2004) Mitochondrial functional interactions between frataxin and Isu1p, the iron–sulfur cluster scaffold protein, in Saccharomyces cerevisiae. FEBS Lett 557(1–3):215–220
Richards TA, van der Giezen M (2006) Evolution of the Isd11-IscS complex reveals a single alpha-proteobacterial endosymbiosis for all eukaryotes. Mol Biol Evol 23(7):1341–1344
Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P (1997) Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat Genet 17(2):215–217
Rouault TA (2015) Mammalian iron–sulphur proteins: novel insights into biogenesis and function. Nat Rev Mol Cell Biol 16(1):45–55
Rufini A, Cavallo F, Condo I, Fortuni S, De Martino G, Incani O, Di Venere A, Benini M, Massaro DS, Arcuri G, Serio D, Malisan F, Testi R (2015) Highly specific ubiquitin-competing molecules effectively promote frataxin accumulation and partially rescue the aconitase defect in Friedreich ataxia cells. Neurobiol Dis 75:91–99
Rufini A, Fortuni S, Arcuri G, Condo I, Serio D, Incani O, Malisan F, Ventura N, Testi R (2011) Preventing the ubiquitin-proteasome-dependent degradation of frataxin, the protein defective in Friedreich’s ataxia. Hum Mol Genet 20(7):1253–1261
Sacca F, Marsili A, Puorro G, Antenora A, Pane C, Tessa A, Scoppettuolo P, Nesti C, Brescia Morra V, De Michele G, Santorelli FM, Filla A (2013) Clinical use of frataxin measurement in a patient with a novel deletion in the FXN gene. J Neurol 260(4):1116–1121
Schagerlof U, Elmlund H, Gakh O, Nordlund G, Hebert H, Lindahl M, Isaya G, Al-Karadaghi S (2008) Structural basis of the iron storage function of frataxin from single-particle reconstruction of the iron-loaded oligomer. Biochemistry 47(17):4948–4954
Schmucker S, Argentini M, Carelle-Calmels N, Martelli A, Puccio H (2008) The in vivo mitochondrial two-step maturation of human frataxin. Hum Mol Genet 17(22):3521–3531
Schmucker S, Martelli A, Colin F, Page A, Wattenhofer-Donze M, Reutenauer L, Puccio H (2011) Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron–sulfur assembly complex. PLoS ONE 6(1):e16199
Schoenfeld RA, Napoli E, Wong A, Zhan S, Reutenauer L, Morin D, Buckpitt AR, Taroni F, Lonnerdal B, Ristow M, Puccio H, Cortopassi GA (2005) Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum Mol Genet 14(24):3787–3799
Shi R, Proteau A, Villarroya M, Moukadiri I, Zhang L, Trempe JF, Matte A, Armengod ME, Cygler M (2010) Structural basis for Fe–S cluster assembly and tRNA thiolation mediated by IscS protein-protein interactions. PLoS Biol 8(4):e1000354
Soderberg C, Gillam ME, Ahlgren EC, Hunter GA, Gakh O, Isaya G, Ferreira GC, Al-Karadaghi S (2016) The Structure of the Complex between Yeast Frataxin and Ferrochelatase: characterization and pre-steady state reaction of ferrous iron delivery and heme synthesis. J Biol Chem 291(22):11887–11898
Steinkellner H, Singh HN, Muckenthaler MU, Goldenberg H, Moganty RR, Scheiber-Mojdehkar B, Sturm B (2017) No changes in heme synthesis in human Friedreich s ataxia erythroid progenitor cells. Gene 621:5–11
Telot L, Rousseau E, Lesuisse E, Garcia C, Morlet B, Leger T, Camadro JM, Serre V (2018) Quantitative proteomics in Friedreich’s ataxia B-lymphocytes: a valuable approach to decipher the biochemical events responsible for pathogenesis. Biochim Biophys Acta Mol Basis Dis 1864(4 Pt A):997–1009
Tsai CL, Barondeau DP (2010) Human frataxin is an allosteric switch that activates the Fe–S cluster biosynthetic complex. Biochemistry 49(43):9132–9139
Uchida T, Kobayashi N, Muneta S, Ishimori K (2017) The iron chaperone protein CyaY from Vibrio cholerae Is a heme-binding protein. Biochemistry 56(18):2425–2434
Urbina HD, Silberg JJ, Hoff KG, Vickery LE (2001) Transfer of sulfur from IscS to IscU during Fe/S cluster assembly. J Biol Chem 276(48):44521–44526
Vazquez DS, Agudelo WA, Yone A, Vizioli N, Aran M, Gonzalez Flecha FL, Gonzalez Lebrero MC, Santos J (2015) A helix-coil transition induced by the metal ion interaction with a grafted iron-binding site of the CyaY protein family. Dalton Trans 44(5):2370–2379
Vivas E, Skovran E, Downs DM (2006) Salmonella enterica strains lacking the frataxin homolog CyaY show defects in Fe–S cluster metabolism in vivo. J Bacteriol 188(3):1175–1179
Wachnowsky C, Fidai I, Cowan JA (2018) Iron–sulfur cluster biosynthesis and trafficking—impact on human disease conditions. Metallomics 10(1):9–29
Wang T, Craig EA (2008) Binding of yeast frataxin to the scaffold for Fe–S cluster biogenesis, Isu. J Biol Chem 283(18):12674–12679
Webert H, Freibert SA, Gallo A, Heidenreich T, Linne U, Amlacher S, Hurt E, Muhlenhoff U, Banci L, Lill R (2014) Functional reconstitution of mitochondrial Fe/S cluster synthesis on Isu1 reveals the involvement of ferredoxin. Nat Commun 5:5013
Wong A, Yang J, Cavadini P, Gellera C, Lonnerdal B, Taroni F, Cortopassi G (1999) The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum Mol Genet 8(3):425–430
Xia H, Cao Y, Dai X, Marelja Z, Zhou D, Mo R, Al-Mahdawi S, Pook MA, Leimkuhler S, Rouault TA, Li K (2012) Novel frataxin isoforms may contribute to the pathological mechanism of Friedreich ataxia. PLoS ONE 7(10):e47847
Yan R, Kelly G, Pastore A (2014) The scaffold protein IscU retains a structured conformation in the Fe–S cluster assembly complex. ChemBioChem 15(11):1682–1686
Yan R, Konarev PV, Iannuzzi C, Adinolfi S, Roche B, Kelly G, Simon L, Martin SR, Py B, Barras F, Svergun DI, Pastore A (2013) Ferredoxin competes with bacterial frataxin in binding to the desulfurase IscS. J Biol Chem 288(34):24777–24787
Yoon H, Golla R, Lesuisse E, Pain J, Donald JE, Lyver ER, Pain D, Dancis A (2012) Mutation in the Fe–S scaffold protein Isu bypasses frataxin deletion. Biochem J 441(1):473–480
Yoon T, Cowan JA (2003) Iron–sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe–2S] clusters in ISU-type proteins. J Am Chem Soc 125(20):6078–6084
Yoon T, Cowan JA (2004) Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J Biol Chem 279(25):25943–25946
Acknowledgements
This work was supported by the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT PICT2016-2280), the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), the Universidad de Buenos Aires and FARA, Friedreich’s Ataxia Research Alliance.
Notes
The authors declare no competing financial interest. While this chapter was being edited, Fox and et al. presented a cryo-electron microscopy structure (3.2 Å resolution, PDB ID: 6NZU) of the human frataxin-bound iron-sulfur cluster assembly complex, containing two copies of the NFS1/ISD11-ACP/ISCU/FXN hetero-pentamer. A key feature of FXN binding is its simultaneous interactions with both NFS1 protomers of the complex and with ISCU (Fox et al. 2019).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Castro, I.H. et al. (2019). Frataxin Structure and Function. In: Harris, J., Marles-Wright, J. (eds) Macromolecular Protein Complexes II: Structure and Function . Subcellular Biochemistry, vol 93. Springer, Cham. https://doi.org/10.1007/978-3-030-28151-9_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-28151-9_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28150-2
Online ISBN: 978-3-030-28151-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)