
Abstract
Friedreich ataxia (FRDA) is caused by a GAA expansion in the first intron of the FXN gene, which encodes frataxin. Four percent of patients harbor a point mutation on one allele and a GAA expansion on the other. We studied an Italian patient presenting with symptoms suggestive of FRDA, and carrying a single expanded 850 GAA allele. As a second diagnostic step, frataxin was measured in peripheral blood mononuclear cells, and proved to be in the pathological range (2.95 pg/μg total protein, 12.7 % of control levels). Subsequent sequencing revealed a novel deletion in exon 5a (c.572delC) which predicted a frameshift at codon 191 and a premature truncation of the protein at codon 194 (p.T191IfsX194). FXN/mRNA expression was reduced to 69.2 % of control levels. Clinical phenotype was atypical with absent dysarthria, and rapid disease progression. l-Buthionine-sulphoximine treatment of the proband’s lymphoblasts showed a severe phenotype as compared to classic FRDA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Friedreich ataxia (FRDA; MIM#229300) is an autosomal recessive neurodegenerative disorder [1]. The disease is characterized by gait and limb ataxia, dysarthria, usually absent tendon reflexes, bilateral Babinski sign, impairment of position/vibratory senses [2]. The most common molecular defect in FRDA is the homozygous trinucleotide GAA expansion in the first intron of the FXN gene [3]. However, 4 % of patients harbor a point mutation on one allele and a GAA expansion on the other allele. Frataxin is a mitochondrial protein involved in iron–sulphur cluster synthesis, heme biogenesis, iron binding/storage, and iron chaperone activity [4].
To date, 46 mutations have been detected in the FXN gene of FRDA patients (missense, nonsense, frameshift, splice-site and deletion) [2, 5]. Phenotypic features of patients harboring point mutations are often similar to typical FRDA, although slightly different characteristics are sometimes observed, which may cause confusion in the clinical diagnosis [6–8]. We recently proposed frataxin measurement as a second diagnostic step of patients in the suspect of FRDA and heterozygous routine genetic testing [9], and before FXN sequencing. We present clinical, molecular, and cellular features in an additional FRDA patient harboring a new mutation in FXN.
Materials and methods
Patients and controls
We collected blood samples from five classic FRDA patients (cFA) with a homozygous GAA expansion on both alleles, five FRDA GAA carriers, and five healthy controls. The Ethics Committee of our Institution approved the procedure (n. 49/09). All individuals gave written informed consent. The study was performed in accordance to the Declaration of Helsinki.
Genetics
Amplification of the FXN gene (MIM#606829; NM_000144.4), triplet repeat primed PCR for correct sizing of the expansion, and PCR amplification of coding exons and exon–intron boundaries of FXN were performed as reported elsewhere [9]. Sequences were examined using Sequence NavigatoreSeqScape2.6 software. The three-dimensional structures of modeled wild-type and mutant proteins were analyzed using Deep View Swiss PDB viewer.
Scale for the assessment and rating of ataxias (SARA)
The SARA scale is a scale consisting of eight items (gait, stance, sitting, speech disturbance, finger chase, nose-finger test, fast alternating hand movements, heel-shin slide). The scale can score from 0 to 40, with 40 being the worst condition. The scale was validated in FRDA [10].
Frataxin and FXN/mRNA
Lateral flow immunoassay was used to determine frataxin levels as reported elsewhere [11]. Briefly, peripheral blood mononuclear cells (PBMCs) were extracted from whole blood using Leucosep® tubes. PBMCs were lysed and total protein was measured using the bicinchoninic acid assay. Samples were assayed in duplicate and interpolated against recombinant human frataxin (MSF31-MSF24, MitoSciences, Eugene, USA).
Total mRNA from PBMCs was extracted with TRIzol® reagent. Quality was checked with spectrophotometry and agarose-formaldehyde electrophoresis. Total RNA was reversely transcribed (High Capacity RNA-to-cDNA, Life Sciences, Carlsbad, USA), and cDNA amplified using the TaqMan® Gene Expression Master Mix. mRNA was quantified using a Gene Expression Assay for frataxin (Life Sciences, catalog n. Hs00175940_m1) and standardized by quantification of hypoxanthine phosphoribosyl-transferase one as a reference gene. Relative expression was calculated with the efficiency-calibrated model [12].
Cell culture
Lymphoblastoid cell lines (LCLs) were obtained from the proband, one unrelated FRDA patient (genotype 1065/1065 GAA), and one control. A total of 300,000 cells were subcultured in a final volume of 300 μL of experiment medium (RPMI 1640, FBS 10 %, 2 mM l-Glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin), for each replicate (total n = 9). Cells were incubated for 168 h with phosphate buffered saline (PBS) or l-Buthionine-sulphoximine (BSO; range 0.5–10 mM). Baseline was set at 0 h, immediately before BSO or PBS addition. Cell viability was measured with an automated Cell Counter (TC10™, Bio-Rad Inc.).
Statistical analysis
Statistical analysis for frataxin and FXN/mRNA levels was conducted by univariate ANOVA. Post hoc analysis was performed with the Bonferroni multiple-comparisons test. Comparisons of the proband’s FXN/mRNA expression and frataxin was performed with a one-sample t-test, in the attempt to compare the single values of the proband with cFA, carriers, and controls.
BSO experiments were analyzed with the Generalized Linear Model (GLM) for repeated measures. Viability was analyzed without normalization \( \left( {\frac{{{\text{Vital}}\,{\text{cells}}}}{{{\text{Total}}\,{\text{cells}}\;}} \times 100} \right) \). Two models were constructed. In the first, time was considered as a within-subjects factor, and phenotype and BSO treatment were both considered as between-subject factors. In the second model, time was considered as a within-subjects factor, whereas 15 groups were created to consider phenotype (three variables), and BSO treatment (five variables), and were set as the only between-subjects factor. Mauchly’s test of sphericity could not be used because of the presence of only two time points. Therefore, the within-subjects effect was tested with multivariate tests using Pillai’s trace, and with the within-subjects effects using the lower-bound correction. A pairwise comparison, with Bonferroni correction, was performed to identify significant differences among groups. All variables were tested for normal distribution with the Kolmogorov–Smirnov test. P values of less than 0.05 were considered statistically significant. Analysis was performed using SPSS software version 18.0.3 for Mac OSX (IBM, Chicago, USA). Graphs were generated with Prism 5.0c for Mac OSX (Graphpad, La Jolla CA, USA).
Results
Patients and controls
Age, disease duration, GAA1, GAA2, point mutation of all individuals included in the study is shown in Table 1.
Genetics
Amplification of the FXN gene of the patient showed a heterozygous state with 850 GAA repeats in the expanded allele and nine GAA repeats in the non-expanded allele. Frataxin protein was measured, as a second step in the diagnosis of FRDA, and was 2.95 pg/μg total protein (12.7 % of controls). Subsequent sequencing of all five exons of the FXN gene, revealed a novel deletion in exon 5a (c.572delC). The same deletion was found in the patient’s mother together with a non-expanded allele. The patient’s father showed an expanded allele with 1157 GAA repeats.
The mutation predicts a frameshift at codon 191 with a shift from Threonine (T) to Isoleucine (I) and a premature truncation of the protein at codon 194 (p.T191IfsX194; Fig. 1a). Modeled structure of mutated frataxin shows a premature termination at the C-terminal of the α2-helix, missing the terminal random coil (Fig. 1b).

Molecular modeling. a Wild-type human frataxin from PDBsum databse (code 1ekg) and mutant human frataxin with sequence annotated by structure (SAS). A color code identifies the amino acid conservation. b Swiss-PDB ribbon diagram of wild-type human frataxin Compact αβ sandwich (α helices red, β strands green, wild-type residue blue). c Swiss-PDB ribbon diagram of mutated human frataxin (c.572delC) Compact αβ sandwich (α helices red, β strands green, shifted residue blue)
Clinical description
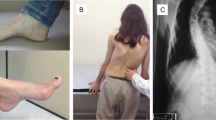
The index case was a 20-year-old Italian Caucasian female. Onset was at the age of 14 years with gait instability. She was the only child of non-consanguineous parents, with negative family history. The first examination, at the age of 20, showed: abnormal gait and stance with the ability to stand and walk only with strong support, square waves in primary position, lateral gaze nystagmus, saccadic smooth pursuit eye movements, upper and lower limbs weakness, absent tendon reflexes, extensor plantar reflexes, mild finger-nose dysmetria, abnormal heel-shin slide, lower limb reduced vibration/position sense, pes cavus, and scoliosis. SARA score was 19, indicating a moderate ataxia. Routine biochemistry showed a subclinical hypothyroidism, slightly reduced vitamin A serum level. Inverted T waves were present at the electrocardiogram. The patient underwent a brain and cervical MRI that showed atrophy of the cervical spinal cord with normal brain imaging. Neurophysiologic study showed a marked axonal degeneration of peripheral sensitive and central corticospinal pathways.
The patient was re-evaluated 2 years later and showed a clear worsening of the clinical picture. She became wheelchair bound, developed slight dysarthria, and showed a worsening of dysmetria. The SARA score was 26, indicating a moderate-severe ataxia.
q-PCR of FXN/mRNA
Relative FXN/mRNA expression was 18.9 % of control values for cFA (range 9.1–27.7), 50.1 % for GAA FRDA carriers (range 44.5–59.9; Fig. 2a). FXN/mRNA levels were significantly different between all groups (p < 0.001). The patient (c.572delC/GAA850) showed 65.7 % of control mRNA, the mother (c.572delC) 77.7 %, the father (GAA1157) 84.6 %. The one-sample t-test showed that the proband’s FXN/mRNA expression was significantly different from cFA (p < 0.001), and controls (p < 0.01), no difference was evident with GAA carriers (p = 0.169).

Functional study patient carrying the 572delC mutation (filled circle), patient’s mother (filled diamond; 9GAA/c.572delC), patient’s father (filled square; 7/1157GAA). a FXN/mRNA expression levels in PMBCs. Box and whiskers plot (min to max) of FXN/mRNA relative expression levels (mean of controls is set as 100 %). b Frataxin protein levels in PMBCs. Box and whiskers plot (min to max) of frataxin levels shown as pg/μg total protein. c Cell viability at 0 and 168 h of untreated cells the c.572delC patient, a cFA patient and control. Viability is shown as not-normalized percentage of live/total cell count. Bars show standard deviation. (asterisk shows statistically significant difference). d Effect of BSO on cell viability after 168 h in the c.572delC patient, a cFA patient and control. Viability is shown as not-normalized percentage of live/total cell count at different concentrations of BSO. Bars show standard error mean. Lines connect repeated measures of the same wells. (*statistically significant difference from controls; °statistically significant difference from controls and cFA)
Frataxin levels
Mean ± SD frataxin in cFA patients was 9.2 ± 2.2 pg/μg total protein (39.4 % of controls; range 5.6–11.6; Fig. 2b). Frataxin in FRDA carriers was 14.6 ± 2.4 pg/μg (62.9 %; 12.0–17.8). Frataxin in controls was 23.2 ± 3.1 pg/μg (20.2–27.9). Frataxin levels were different between all groups (p < 0.001). Frataxin was 2.95 pg/μg for the proband (12.7 % of controls), 10.05 pg/μg for the mother, and 12.35 pg/μg for the father. The one-sample t-test showed significantly different levels between the proband’s frataxin levels and cFA (p < 0.01), FRDA carriers (p < 0.001), and controls (p < 0.001).
Lymphoblastoid cell line (LCL) viability after BSO treatment
At baseline viability for untreated LCLs was 70.0 % for the control, 70.8 % for the cFA, and 48.1 % for the proband. After 7 days untreated cells showed an absolute viability of 39.9, 38.1, and 33.0 % (Fig. 2c). This resulted in a viability difference in the 0–168 h time frame of 14.4 % between the proband and control (p < 0.0001), and 13.9 % between the proband and cFA (p < 0.0001). BSO significantly reduced viability, as compared to PBS, in a concentration dependent manner with a different effect depending on the phenotype (p < 0.0001; Fig. 2d). The proband suffered a stronger effect of BSO as compared to control and to cFA for all concentrations (p < 0.001). cFA showed a stronger effect of BSO as compared to control for concentrations of 5 mM (p < 0.01), and 10 mM (p < 0.001).
Discussion
We describe a novel deletion in exon 5a of the FXN gene. The clinical phenotype was different from classic FRDA for the absence of dysarthria in the first disease stage, and a rapid disease progression. The patient became wheelchair-bound 8 years after disease onset, and worsened seven points in the SARA scale during the last 2 years of observation. This is clearly different from typical milestones in FRDA progression (classic phenotype), where the use of a wheelchair takes place 13.1 years after onset [13], and where typical progression in the SARA score is 1.36 points/year [14].
Frataxin levels are reduced in cFA, and to a higher extent in point mutation FRDA patients. We previously proposed frataxin measurement as and intermediate step in the diagnosis of suspect FRDA where the routine genetic test reveals a heterozygous state [9]. The method proved to be useful in clinical practice and lead us to the diagnosis of the novel mutation.
Taking into account the limitations of our sample size and the use of the one-sample t-test, the proband showed a reduced expression of FXN/mRNA of similar degree to that of GAA expansion carriers. This may be explained by the effect of the GAA expansion on one allele, whereas the c.572delC on the other allele seems not to affect transcription, as it does not further reduce FXN/mRNA expression. On the contrary, frataxin protein level was extremely low in the patient. These results are consistent with the reported effects on expression and protein analysis of previously reported point mutations [9, 15].
The new deletion crops the C-terminal of frataxin, which does not appear to be well conserved through different species (Fig. 1a). Nevertheless, three-dimensional models of frataxin have shown that the hydrophobic side chains of Leu-198, Leu-200 and Leu-203, all truncated in our patient, are essential for frataxin stability [16, 17].
BSO inhibits γ-glutamyl cysteine synthase, the rate-limiting enzyme, involved in the de novo synthesis of glutathione, leading to its depletion. Glutathione is involved in a variety of cellular functions: protection against oxidative damage by reacting with reactive oxygen species, maintenance of the mitochondrial structure and membrane integrity, cell differentiation and development [18]. Cell viability after BSO exposure has been previously used as a phenotypic model of FRDA, using control and patient fibroblasts [19]. We were able to reproduce a similar effect using LCLs, that are easier to culture than fibroblasts, can be obtained non-invasively, and have a longer life span. Untreated LCLs of the patient showed a lower viability at baseline and appeared to survive less than control and cFA cells under starving conditions. The c.572delC deletion caused a severe phenotype after BSO treatment, allowing us to phenotypically separate our patient over cFA and controls. It is interesting to note that low concentrations of BSO, such as 0.5 mM, were able to differentiate the proband and cFA, thus supporting the idea that the mutation causes an increased sensitivity to oxidative stress. We can speculate that altered protein stability may contribute to the higher sensitivity of LCLs carrying the deletion. This, in turn, could lead to a rapid degenerative process and be responsible of the observed rapid disease progression.
In conclusion, we report a novel deletion in the FXN gene, with slightly altered transcription, severely reduced frataxin and higher oxidative stress sensitivity compared to classic FRDA patients. The clinical picture is atypical with a rapidly progressive phenotype.
References
Harding AE (1981) Friedreich’s ataxia: a clinical and genetic stud of 90 families with analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 104:589–620
Santos R, Lefevre S, Sliwa D et al (2010) Friedreich Ataxia: molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid Redox Signal 13:651–690
Campuzano V, Montermini L, Moltò MD et al (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271:1423–1427
Adinolfi S, Iannuzzi C, Prischi F et al (2009) Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat Struct Mol Biol 16:390–396
Anheim M, Mariani LL, Calvas P et al (2012) Exonic deletions of FXN and early-onset Friedreich ataxia. Arch Neurol 69:912–916
De Michele G, Filla A, Cavalcanti F et al (2000) Atypical Friedreich ataxia phenotype associated with a novel missense mutation in the X25 gene. Neurology 54:496–499
Cossée M, Dürr A, Schmitt M et al (1999) Friedreich’s ataxia: point mutations and clinical presentation of compound heterozygotes. Ann Neurol 45:200–206
McCormack ML, Guttmann RP, Schumann M et al (2000) Frataxin point mutations in two patients with Friedreich’s ataxia and unusual clinical features. J Neurol Neurosurg Psychiatry 68:661–664
Saccà F, Puorro G, Antenora A et al (2011) A combined nucleic acid and protein analysis in Friedreich ataxia: implication for diagnosis, pathogenesis and clinical trial design. PLoSOne 6:e17627
Schmitz-Hübsch T, du Montcel ST, Baliko L et al (2006) Scale for the assessment and rating of ataxia: development os a new clinical scale. Neurology 66:1717–1720
Saccà F, Piro R, De Michele G et al (2011) Epoetin alfa increases Frataxin production in Friedreich’s Ataxia without affecting hematocrit. Mov Disord 26:739–742
Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30:e36
Filla A, De Michele G, Cavalcanti F et al (1996) The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich Ataxia. Am J Hum Genet S9:S54–S60
Marelli C, Figoni J, Charles P et al (2012) Annual change in Friedreich’s ataxia evaluated by the Scale for the Assessment and Rating of Ataxia (SARA) is independent of disease severity. Mov Disord 27:135–138
Deutsch EC, Santani AB, Perlman SL, Farmer JM, Stolle CA et al (2010) A rapid, noninvasive immunoassay for frataxin: utility in assessment of Friedreich ataxia. Mol Genet Metab 101:238–245
Musco G, Stier G, Kolmerer B et al (2000) Towards a structural understanding of Friedreich’s ataxia: the solution structure of frataxin. Structure 8:695–707
Dhe-Paganon S, Shigeta R, Chi YI, Ristow M, Shoelson SE (2000) Crystal structure of human frataxin. J Biol Chem 275:30753–30756
Hammond CL, Lee TK, Ballatori N (2001) Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. J Hepatol 34:946–954
Jauslin ML, Wirth T, Meier T, Schoumacher F (2002) A cellular model of Friedreich Ataxia reveals small-molecule glutathione peroxidase mimetics as novel treatment strategy. Hum Mol Genet 11:3055–3063
Acknowledgments
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. We thank all the patients and their families for their willingness to take part to this research project.
Conflicts of interest
The authors declare they have no conflict of interest for the present manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Saccà, F., Marsili, A., Puorro, G. et al. Clinical use of frataxin measurement in a patient with a novel deletion in the FXN gene. J Neurol 260, 1116–1121 (2013). https://doi.org/10.1007/s00415-012-6770-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-012-6770-5