
Abstract
RNA-binding hot spots are dominant and fundamental residues that contribute most to the binding free energy of protein-RNA interfaces. As experimental methods for identifying hot spots are expensive and time-consuming, high-efficiency computational approaches are required in predicting hot spots on a large scale. In this work, we proposed a sequence-based machine learning method to predict hot spots in protein-RNA complexes. We extracted 83 relative independent physicochemical features from a set of the 544 properties in AAindex1. Each physicochemical feature was combined with the predicted relative accessible surface area (RASA) and substitution probability feature from Blocks Substitution Matrix (BLOSUM) for training models by support vector machine (SVM) and k-nearest neighbor algorithm (k-NN). The combinations of the 166 individual models were explored and 33 top-performance models were selected to construct the final ensemble classifier by a majority voting technique. The ensemble classifier outperformed the state-of-the-art computational methods, yielding F1 score of 0.742 and AUC of 0.824 on the independent test set.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
During most biological processes, proteins work by interacting with other molecules through their interfaces, where protein-RNA interactions play key roles in regulating gene expression and structural recognition [1, 2]. Many previous studies have revealed that the binding free energy of proteins is not substantially distributed over the interaction surfaces [3, 4]. Only a small and complementary set of interface residues termed hot spots contribute mainly to the binding free energy. As experimental technique for identifying hot spots is slow and labor-intensive, there is a need for researchers to develop alternative approaches to predict hot spots in silico.
Due to the crucial role played by hot spots, many studies have been developed to predict RNA-binding sites [5,6,7,8,9]. However, few efforts were made to identify hot spots in protein-RNA complexes. Previously, Barik et al. [10] used evolutionary conservation along with structural and physicochemical attributes to discriminate the binding hot spots at protein-RNA recognition sites, which is the pioneering work in this field. For the lack of public available database to analysis protein-RNA binding hot spots collected from mutagenesis experiments, Pan et al. [11] created a reference protein-RNA hot spots dataset which was manually curated from the literature. In addition, they proposed a feature-based machine-learning method called PrabHot to identify RNA-binding hot spots. Both two methods require structural information of proteins.
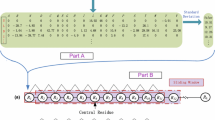
In this work, we developed a sequence-based prediction tool, to predict RNA-binding hot spots. In our model, only protein sequences were used as input information. We got 83 physicochemical descriptors from AAindex1 [12] and encoded sequences with each descriptor. Moreover, relative accessible surface area (RASA) and blocks substitution matrix were integrated with each descriptor to encode amino acid residues. Two algorithms radial basis function (RBF)-based support vector machine (SVM) and k-nearest neighbor algorithm (k-NN), were then employed to construct classifiers with each descriptor, and we got 166 individual classifiers. In order to improve the prediction performance, 33 optimal classifiers were selected with the highest AUC ranks. Finally, we built an ensemble vote classifier based on these 33 classifiers. The better prediction performance than the state-of-the-art methods indicates that our sequence-based approach can achieve a comparable prediction accuracy.
2 Method
2.1 Dataset
In this study, we used 47 protein-RNA complexes as our datasets which come from Pan et al.’s work [11]. Among 47 complexes, 32 were selected randomly as the training dataset including 79 hot spots and 72 non-hot spots and the other 15 complexes were used as the test dataset with 28 hot spots and 30 non-hot spots.
2.2 Sequence-Based Features
Physicochemical Features
We employed AAindex1 [12] which contains 544 physicochemical characteristics to encode every residue of our protein sequences. Owing to the highly correlated characteristics may decrease the performance of predictors with bias, we removed a subset of characteristics to ensure the correlation coefficient between any pair of characteristics was less than 0.6, which resulted in 83 descriptors [13]. Next, Hu’s encoding approach called the sequence order effect was used for reference to derive physicochemical features for each residue [14], which is original from Chou et al. [15]. To improve prediction quality, both sequence order information and amino acid composition were used. The sequence order effect can be formulized as:
where L is the length of sliding window, R represents a residue of protein sequences, k is the distance between two residues and k is less than L − 1, and \( \varPhi \left( {R_{i} } \right) \) shows the numeralization of 83 AAindex1 descriptors on i-th residue. In Eq. (1), \( \theta_{1} \) is called the first-tier correlation factor when the step-size between two residues is 1, \( \theta_{2} \) is the second-tier correlation factor which indicates the sequence order correlation between residues are the second nearest, \( \theta_{3} \) is the third-tier correlation factor that reflects the third nearest sequence order correlation, and so forth. According to Eq. (1), each residue is set as the central residue of sliding window and we calculate properties of them. If the number of left or right-hand neighbors is less than L/2, zero is given to these void places. In this work, we applied 11 residues as the length of sliding window, that is to say, 10 properties can be obtained to encode every central residue with each descriptor from AAindex1.
Relative Accessible Surface Area (RASA)
Relative accessible surface area (RASA) of a protein residue is a measure of residue solvent exposure. Here, we applied a sequence-based tool, NetSurfP [16], to calculate RASA.
Blocks Substitution Matrix (BLOSUM)
BLOSUM is a substitution matrix used to score alignments between evolutionarily divergent protein sequences [17]. We obtained BLOSUM62 from AAindex2 database [12] as the encoding feature used in our model.
2.3 Ensemble Classifier Construction
The ensemble vote classifier (EVC) is a meta-classifier for combining similar or conceptually different machine learning classifiers for classification via majority or plurality voting. The sample labels were predicted using average probability values. Allowing for 83 descriptors from AAindex1 were included in our study, we utilized two well-known machine-learning algorithms as follow: RBF-based SVM [18] and k-NN algorithm. To construct EVC, top n classifiers were chosen in terms of AUC performance generated on the training set using 10-fold cross-validation.
2.4 Evaluation Criteria
To assess classification performance of our model, we adopted a few commonly used measures, including sensitivity (SEN), specificity (SPE), precision (PRE), F1 score (F1), Matthew’s correlation coefficient (MCC) and accuracy (ACC). These measurements were defined as:
where TP, FP, TN, FN represent the number of true positive (correctly predicted hot spot residue), false positive (non-hot spot residue incorrectly predicted as hot spot), true negative (correctly predicted non-hot spot residue) and false negative (hot spot residue incorrectly predicted as non- hot spot), respectively. We also adopted the ROC curve as the evaluation measure in this work. From the ROC curve, we calculated the area under the ROC curve (AUC).
3 Results and Discussion
3.1 Features Evaluation
In our work, three groups of sequence features were used, which are physicochemical properties, RASA, and BLOSUM62. Experientially, RASA is deemed as a useful feature which has been commonly used to investigate hot spot residues in protein-protein compounds [15, 19, 20]. Here, we evaluated the performance of each feature group to find out how they impact on our model. The comparison results on the training set measured by AUC score can be seen in Fig. 1. When BLOSUM62 is removed, the AUC value decreases from 0.857 to 0.722 (∆AUC = 0.135), and the performance is worse when RASA is excluded, with the AUC score 0.699 (∆AUC = 0.158). The results show that RASA is an indispensable and discriminative feature to define hot spots.

Importance evaluation of features used in our method.
3.2 Compared with Other Methods
Recently, two computational methods PrabHot and HotSPRing were published to predict RNA-binding hot spots. In order to make a fair comparison among three methods, we used 10-fold cross-validation to evaluate predictor performance on 32 complexes (training set). To precisely evaluate the model, Pan et al. iterated 10-fold cross-validation for 50 times and this resulted in PrabHot-50. For the same purpose, we used the average performance of 10-fold cross-validation across 50 runs. Meanwhile, we set the prediction threshold as 0.561 to get a maximized F1 score. As we can see in Table 1, our method shows similar success rate in comparison with PrabHot. The F1 score, MCC and AUC of our model on the training set is 0.812, 0.589 and 0.857 respectively, while PrabHot correctly predicts hot spots with F1 score = 0.754, MCC = 0.513 and AUC = 0.860. HotSPRing, which is the first prediction tool in this field, correctly identifies RNA-binding hot spots with F1 score = 0.667, MCC = 0.280 and AUC = 0.699. From the high sensitivity score we can see that our method performs better in identifying hot spots than non-hot spots on the training set.
To further confirm the robustness of our model, we compared three prediction methods on the independent test set. Performance in Table 2 illustrates that our predictor displays better discernibility than PrabHot (F1 score = 0.742, MCC = 0.453, and AUC = 0.804) on the same dataset by achieving an F1 score of 0.742, MCC of 0.461 and AUC of 0.824. Because there were no predicted values returned from HotSPRing web server after submitting the complex files, the measures of HotSPRing were all from PrabHot [11]. Our method was designed to stress the point that sequence-based method owns as good prediction ability as structure-based method in identifying hot spots.
3.3 Case Study: Human HnRNP A2/B1 Protein Bound to 10-mer RNA Complex
Heterogeneous nuclear ribonucleoproteins (hnRNPs) play a vital role in RNA splicing, modification, location and translation. Each hnRNP contains at least one RNA-binding domain (RBD). For this complex (PDB ID: 5HO4), experiments identified two hot spots and four non-hot spots on the protein chain (Fig. 2A). The prediction results of our method and PrabHot can be found in Fig. 2B and C. It is obvious that two non-hot spots (H108 and F24) were wrongly predicted by PrabHot and only one non-hot spot (F24) was wrongly identified by our method.

The hot spot residues identified by experiments (A) compared with the prediction results of two methods, our method (B) and PrabHot (C). The lightblue chain represents the protein sequence (PDB ID: 5HO4, chain A) while the yellow chain represents the RNA sequence (PDB ID: 5HO4, chain B). Red residues in spheres denote experimentally identified hot spots, blue residues in spheres denote experimentally identified non-hot spots and green residues in B and C denote mistakenly predicted residues. (Color figure online)
4 Conclusion
In this article, we proposed a sequence-based method to distinguish hot spots and non-hot spots on protein-RNA interfaces. To develop our model, a total of 83 descriptors were extracted from AAindex1 database. Every residue was encoded by a physicochemical descriptor using 11-length sliding window, which results in 10 features. Combined with RASA and BLOSUM62, a total of 12-dimensional features were extracted for each residue. Two algorithms RBF-based SVM and k-NN were then used to build the ensemble classifier based on 33 selected optimal classifiers. The prediction results show that the sequence-based approach we proposed can yield a good performance which is comparable to structural methods.
Apart from the distinguished performance of our predictor, there is still room to improve our method. As nowadays most explored features are related to protein and amino acid, we will excavate more RNA-related features and put them into our model. Meanwhile, we aim to construct an optional model for both sequence and structure information provided so that this flexible model can predict potential hot spots and non-hot spots with or without structural information.
References
König, J., et al.: Protein-RNA interactions: new genomic technologies and perspectives. Nat. Rev. Genetics 13(2), 77 (2012)
Ellis, J.J., Broom, M., Jones, S.: Protein-RNA interactions: structural analysis and functional classes. Proteins: Struct. Funct. Bioinf. 66(4), 903–911 (2007)
Clackson, T., Wells, J.A.: A hot spot of binding energy in a hormone-receptor interface. Science 267(5196), 383–386 (1995)
Moreira, I.S., Fernandes, P.A., Ramos, M.J.: Hot spots-A review of the protein-protein interface determinant amino-acid residues. Proteins: Struct. Funct. Bioinf. 68(4), 803–812 (2007)
Kumar, M., Gromiha, M.M., Raghava, G.: Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins: Struct. Funct. Bioinf. 71(1), 189–194 (2008)
Liu, Z.-P., et al.: Prediction of protein-RNA binding sites by a random forest method with combined features. Bioinformatics 26(13), 1616–1622 (2010)
Tang, Y., et al.: A boosting approach for prediction of protein-RNA binding residues. BMC Bioinf. 18(13), 465 (2017)
Walia, R.R., et al.: RNABindRPlus: a predictor that combines machine learning and sequence homology-based methods to improve the reliability of predicted RNA-binding residues in proteins. PLoS One 9(5), e97725 (2014)
Yang, X., et al.: SNBRFinder: a sequence-based hybrid algorithm for enhanced prediction of nucleic acid-binding residues. PLoS One 10(7), e0133260 (2015)
Barik, A., et al.: Probing binding hot spots at protein-RNA recognition sites. Nucleic Acids Res. 44(2), e9 (2015)
Pan, Y., et al.: Computational identification of binding energy hot spots in protein-RNA complexes using an ensemble approach. Bioinformatics 34(9), 1473–1480 (2017)
Shuichi, K., et al.: AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 36(Database issue), D202–D205 (2008)
Chen, P., et al.: Accurate prediction of hot spot residues through physicochemical characteristics of amino acid sequences. Proteins: Struct. Funct. Bioinf. 81(8), 1351–1362 (2013)
Hu, S.-S., et al.: Protein binding hot spots prediction from sequence only by a new ensemble learning method. Amino Acids 49(10), 1773–1785 (2017)
Chou, K.C.: Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins: Struct. Funct. Bioinf. 43(3), 246–255 (2001)
Morten, N., et al.: A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 9(1), 51 (2009)
Henikoff, S., Henikoff, J.G.: Amino acid substitution matrices from protein blocks. Proc. Nat. Acad. Sci. 89(22), 10915–10919 (1992)
Chang, C.-C., Lin, C.-J.: LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. (TIST) 2(3), 27 (2011)
Xia, J.-F., et al.: APIS: accurate prediction of hot spots in protein interfaces by combining protrusion index with solvent accessibility. BMC Bioinf. 11(1), 174 (2010)
Zhu, X., Mitchell, J.C.: KFC2: a knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins: Struct. Funct. Bioinf. 79(9), 2671–2683 (2011)
Acknowledgement
This work was supported by the National Natural Science Foundation of China (61672037, 21601001, and 11835014), the Anhui Provincial Outstanding Young Talent Support Plan (gxyqZD2017005), the Young Wanjiang Scholar Program of Anhui Province, the Recruitment Program for Leading Talent Team of Anhui Province (2019-16), the China Postdoctoral Science Foundation Grant (2018M630699) and the Anhui Provincial Postdoctoral Science Foundation Grant (2017B325).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Zhao, L., Zhang, S., Xia, J. (2019). Sequence-Based Prediction of Hot Spots in Protein-RNA Complexes Using an Ensemble Approach. In: Huang, DS., Bevilacqua, V., Premaratne, P. (eds) Intelligent Computing Theories and Application. ICIC 2019. Lecture Notes in Computer Science(), vol 11643. Springer, Cham. https://doi.org/10.1007/978-3-030-26763-6_55
Download citation
DOI: https://doi.org/10.1007/978-3-030-26763-6_55
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-26762-9
Online ISBN: 978-3-030-26763-6
eBook Packages: Computer ScienceComputer Science (R0)